

Abstract
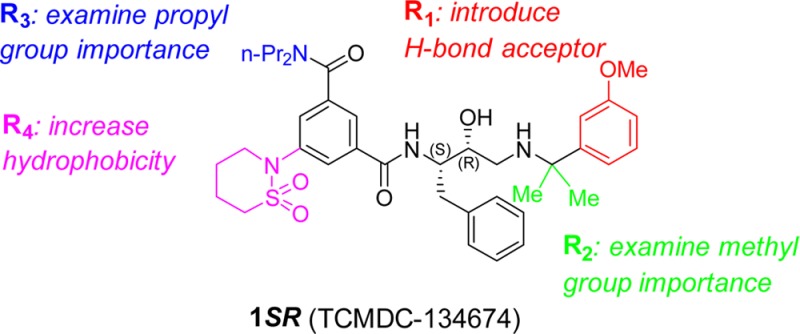
Antimalarial hit 1SR (TCMDC-134674) identified in a GlaxoSmithKline cell based screening campaign was evaluated for inhibitory activity against the digestive vacuole plasmepsins (Plm I, II, and IV). It was found to be a potent Plm IV inhibitor with no selectivity over Cathepsin D. A cocrystal structure of 1SR bound to Plm II was solved, providing structural insight for the design of more potent and selective analogues. Structure-guided optimization led to the identification of structurally simplified analogues 17 and 18 as low nanomolar inhibitors of both, plasmepsin Plm IV activity and P. falciparum growth in erythrocytes.
Keywords: Malaria, Plasmodium falciparum, plasmepsins, Cathepsin D, inhibition, structure-guided optimization, hydroxyethylamine
Despite extensive eradication campaigns, malaria caused by Plasmodium parasites remains a devastating disease with an estimated 219 million cases and 660 thousand lethal outcomes in 2010.1 Widespread resistance to practically all currently used drugs has activated the search for antimalarials with novel mechanisms of action.2−4 Low profit potential of antimalarial drugs has promoted collaboration between academic, private, and charitable organizations to establish novel drug discovery programs. As a part of an antimalarial initiative, pharmaceutical companies contribute with their unique resources to the development of antimalarials, making their data publicly available.5 Recently, researchers at GlaxoSmithKline (GSK) published the structures of 13 533 hits from the screening of nearly 2 million compounds that inhibited malaria parasite growth by at least 80% at 2 μM concentration.6 These hits were further analyzed using cheminformatics to identify 47 series of high-quality starting points for lead optimization.7 The series 3 included 74 compounds based on a hydroxyethylamine scaffold that is characteristic for plasmepsin inhibitors.8−11 Given the interest in the digestive vacuole plasmepsins (Plm I, II, and IV) as targets for antimalarial drug discovery, we selected the most active compound from this series, 1SR (TCMDC-134674), for investigation of its Plm I, II, and IV inhibitory activity (Chart 1).12
Chart 1. Structure of GSK Cell Based HTS Antimalarial Hit.

Compound 1SR was resynthesized (see Supporting Information) and tested in enzymatic assays, which showed that it is a potent Plm IV inhibitor (IC50 = 29 nM) while being a less effective inhibitor of Plm II (IC50 = 0.15 μM) and Plm I (IC50 = 0.70 μM) (Table 1). Selectivity studies of compound 1SR showed that it was not a selective inhibitor of plasmesins over the human aspartic protease Cathepsin D (CatD, IC50 of 43 nM, Table 1).
Table 1. SAR of R1 Group.
