

Abstract
Herein, novel hepatitis C virus NS3/4A protease inhibitors based on a P2 pyrimidinyloxyphenylglycine in combination with various regioisomers of an aryl acyl sulfonamide functionality in P1 are presented. The P1′ 4-(trifluoromethyl)phenyl side chain was shown to be particularly beneficial in terms of inhibitory potency. Several inhibitors with Ki-values in the nanomolar range were developed and included identification of promising P3-truncated inhibitors spanning from P2–P1′. Of several different P2 capping groups that were evaluated, a preference for the sterically congested Boc group was revealed. The inhibitors were found to retain inhibitory potencies for A156T, D168V, and R155K variants of the protease. Furthermore, in vitro pharmacokinetic profiling showed several beneficial effects on metabolic stability as well as on apparent intestinal permeability from both P3 truncation and the use of the P1′ 4-(trifluoromethyl)phenyl side chain.
Keywords: HCV, NS3, protease inhibitors, peptidomimetics, phenylglycine
Hepatitis C is an inflammatory disease that affects around 2.2–3.0% of the world’s population and is caused by the blood-borne hepatitis C virus (HCV).1 The infection resides mainly in hepatocytes and becomes chronic in approximately 80% of infected individuals. Progression of the disease often develops into various stage of liver disease, such as cirrhosis or liver cancer, within a time period of 30 years.2 There are several potential targets for anti-HCV therapy, of which the protease part of the bifunctional nonstructural protein 3 (NS3) is one of the most well-studied.3 Proof-of-concept, that inhibition of the NS3/4A protease leads to reduction in plasma HCV RNA loads in infected patients, was established with ciluprevir (BILN 2061, Figure 1).4 Thereafter, several NS3/4A protease inhibitors have been developed and entered into clinical trials, some of which are shown in Figure 1.5 Two of the them, Incivek (Vertex) and Victrelis (Merck and Co.), have recently been launched on the market and are now used in combination with the standard therapy of pegylated interferon-α and ribavirin for treatment of HCV genotype 1.6
Figure 1.

Selection of HCV NS3/4A protease inhibitors that have entered clinical trials.5
The above-mentioned inhibitors of HCV NS3/4A (as exemplified in Figure 1) share some structural similarities. They are peptide-based and share a proline or a proline mimic in the P2 position. Although they possess excellent antiviral capacities, the emergence of resistant strains of the protease is disquieting.6,7 The mutations that have emerged in in vitro and in vivo studies of the viral genome, under a selective pressure of HCV NS3/4A protease inhibitors, are often related to amino acids close to the S2 position of the protease, thus accounting for cross-resistance issues.6−9 We have therefore searched for alternatives to the commonly employed P2 proline residue in HCV NS3/4A protease inhibitors. A promising alternative is the use of a phenylglycine as a P2 moiety.10,11 In addition, P2 phenylglycine-based inhibitors have been shown to retain activity against mutated strains of the protease.11−13 Further development and optimization are still necessary in order to improve antiviral activities. During our initial studies, we discovered that a 2-phenyl-6-methoxypyrimidine based P2 substituent was beneficial in combination with a vinylated phenylglycine, an aromatic P1 moiety, and alkenylic P1′ substituents (1, Figure 2). We also discovered that a P3 truncated inhibitor (2) showed reasonable inhibitory potency (Ki = 190 nM, Figure 2)
