

Abstract

We have previously reported the discovery of our P2–P4 macrocyclic HCV NS3/4a protease inhibitor MK-5172, which in combination with the NS5a inhibitor MK-8742 recently received a breakthrough therapy designation from the US FDA for treatment of chronic HCV infection. Our goal for the next generation NS3/4a inhibitor was to achieve pan-genotypic activity while retaining the pharmacokinetic profile of MK-5172. One of the areas for follow-up investigation involved replacement of the quinoxaline moiety in MK-5172 with a quinoline and studying the effect of substitution at 4-position of the quinoline. The rationale for this effort was based on molecular modeling, which indicated that such modifications would improve interactions with the S2 subsite, in particular with D79. We wish to report herein the discovery of highly potent inhibitors with pan-genotypic activity and an improved profile over MK-5172, especially against gt-3a and A156 mutants.
Keywords: Antiviral, HCV, NS3/4a, macrocycle, pan-genotypic, genotype 3a
An estimated 180 million people worldwide are chronically infected with the hepatitis C virus (HCV) and about 20% of this population is at a risk of developing liver cirrhosis, which can lead to end stage liver disease and hepatocellular carcinoma.1−3 The majority of infections in the developed world are caused by HCV genotypes 1, 2, and 3. While standard of care treatment with pegylated IFN-α (P) and ribavirin (R) results in a cure rate of ∼70–90% in patients infected with genotypes 2 and 3, the cure rate is only ∼45% for genotype 1 infected patients.4 The addition of direct acting HCV serine protease NS3/4a inhibitors such as boceprevir or telaprevir to PR treatment regimens has significantly improved the sustained virological response (SVR) rate to up to 75% for naïve HCV genotype 1 patients (representing more than 70% of all cases of chronic HCV infection in the United States).5,6 Furthermore, side effects associated with PR and first generation NS3/4a inhibitors, the rapid emergence of drug resistance, and suboptimal SVR have led to the development of more potent NS3/4a protease inhibitors with a higher barrier to resistance. These drug candidates in combination with compounds from additional classes of HCV direct acting antivirals offer promising all-oral interferon sparing treatment regimens.7
We have previously reported the discovery of our P2–P4 macrocyclic HCV NS3/4a protease inhibitor MK-5172 (Figure 1), which is currently undergoing clinical trials.8,9 Preclinically, MK-5172 demonstrated broad genotype and mutant enzyme potency and cellular activity. One of the areas for follow-up investigation involved replacement of the quinoxaline moiety in MK-5172 with a quinoline and studying the effect of substitution at 4-position of the quinoline. The rationale for this effort was based on molecular modeling, which indicated that such modifications would improve interactions with the S2 subsite, in particular with D79 (Figure 2). Although in the NS3 protease structure of the catalytic domain D79 points toward the solvent, in the full-length enzyme (pdb 1cu1) it is at the interface of NS3 protease and helicase domains.10,11 We hypothesized that having a basic group in this region would offer charge complementarity to D79 and improve inhibitor binding energetics. In addition, from sequence alignment D79 is conserved across most of the known genotypes, and therefore, we rationalized that targeting this interaction would be beneficial to maintain/improve the overall gt profile. On the basis of some of our previous SAR we also knew that replacement of the t-butyl group in the P3 region of MK-5172 was tolerated, and the cyclopropyl group in the sulfonamide region could be replaced with a methylcyclopropyl group without losing activity; these modifications had resulted in better rodent pharmacokinetics. This letter will describe our SAR findings in the series represented by macrocycle I (Figure 1), where the R group contains a basic side-chain capable of interacting with D79 (Figure 2).
Figure 1.

Structures of MK-5172 and quinoline-based P2–P4 macrocyclic core.
Figure 2.