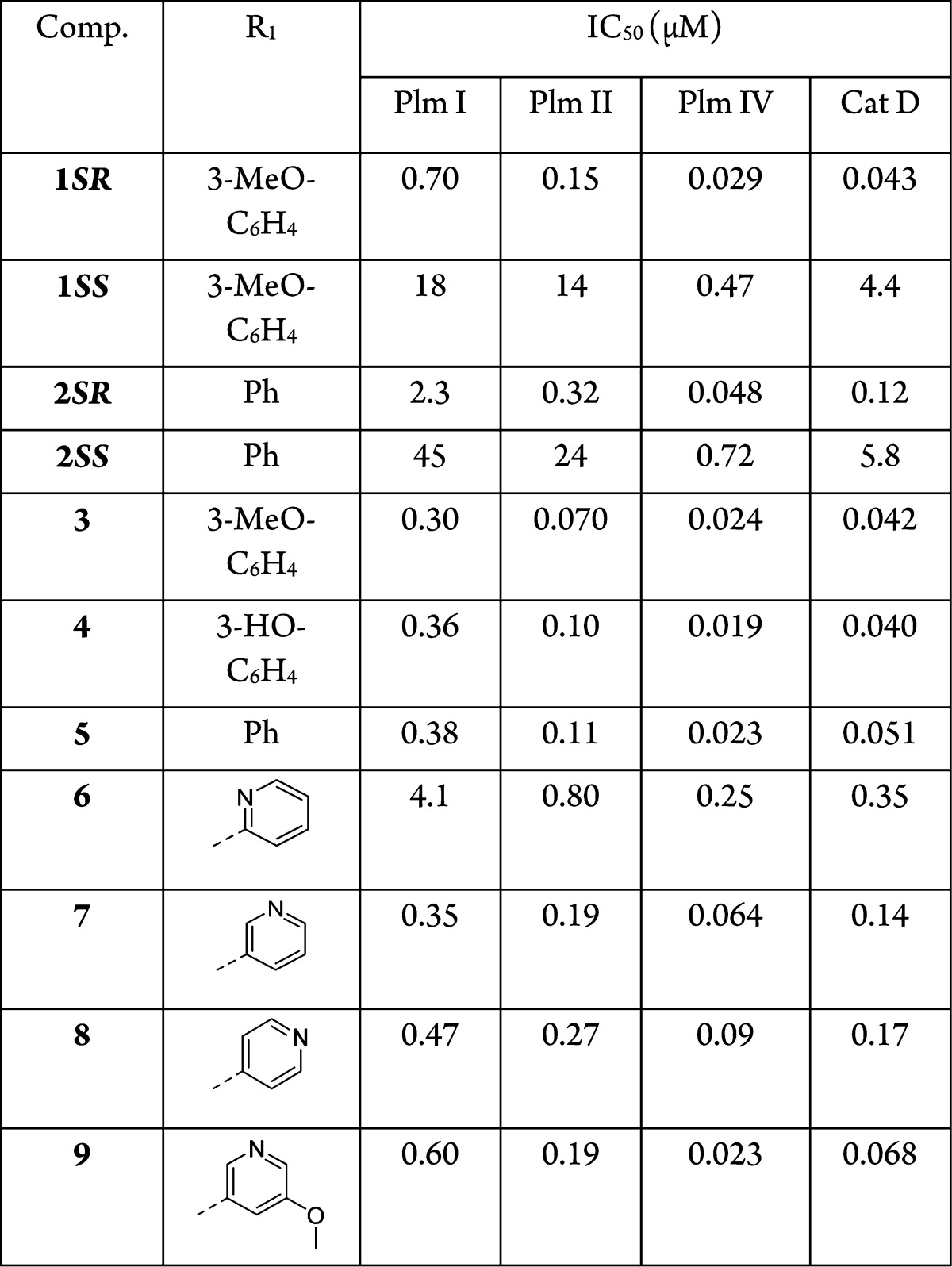
While a number of digestive vacuole plasmepsin inhibitors have been previously identified, to the best of our knowledge none of them displayed low nanomolar activity in cell based models. Therefore, it was an exciting finding that compound 1SR, which was reported as highly active against intraerythrocytic P. falciparum cell growth (IC50 of 30 nM),6 is a potent Plm IV inhibitor. The critical role of Plm IV is still unclear, ; however, gene knockout studies have revealed that out of all individual digestive vacuole plasmepsin knockouts only Δpfpm4 produced a statistically significant reduction in hemozoin accumulation indicating impaired hemoglobin digestion.13 In addition, the quadruple-plasmepsin knockout mutant (Δpfpm1–4) showed a significantly slower rate of growth in contrast to a triple plasmepsin knockout mutant (Δpfpm1–3).14 On the basis of the available data, it was intriguing to speculate that inhibition of Plm IV could be responsible for the activity of compound 1SR in the cell based assay. Nevertheless, it cannot be excluded that any other of the nondigestive plasmepsins structurally similar to PlmIV could be the target (or an additional target) for compound 1SR.15,16
To provide a structural basis for the design of structurally simplified, potent and selective analogues of compound 1SR, the X-ray crystal structure of Plm II in complex with compound 1SR was solved (Chart 2). Plm II was chosen for the crystallization studies because it was easily obtainable in milligram quantities, and its crystallization conditions have been described previously.17 Plm II mutant M205S was used, which displays enhanced resistance to self-cleavage compared to the wild type enzyme.18 The obtained crystals diffracted to 1.85 Å resolution and belonged to space group C2 with six molecules in the asymmetric unit. The electronic density of the inhibitor was very similar in all six subunits and did not improve significantly upon averaging. As shown in Chart 2a, electron density was good for the central part of the inhibitor but was weak or lacking for aliphatic propyl groups, the methoxy moiety and part of the 1,2-thiazinane-1,1-dioxide ring. As expected, the structure shows an interaction between the hydroxyethylamine core of 1SR and the Plm II catalytic dyad Asp34–Asp214 (Chart 2b). However, in contrast to other structures of transition state analogue–Plm II complexes,17,19,20 the hydroxyl group is not within the characteristic hydrogen-bond distances to both carboxylic acids, but only Asp34 (O–O distance of 2.69 