Figure 2.

P3–P1′ spanning HCV NS3/4A protease inhibitor (1), its P3 truncated analogue (2), and a proline-based HCV NS3/4A protease inhibitor (3).14
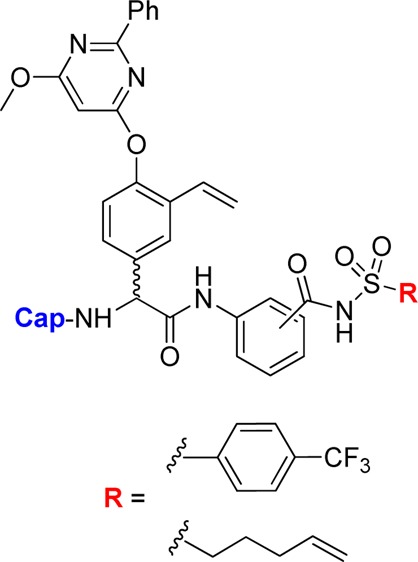
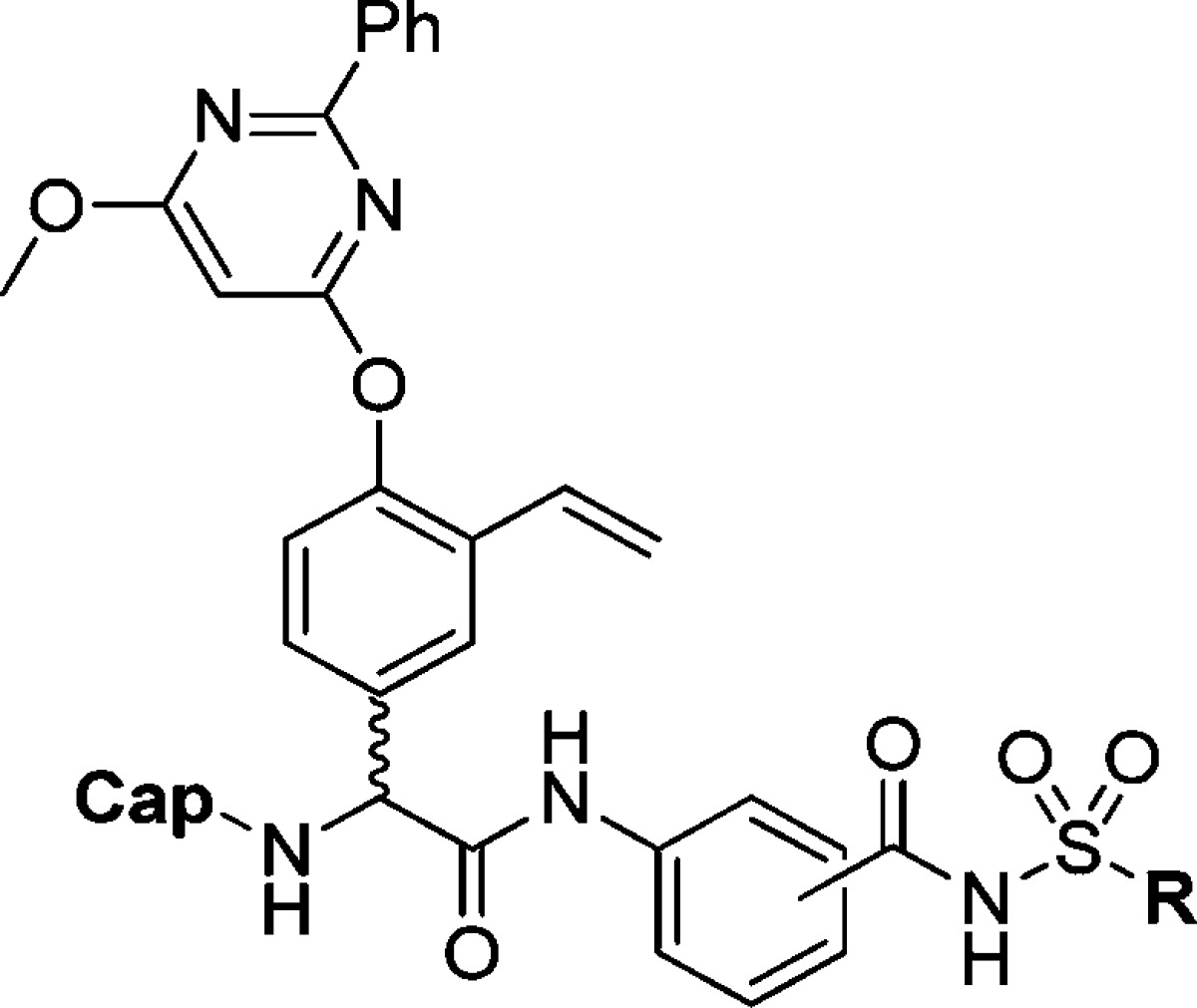
We have investigated the use of an aromatic P1 moiety in proline-based inhibitors previously (e.g., 3 in Figure 2).14 Because the aromatic P1 enables synthesis of regioisomers, it was appealing to us to utilize this property to examine how acyl sulfonamides at different positions around the aromatic P1 would influence inhibitory potency for phenylglycine-based inhibitors. Furthermore, we felt prompted to investigate the possibility to develop smaller, less peptide-like but more drug-like inhibitors with only one α-amino acid and therefore designed P3 truncated inhibitors (exemplified by 2). Thus, we herein present the synthesis and biochemical evaluation of a series of P3–P1′ and P2–P1′ spanning HCV NS3/4A protease inhibitors comprising a vinylated hydroxy phenylglycine substituted with a 2-phenyl-6-methoxy-pyrimidine, combined with an ortho (o-), meta (m-), or para (p-)-regioisomer of a P1 aryl acyl sulfonamide with either the pent-4-enyl side chain or the 4-(trifluoromethyl)phenyl side chain in P1′-position. For the P3 truncated inhibitors, a scan of various P2 N-capping groups was also performed to establish any important inhibitor-protease interactions at the S3–S2 border. Since we are aiming for inhibitors with a unique binding mode, the ability to inhibit known drug resistant enzyme variants was evaluated, as well as their in vitro pharmacokinetic properties.
The synthesis of the P2 building block 6 is described in Scheme 1. Compound 6 was synthesized from a 3-bromo, 4-hydroxy-phenylglycine derivative via a substitution reaction followed by a Suzuki reaction to give the partially racemic compound 6. The racemization is a known side-effect during Suzuki couplings with phenylglycine.15
Scheme 1. Synthetic Route.

Reagents: (a) 4-chloro-6-methoxy-2-phenylpyrimidine, KOtBu, DMSO, 64 °C (87%); (b) 2,4,6-trivinylcycloboroxane pyridine complex, Pd(OAc)2, [(tBu)3PH]BF4, K2CO3, DME, H2O, microwave irradiation at 100 °C for 15 min (82%).
In a previous study, we found that a 4-(trifluoromethyl)benzene sulfonamide was beneficial as a P1′ substituent in combination with an aromatic P1.14 For the synthesis of the P1–P1′ building blocks, a microwave-assisted carbonylative approach including bromoaniline, trans-bis(acetato)bis[o-(di-o-tolylphosphino)benzyl]-dipalladium(II) (Herrmann’s palladacycle), [(tBu)3PH]BF4, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), and Mo(CO)6 as a solid carbon monoxide source was applied (Scheme 2).16 Using this method, the P1–P1′ building blocks as o-, m- and p-isomers 7–9 could be synthesized in only 25 min, without need for amine protection. The P1–P1′ building blocks containing a pent-4-enyl sulfonamide substituent were synthesized in the standard way by coupling of Boc-protected m- and p-aminobenzoic acids with pent-4-ene sulfonamide using carbonyldiimidazole (CDI) in dry THF with DBU as base (Scheme 2).
