

Abstract
Introduction of an aliphatic side chain to a key position of a novel piperidine-based HDM2 inhibitor scaffold resulted in significant potency gains, enabling further series progression.
Keywords: HDM2, MDM2, p53, substituted piperidine, protein−protein interaction, small molecule inhibitor
The tumor suppressor protein p53 plays a central role in maintaining the integrity of the genome in a cell. It regulates the expression of a diverse array of genes responsible for DNA repair, cell cycle and growth arrest, and apoptosis.1−5 The oncogene HDM2 is a ubiquitin protein ligase that negatively regulates tumor suppressor p53 and lessens its transcriptional activity as well as promoting p53 protein degradation.6−9 Antagonists of the tumor suppressor p53-HDM2 interaction represent an attractive and novel mechanism in oncological therapeutics. Disrupting this HDM2-p53 protein–protein interaction with a small molecule is hypothesized to increase intracellular levels of p53 leading to decreased proliferation and increased apoptosis of tumor cells, which offers great therapeutic potential for the treatment of cancers.10−13 Crystallographic studies of MDM2- (the murine homologue of human HDM2) and p53-derived peptides bound together have revealed a deep hydrophobic binding pocket involving residues Phe19, Trp23, and Leu26 of p53.14 Since the discovery of Roche’s Nutlin imidazoline series, which antagonizes this HDM2–p53 interaction,15 several potent low molecular weight inhibitors have also been described.16,17 Recently, we have reported the discovery of a novel series of substituted piperidines as small molecule antagonists of HDM2, which selectively inhibit proliferation of wild-type p53 cancer cell lines (Figure 1).18,19 It has historically been challenging to overcome the large surface contacts of protein–protein interactions with a small molecule inhibitor. Herein we report a breakthrough made in our efforts to further optimize this substituted piperidine series through chemical modifications at the piperidine 2-position.
Figure 1.

Novel piperidine-based HDM2 inhibitors.
Chemistry
The synthesis of this series of 2-substituted piperidine-based p53-HDM2 inhibitors was carried out in a fashion analogous to that described previously for 2-unsubstituted compounds 1 and 2 (Figure 1).18,19 Beginning with N-benzyl-3-piperidone 3, which was readily converted to the corresponding methyl carbamate 4 in a single step under conditions described by Brubaker,20 the resulting ketone was subjected to an enamine alkylation with allyl bromide to provide 5 (Scheme 1). The requisite quaternary center was then introduced through a Bargellini reaction18,21,22 with 4-trifluoromethylphenol to give racemic carboxylic acid 6, which was converted to corresponding methyl ester for ease in handling. Chiral HPLC conditions were developed to resolve 7 to a single enantiomer (refer to Supporting Information for details). The carbamate protecting group on 7 was removed through the action of trimethylsilyliodide, and the resulting free amine was subjected to standard amidation conditions with 4-trifluoromethylnicotinic acid to give intermediate 8a. Ester hydrolysis of 8a provided carboxylic acid 8b, which underwent amidation with the corresponding piperazine building block to give final compound 9, the absolute stereochemistry of which was confirmed by analysis of its X-ray cocrystal structure with HDM2.
Scheme 1. Synthesis of the 2-Substituted Piperidine Core and Key Analogues.

Reagents and conditions: (a) methyl chloroformate, Et3N, CHCl3 0 °C to r.t., 57%; (b) 1. pyrrolidine, benzene reflux; 2. allyl bromide, MeCN, 70 °C, 67%; (c) 4-trifluoromethylphenol, NaOH, CHCl3, 0–40 °C; (d) TMS-CHN2, MeCN, MeOH, 0 °C to r.t., ∼40% overall yield from 5; (e) chiral HPLC (see Supporting Information for the condition), 27%; (f) iodotrimethylsilane, CH2Cl2, r.t., >95%; (g) 4-trifluoronicotinic acid, HATU, Et2NiPr, CH2Cl2, rt to 45 °C, >85%; (h) KOH, EtOH, H2O, 65 °C, 88%; (i) HATU, 1-[2-(2-methoxy–ethoxy)-phenyl]-piperazine, Et2NiPr, CH2Cl2, r.t. to 40 °C, 53%.
The allyl substituent in the 2-position of the piperidine ring in compound 9 served as a synthetic handle for further elaboration at this position (Scheme 2). Reduction of the olefin in 9 by hydrogenation led to compound 10. Polarity was introduced at this position in the form of two hydroxyl groups through dihydroxylation of the double bond in 9 to give diols 11, which in turn were converted to alcohol 12 through in situ sodium periodate-promoted cleavage followed by reduction of the resulting aldehyde.
Scheme 2. Modification of the Olefin.

