

Abstract
The identification and in vitro and in vivo characterization of a potent SHI-1:2 are described. Kinetic analysis indicated that biaryl inhibitors exhibit slow binding kinetics in isolated HDAC1 and HDAC2 preparations. Delayed histone hyperacetylation and gene expression changes were also observed in cell culture, and histone acetylation was observed in vivo beyond disappearance of drug from plasma. In vivo studies further demonstrated that continuous target inhibition was well tolerated and efficacious in tumor-bearing mice, leading to tumor growth inhibition with either once-daily or intermittent administration.
Keywords: Histone acetylation, histone deacetylase inhibitor, isoform selectivity, kinetics
Histone deacetylases (HDACs) catalyze the removal of acetyl groups from lysine residues of proteins, most notably nucleosomal histones as well as a variety of nonhistone proteins (e.g., p53, E2Fs, nuclear receptors, NFκB, HSP90, α-tubulin, KU70).1,2 Consequently, small molecule HDAC inhibitors have been shown to regulate gene transcription, cell cycle progression, and induce differentiation and/or apoptosis in cancer cells.3−6 HDACs 1 and 2 in particular are both overexpressed in human cancers and knockdown leads to increased apoptosis in certain cellular contexts.7−10 They share 92% sequence homology and 82% sequence identity. Moreover, emerging preclinical studies suggest roles for HDACs in inflammatory and neurodegenerative diseases.11
In our previous communication, we described the discovery of spirocyclic nicotinamide 1, a potent and efficacious selective SHI-1:2 (Figure 1).12 Unfortunately, the safety profile of 1 was unacceptable due to hERG binding.
Figure 1.

Early HDAC1/HDAC2-selective biaryl spirocyclic nicotinamide inhibitor.
Additional studies demonstrated that the incorporation of a cyclic carbamate functional group, to afford compound 2, was well tolerated and that this modification attenuated hERG activity (Table 1). Since 2 was inactive in an hERG binding assay, we varied the biaryl substituent for further HDAC inhibition potency optimization (Table 1). While the thiophen-3-yl analogue 3 was potent and devoid of hERG binding, we found that 4-methylthiophen-2-yl, phenyl, and 4-flourophenyl analogues 4–6 did bind to hERG, indicating that the spirocyclic carbamate moiety was not a general solution to the hERG problem in this series. As seen in the SAR profile of this series, polar biaryls containing pyrazole or imidazole moieties (7 and 8) had promising biochemical potency, but poor cell potency. Removal of the internal cavity binding motif, such as simple benzamide 9, diminished potency considerably. Remarkable in vitro and in vivo properties of key inhibitor 2 were identified, which we believe represent the biaryl benzamides as a whole, and warrants this separate communication.