Scheme 2. Synthetic Route to P1–P1′ Building Blocks.

Reagents: (a) Herrmann’s palladacycle, [(tBu)3PH]BF4, Mo(CO)6, DBU, 1,4-dioxane, microwave irradiation at 140 °C for 25 min (7, 50%; 8, 64%; 9, 62%); (b) CDI, DBU, THF, 60 °C → rt (10, 71%; 11, 69%); (c) 4.0 M HCl in 1,4-dioxane (quantitative).
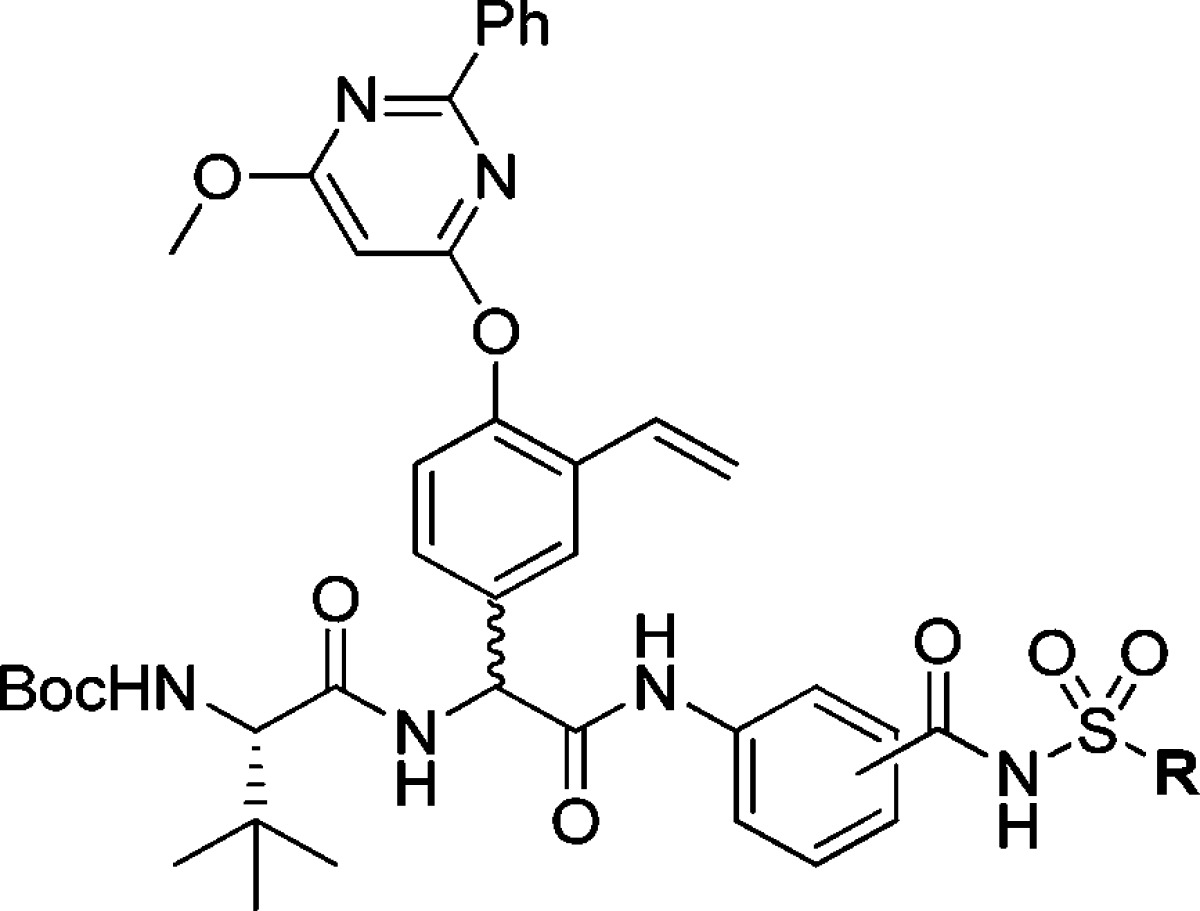
Amide bond formation between the P2 building block 6 and the poorly nucleophilic P1–P1′ moieties 7–11 was employed using HATU and N,N-diisopropylethylamine (DIEA) in dry CH2Cl2 at 45 °C and gave P2–P1′spanning compounds 12–16 in reasonable yields. The P3–P1′ spanning analogues 17L–21D were obtained after standard HATU promoted couplings of P3 Boc-l-tLeu and RP-HPLC purification. Only the l-Phg isomers could be isolated for compounds 17 and 18 (Scheme 3).
Scheme 3. Synthetic Route.

Reagents: (a) HATU, DIEA, CH2Cl2, 45 °C (12, 83%; 13, 50%; 14, 56%; 15, 74%; 16, 73%); (b) 4.0 M HCl in 1,4-dioxane; (c) HATU, DIEA, DMF, rt (17L, 31%; 18L, 29%; 19L, 16%; 19D, 5%; 20L, 34%; 20D, 10%; 21L, 22%; 21D, 12%).