Å) and Asp214 is hydrogen-bonded with the secondary amino group (N–O distance of 2.47 Å, Chart 2c). The secondary amine is linked to the isopropyl-2-(3-methoxyphenyl) moiety that occupies the S1′ and part of the S2 pockets. The other side of the hydroxyethylamine core is linked via a methine bridge to the benzyl group positioned in the S1 pocket and the 3,5-disubstituted benzamide moiety, whose N,N-dipropylamide and 2-(1,2-thiazinane-1,1-dioxide)-substituents bind in the S3 and S4 pockets, respectively. The interactions in the S1–S4 and S1′ pockets are predominantly hydrophobic, while a few H-bond interactions are formed with the more central groups of the inhibitor, i.e., between the benzamide and Gly216 and the N,N-dipropylamide and Ser218 (Chart 2c). Additionally, the secondary amino group seems to interact simultaneously with the carboxylate of Asp214 and the carbonyl of Gly36. Detailed inspection of the structure highlighted three sites of potentially repulsive interactions between the inhibitor and Plm II active-site residues (Chart 2c). First, the sulfonyl group of the 2-(1,2-thiazinane-1,1-dioxide) moiety is only 3.0 Å from the hydrophobic Val78 side chain. This repulsion seems to interfere with flap closing, making the binding cavity more open than in other Plm II–inhibitor complexes.21 Second, the 6-C atom of the 3-methoxyphenyl group lies 3.3 Å from the hydrophilic side chains of Thr217 and Asp214, and third, one methyl group of the isopropyl-2-(3-methoxyphenyl) moiety is 3.6 Å from the carbonyl of Gly36. Importantly, these interactions may also be present in complexes with Plm I and IV as the interacting residues are conserved among the food vacuole plasmepsins, except Val78, which is replaced by a Gly in Plm IV. It was also apparent that the aliphatic chains of the N,N-dipropylamide moiety make only weak contacts with the protein as their electron density was indiscernible (Chart 1a).
Chart 2. Binding of 1 (TCMDC-134674)6 within the Active Site of Plm IIa.
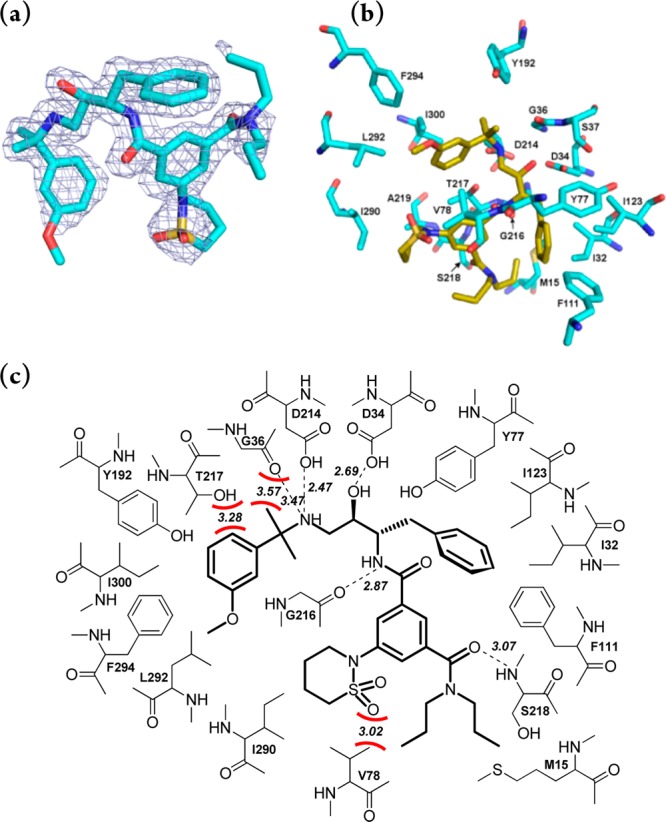
a (a) X-ray crystal structure showing the ligand and residues located within 4 Å from it as stick models (colored in yellow and cyan, respectively). (b) 2Fo – Fc electron density of the bound ligand contoured at 1σ. The figure was generated using PyMOL.22 (c) Ligand interaction diagram. Hydrogen bonds are shown as dashed lines, and distances between heavy atoms are indicated. Potentially repulsive interactions between the ligand and protein atoms are shown as red curves, and distances between the closest heavy atoms are indicated in Å.