Table 1. SAR Profile of the Spirocyclic Carbamate Nicotinamides.
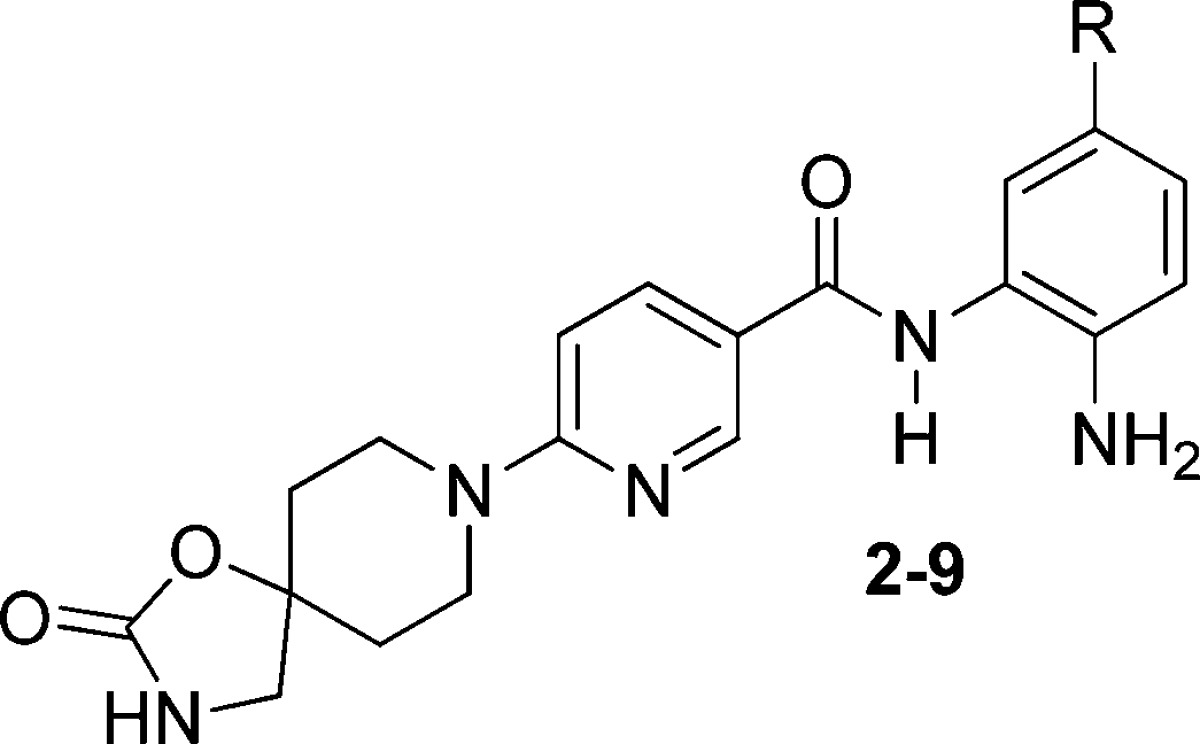
| compound: substituent | HDAC1 IC50 (nM) | HCT–116 Gl50 (nM) | heRG Ki (μM) |
|---|---|---|---|
| 2: thiophen-2-yl | 6 | 97 | >30 |
| 3: thiophen-3-yl | 11 | 270 | >30 |
| 4: 4-methylyhiophen-2-yl | 14 | 890 | 2.0 |
| 5: phenyl | 17 | 490 | 3.4 |
| 6: 4-fluorophenyl | 20 | 710 | 0.62 |
| 7: 1-methylpyrazol-4-yl | 18 | 2120 | >10 |
| 8: 1-methylpyrazol-4-yl | 51 | 4650 | >10 |
| 9: hydrogen | 580 | 5080 | >10 |
In vitro HDAC potency and selectivity were measured by monitoring the deacetylation of a synthetic acetyl-lysine-containing peptide by subtype-specific HDAC complexes purified from mammalian cells overexpressing recombinant human enzyme. The in vitro profile of 2 is summarized in Figure 2.13 Compound 2 was a potent inhibitor of both HDAC1 (IC50 6 nM) and HDAC2 (IC50 45 nM). Characteristic to all SHI-1:2 biaryl benzamides,14−16 it was also highly selective versus HDAC3 (IC50 7.0 μM), which itself may be important for cardiac function.17 Inhibition of HDAC3 also produces mitotic defects.18 It was a weak inhibitor of HDACs 8 and 11 (IC50 20–25 μM), and inactive in HDACs 4–7 assays.
Figure 2.
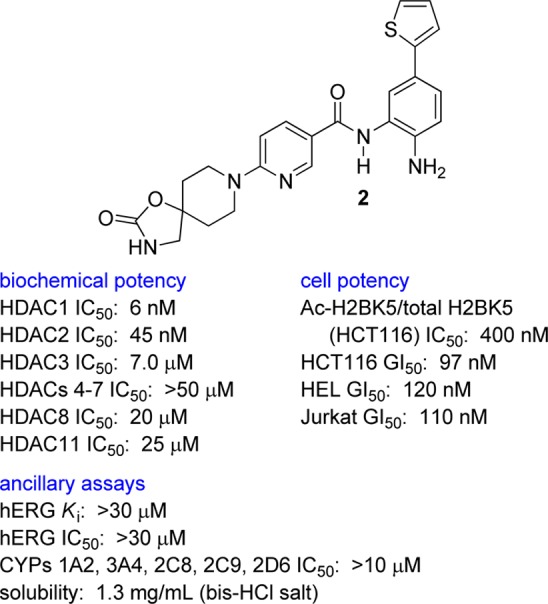
In vitro profile of key HDAC1/HDAC2 inhibitor 2. HCT116, human colon carcinoma; HEL, human erythroleukemia; Jurkat, T cell lymphoma.
In vitro kinetic analysis indicated that 2 was a time-dependent, substrate-competitive, and reversible inhibitor of HDAC1 and HDAC2 with relatively slow binding kinetics.19 While the IC50 values reported in Figure 2 were obtained after 10 min of preincubation time, compound 2 required ≥4 h to maximally inhibit HDAC1 and HDAC2 enzymatic activities (Figure 3). Additional kinetic experiments indicated that 2 dissociated from HDAC1 with an apparent half-life exceeding 100 min; significantly slower than that observed with unselective hydroxamic acid inhibitors that typically have a dissociation half-life of ∼1 min.20 A similar observation was reported by Gottesfeld using a simple nonbiaryl benzamide in a HDAC3 assay.21 Compound 2 maintained excellent selectivity versus HDAC3 even with longer preincubation times. We hypothesize that protein movement required to accommodate the biaryl into the internal cavity of HDAC1 and HDAC2 gives rise to the observed slow binding kinetics. A model of the boot-like active site and proposed binding into the internal cavity is described in greater detail in ref (15).
Figure 3.
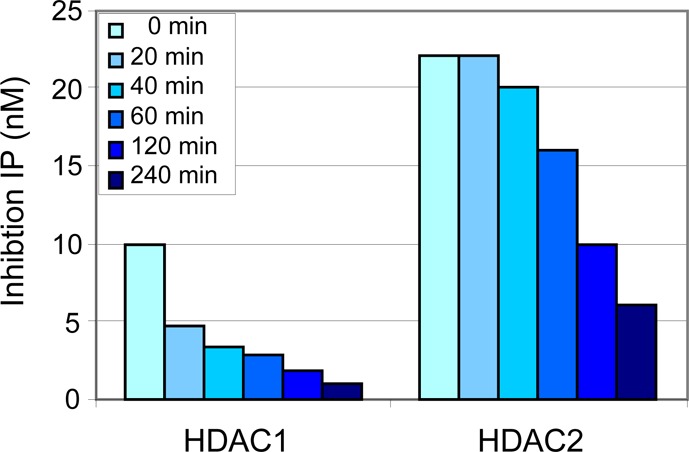
Time-dependent inhibition of HDACs 1 and 2 with 2. Preincubation time (0–240 min) of enzyme and 2 prior to initiating deacetylase reactions with peptide substrate.