To evaluate the effect of the N-capping group, compounds 13–15 were N-deprotected and reacted with t-butylisocyanate, acetyl chloride, 4-morpholinecarbonyl-chloride, nicotinoylchloride hydrochloride, or cyclopentyl chloroformate, giving compounds 23–29 (Scheme 4).
Scheme 4. Synthetic Route.

Reagents: (a) 4.0 M HCl in dioxane (100%); (b) CH2Cl2 or THF, Et3N/DMF, DIEA, t-butylisocyanate (23, 55%), acetyl chloride (24, 34%), 4-morpholinecarbonylchloride (25, 17%), nicotinoylchloride × HCl (26, 39%), or cyclopentyl chloro-formate (27, 48%; 28, 56%; 29, 51%).
For compounds 1, 2, and 12–29, Ki values were determined in a biochemical inhibition assay using full length NS3 protein of genotype 1a (see Supporting Information) and a 16 amino acid peptide corresponding to the activating region of NS4a (Tables 1 and 2).
Table 1. Inhibition Constants for NS3/4A Proteasea.
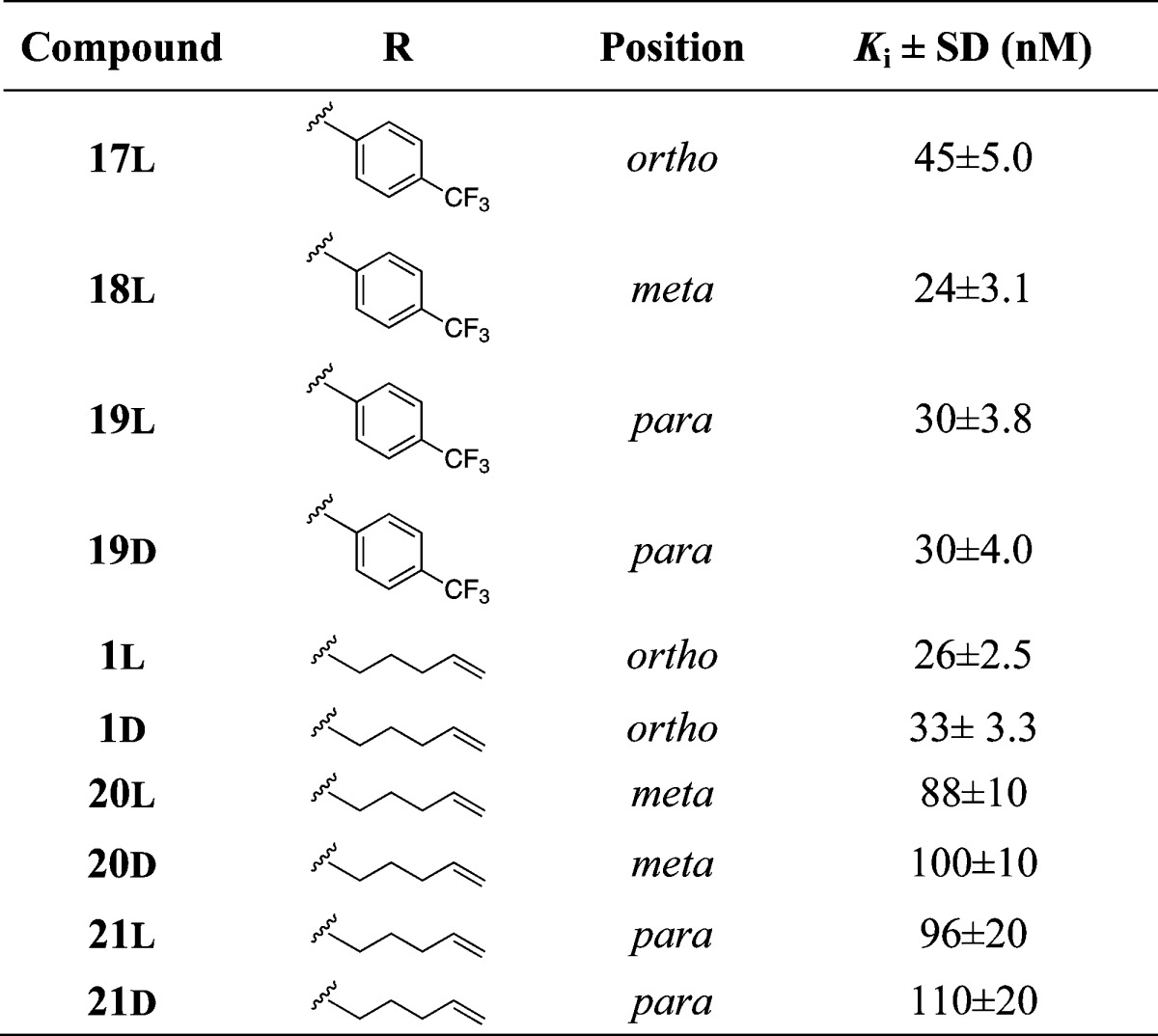
Ki values of P3–P1′ spanning inhibitors were measured with full-length NS3/4A protein from genotype 1a.
Table 2. Inhibition Constants for NS3/4A Proteasea.
Ki values of truncated inhibitors were measured with full-length NS3/4A protein from genotype 1a.