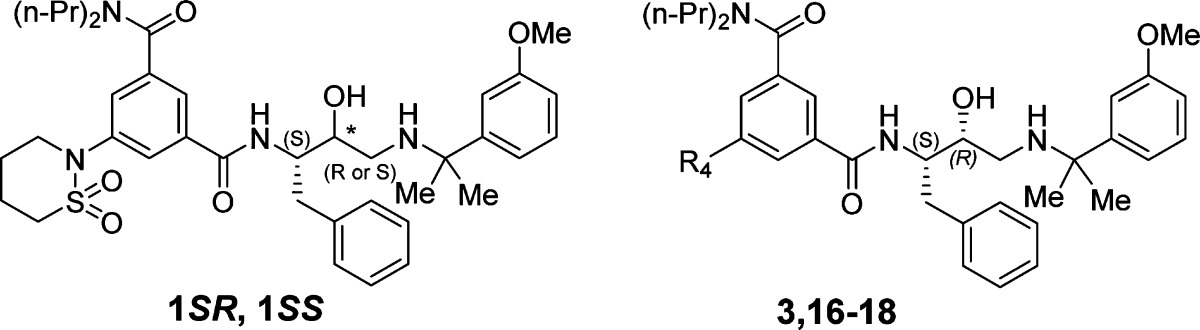
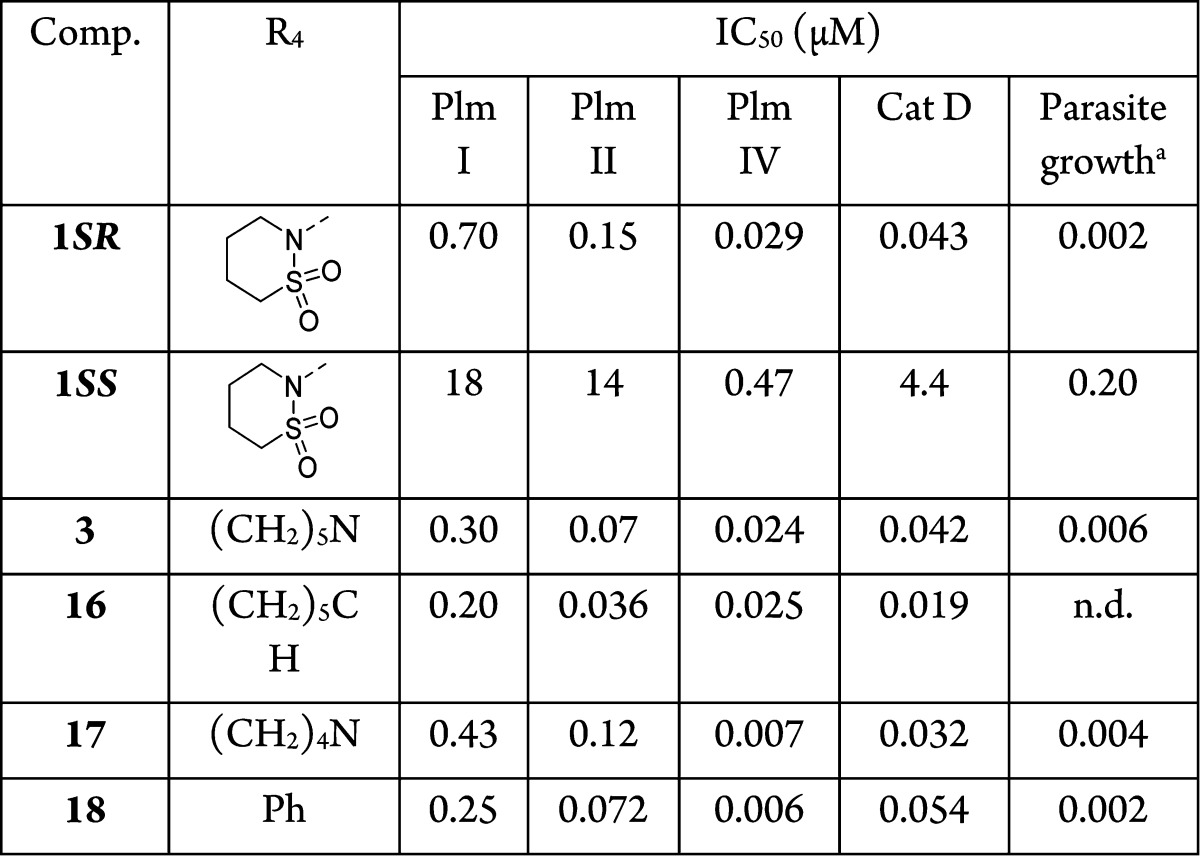
Guided by these insights from the crystal structure, we started our SAR study of 1SR and defined four positions, where appropriate chemical modifications were likely to improve intermolecular interactions (Tables 1–4, R1 to R4). Modifications of R1, R2, and R4 were also expected to contribute to the selectivity versus CatD as these moieties are located in the S1′ and S2 and S4 pockets that are most different between the two enzymes.21 Keeping R4 substituent (2-(1,2-thiazinane-1,1-dioxide)) constant while optimizing substituents R1–R3 proved to be impractical due to the very lengthy synthesis. Therefore, we preferred to use the more accessible piperidinyl group as the R4 substituent for optimization around R1–R3. To confirm that this modification does not significantly alter the bound conformation observed in the crystal structure, a few analogues were synthesized with both piperidinyl and 1,2-thiazinane-1,1-dioxide groups. Comparison of their activities (compound pairs 2SR and 5, 10, and 12) led to similar conclusions about changes in inhibition activity suggesting that the piperidinyl analogue binds similarly to compound 1SR.