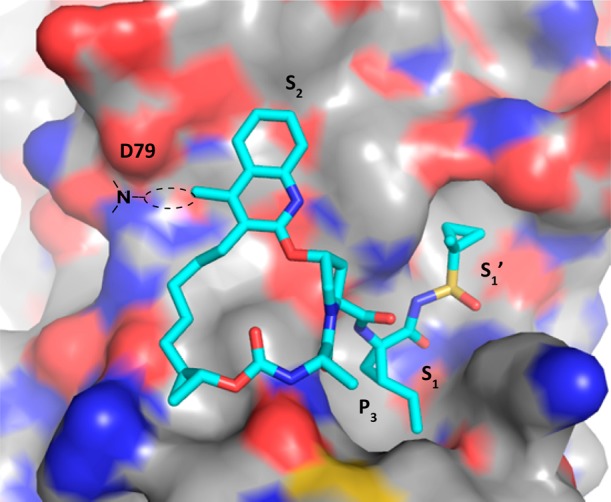
Model of quinoline-based P2–P4 macrocycle bound to the HCV NS3 gt-1b protease active site. The protein is shown as the surface (white carbon) and the inhibitor as a stick (cyan carbon).
On the basis of our modeling rationale, we explored a series of amine analogues linked to the quinoline via different spacers. These analogues were prepared from a common 4-hydroxyquinoline-based macrocyclic core, which was synthesized as shown in Scheme 1. Treatment of (R),(R)-alkynol 1 with N,N′-discuccinimidyl carbonate (DSC) and coupling the resulting DSC-adduct with P3 cyclohexyl-glycine 2 provided the alkyne-carboxylic acid 3.12 Mitsunobu reaction between the commercially available hydroxyproline derivative 4 and bromoquinolinone 5(12) furnished the proline-containing bromoquinoline fragment 6, which underwent Sonagashira coupling with alkyne 3 to provide intermediate 7. Hydrolysis of the NBoc group followed by an intramolecular amide coupling resulted in macrocycle 8, and subsequent hydrogenation provided the macrocyclic intermediate 9, which was utilized for further derivatization as shown in Scheme 2. Hydrolysis of the methyl ester in 9 followed by an amide coupling with the acylsulfonamide-containing amine 10(12) gave 11, which upon alkylation with 1,3-dibromopropane resulted in bromide 12. Displacement of the bromide in 12 with amines of varying basicity gave the first set of analogues (e.g., 13–18) in which the basic group was separated from the quinoline moiety by a propyloxy linker. Enzyme inhibition data against genotypes 1b, 3a, and relevant gt-1b mutants is summarized in Table 1.13
Scheme 1. Synthesis of an Advanced P2–P4 Macrocyclic Intermediate.

Reagents and conditions: (a) DSC, pyridine, DMAP, MeCN, 40 °C; (b) 2, Et3N, MeCN; (c) DIAD, Ph3P, THF; (d) 3, PdCl2(MeCN)2tBu3P·BF4, K2CO3, Bn2NH, MeCN, 75 °C; (e) TFA, CH2Cl2; (f) HATU, DIPEA, DMF; (g) 10% Pd–C, H2, MeOH, THF, 1 atm.
Scheme 2. Synthesis of Amine-Based P2–P4 Macrocyclic NS3/4a Protease Inhibitors.

Reagents and conditions: (a) LiOH·H2O, THF-MeOH; (b) 10, HATU, DIPEA, DMF; (c) 1,3-dibromopropane, Cs2CO3, DMF, 50 °C; (d) DIPEA/Et3N, KI, HNR1R2; (e) DIAD, Ph3P, 4-hydroxy-1-methylpiperidine or N-Boc-4-hydroxypiperidine, THF, 40 °C (f) TFA, CH2Cl2; (g) RX, DIPEA/Et3N, KI, MeCN or DMF, 100 °C or (methylsulfonyl)ethene, DIPEA, CH2Cl2 for synthesis of 28.
Table 1. SAR of Extended Amine Analogues.