Gratifyingly, the use of a P1′ 4-(trifluoromethyl)phenyl side chain was shown to be beneficial in terms of inhibitory potency giving several inhibitors in the nanomolar range (13, 14, 17–19L, 19D, 27, and 28, Ki ≈ 20–70 nM, Tables 1 and 2). The aromatic P1′ side chain was especially advantageous for the truncated inhibitors 13 and 14 (Ki = 58 and 46 nM) as compared to their P1′ pent-4-enylic counterparts 15 and 16 (Ki ≈ 200 nM). Biochemical evaluation of truncated inhibitors comprising P1 regioisomers 12 (o-, Ki = 240 nM), 13 (m-, Ki = 58 nM), and 14 (p-, Ki = 46 nM), with a 4-(trifluoromethyl)phenyl P1′ substituent, revealed a preference for the m- and p-isomers.
The same comparison of the P1′ pent-4-enyl regioisomers 15 (m-, Ki = 210 nM) and 16 (p-, Ki = 220 nM) reveals that these are equipotent to o-compound 2 (Ki = 190 nM), although with a slightly decreased inhibitory potency compared to the P1′ 4-(trifluoromethyl)-phenyl containing inhibitors. Coupling of 12–14 with the P3 Boc-l-tLeu gave P3–P1′ spanning inhibitors 17L (o-, Ki = 45 nM), 18L (m-, Ki = 24 nM), 19L (p-, Ki = 30 nM), and 19D (p-, Ki = 30 nM). Again, a slight preference was shown for the m- and p-isomers, although the contribution to inhibitory potency for the m- and p-isomers from the P3 substituent was only 1.5- and 2.5-fold, respectively, as compared with 5-fold for the o-isomer. The same increase in potency was noted for the pent-4-enyl containing inhibitors after coupling with the P3 Boc-L-tLeu. Here, it was revealed that the m-compounds (20L, Ki = 88 nM; 20D, Ki = 100 nM) and p-compounds (21L, Ki = 96 nM; 21D, Ki = 110 nM) were less potent than the o-compounds (1L, Ki = 26 nM; 1D, Ki = 33 nM). Moreover, no apparent difference was seen between the stereoisomers of these inhibitors. Noteworthy, a comparison with the proline-based aromatic P1 inhibitor 3 (Figure 2) reveals that the aromatic P1 moiety is better suited for combination with phenylglycine than with proline. Both the 4-(trifluoromethyl)phenyl and the pent-4-enyl P1′side chain combined with a P2 2-(4-((6-methoxy-2-phenylpyrimidin-4-yl)oxy)-3-vinylphenylglycine (compounds 17L, 1L, and 1D) gives inhibitors with ca. 10-fold better inhibitory potency than to the corresponding proline inhibitor 3 (Figure 2).
A further evaluation of the N-capping group of the P3 truncated m-isomer 13 showed that removal of the Boc-protecting group (compound 22, Ki = 1200 nM) caused a 20-fold impairment in the inhibitory activity. The replacement of the Boc-group with an N-acetyl capping group reduced the inhibitory potency 14-fold (24, Ki = 700 nM) possibly as a result of the reduced bulk. The larger 4-morpholinecarbonyl group (compound 25, Ki = 590 nM) was slightly better than the N-acetyl capping group but was still 12 times less potent compared to inhibitors bearing the Boc-group. The introduction of an N-nicotinoyl capping group (compound 26, Ki = 240 nM) gave a 2-fold improvement compared to the 4-morpholinecarbonyl capping group but was still 4 times less favorable than the Boc-group. The N-t-butyl carbamoyl (23Ki = 140 nM) and the N-cyclopentyloxy carbonyl (27, Ki = 72 nM) capping groups proved to be the best alternatives to the Boc-group. Thus, the steric bulk of the capping group is clearly of importance. Indeed, exchange of the Boc group on the P3 truncated p-compound 14 (Ki = 46 nM) by a cyclopentyl carbamate gave the equipotent inhibitor 28 (Ki = 55 nM). Substitution of Boc by a cyclopentyl carbamate on m-compound 15 (Ki = 210 nM) gave compound 29 (Ki = 130 nM) with a slightly improved potency.
Additionally, compounds 13 and 18L were evaluated toward the drug resistant A156T, D168V, and R155K variants of the protease (Table 3). Vitality values were calculated to normalize the inhibitory effects of the inhibitors with respect to the effects of amino acid substitutions on catalytic efficiency (kcat/Km) of the enzyme variants. A vitality value less than 1 indicates a more efficient inhibitor against the mutated variant compared to the wild-type enzyme, whereas a value larger than 1 shows that the inhibitor is less efficient against the mutated virus.12 The possibility to endure antiviral activity on mutated strains of the protease differs among compounds that have entered clinical trials. For example, Incivek (Figure 1) is favored by the D168V substitution17 (V = 0.4) but highly affected by the A156T substitution (V = 480).12 Ciluprevir is also highly affected by both the A156T (V = 1600) and D168V (V = 3200) substitutions.12 Herein, the vitality values for compounds 13 and 18L shows that the inhibitors have the capacity to retain most of their inhibitory activity on the A156T, D168V, and R155K variants of the protease (vitality values around 1). Compound 18L is equally affected by the A156T, D168V, and R155K substitutions (V = 1.9, 1.9, and 1.6), while compound 13, lacking a P3 substituent, is slightly less affected by the D168V substitution (V = 1.3) than the A156T and R155K (V = 1.6 and 1.8) substitutions.