Table 4. SAR of R4 Group.
3D7 strain of P. falciparum
Previous SAR studies of statine- and hydroxyethylamine-based Plm II inhibitors have indicated a preference for the S-configuration at the central hydroxyl group;21,23 therefore, we were surprised that compound 1 has the R-configuration at this stereogenic center. We prepared both diastereoisomers of two of the studied compounds (1 and 2) and tested their inhibitory activity against Plm I, II, and IV. The stereoisomers 1SS and 2SS showed 15–90 -fold lower activity than the isomers 1SR and 2SR indicating that the R-configuration of the central hydroxyl-bearing stereo center is critical for plasmepsin inhibition (see Table 1) opposed to statine based inhibitors.21 This may be explained by the different binding mode of the hydroxyethylamine core of 1SR in the Plm II catalytic center as was observed in the crystal structure of the complex and discussed above.
SAR studies on R1 (Table 1) were performed to evaluate the importance of the 3-methoxy group as well as to explore the possibility of introducing an H-bond acceptor for the adjacent hydroxyl group of Thr217. After preparing compound 2, the unsubstituted phenyl analogue of 1, we decided to perform further studies with piperidinyl group as the R4 substituent for the reasons described above and synthesized compounds 3–5. It should be noted that this R4 substitution yielded 2-fold higher activity against Plm I and II, while the activity increase against Plm IV was negligible. Replacing the methoxy group with a hydroxyl functionality (compound 4) did not change its activity. The phenyl-substituted analogue (compound 2) of 1 was 2- to 3-fold less active, while the corresponding analogue (compound 5) of 3 was of similar potency, indicating some cooperativity between the R1 and R4 substituents in the case of 5. In order to explore possible H-bonding interactions with the hydroxyl group of Thr217, we prepared compounds 6–8 by replacing the 3-methoxyphenyl group with 2-, 3-, and 4-pyridyl, respectively, hoping that the nitrogen atom might act as the H-bond acceptor. However, this did not result in improved activity, possibly, due to protonation of the pyridine nitrogen atom at the pH of the assay (pH 4.6). The 3-pyridyl-substitution (compound 7) was tolerated best from this series and in combination with the 5-methoxy group yielded compound 9 with similar activity (IC50 of 23 nM) against Plm IV as 3 (IC50 of 24 nM).
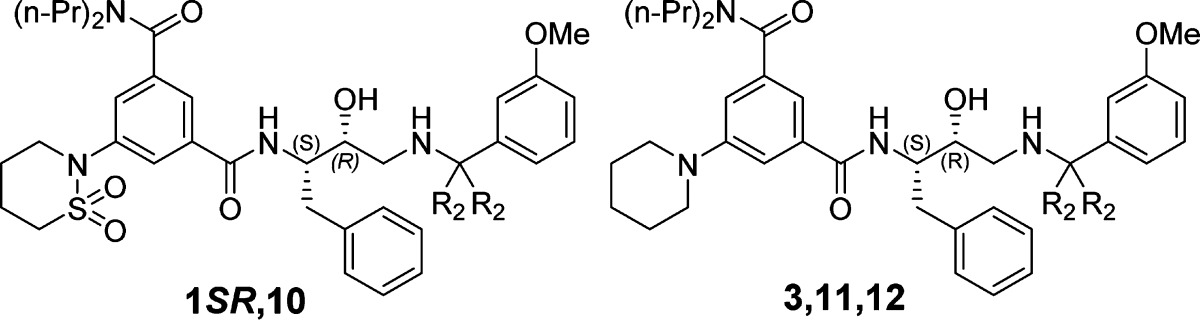
Next, we investigated SARs associated with R2 (Table 2). Although the crystal structure showed that one methyl group is located in a polar environment while the other one lies in a hydrophobic environment, we synthesized symmetrically substituted analogues to avoid the complication associated with an additional stereogenic center. Removal of both methyl groups (compounds 10 and 12) resulted in a significant decrease in activity, whereas a cyclopropyl functionality (compound 11) was well tolerated at this position and at the same time slightly enhanced selectivity for Plm IV over Cat D.