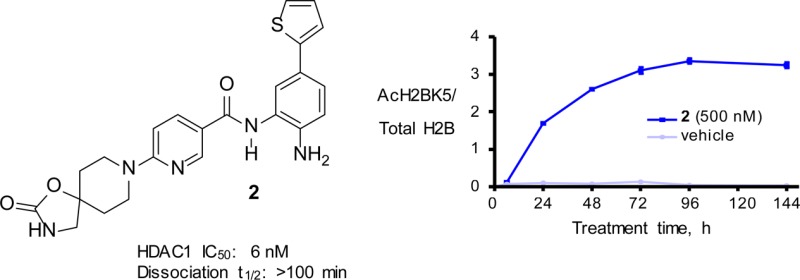
Histones are classic substrates for several HDAC subtypes, including HDAC1 and HDAC2, representing proximal pharmacodynamic biomarkers for target engagement. Treatment of HCT116 colon carcinoma cells with 500 nM of 2 followed by cell lysis indicated a time-dependent hyperacetylation of several histone residues, including lysine 5 of histone H2B (H2BK5). H2BK5 hyperacetylation becomes apparent within 4–8 h of treatment and plateaus at 96 h (Figure 4). At 24 h, the half maximal response was observed at a 400 nM concentration of 2. It also inhibited HCT116 cell growth with a GI50 of 97 nM with concomitant apoptosis (72 h assay).
Figure 4.
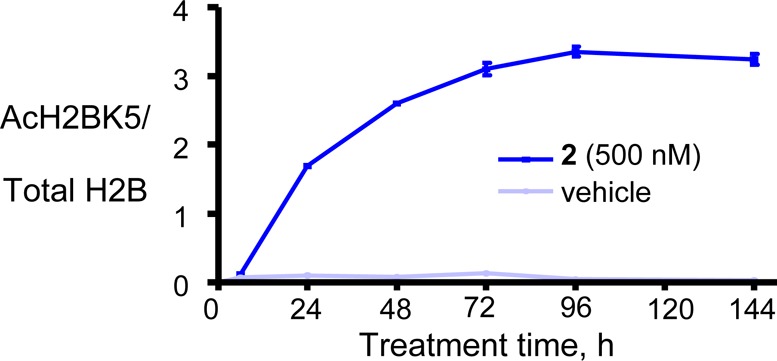
Treatment of HCT116 cells with 2 (500 nM) led to a delayed increase in histone H2BK5 acetylation.
In separate cell proliferation experiments, continuous exposure to 2 was shown to be more efficacious than intermittent exposure. For example, continuous treatment of HCT116 cells with 3 μM of 2 (24 h/day) for four days gave >90% reduction in cell viability, while treatment with 3 μM of 2 for 8 h/day (4 days) gave only 50% reduction in cell viability.
A radioligand-based HDAC binding assay indicated that the binding affinity of 2 for HDAC binding sites in intact HCT116 cells was 16 nM; close to the biochemical activity assay IC50 of 6 nM. At 97 nM, the GI50 value with HCT116 cells, approximately 80% occupancy of HDAC binding sites was achieved. This was consistent with RNAi-based observations that HDAC1 knockdown must approach 80–90% before significant cell death is observed in tumor cell lines.
Following treatment with 500 nM of 2, 97% of HCT-116 colon carcinoma cells were TUNEL positive, indicating that 2 had induced significant apoptosis after 72 h treatment. After 48 h of continuous treatment with 500 nM of 2, HCT116 cells undergo a significant decrease in the percentage of cells in S-phase from 29% in vehicle-treated cells to 6% in the group treated with 2, indicating that fewer cells are undergoing DNA replication. Concomitant with the decrease in the S phase cell population was a modest increase in both the G1 and G2 cell populations. After 96 h of treatment a significant proportion of cells (61%) accumulated in the subG1 fraction, consistent with the induction of apoptosis.
Since histone acetylation alters gene expression, we evaluated whether gene expression changes induced by 2 occur at biologically relevant concentrations. Two genes that were previously identified from siRNA gene profiling experiments as biomarkers of HDAC1 inhibition are DHRS2 and FAM46c. FAM46c encodes a gene of unknown function, while DHRS2 is an NADPH-dependent dicarbonyl reductase that is reported to be upregulated in response to cellular stress. Indeed, robust dose- and time-dependent increases in the expression of both DHRS2 and FAM46c were noted following treatment of HCT116 cells with 2 (Figure 5). These results indicated that at times and concentrations that inhibit cell proliferation and induce apoptosis, changes were also occurring at the level of gene expression.
Figure 5.