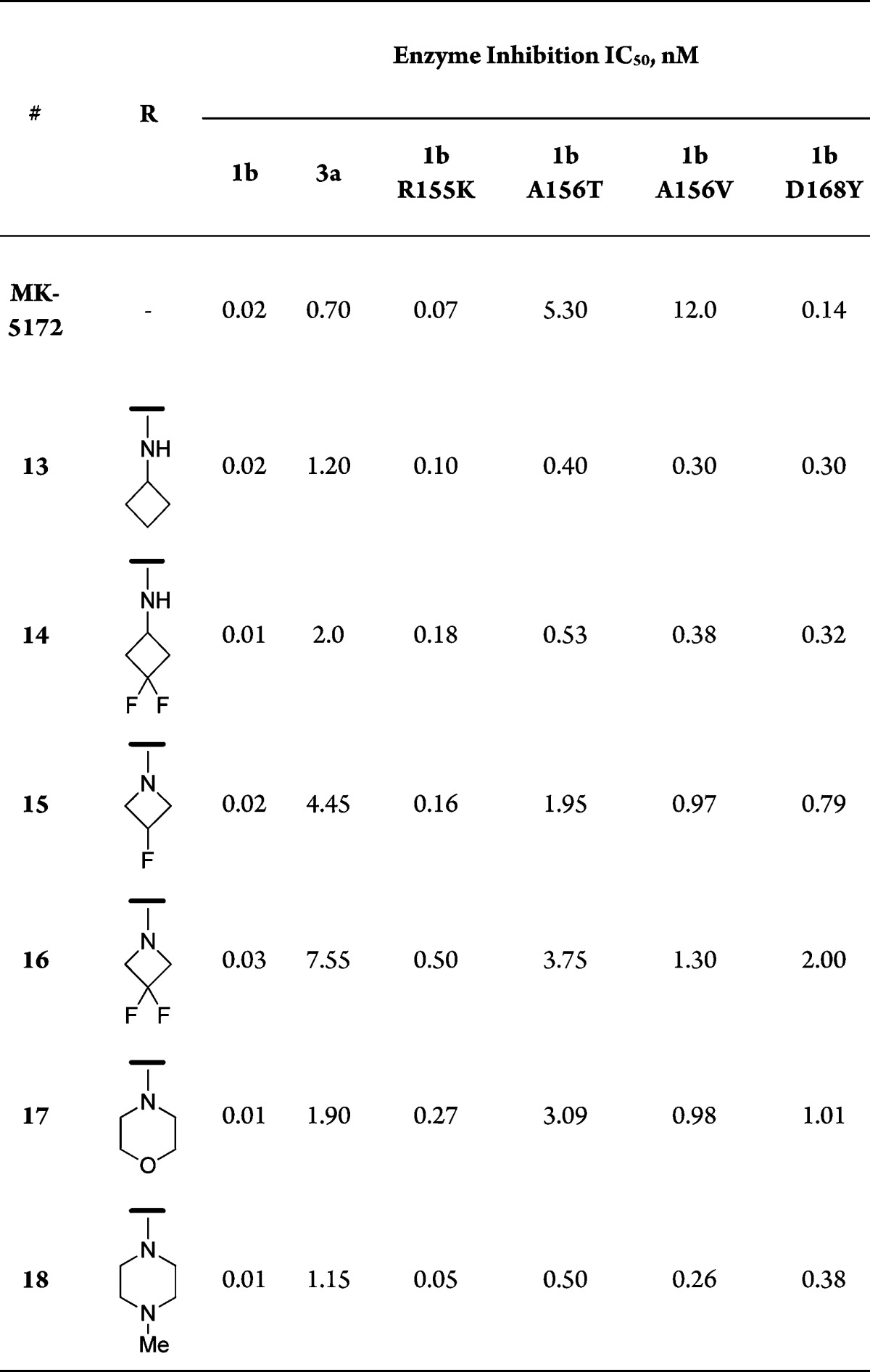
Data summarized in Table 1 shows that replacing the quinoxaline group in MK-5172 with a quinoline and incorporating alkoxy-linked amines at 4-position are tolerated. Similar to MK-5172, most compounds displayed excellent activity against gt-1b and mutants R155K and D168Y. An important differentiation point, however, was the improvement in activity against the key A156 mutants (A156T and A156V) for several compounds in this series; activity against gt-3a was variable, although not an improvement over MK-5172. The more promising compounds, however, were associated with poor rat pharmacokinetic properties (e.g., 18, AUC = 0.1 μM·h, 5 mpk po).14
On the basis of the promising activity against A156T and A156V mutants observed with the 3 carbon-linked amine analogues, we decided to extend this SAR by cyclizing the linker and exploring some piperidine-based derivatives. These targets were synthesized as shown in Scheme 2. Mitsunobu reaction between quinol 9 and N-substituted 4-hydroxypiperidine gave the corresponding ether derivatives (19 and 20). The acyl sulfonamide motif was then introduced as described earlier for the synthesis of 11. Hydrolysis of the piperidine NBoc group followed by N-derivatization gave the desired targets (Table 2). In the piperidine-ether series (Table 2), the nonbasic NBoc analogue 22 resulted in a significant drop in activity; a similar trend was observed with the corresponding sulfonamide and amide derivatives (data not shown). The free piperidine and some of the N-alkylated analogues, however, exhibited excellent overall activity. Unlike the earlier acyclic-spacer series, the piperidine analogues 21 and 23–25 displayed subnanomolar IC50 activities not only against the A156 mutants but also against the critical genotype 3a. Increasing the size of the alkyl group had a detrimental effect on activity across all genotypes and mutants (e.g., 26, 27, and 28), and rendering the piperidine nitrogen less basic (e.g., 29) resulted in a significant loss of activity. We were able to regain the potency by fine-tuning the piperidine basicity through incorporation of fewer fluorine atoms; however, these compounds (e.g., 30) did not offer additional advantage over other leads. Finally, the reduction in activity for the pyran analogue 31 confirmed the importance of a basic nitrogen to make a favorable interaction with D79 residue in this region.