Table 3. Inhibition Constants and Vitality Values Evaluated with A156T, D168V, and R155K Variants of NS3.
| A156T |
D168V |
R155K |
||||
|---|---|---|---|---|---|---|
| compd | Ki ± SD (nM) | Va | Ki ± SD (nM) | Va | Ki ± SD (nM) | Va |
| 13 | 110 | 1.6 | 180 | 1.3 | 580 | 1.8 |
| 18L | 53 | 1.9 | 110 | 1.9 | 210 | 1.6 |
Vitality values (V) were calculated using the equation V = [Ki × (kcat/Km)]variant/[Ki × (kcat/Km)]wild-type.12
Compounds 1 (racemate), 13, and 18L were also tested for EC50-values in a replicon Huh-7 cell line containing subgenomic HCV RNA genotype 1b replicon with firefly luciferase.18 Compound 1 and the P2–P1′ inhibitor 13 showed EC50 values of 12.5 and 27.5 μM (CC50 ≥ 50 μM; in Huh-7 cells), respectively, whereas the P3-comprising inhibitor 18L had a EC50 value of >50 μM (CC50 ≥ 50 μM). In analogy with acyl sulfonamide based NS3/4A protease inhibitors of submicromolar potency in general there is a significant discrimination between cellular and enzymatic potencies.19 Clearly, there is room for optimization of the overall potencies, down to low or subnanomolar, for these type of inhibitors.
Compounds 12 (o), 13 (m), and 14 (p) were modeled into the binding site of the full-length NS3/4A protein after removal of the cocrystallized macrocyclic inhibitor.20 The modeling showed that compound 13 (m) finds key interactions with amino acid backbone of the protease (R155 and A157) and that the P1 carbonyl interacts with the oxyanion hole (G137 and S139) (Figure 3). A possible π stacking interaction is seen between the 4-(trifluoromethyl)phenyl substituent and Q41. This type of interaction is seen for the original cocrystallized macrocyclic inhibitor, as well as for a prime side extended α-keto amide inhibitor.20,21 Additionally, hydrogen bond interaction with the helicase (Q526) is captured by compound 13 (m), in analogy with the cocrystallized structure used in the molecular modeling. The p-regioisomer 14 showed a similar binding mode in the protein active site. However, the o-regioisomer 12 did not fit well in the protein active site. In the docking suggested conformations, several of the significant interactions with the protein were missing, supporting the preference of m- and p-isomers observed for this type of inhibitors.
Figure 3.
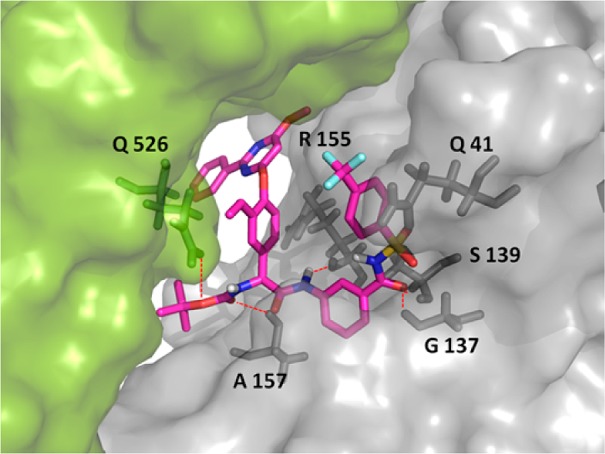
Compound 13 (m) docked in the binding site of the full-length NS3/4A protein (PDB code: 4A92).20 The protease domain is shown in gray; the helicase domain is shown in green. Hydrogen bonds are highlighted as red dashed lines.
To further address the potential of this series of inhibitors and the P3 truncated inhibitors in particular, drug-like properties were evaluated for the various regioisomers of the most potent P1′ 4-(trifluoromethyl)phenyl containing inhibitors with the Boc capping group (12–14), using in vitro pharmacokinetic profiling (Table 4). One inhibitor each of the corresponding urea- (23), P3 t-leucine- (18L), and the P1′ pent-4-enyl (2) analogues were included for comparison. Although the predicted log D values were in the same range, the compounds showed relatively large variation in solubility. Whereas the inhibitor comprising the P1′ pent-4-enyl side chain (2) showed excellent solubility (>100 μM), the corresponding P1′ 4-(trifluoromethyl)phenyl based inhibitors exhibited lower solubility. The ortho analogue (12) displayed a poor solubility of 5 μM, while the p-, P3-comprising-, and the urea analogue showed acceptable (>20 μM) solubilities in the range of 30–40 μM.
Table 4. In Vitro and in Silico Determined Properties.
| in vitro |
in silico |
|||||
|---|---|---|---|---|---|---|
| compd | solubility (μM) pH 7.4 | Caco-2 permeability Papp (10–6 cm/s)a a–bb | Clint (μL/min/mg)c | t1/2d (min) | pKae | log D7.4 |
| 12 | 5 | 4.0 | 16 | 88 | 4.8 | 4.4 |
| 13 | n.d.f | 1.0 ± 0.1 | 12 | 117 | 4.1 | 3.8 |
| 14 | 37 | 5.7 ± 0.5 | 25 | 55 | 4.4 | 3.9 |
| 18L | 43 | 9.7 ± 0.9 | 80 | 17 | 4.3 | 4.5 |
| 23 | 31 | 0.36 ± 0.3 | 7 | 194 | 4.2 | 4.1 |
| 2 | 101 | 1.0 ± 0.7 | 66 | 21 | 5.8 | 4.0 |
Papp = apparent permeability coefficient.
a–b = apical to basolateral.