Reagents and conditions: (a) H2, Pd/C, EtOAc, 87%; (b) OsO4, NMO, THF, H2O, r.t., ∼18% for combined two isomers; (c) 1. OsO4, NaIO4, 2,6-lutidine, dioxane, H2O, r.t.; 2. NaBH4, MeOH, r.t., 25%.
Results and Discussion
While it was anticipated that improvements to the in vitro potency and concomitant cell-based activity of C2-unsubstituted compounds such as 13(18) could be made through modifications of the largely hydrophobic binding interactions with the HDM2 protein, optimization of the overall DMPK profile was expected to require the introduction of polar substituents to the inhibitor in such a way as to not interfere with binding to HDM2. The addition of an allyl group as a starting point for synthetic diversification as described in the previous paragraph was targeted to do just this; however, introduction of this simple aliphatic side chain itself provided an unexpected yet significant impact on binding efficiency. Compared to unsubstituted analogue 13, the addition of allyl (9) and n-propyl (10) groups at C2 of the piperidine core provided a 4- and 7-fold improvement, respectively, in the biochemical fluorescence polarization (FP) peptide displacement assay23 (Table 1). As a result of this intrinsic potency gain, activity of these compounds in the cellular viability assay with osteosarcoma SJSA-1 cells translated to similar levels of improvement with both 9 and 10 displaying IC50 values around 1 μM. As anticipated, the incorporation of more polar functional groups such as diols 11a/b and the primary alcohol 12 did not negatively impact the binding affinity with biochemical IC50 values from 16 to 24 nM.
Table 1. Biochemical and Cellular Activities of Compounds 9–14a.
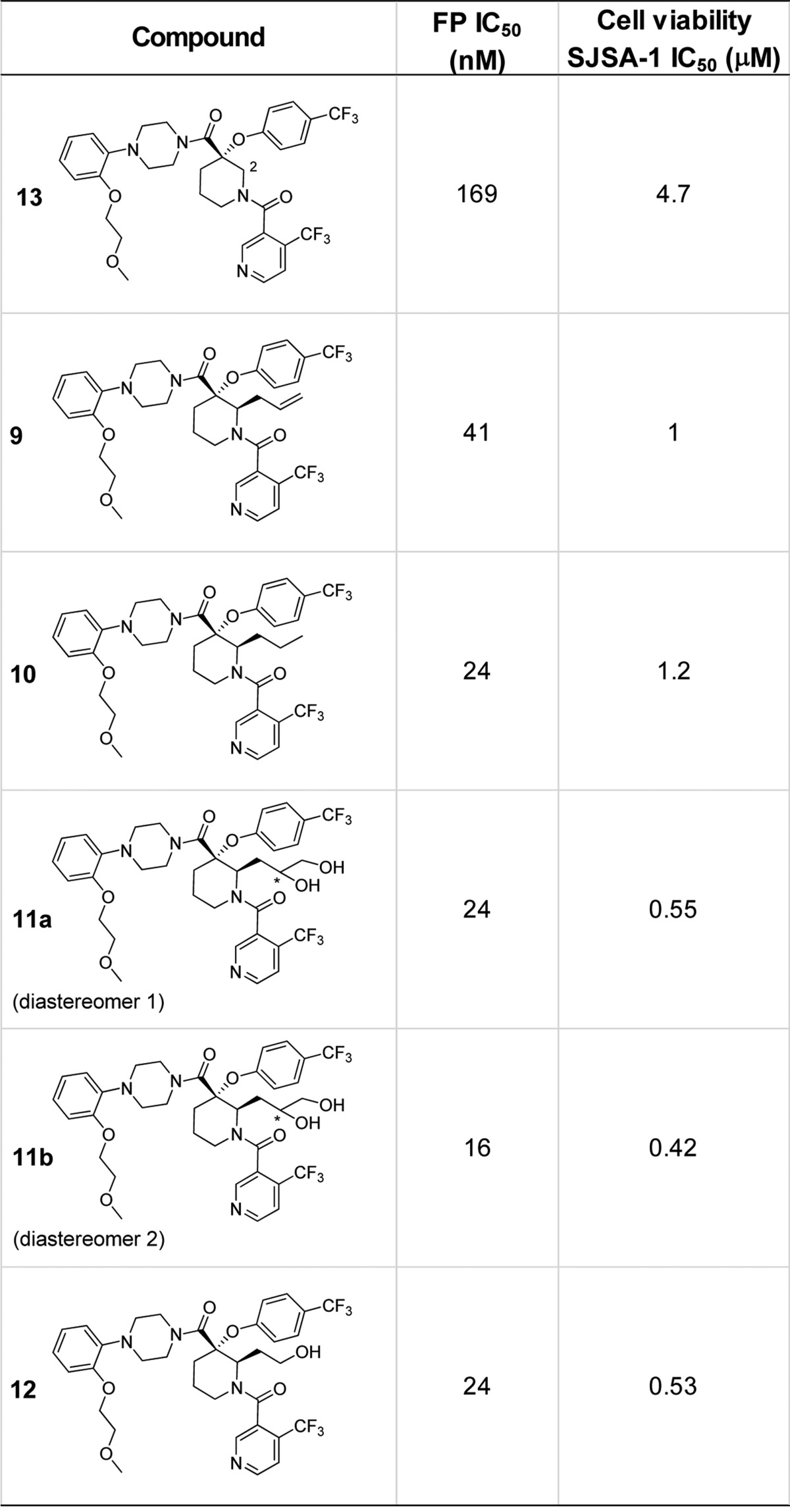
All compounds were tested as HCl salts. Fluorescent polarization was measured by reading the plate using the Analyst AD (Molecular Device). IC50 was determined as described in Zhang et al.23
In the absence of binding activities with the protein, the piperidine core as a six-membered cyclic ring should have several conformations coexisting in solution; however, only one of them was found to fit into HDM2’s p53-binding pocket. The energy required for the 2-unsubstituted piperidine 13 to adopt the chair conformation necessary for binding to HDM2 (17.5 kcal/mol) was equivalent to that of the