Table 2. SAR of Piperidine-Ether Analogues.
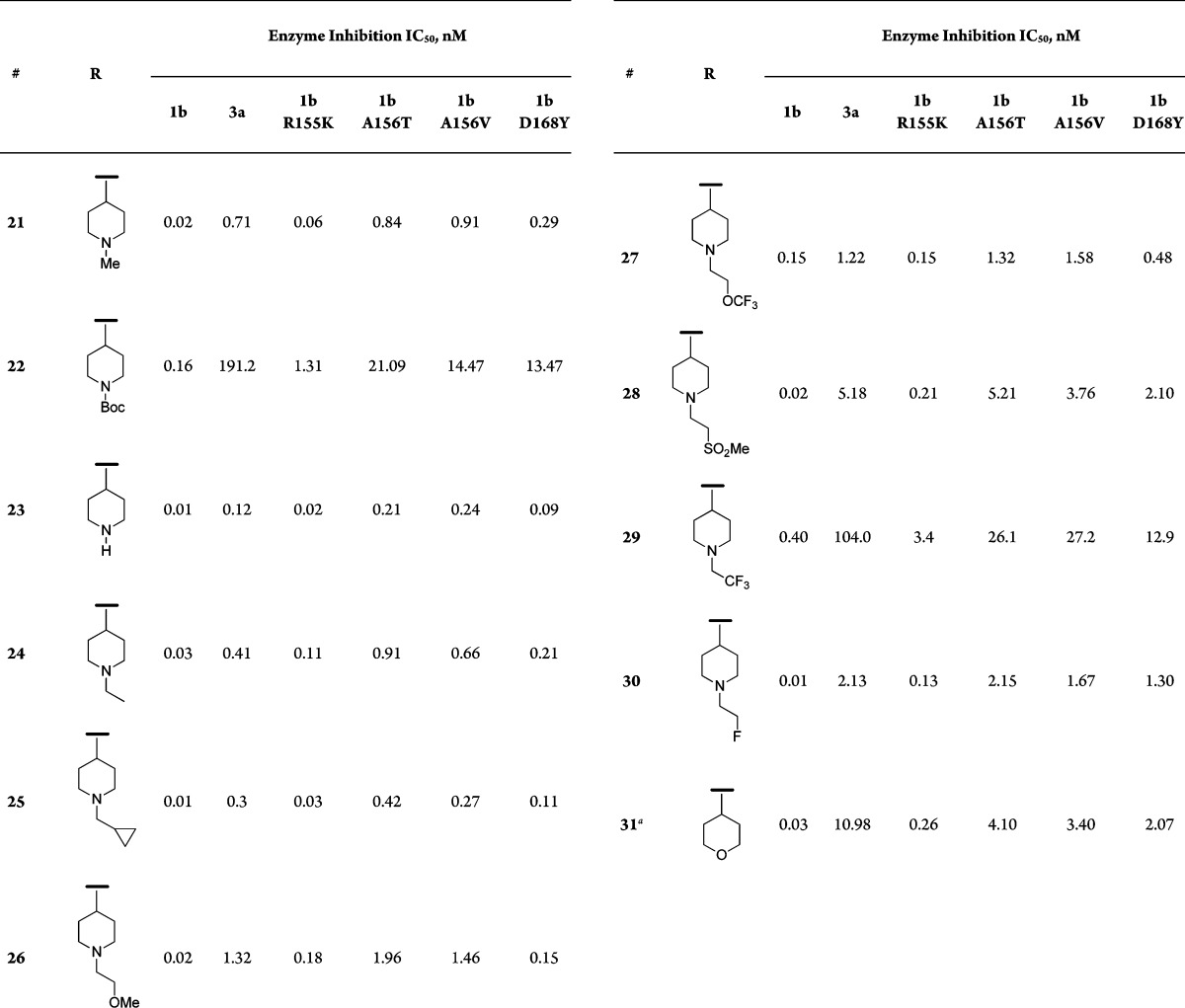
Compound 31 was synthesized in a similar manner as compound 21; 4-hydroxy-1-methylpiperidine was replaced by tetrahydro-2H-pyran-4-ol.