Table 2. SAR of R2 Group.
| IC50 (μM) |
|||||
|---|---|---|---|---|---|
| compd | R2 | Plm I | Plm II | Plm IV | CatD |
| 1SR | CH3 | 0.70 | 0.15 | 0.029 | 0.043 |
| 10 | H | 18 | 2.7 | 0.20 | 0.36 |
| 3 | CH3 | 0.30 | 0.070 | 0.024 | 0.042 |
| 11 | –CH2– | 0.33 | 0.15 | 0.025 | 0.10 |
| 12 | H | 6.1 | 0.6 | 0.18 | 0.20 |
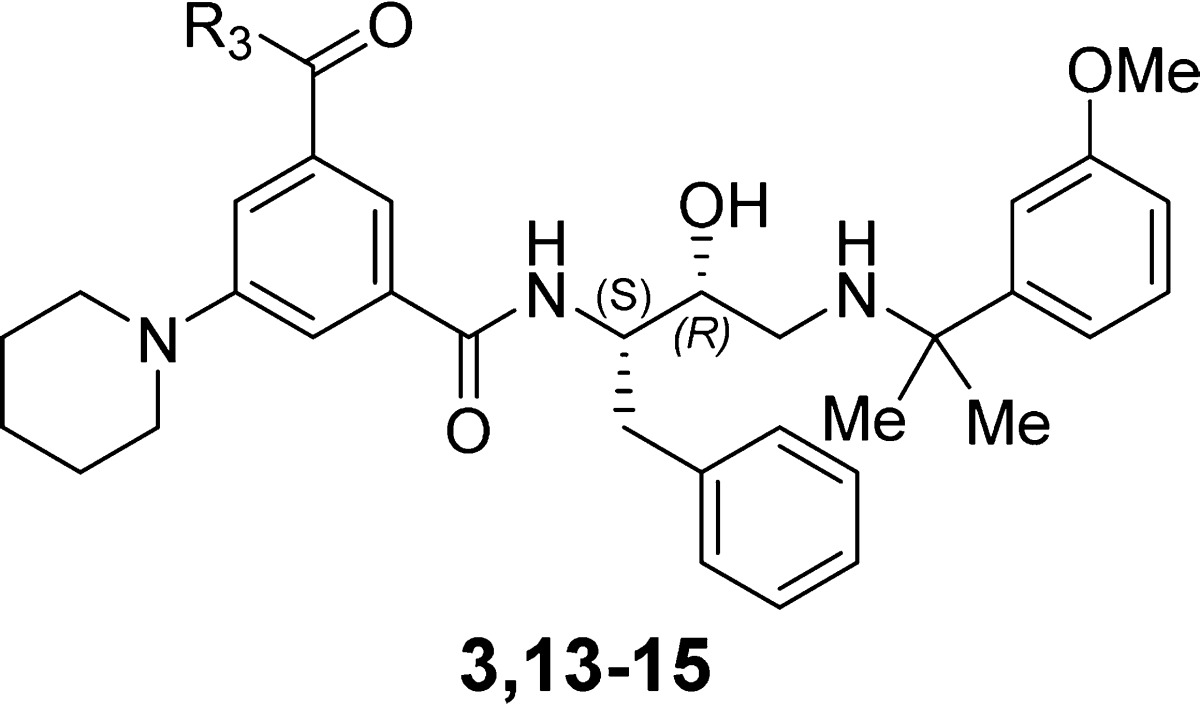
Further we examined the importance of the propyl groups in the N,N-dipropylamide moiety (R3) that showed poor electron density in the crystal structure. Removal of one propyl group (compound 13) resulted in 2- to 7-fold decreased activity. A decrease in activity was also observed upon changing the N,N-dipropyl for a pyrrolidinyl group (compound 14) (Table 3). Removal of both propyl groups (compound 15) resulted in substantial activity loss suggesting that the propyl chains may make transient contacts with the protein that are important for binding affinity.
Table 3. SAR of R3 Group.
| IC50 (μM) |
|||||
|---|---|---|---|---|---|
| compd | R3 | Plm I | Plm II | Plm IV | CatD |
| 3 | N(Pr)2 | 0.30 | 0.070 | 0.024 | 0.042 |
| 13 | NHPr | 0.75 | 0.50 | 0.038 | 0.11 |
| 14 | N(CH2)4 | 1.6 | 0.50 | 0.11 | 0.038 |
| 15 | NH2 | 7.6 | 4.4 | 0.17 | 0.50 |
Initial optimization of R4 was achieved by replacing the 2-(1,2-thiazinane-1,1-dioxide) moiety with the piperidinyl group to ease the synthesis of new analogues. This resulted in a 2-fold higher activity against Plm I and II, but only a small improvement against Plm IV (Table 4). A similar, but more pronounced trend was observed also by changing R4 to a cyclohexyl moiety (compound 16). In contrast, changing R4 to pyrrolidinyl (compound 17) and phenyl (compound 18) resulted in more than 4-fold higher activity against Plm IV, while the improvement against Plm I and II was up to 2-fold. It implies that nonplanar hydrophobic groups such as piperidinyl and cyclohexyl at this position are better accommodated by Plm I and II because of the favorable interaction with Val78, while planar and more compact groups are preferred by Plm IV, possibly, due to favorable flap-closing interactions. Importantly, inhibitory activity of CatD for compounds 17 and 18 remained at the level of starting hit 1, which improved selectivity for Plm IV versus CatD inhibition (ratios of 4.6 and 9, respectively) as compared to all other analogues. This indicates that interactions with flap residues are important for Plm IV selectivity.