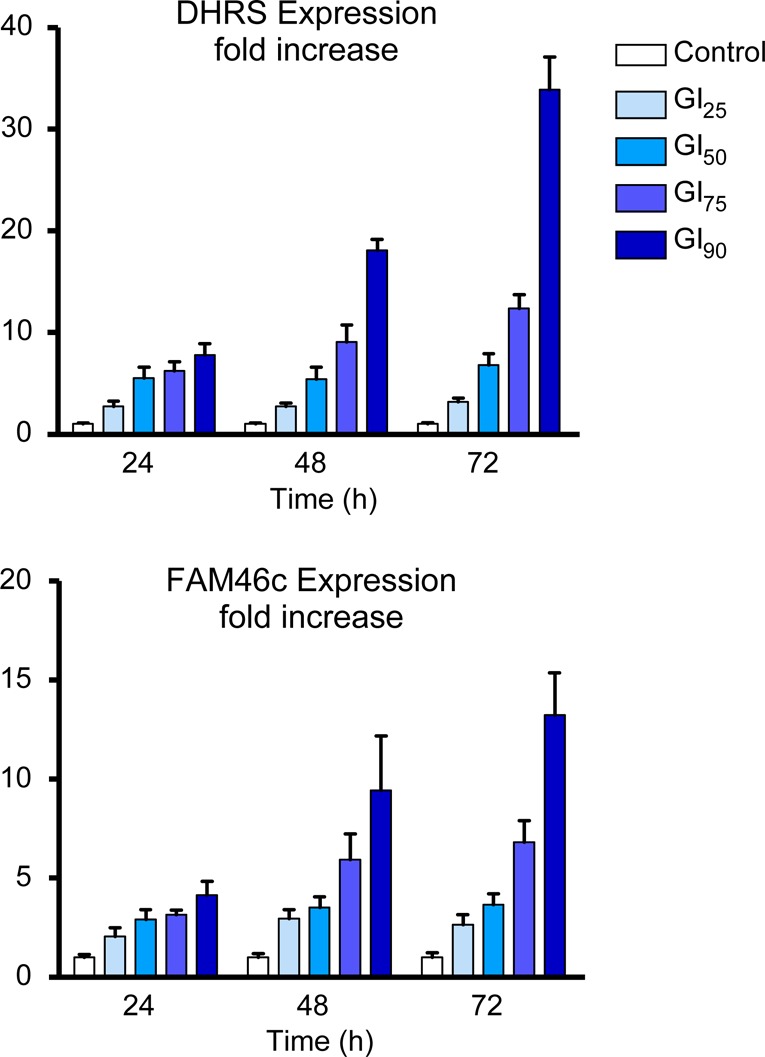
Effects of 2 on gene expression of DHRS2 and FAM46c in HCT116 colon carcinoma cells. TaqMan real-time PCR was used to evaluate time and dose-dependent regulation of DHRS2 and FAM46c in vitro. The concentrations of 2 used represent GI25 (50 nM), GI50 (97 nM), GI75 (188 nM), and GI90 (367 nM) values for inhibition of HCT116 cell proliferation in a 72 h proliferation assay.
Given that 2 potently inhibited the growth of other cell lines, such as HEL human erythroleukemia and Jurkat T cell lymphoma, we proceeded to screen it in a cancer cell proliferation assay panel (Figure 6). Potent antiproliferative activity was observed with numerous multiple myeloma, colon, nonsmall cell lung, breast, and small cell lung cancer cell lines.
Figure 6.
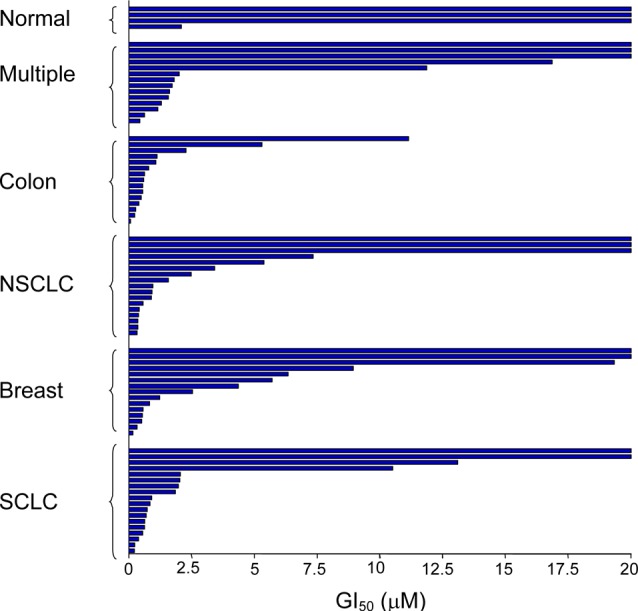
Cancer cell proliferation assay panel in the presence of 2. NSCLC, nonsmall cell lung cancer; SCLC, small cell lung cancer.
Compound 2 was inactive in the hERG binding assay (Ki >30 μM) as well as the potassium channel voltage-clamp assay in human embryonic kidney cells stably expressing Ikr (IC50 > 30 μM). Importantly, there was no effect on electrocardiographic intervals in anesthetized dogs up to 150 μM.