Constrained bicyclic piperidine analogues were explored next as an extension to the above findings. Compounds shown in Table 3 were prepared following the general synthetic route shown in Schemes 1 and 2; the corresponding bicyclic alcohols were either purchased through commercial sources or synthesized according to known literature procedures.15 The bicyclic piperidine SAR (Table 3) was conducted first by preparing nortropane analogues in the P3 t-Bu series, and it was quickly realized that the exo isomer was better tolerated than the endo isomer (e.g., 33 vs 32, and 35 vs 34); 33 and 35 demonstrated excellent gt-3a and A156 mutant profiles. The same trend was observed in the P3 cyclohexyl series where several pan-genotypic compounds were identified (e.g., 36–38).16
Table 3. SAR of Bicyclic Piperidine Analogues.
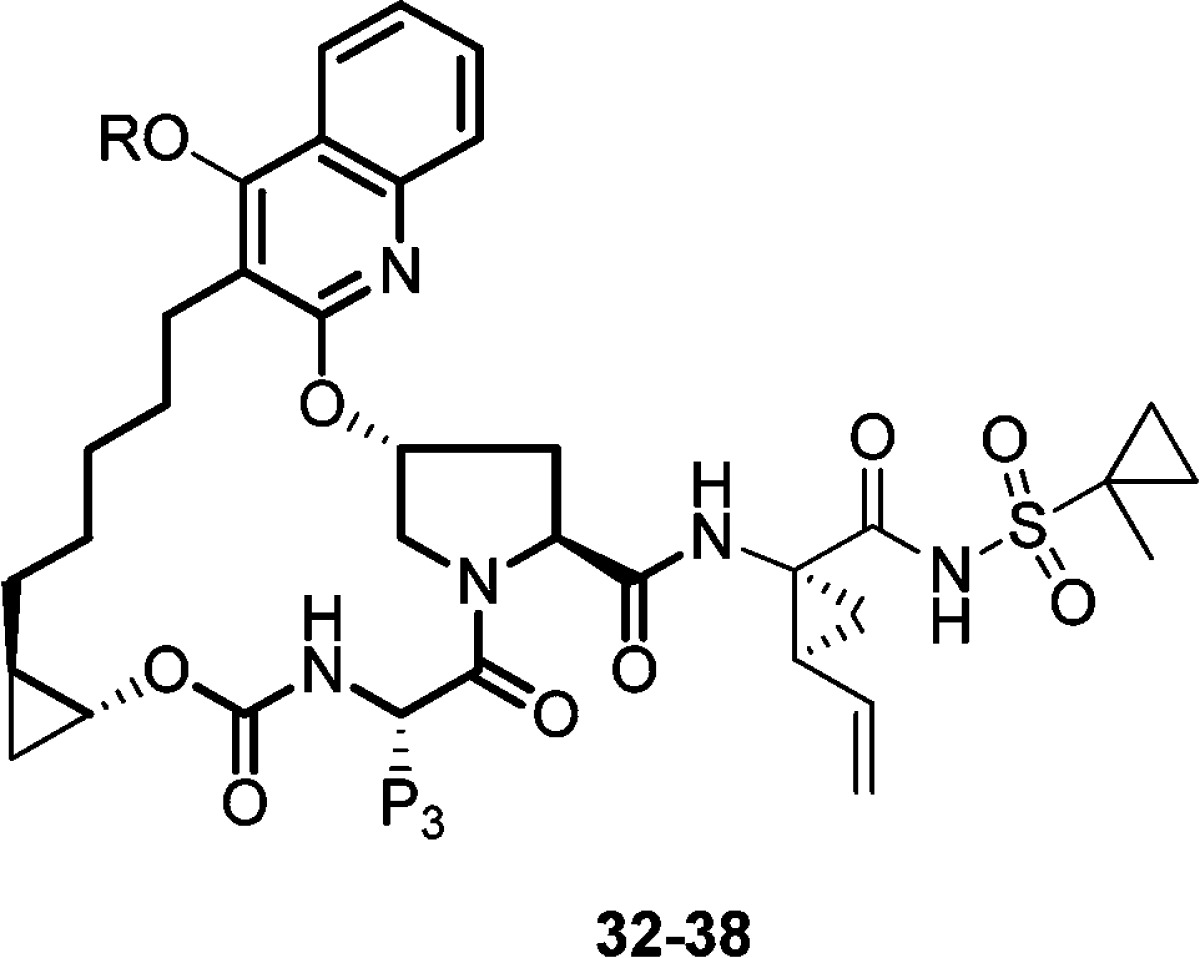
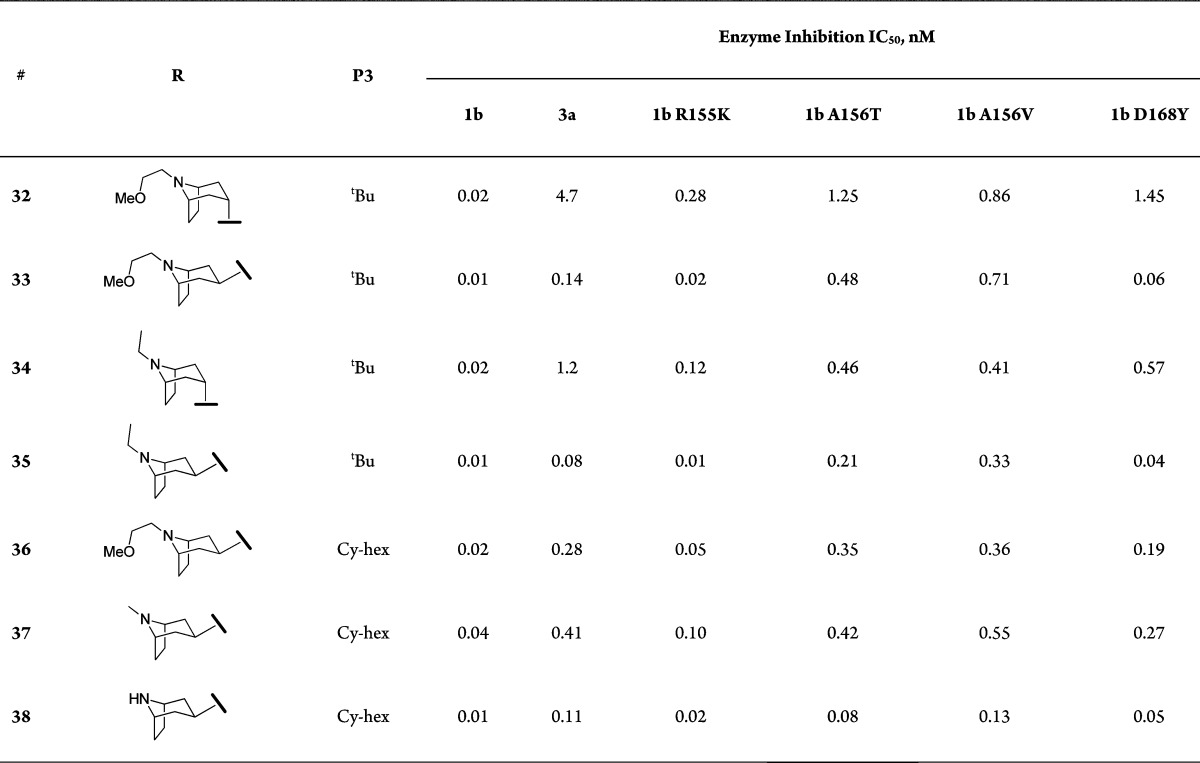
Table 4 shows gt-1b and gt-3a replicon potency and rat AUCs for selected compounds in the piperidine-ether series. There was a general improvement in gt-1b replicon activity for these analogues compared to MK-5172. With regards to gt-3a activity, compound 37 was highly potent and demonstrated a ∼14-fold improvement in activity over MK-5172. This compound also exhibited an oral rat pharmacokinetic profile similar to MK5172. Other compounds in the P3 cyclohexyl series, with an exception of the free amine 23, also exhibited good oral AUC in rats (e.g., 21, 24, and 25); compound 35 in the P3 t-Bu series, however, was associated with inferior rat PK.