Selected compounds exhibiting high Plm IV inhibitory activity were tested for inhibition of P. falciparum parasite growth in erythrocytes using a SYBR green assay (Table 4). Compound 1SR showed somewhat higher activity to inhibit the growth 3D7 parasite strain in erythrocytes compared to GSK LDH assay.6 This deviation can be due to the different assays used to monitor parasite growth. The results of parasite growth inhibition of compounds 1SS, 1SS, 3, and 16–18 again showed a clear correlation between parasite growth suppression and Plm IV inhibition, and not with Plm I or Plm II inhibition. Compounds 17 and 18, which were high nanomolar inhibitors of Plm I and Plm II, were still very active in the cell based assay. In addition, diastereomer 1SS, which was a micromolar inhibitor of PlmI and II but a high nanomolar inhibitor of Plm IV, exhibited considerable activity in the cell based assay (Table 4).
In conclusion, we have identified Plm IV as the putative target of 1SR (TCMDC-134674) in the malaria parasite P. falciparum. A cocrystal structure of 1SR bound to Plm II was solved and provided structural insight for the design of more potent and selective analogues. Structure-guided optimization then led to the identification of simplified analogues 17 and 18 as a low nanomoalr inhibitors of Plm IV activity and P. falciparum growth in erythrocytes. Selectivity of PlmIV versus CatD inhibition for these compounds was improved by increasing their PlmIV inhibitory activity. Structure guided optimization studies provide a base to further optimize drug-like properties of this series of compounds.
Acknowledgments
We would like to thank Prof. B. Dunn for providing Plm II-transformed BL21(DE3)pLysS cells. We also thank the personnel at MAX-Lab for their help during our stay at the synchrotron.
Glossary
ABBREVIATIONS
- GSK
GlaxoSmithKline
- Plm I
plasmepsin I
- Plm II
plasmepsin I
- Plm IV
plasmepsin IV
- Cat D
cathepsin D
Supporting Information Available
Description of the synthesis of compounds 1–18, description of crystallographic studies, description of PlmI, II, IV, and CatD enzymatic assays, and parasite growth inhibition assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Financial support from ERAF 2010/2DP/2.1.1.1.0./10/APIA/VIAA/074 is gratefully acknowledged.
The authors declare no competing financial interest.
Supplementary Material
References
- WHO. World Malaria Report 2012; World Health Organization: Geneva, Switzerland, 2012; p 59. [Google Scholar]
- Hyde J. E. Drug-resistant malaria: an insight. FEBS J. 2007, 274, 4688–4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S. R.; Mukherjee P.; Avery M. A. The fight against drug-resistant malaria: novel plasmodial targets and antimalarial drugs. Curr. Med. Chem. 2008, 15, 161–171. [DOI] [PubMed] [Google Scholar]
- Wells T. N.; Alonso P. L.; Gutteridge W. E. New medicines to improve control and contribute to the eradication of malaria. Nat. Rev. Drug Discovery 2009, 8, 879–891. [DOI] [PubMed] [Google Scholar]
- ChEMBL-NTD. https://www.ebi.ac.uk/chemblntd (accessed Aug 16, .2013).
- Gamo F. J.; Sanz L. M.; Vidal J.; de Cozar C.; Alvarez E.; Lavandera J. L.; Vanderwall D. E.; Green D. V.; Kumar V.; Hasan S.; Brown J. R.; Peishoff C. E.; Cardon L. R.; Garcia-Bustos J. F. Thousands of chemical starting points for antimalarial lead identification. Nature 2010, 465, 305–310. [DOI] [PubMed] [Google Scholar]
- Calderón F.; Barros D.; Bueno J. M.; Coterón J. M.; Fernández E.; Gamo F. J.; Lavandera J. L.; León M. L.; Macdonald S. J. F.; Mallo A.; Manzano P.; Porras E.; Fiandor J. M.; Castro J. An invitation to open innovation in malaria drug discovery: 47 quality starting points from the TCAMS. ACS Med. Chem. Lett. 2011, 2, 741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haque T. S.; Skillman A. G.; Lee C. E.; Habashita H.; Gluzman I. Y.; Ewing T. J.; Goldberg D. E.; Kuntz I. D.; Ellman J. A. Potent, low-molecular-weight non-peptide inhibitors of malarial aspartyl protease plasmepsin II. J. Med. Chem. 1999, 42, 1428–1440. [DOI] [PubMed] [Google Scholar]