Given the overall favorable profile of 2, we profiled the PK properties of the compound in male CD-1 mouse, male Sprague–Dawley rat, beagle dog, and rhesus monkey (Table 2). In mouse, we observed moderate clearance and half-life (2.0 h) with excellent bioavailability giving a moderate oral exposure (AUCN 1.1 μM·h·kg/mg), which would facilitate characterization in our mouse models. In rat plasma, lower clearance was masked by higher plasma protein binding. The PK in dog and monkey was characterized by moderate plasma clearance with particularly good bioavailability in monkey.
Table 2. Pharmacokinetic Profile of 2.
| mouse | rat | dog | monkey | |
|---|---|---|---|---|
| iv dose(mg/kg) | 5 | 2 | 0.5 | 0.5 |
| Clp (mL/min/kg) | 27 | 3.6 | 22 | 27 |
| Vdss (L/kg) | 2.1 | 0.7 | 2.2 | 7.8 |
| t1/2 (h) | 2.0 | 4.2 | 1.4 | 7.1 |
| po dose(mg/kg) | 10 | 4 | 1 | 1 |
| AUCN (μM·h·kg/mg) | 1.1 | 3.0 | 0.3 | 1.8 |
| F (%) | 82 | 28 | 17 | 100 |
| plasma protein binding (%, 2 μM) | 98.5 | 99.8 | 97.6 | 98.4 |
Clearance of 2 in rats was primarily through metabolism, including hydrolysis of the anilide bond (major), oxidation of the thienyl group to form a monooxidized metabolite and glucuronidation of monooxidized metabolite; these were consistent with observations in rat hepatocytes. In dog hepatocytes, the major metabolic pathway for 2 was hydrolysis of the oxazolidinone group. In human hepatocytes, the major metabolite was a direct glucuronide conjugate. Additional metabolites in human hepatocytes included a carboxylic acid product formed by hydrolysis of anilide bond, a product derived from oxidation of the piperidine ring and a glucose conjugate. Compound 2 did not undergo extensive oxidative metabolism in liver microsomes in the presence of NADPH, suggesting that 2 was not significantly metabolized by CYP isoforms.
A mouse HCT116 xenograft tumor model was used to corroborate the compelling in vitro data. HCT116 cells (5 × 106 per mouse) were injected subcutaneously into the right flank of CD1 nu/nu mice. When the tumors reached ∼325 mm3, the mice were randomized into groups of 12 animals and treated with oral doses of 2 at 67 mg/kg tid, 100 mg/kg bid, and 200 mg/kg qd. At 24, 48, and 72 h postdose, tumors were harvested and histones isolated. Histone 2B lysine 5 (H2BK5) acetylation was measured by Elisa (Figure 7). After 24 h, ≥6-fold increase in histone acetylation was observed, which was maintained by ∼4-fold after 72 h; indicating that 2 exhibited an extended duration of target inhibition. While the three times per day regimen gave the greatest histone hyperacetylation at 24 h, by 72 h all three regimens gave comparable response. Hence, in subsequent efficacy experiments, we chose once daily administration. Interestingly, histone hyperacetylation was observed at plasma levels as low at 0.2 μM. This was in sharp contrast to the nonselective hydroxamic acid-containing inhibitor class that ellicit only a short duration histone hyperacetylation (<12 h) that mirrors compound exposure. This behavior was consistent with 2 exhibiting slow-off rate binding kinetics. A similar observation was reported using MGCD0103 in human whole blood.22−24
Figure 7.
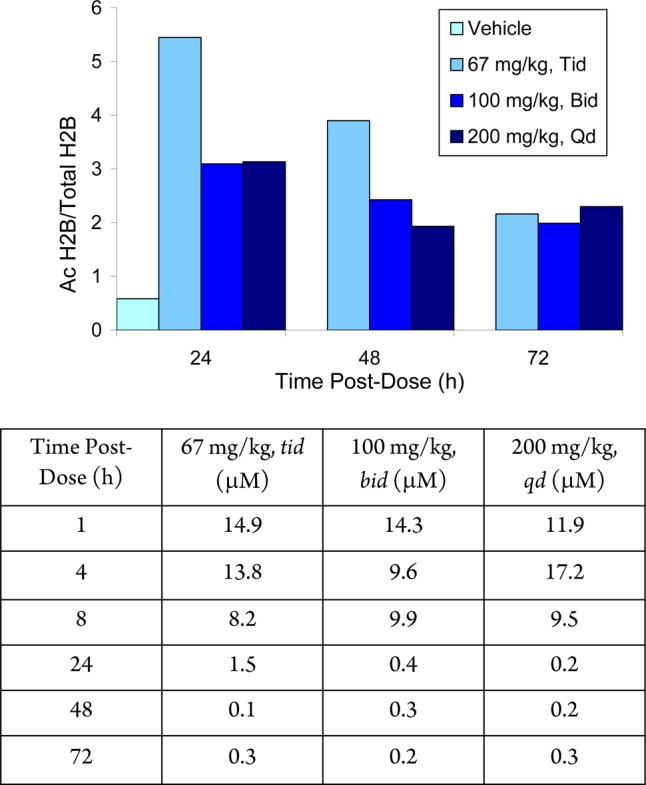
Acute oral administration of 2 induced significantly elevated and prolonged tumor histone H2BK5 acetylation in the HCT116 tumor model (above) and associated average plasma levels (below).