Table 4. Cellular Potencya and Rat Pharmacokinetic Profileb for Selected Piperidine-Ether Compounds.
| compd | MK-5172 | 21 | 23 | 24 | 25 | 35 | 37 |
|---|---|---|---|---|---|---|---|
| 1b EC50, nM | 1.5 | 0.25 | 0.21 | 0.53 | 0.73 | 0.48 | 0.62 |
| 3a EC50, nM | 13.0 | 3.1 | 7.8 | 5.2 | 13.5 | 1.06 | 0.26 |
| rat AUC, μM·h | 0.7 | 2.7 | 0.01 | 2.3 | 2.8 | 0.01 | 1.2 |
Cell-based replicon assays conducted in presence of 10% fetal-bovine serum (FBS); EC50 determination was conducted using a TaqMan-based assay.
5 mg/kg, po, methyl cellulose (PEG400 for MK5172).
In summary, we have discovered a new series of HCV NS3/4a protease inhibitors based on the P2–P4 macrocyclic scaffold of our clinical compound MK-5172. Replacing the quinoxaline moiety with quinoline and incorporating moderately basic groups to provide an interaction with D79 within the S2 subsite, as determined through molecular modeling, provided highly potent inhibitors. The bicyclic piperidine analogue 37 was identified as the lead compound, which displayed pan-genotypic activity and an improved profile over MK-5172, especially against gt-3a and A156 mutants. This compound also displayed acceptable pharmacokinetic properties in rats upon oral dosing. Further SAR development and advancement of lead compounds will be disclosed in the near future.
Acknowledgments
The authors would like to thank Dr. Rebecca Ruck for providing advanced intermediates for SAR exploration. We also thank Dr. Mike Rowley for providing valuable input during the preparation of this manuscript.
Supporting Information Available
Synthetic experimental details including 1H NMR, 13C NMR, and mass spectral data for selected compounds, and description of primary biological assays. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Armstrong G. L.; Wasley A.; Simard E. P.; McQuillan G. M.; Kuhnert W. L.; Alter M. J. The Prevalence of Hepatitis C Virus Infection in the United States, 1999 Through 2002. Ann. Intern. Med. 2006, 144, 705–714. [DOI] [PubMed] [Google Scholar]
- Lavanchy D. The Global Burden of Hepatitis C. Liver Int. 2009, 29Suppl 174–81. [DOI] [PubMed] [Google Scholar]
- Deuffic-Burban S.; Poynard T.; Sulkowski M. S.; Wong J. B. Estimating the Future Health Burden of Chronic Hepatitis C and Human Immunodeficiency Virus Infections in the United States. J. Viral Hepatitis 2007, 14, 107–115. [DOI] [PubMed] [Google Scholar]
- Ghany M. G.; Strader D. B.; Thomas D. L.; Seeff L. B. Diagnosis, Management, and Treatment of Hepatitis C: An Update. Hepatology 2009, 49, 1335–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poordad F.; McCone J. Jr.; Bacon B. R.; Bruno S.; Manns M. P.; Sulkowski M. S; Jacobson I. M.; Reddy K. R.; Goodman Z. D.; Boparai N.; DiNubile M. J.; Sniukiene V.; Brass C. A.; Albrecht J. K.; Bronowicki J. P. SPRINT-2 Investigators. Boceprevir for Untreated Chronic HCV Genotype 1 Infection. N. Engl. J. Med. 2011, 364, 1195–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson I. M.; McHutchison J. G.; Dusheiko G.; Di Bisceglie A. M.; Reddy K. R.; Bzowej N. H.; Marcellin P.; Muir A. J.; Ferenci P.; Flisiak R.; George J.; Rizzetto M.; Shouval D.; Sola R.; Terg R. A.; Yoshida E. M.; Adda N.; Bengtsson L.; Sankoh A. J.; Kieffer T. L.; George S.; Kauffman R. S.; Zeuzem S. ADVANCE Study Team. Telaprevir for Previously Untreated Chronic Hepatitis C Virus Infection. N. Engl. J. Med. 2011, 364, 2405–2416. [DOI] [PubMed] [Google Scholar]