Clint = in vitro intrinsic clearance.
t1/2 = in vitro half-life.
Acidic pKa (acyl sulfonamide).
n.d. = not determined.
In analogy, several of the inhibitors with the higher aromatic ring count penetrated the intestinal epithelial cells very well (Papp = (4–9) × 10–6 cm/s, compounds 12, 14, and 18L). The m-isomers showed moderate permeability, and when an additional hydrogen bond donor is introduced as in the urea group, this appears to have a negative impact on the transport over cell membranes as shown from the decreased Papp value (0.4 × 10–6 cm/s) of compounds 23 as compared to the corresponding carbamate 13 (Papp value = 1.0 × 10–6 cm/s), although the difference is not statistically significant. Gratifyingly, all the truncated inhibitors with a P1′ 4-(trifluoromethyl)phenyl side chain (12–14 and 23) showed a very low risk for high oxidative metabolism in microsomes (Clint = 7–25 μL/min/mg). These compounds clearly benefitted from the removal of the P3 amino acids since the P3–P1′ spanning inhibitor 18L showed a significantly higher intrinsic clearance value (80 μL/min/mg). Additionally, the inhibitor with a P1′ pent-4-enyl side chain (2, Clint = 66 μL/min/mg) was more susceptible for oxidative metabolism in microsomes compared to its aromatic counterparts.
In summary, truncated phenylglycine based HCV NS3/4A protease inhibitors of nanomolar potency have been discovered. The aryl acyl sulfonamide moiety in P1 position was preferably combined with a 4-(trifluoromethyl)phenyl P1′ side chain in m- and p-positions, and the sterically congested Boc group turned out to be the preferred P2 capping group. In general, the P2–P1′spanning inhibitors comprising the Boc and 4-(trifluoromethyl)phenyl groups exhibited very low risk for high first pass metabolism, good apparent intestinal permeability, and moderate solubility under in vitro settings. Further optimization should be devoted to improving the cell-based inhibitory potency. This could probably be achieved by further optimization of the inhibitory potency against the protease and by improving solubility, while maintaining good cell-permeability and metabolic stability. Special attention should be dedicated to drug resistance variants in the lead optimization process since the inhibitors displayed promising inhibition of the A156T, D168V, and R155K variants of the protease.
Acknowledgments
This work was supported by grants from the Swedish Research Council (Grants 9478 and 21386 and a grant to the Chemical Biology Consortium Sweden). We gratefully acknowledge support from Knut and Alice Wallenberg’s foundation and Science for Life Laboratory. We also thank Simulations Plus for providing access to the ADMET Predictor software and Dr Aleh Yahorau, Department of Pharmaceutical Biosciences, Uppsala University, for help with HRMS analyzes and Medivir AB, Huddinge, Sweden for EC50-determinations and financial support.
Glossary
Abbreviations
- HCV
hepatitis C virus
- NS
nonstructural
- Phg
phenylglycine
- Herrmann’s palladacycle
trans-bis(acetato)bis[o-(di-o-tolylphosphino)-benzyl]-dipalladium(II)
- DBU
1,8-diazabicyclo[5.4.0]undec-7-ene
- HATU
N-[(dimethylamino)-1H-1,2,3-triazolo-[4,5-b]pyridin-1-ylmethylene]-N-methylmethanaminium hexa-fluorophosphate N-oxide
- CDI
carbonyldiimidazole
- DIEA
N,N-diisopropylethylamine
- Clint
intrinsic clearance
- Papp
apparent permeability coefficient
Supporting Information Available
All experimental details such as synthetic procedures and characterization of compounds 4–29, enzymatic inhibition assays, molecular modeling procedures, in silico prediction, and in vitro pharmacokinetic assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Lavanchy D. The global burden of hepatitis C. Liver Int. 2009, 29, 74–81. [DOI] [PubMed] [Google Scholar]
- Lauer G. M.; Walker B. D. Medical progress: Hepatitis C virus infection. New Engl. J. Med. 2001, 345, 41–52. [DOI] [PubMed] [Google Scholar]
- Rönn R.; Sandström A. New developments in the discovery of agents to treat hepatitis C. Curr. Top. Med. Chem. 2008, 8, 533–562. [DOI] [PubMed] [Google Scholar]
- Lamarre D.; Anderson P. C.; Bailey M.; Beaulieu P.; Bolger G.; Bonneau P.; Bös M.; Cameron D. R.; Cartier M.; Cordingley M. G.; Faucher A. M.; Goudreau N.; Kawai S. H.; Kukolj G.; Lagacé L.; Laplante S. R.; Narjes H.; Poupart M. A.; Rancourt J.; Sentjens R. E.; St George R.; Simoneau B.; Steinmann G.; Thibeault D.; Tsantrizos Y. S.; Weldon S. M.; Yong C. L.; Llinàs-Brunet M. An NS3 protease inhibitor with antiviral effects in humans infected with hepatitis C virus. Nature 2003, 426, 186–189. [DOI] [PubMed] [Google Scholar]
- For review, seeChatel-Chaix L.; Baril M.; Lamarre D. Hepatitis C virus NS3/4A protease inhibitors: A light at the end of the tunnel. Viruses 2010, 2, 1752–1756. [DOI] [PMC free article] [PubMed] [Google Scholar]; Original references for each inhibitor can be found in the Supporting Information.