We next examined the effect of three-week oral administration of 2 on tumor size, as estimated using caliper measurements (Figure 8). Tumor growth inhibition was observed in a dose-dependent manner. At 150 mg/kg/day a 78% tumor growth inhibition was achieved without body weight loss. Higher doses, however, did not provide greater tumor growth inhibition. With 150 mg/kg/day, plasma levels of 2 reached 16 μM (Cmax) with a trough level of 3.2 μM (C24h), and a 0–24 h exposure of 208 μM·h. On the basis of the protein binding in mouse, the unbound Cmax and C24h concentrations were 240 and 48 nM, respectively. This correlates well with the in vitro hyperacetylation of H2BK5 in HCT116 cells, with an IC50 of 400 nM. Tumor histone hyperacetylation increased in a dose-dependent fashion up to a plateau of 4-fold, although maximal efficacy was achieved with a 2-fold increase in acetylation. The ∼78% tumor growth inhibition plateau may be a result of limited tumor penetration or reflect a limited role of protein acetylation in the proliferation of the HCT116 cell line.
Figure 8.

Top: 21-Day treatment of HCT116 xenograft mice with once daily (qd) or biweekly (biw) 2. Middle: Tumor histone H2BK5 acetylation after a single dose of 2. Bottom: Xenograft model summary and plasma levels of 2. Dosing formulation was 40% v/v PEG-400 and 25% w/v HPβCD, 20 μL/g.
In order to better understand the impact of prolonged histone hyperacetylation observed beyond 72 h (see Figure 7), biweekly oral administration of 2 was explored to assess the relationship between tumor growth inhibition and histone acetylation induction. In this study, a dose-dependent tumor growth inhibition was observed, with 525 mg/kg biweekly providing 59% tumor growth inhibition, although increasing the dose to 875 mg/kg biweekly did not lead to improved growth inhibition. These efficacious doses of 2 were well-tolerated, with minimal effects on body weight loss, platelet, or lymphocyte count in mice. In separate studies, inhibitor 2 was also efficacious and well tolerated in a 32-day transgenic NeuT breast model (86% tumor growth inhibition with oral 150 mg/kg qd). In addition, 2 exhibited tumor growth inhibition when dosed orally in several xenograft models of small and nonsmall cell lung cancer (e.g., A549, NCI-H69, NCI-H2122; data not shown).
In connection with earlier in vitro HCT116 proliferation experiments, which suggest continuous exposure to 2 was more efficacious than intermittent exposure, we sought to address whether a Cmax threshold was required or a continuous Ctrough was sufficient for tumor growth inhibition. Tumor bearing mice were implanted subcutaneously (sc) with an osmotic minipump that slowly released compound 2. A dose of 20 mg/mL maintained a continuous plasma level of 2.6 μM, was well-tolerated, and provided a 43% tumor growth inhibition over a four-week treatment period. When this result is compared to the 50 mg/kg qd dose group in Figure 8, which was not efficacious reaching a comparable Cmax of 3.5 μM, it is clear that continuous exposure of 2 is optimal in vivo, as predicted from in vitro studies.
A synthesis of 2 is outlined in Scheme 1.25 4-Bromo-2-nitroaniline was protected and coupled with thiophene-2-boronic acid using Pd(PPh3)4 and aqueous K2CO3. The nitro group was reduced and the resulting aniline coupled to give chloronicotinamide 11. Separately, the spirocycle was formed by reduction of nitrile 12 and the resulting amino alcohol cyclized with carbonyl diimidazole. Precursors 11 and 13 were coupled at elevated temperature and the resulting amide deprotected giving 2.
Scheme 1. Synthesis of Compound 2.

Reagents and conditions: (a) Boc2O, NEt3, DMAP, DCM, 62%; (b) thiophen-2-yl boronic acid, Pd(PPh3)4, K2CO3, dioxane, water, 90 °C, 92%; (c) 1 atm H2, Pd/C, EtOAc, 76%; (d) 6-chloronicotinoyl chloride, pyridine, 96%; (e) LiAlH4, THF; (f) CDI, DCM, 51% two steps; (g) 1 atm H2, Pd(OH)2/C, EtOAc, MeOH, 100%; (h) 11, 13 (2.5 equiv), NEt3, DMA, 90 °C, 70%; (i) TFA, DCM, 87%.
In summary, characterization of the HDAC1/HDAC2 inhibitor 2 revealed that this selective biaryl SHI-1:2 offers unique slow in vitro HDAC binding kinetics, as well as delayed and prolonged cell hyperacetylation.26 These results were consistent with the extended histone hyperacetylation for >72 h found in vivo, which continues even after the disappearance of drug from plasma. It is plausible that the prolonged PD is a result of the intrinsic slow binding kinetics of 2 to HDAC1/HDAC2 enzymes. These studies also demonstrated that continuous target inhibition is well tolerated in mice and may offer enhanced antitumor efficacy in the clinic versus the short-acting hydroxamic acid-based inhibitors.