- Kiser J. J.; Flexner C. Direct-Acting Antiviral Agents for Hepatitis C Virus Infection. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 427–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summa V.; Ludmerer S. W.; McCauley J. A.; Fandozzi C.; Burlein C.; Claudio G.; Coleman P. J.; Dimuzio J. M.; Ferrara M.; DiFilippo M.; Gates A. T.; Graham D. J.; Harper S.; Hazuda D. J.; McHale C.; Monteagudo E.; Pucci V.; Rowley M.; Rudd M. T.; Soriano A.; Stahlhut M. W.; Vacca J. P.; Olsen D. B.; Liverton N. J.; Carroll S. S. MK-5172, a Selective Inhibitor of Hepatitis C Virus NS3/4a Protease With Broad Activity Across Genotypes and Resistant Variants. Antimicrob. Agents Chemother. 2012, 5684161–4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper S.; McCauley J. A.; Rudd M. T.; Ferrara M.; DiFilippo M.; Crescenzi B.; Koch U.; Petrocchi A.; Holloway M. K.; Butcher J. W.; Romano J. J.; Bush K. J.; Gilbert K. F.; McIntyre C. J.; Nguyen K. T.; Nizi E.; Carroll S. S.; Ludmerer S. W.; Burlein C.; DiMuzio J. M.; Graham D. J.; McHale C. M.; Stahlhut M. W.; Olsen D. B.; Monteagudo E.; Cianetti S.; Giuliano C.; Pucci V.; Trainor N.; Fandozzi C. M.; Rowley M.; Coleman P. J.; Vacca J. P.; Summa V.; Liverton N. J. Discovery of MK-5172, a Macrocyclic Hepatitis C Virus NS3/4a Protease Inhibitor. ACS Med. Chem. Lett. 2012, 34332–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings M. D.; Lindberg J. D.; Lin T.-I.; de Kock H.; Lenz O.; Lilja E.; Fellander S.; Baraznenok V.; Nystrom S.; Nilsson M.; Vrang L.; Edlund M.; Rosenquist A.; Samuelsson B.; Raboisson P.; Simmen K. Induced-Fit Binding of the Macrocyclic Noncovalent Inhibitor TMC435 to its HCV NS3/NS4A Protease Target. Angew. Chem., Int. Ed. 2010, 49, 1652–1655. [DOI] [PubMed] [Google Scholar]
- Yao N.; Reichert P.; Taremi S. S.; Prosise W. W.; Weber P. C. Molecular Views of Viral Polyprotein Processing Revealed by the Crystal Structure of the Hepatitis C Virus Bifunctional Protease-helicase. Struct. Folding Des. 1999, 7, 1353–1363. [DOI] [PubMed] [Google Scholar]
- Rudd M. T.; McCauley J.; Liverton N.; Grise-Bard C.; Brouchu M.-C.; Charron S.; Aulakh V.; Bachand B.; Beaulieu P.; Zaghdane H.; Han Y.; Ferrara M.; Harper S.; Summa V.; Chackalamannil S.; Venkatraman S.; Shah U.; Velazquez F.. Preparation of Macrocyclic Quinoline and Naphthyridine Peptides as HCV NS3 Protease Inhibitors Useful in the Treatment of Hepatitis C Infection. WO Pat. Appl. 2013074386, 2013.
- The HCV NS3 protease inhibitory activity was measured using the protease time-resolved fluorescence (TRF) assay described in WO Pat. Appl. 2006102087. Also see Supporting Information section for detailed description of the assay.
- Additional SAR work around the spacer length demonstrated that the corresponding 2C-linked analogues were somewhat inferior to their 3C-linked counterparts.
- Shah U. G.; Boyle C. D.; Chackalamannil S.; Neustadt B. R.; Harris J. M.; Stamford A.; Greenlee W. J.; Neelamkavil S. F.. Preparation of Pyrimidinylazabicyclooctane Derivatives and Analogs for Use as GPCR Modulators. WO Pat. Appl. 2010075273, 2010.
- Changing the bridge to an oxaza bicyclic system or moving the bridge away from the nitrogen resulted in compounds with inferior activity, especially against gt-3a.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.