- Noteberg D.; Hamelink E.; Hulten J.; Wahlgren M.; Vrang L.; Samuelsson B.; Hallberg A. Design and synthesis of plasmepsin I and plasmepsin II inhibitors with activity in Plasmodium falciparum-infected cultured human erythrocytes. J. Med. Chem. 2003, 46, 734–746. [DOI] [PubMed] [Google Scholar]
- Noteberg D.; Schaal W.; Hamelink E.; Vrang L.; Larhed M. High-speed optimization of inhibitors of the malarial proteases plasmepsin I and II. J. Comb. Chem. 2003, 5, 456–464. [DOI] [PubMed] [Google Scholar]
- Muthas D.; Noteberg D.; Sabnis Y. A.; Hamelink E.; Vrang L.; Samuelsson B.; Karlen A.; Hallberg A. Synthesis, biological evaluation, and modeling studies of inhibitors aimed at the malarial proteases plasmepsins I and II. Bioorg. Med. Chem. 2005, 13, 5371–5390. [DOI] [PubMed] [Google Scholar]
- Recently, hydroxyethyl-amine based compounds similar to 1SR were reported as inhibitors of P.falciparum growth in red blood cell:Ciana C.-L.; Siegrist R.; Aissaoui H.; Marx L.; Racine S.; Meyer S.; Binkert C.; de Kanter R.; Fischli C.; Wittlin S.; Boss C. Novel in vivo active anti-malarials based on a hydroxy-ethyl-amine scaffold. Bioorg. Med. Chem. Lett. 2013, 23, 658–662. [DOI] [PubMed] [Google Scholar]
- Bonilla J. A.; Moura P. A.; Bonilla T. D.; Yowell C. A.; Fidock D. A.; Dame J. B. Effects on growth, hemoglobin metabolism and paralogous gene expression resulting from disruption of genes encoding the digestive vacuole plasmepsins of Plasmodium falciparum. Int. J. Parasitol. 2007, 37, 317–327. [DOI] [PubMed] [Google Scholar]
- Bonilla J. A.; Bonilla T. D.; Yowell C. A.; Fujioka H.; Dame J. B. Critical roles for the digestive vacuole plasmepsins of Plasmodium falciparum in vacuolar function. Mol. Microbiol. 2007, 65, 64–75. [DOI] [PubMed] [Google Scholar]
- Meyers M. J.; Goldberg D. E. Recent advances in plasmepsin medicinal chemistry and implications for future antimalarial drug discovery efforts. Curr. Top. Med. Chem. 2012, 12, 445–455. [DOI] [PubMed] [Google Scholar]
- As pointed by one of the referees, compound 1 and its analogues may be impacting on the activity of Plm V, IX, and X, which could be more crucial then Plm IV inhibition. However, these enzymes are not yet described in the form suitable for inhibitory assays.
- Asojo O. A.; Gulnik S. V.; Afonina E.; Yu B.; Ellman J. A.; Haque T. S.; Silva A. M. Novel uncomplexed and complexed structures of plasmepsin II, an aspartic protease from Plasmodium falciparum. J. Mol. Biol. 2003, 327, 173–181. [DOI] [PubMed] [Google Scholar]
- Gulnik S. V.; Afonina E. I.; Gustchina E.; Yu B.; Silva A. M.; Kim Y.; Erickson J. W. Utility of (His)6 tag for purification and refolding of proplasmepsin-2 and mutants with altered activation properties. Protein Expr. Purif. 2002, 24, 412–9. [DOI] [PubMed] [Google Scholar]
- Asojo O. A.; Afonina E.; Gulnik S. V.; Yu B.; Erickson J. W.; Randad R.; Medjahed D.; Silva A. M. Structures of Ser205 mutant plasmepsin II from Plasmodium falciparum at 1.8 A in complex with the inhibitors rs367 and rs370. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2002, 58, 2001–2008. [DOI] [PubMed] [Google Scholar]
- Liu P.; Marzahn M. R.; Robbins A. H.; Gutierrez-de-Teran H.; Rodriguez D.; McClung S. H.; Stevens S. M. Jr.; Yowell C. A.; Dame J. B.; McKenna R.; Dunn B. M. Recombinant plasmepsin 1 from the human malaria parasite plasmodium falciparum: enzymatic characterization, active site inhibitor design, and structural analysis. Biochemistry 2009, 48, 4086–4099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ersmark K.; Samuelsson B.; Hallberg A. Plasmepsins as potential targets for new antimalarial therapy. Med. Res. Rev. 2006, 26, 626–666. [DOI] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, version 1.5.0.1.; Schrödinger, LLC.: New York, 2012. [Google Scholar]
- Carroll C. D.; Patel H.; Johnson T. O.; Guo T.; Orlowski M.; He Z. M.; Cavallaro C. L.; Guo J.; Oksman A.; Gluzman I. Y.; Connelly J.; Chelsky D.; Goldberg D. E.; Dolle R. E. Identification of potent inhibitors of Plasmodium falciparum plasmepsin II from an encoded statine combinatorial library. Bioorg. Med. Chem. Lett. 1998, 8, 2315–2320. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.