Supporting Information Available
Experimental procedures for assay protocols, in vivo studies, and synthesis of compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Zhang Y.; Fang H.; Jiao J.; Xu W. The structure and function of histone deacetylases: the target for anti-cancer therapy. Curr. Med. Chem. 2008, 15, 2840–2849. [DOI] [PubMed] [Google Scholar]
- Yang X.-J.; Seto E. The Rpd3/Hda1 family of lysine deacetylases: from bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008, 9, 206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller T. A.; Witter D. J.; Belvedere S. Histone deacetylase inhibitors. J. Med. Chem. 2003, 46, 5097–5116. [DOI] [PubMed] [Google Scholar]
- Paris M.; Porcelloni M.; Binaschi M.; Fattori D. Histone deacetylase inhibitors: from bench to clinic. J. Med. Chem. 2008, 51, 1505–1529. [DOI] [PubMed] [Google Scholar]
- Bieliauskas A. V.; Pflum M. K. H. Isoform-selective histone deacetylase inhibitors. Chem. Soc. Rev. 2008, 37, 1402–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegel S.; Milstien S.; Grant S. Endogenous modulators and pharmacological inhibitors of histone deacetylases in cancer therapy. Oncogene 2011, 31, 537–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoepfler P. S.; Eisenman R. N. Sin meets NuRD and other tails of repression. Cell 1999, 99, 447–450. [DOI] [PubMed] [Google Scholar]
- Lagger G.; O’Carroll D.; Rembold M.; Khier H.; Tischler J.; Weitzer G. Essential function of histone deacetylase 1 in proliferation control and CDK inhibitor repression. EMBO J. 2002, 21, 2672–2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang B. H.; Laban M.; Leung C. H.; Lee L.; Lee C. K.; Salto-Tellez M. Inhibition of histone deacetylase 2 increases apoptosis and p21Cip1/WAF1 expression, independent of histone deacetylase 1. Cell Death Differ. 2005, 12, 395–404. [DOI] [PubMed] [Google Scholar]
- Weihert W.; Roske A.; Gekeler V.; Beckers T.; Stephan C.; Jung K.; Fritzsche F. R.; Niesporek S.; Denkert C.; Dietel M.; Kristiansen G. Histone deacetylases 1, 2 and 3 are highly expressed in prostate cancer and HDAC2 expression is associated with shorter PSA relapse time after radical prostatectomy. Br. J. Cancer 2008, 98, 604–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfils C.; Walkinshaw D. R.; Besterman J. M.; Yang X.-J.; Li Z. Pharmacological inhibition of histone deacetylases for the treatment of cancer, neurodegenerative disorders and inflammatory diseases. Expert Opin. Drug Discovery 2008, 3, 1041–1065. [DOI] [PubMed] [Google Scholar]
- Methot J. L.; Hamblett C. L.; Mampreian D. M.; Jung J.; Harsch A.; Szewczak A. A.; Dahlberg W. K.; Middleton R. E.; Hughes B.; Fleming J. C.; Wang H.; Kral A. M.; Ozerova N.; Cruz J. C.; Haines B.; Chenard M.; Kenific C. M.; Secrist J. P.; Miller T. A. SAR profiles of spirocyclic nicotinamide derived selective HDAC1/HDAC2 inhibitors (SHI-1:2). Bioorg. Med. Chem. Lett. 2008, 18, 6104–6109. [DOI] [PubMed] [Google Scholar]
- For enzyme and cell assay protocols, seeHamblett C. L.; Methot J. L.; Mampreian D. M.; Sloman D. L.; Stanton M. G.; Kral A. M.; Fleming J. C.; Cruz J. C.; Chenard M.; Ozerova N.; Hitz A. M.; Wang H.; Deshmukh S. V.; Nazef N.; Harsch A.; Hughes B.; Dahlberg W. K.; Szewczak A. A.; Middleton R. E.; Mosley R. T.; Secrist J. P.; Miller T. A. The discovery of 6-amino nicotinamides as potent and selective histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 5300–5309. [DOI] [PubMed] [Google Scholar]
- Witter D. J.; Harrington P.; Wilson K. J.; Fleming J. C.; Kral A. M.; Secrist J. P.; Miller T. A. Optimization of biaryl selective HDAC1&2 inhibitors (SHI-1:2). Bioorg. Med. Chem. Lett. 2008, 18, 726–731. [DOI] [PubMed] [Google Scholar]
- Methot J. L.; Chakravarty P. K.; Chenard M.; Close J.; Cruz J. C.; Dahlberg W. K.; Fleming J.; Hamblett C. L.; Hamill J. E.; Harrington P.; Harsch A.; Heidebrecht R.; Hughes B.; Jung J.; Kenific C. M.; Kral A. M.; Meinke P. T.; Middleton R. E.; Ozerova N.; Sloman D. L.; Stanton M. G.; Szewczak A. A.; Tyagarajan S.; Witter D. J.; Secrist J. P.; Miller T. A. Exploration of the internal cavity of histone deacetylase (HDAC) with selective HDAC1/HDAC2 inhibitors (SHI-1:2). Bioorg. Med. Chem. Lett. 2008, 18, 973–978. [DOI] [PubMed] [Google Scholar]