ring-flipped conformation, within the resolution (5 kcal/mol) of the Sybyl software used to perform the calculations (Figure 2). However, the binding conformation of the 2-allyl analogue 9 was calculated to be more than 10 kcal/mol more favorable than the nonbinding conformer (17.6 vs 27.7 kcal/mol), likely due to the unfavorable steric interactions of the two amides with the adjacent axial allyl substituent in the latter. As a result, by introducing the side chain at the piperidine-2 position, exchange between ring conformations has been significantly slowed down with the preferred binding conformation existing predominantly in the absence of HDM2. Locking the orientation of the substituents on the piperidine core in this way improved the binding affinity of 2-substituted compounds such as 9 considerably.
Figure 2.

Impact of side chain substitution on the piperidine ring conformations.
As potent HDM2 inhibitors, this series of compounds exhibited a ∼3× improvement in the cell Titer-Glo assay with SJSA-1 osteosarcoma cells compared to well-studied Nutlin-3a (Figure 3).15,16 As with Nutlin-3a, these compounds were only active against wild-type (wt)-p53 cell lines like SJSA-1 with no activity observed in the mutant-p53 cell lines, such as the endometrial cancer cell line SKUT-1, indicating on-target mechanism-based antiproliferative activity. Thus, a ∼30× toxicity window was achieved between the wt-p53 and mutant-p53 cancer cell lines.
Figure 3.
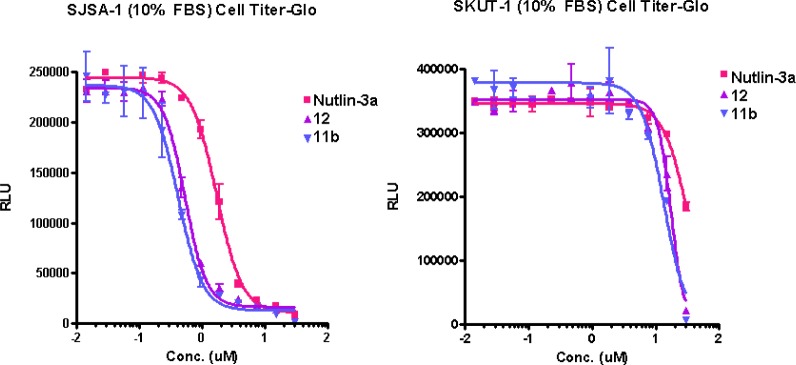
Novel HDM2 inhibitors selectively inhibit wt-p53 cancer cell lines.
As anticipated, DMPK analysis of compound 9 indicated a similar cytochrome P450 (CYP) profile to that of the 2-unsubstituted compound 13 as both lipophilic molecules did not inhibit CYP 2D6 and 2C9 isoforms but exhibited both direct (coincubation IC50 = 2.1–4.9 μM) and metabolism/mechanism-based CYP 3A4 inhibition (coincubation IC50/preincubation IC50 ≈ 7–16-fold) (Table 2). The introduction of one (12) or two (11a) polar hydroxyl groups decreased the direct inhibition of CYP 3A4 with IC50 values of 13.3 and >30 μM, respectively. However, both compounds displayed CYP 3A4 metabolism/mechanism-based inhibition (coincubation IC50/preincubation IC50 ≈ 4–15-fold), which led to a more thorough investigation that will be the subject of a future publication.