- Pearlman B. L. Protease inhibitors for the treatment of chronic hepatitis C genotype-1 infection: the new standard of care. Lancet Infect. Dis. 2012, 12, 717–728. [DOI] [PubMed] [Google Scholar]
- Koev G.; Kati W. The emerging field of HCV drug resistance. Expert Opin. Investig. Drugs 2008, 17303–319. [DOI] [PubMed] [Google Scholar]
- Courcambeck J.; Bouzidi M.; Perbost R.; Jouirou B.; Amrani N.; Cacoub P.; Pepe G.; Sabatier J.-M.; Halfon P. Resistance of Hepatitis C virus to NS3–4A protease inhibitors: mechanisms of drug resistance induced by R155Q, A156T, D168A and D168V mutations. Antiviral Ther. 2006, 11, 847–855. [PubMed] [Google Scholar]
- Romano K. P.; Ali A.; Royer W. E.; Schiffer C. A. Drug resistance against HCV NS3/4A inhibitors is defined by the balance of substrate recognition versus inhibitor binding. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 20986–20991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Örtqvist P.; Peterson S. D.; Åkerblom E.; Gossas T.; Sabnis Y. A.; Fransson R.; Lindeberg G.; Danielson U. H.; Karlén A.; Sandström A. Phenylglycine as a novel P2 scaffold in hepatitis C virus NS3 protease inhibitors. Bioorg. Med. Chem. 2007, 15, 1448–1474. [DOI] [PubMed] [Google Scholar]
- Lampa A.; Ehrenberg A.; Gustafsson E.; Vema S.; Åkerblom A.; Lindeberg E.; Karlén G.; Danielson A.; Sandström U. H. A. Improved P2 phenylglycine-based hepatitis C virus NS3 protease inhibitors with alkenylic prime-side substituents. Bioorg. Med. Chem. 2010, 18, 5413–24. [DOI] [PubMed] [Google Scholar]
- Dahl G.; Sandström A.; Åkerblom E.; Danielson U. H. Resistance profiling of hepatitis C virus protease inhibitors using full-length NS3. Antiviral Ther. 2007, 12, 733–740. [PubMed] [Google Scholar]
- Örtqvist P.; Vema A.; Ehrenberg A. E.; Dahl G.; Rönn R.; Åkerblom E.; Karlén A.; Danielson U. H.; Sandström A. Structure–activity relationships of HCV NS3 protease inhibitors evaluated on the drug-resistant variants A156T and D168V. Antiviral Ther. 2010, 15, 841–852. [DOI] [PubMed] [Google Scholar]
- Rönn R.; Lampa A.; Peterson S. D.; Gossas T.; Åkerblom E.; Danielson U. H.; Karlén A.; Sandström A. Hepatitis C virus NS3 protease inhibitors comprising a novel aromatic P1 moiety. Bioorg. Med. Chem. 2008, 16, 2955–2967. [DOI] [PubMed] [Google Scholar]
- Prieto M.; Mayor S.; Rodriguez K.; Lloyd-Williams P.; Giralt E. Racemization in Suzuki couplings: a quantitative study using 4-hydroxyphenylglycine and tyrosine derivatives as probe molecules. J. Org. Chem. 2007, 72, 1047–1050. [DOI] [PubMed] [Google Scholar]
- Wu X. Y.; Rönn R.; Gossas T.; Larhed M. Easy-to-execute carbonylations: Microwave synthesis of acyl sulfonamides using Mo(CO)6 as a solid carbon monoxide source. J. Org. Chem. 2005, 70, 3094–3098. [DOI] [PubMed] [Google Scholar]
- Lin C.; Gates C. A.; Rao B. G.; Brennan D. L.; Fulghum J. R.; Luong Y.-P.; Frantz J. D.; Lin K.; Ma S.; Wei Y.-Y.; Perni R. B.; Kwong A. D. In vitro studies of cross-resistance mutations against two Hepatitis C Virus serine protease inhibitors, VX-950 and BILN 2061. J. Biol. Chem. 2005, 280, 36784–36791. [DOI] [PubMed] [Google Scholar]
- Lohmann V.; Korner F.; Koch J. O.; Herian U.; Theilmann L.; Bartenschlager R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999, 285, 110–113. [DOI] [PubMed] [Google Scholar]
- Rönn R.; Sabnis Y. A.; Gossas T.; Åkerblom E.; Danielson U. H.; Hallberg A.; Johansson A. Exploration of acyl sulfonamides as carboxylic acid replacements in protease inhibitors of the Hepatitis C virus full-length NS3. Bioorg. Med. Chem. 2006, 14, 544–559. [DOI] [PubMed] [Google Scholar]
- Schiering N.; D’Arcy A.; Villard F.; Simic O.; Kamke M.; Monnet G.; Hassiepen U.; Svergun D.; Pulfer R.; Eder J.; Raman P.; Bodendorf U. A macrocyclic HCV NS3/4A protease inhibitor interacts with protease and helicase residues in the complex with its full-length target. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 21052–21056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y. Y.; Stoll V. S.; Richardson P. L.; Saldivar A.; Klaus J. L.; Molla A.; Kohlbrenner W.; Kati W. M. Hepatitis C NS3 protease inhibition by peptidyl-alpha-ketoamide inhibitors: kinetic mechanism and structure. Arch. Biochem. Biophys. 2004, 421, 207–216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.