- Moradei O. M.; Mallais T. C.; Frechette S.; Paquin I.; Tessier P. E.; Leit S. M.; Fournel M.; Bonfils C.; Trachy-Bourget M.-C.; Liu J.; Yan T. P.; Lu A.-H.; Rahil J.; Wang J.; Lefebvre S.; Li Z.; Vaisburg A. F.; Besterman J. M. Novel aminophenyl benzamide-type histone deacetylase inhibitors with enhanced potency and selectivity. J. Med. Chem. 2007, 50, 5543–5546. [DOI] [PubMed] [Google Scholar]
- Montgomery R. L.; Potthoff M. J.; Haberland M.; Qi X.; Matsuzaki S.; Humphries K. M.; Richardson J. A.; Bassel-Duby R.; Olson E. N. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J. Clin. Invest. 2008, 118, 3588–3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrener R.; Chia K.; Warren W. D.; Brooks K.; Gabrielli B. Inhibition of histone deacetylase 3 produces mitotic defects independent of alterations in histone H3 lysine 9 acetylation and methylation. Mol. Pharmacol. 2010, 78, 384–393. [DOI] [PubMed] [Google Scholar]
- Chenard M.; Close J.; Cruz J.; Deshmukh S.; Fleming J.; Grimm J.; Haines B.; Hamblett C.; Harrington P.; Harsch A.; Heidebrecht R.; Hitz A.; Hubbs J.; Hughes B.; Jung J.; Kattar S.; Kral A. M.; Kenific C.; Mampreian D.; Methot J.; Middleton R.; Otte K.; Ozerova N.; Siliphaivanh P.; Sloman S.; Stanton M.; Surdi L.; Szewczak A.; Wang H.; Wilson K.; Witter D.; Secrist P.; Miller T.. Prolonged Histone Hyperacetylation with a Novel Class of HDAC1/2 Selective Inhibitors, Abstract 5433; American Association for Cancer Research, Washington, D.C., April 17–21, 2010; [Google Scholar]
- Details of the HDAC1 kinetic studies providing kon and koff for compound 2 are forthcoming in a subsequent publication.
- Chou C. J.; Herman D.; Gottesfeld J. M. Pimelic diphenylamide 106 is a slow, tight-binding inhibitor of class I histone deacetylases. J. Biol. Chem. 2008, 283, 35402–35409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonfils C.; Kalita A.; Dubay M.; Siu L. L.; Carducci M. A.; Reid G.; Martell R. E.; Besterman J. M.; Li Z. Evaluation of the pharmacodynamic effects of MGCD0103 from preclinical models to human using a novel HDAC enzyme assay. Clin. Cancer Res. 2008, 14, 3441–3449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournel M.; Bonfils C.; Hou Y.; Yan P. T.; Trachy-Bourget M.-C.; Kalita A.; Liu J.; Lu A.-H.; Zhou N. Z.; Robert M.-F.; Gillespie J.; Wang J. J.; Ste-Croix H.; Rahil J.; Lefebvre S.; Moradei O.; Delorme D.; MacLeod A. R.; Besterman J. M.; Li Z. MGCD0103, a novel isotype-selective histone deacetylase inhibitor, has broad spectrum antitumor activity in vitro and in vivo. Mol. Cancer Ther. 2008, 7, 759–768. [DOI] [PubMed] [Google Scholar]
- Zhou N.; Moradei O.; Raeppel S.; Leit S.; Frechette S.; Gaudette F.; Paquin I.; Bernstein N.; Bouchain G.; Vaisburg A.; Jin Z.; Gillespie J.; Wang J.; Fournel M.; Yan P. T.; Trachy-Bourget M.-C.; Kalita A.; Lu A.; Rahil J.; MacLeod A. R.; Li Z.; Besterman J. M.; Delorme D. Discovery of N-(2-aminophenyl)-4-[(4-pyridin-3-ylpyrimidin-2-ylamino)methyl]benzamide (MGCD0103), an orally active histone deacetylase inhibitor. J. Med. Chem. 2008, 51, 4072–4075. [DOI] [PubMed] [Google Scholar]
- Berk S. C.; Close J.; Hamblett C.; Heidebrecht R. W.; Kattar S. D.; Kliman L. T.; Mampreian D. M.; Methot J. L.; Miller T.; Sloman D. L.; Stanton M. G.; Tempest P.; Zabierek A. A.. Spirocyclic compounds as HDAC inhibitors. WO 2007/061978.
- A comparative in vitro and in vivo analysis of vorinostat and compound 2 (also known as MRLB-223) has recently been reported by Johnstone et al. SeeNewbold A.; Matthews G. M.; Bots M.; Cluse L.; Clarke C. J. P.; Banks K.-M.; Cullinane C.; Bolden J. E.; Christiansen A. J.; Dickins R. A.; Miccolo C.; Chiocca S.; Kral A. M.; Ozerova N. D.; Miller T. A.; Methot J. L.; Richon V.; Secrist J. P.; Minucci S.; Johnstone R. W. Molecular and biological analysis of histone deacetylase inhibitors with diverse specificities. Mol. Cancer Ther. 2013, 12, 2709–2721. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.