Table 2. Cytochrome P450 Inhibition of Select HDM2 Inhibitors.
| cytochrome
P450 inhibition |
||||||
|---|---|---|---|---|---|---|
| coincubation (IC50, μM) |
preincubation
(IC50, μM) |
|||||
| compound | 3A4 | 2D6 | 2C9 | 3A4 | 2D6 | 2C9 |
| 13 | 4.9 | >30 | >30 | 0.3 | >30 | >30 |
| 9 | 2.1 | >30 | >30 | <0.3 | >30 | >30 |
| 11a | >30 | >30 | >30 | 7.5 | >30 | >30 |
| 12 | 13.3 | >30 | >30 | 0.9 | >30 | >30 |
In summary, while continuing to optimize the substituted piperidine as a novel series of HDM2 inhibitors, significant potency enhancement was achieved by introducing an aliphatic side chain at the piperidine 2-position. As a result, corresponding cell-based activities were improved while maintaining a favorable toxicity window between wt-p53 and mutant-p53 cell lines for this new series inhibitor. Introduction of this side chain afforded a great opportunity for further optimization of activity and DMPK profiles toward the discovery of an improved small molecule HDM2 antagonist.
Acknowledgments
Authors would like to acknowledge Diane Rindgen, Hui Wan, Anthony Soares, and Shiying Chen for processing and interpreting CYP data.
Supporting Information Available
Experimental procedures and LC–MS characterization data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Levine A. J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [DOI] [PubMed] [Google Scholar]
- Vousden K. H.; Lu X. Live or let die: the cell’s response to p53. Nat. Rev. Cancer 2002, 2, 594–604. [DOI] [PubMed] [Google Scholar]
- Oren M. Decision making by p53: life, death and cancer. Cell Death Differ. 2003, 10, 431–442. [DOI] [PubMed] [Google Scholar]
- Fridman J. S.; Lowe S. W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [DOI] [PubMed] [Google Scholar]
- Vazquez A.; Bond E. E.; Levine A. J.; Bond G. L. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat. Rev. Drug Discovery 2008, 7, 979–987. [DOI] [PubMed] [Google Scholar]
- Chen J.; Wu X.; Lin J.; Levine A. J. mdm-2 inhibits the G1 arrest and apoptosis functions of the p53 tumor suppressor protein. Mol. Cell. Biol. 1996, 16, 2445–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momand J.; Zambetti G. P.; Olson D. C.; George D.; Levine A. J. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992, 69, 1237–1245. [DOI] [PubMed] [Google Scholar]
- Momand J.; Wu H. H.; Dasgupta G. MDM2: master regulator of the p53 tumor suppressor protein. Gene 2000, 242, 15–29. [DOI] [PubMed] [Google Scholar]
- Oliner J. D.; Pietenpol J. A.; Thiagalingam S.; Gyuris J.; Kinzler K. W.; Vogelstein B. Oncoprotein MDM2 conceals the activation domain of tumor suppressor p53. Nature 1993, 362, 857–860. [DOI] [PubMed] [Google Scholar]
- Momand J.; Jung D.; Wilczynski S.; Niland J. The MDM2 gene amplification database. Nucleic Acids Res. 1998, 26, 3453–3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polsky D.; Bastian B. C.; Hazan C.; Melzer K.; Pack J.; Houghton A.; Busam K.; Cordon-Cardo C.; Osam I. HDM2 protein overexpression, but not gene amplification, is related to tumorigenesis of cutaneous melanoma. Cancer Res. 2001, 61, 7642–7646. [PubMed] [Google Scholar]
- Chene P. Inhibiting the p53-MDM2 interaction: an important target for cancer therapy. Nat. Rev. Cancer 2003, 3, 102–109. [DOI] [PubMed] [Google Scholar]
- Bottger A.; Bottger V.; Sparks A.; Liu W.-L.; Howard S. F.; Lane D. P. Design of a synthetic MDM2-binding mini protein that activates the p53 response in vivo. Curr. Biol. 1997, 7, 860–869. [DOI] [PubMed] [Google Scholar]
- Kussie P. H.; Gorina S.; Marechal V.; Elenbaas B.; Moreau J.; Levine A. J.; Pavletich N. P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [DOI] [PubMed] [Google Scholar]
- Vassilev L. T.; Vu B. T.; Graves B.; Carvajal D.; Podlaski F.; Filipovic Z.; Kong N.; Kammlott U.; Lukacs C.; Klein C.; Fotouhi N.; Liu E. A. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004, 303, 844–848. [DOI] [PubMed] [Google Scholar]
- Hu C.-Q.; Hu Y.-Z. Small molecule inhibitors of the p53-MDM2. Curr. Med. Chem. 2008, 15, 1720–1730. [DOI] [PubMed] [Google Scholar]
- Patel S.; Player M. R. Small-molecule inhibitors of the p53-HDM2 interaction for the treatment of cancer. Expert Opin. Invest. Drugs 2008, 17, 1865–1882. [DOI] [PubMed] [Google Scholar]
- Ma Y.; Lahue B.; Shipps G.; Brookes J.; Wang Y. Substituted piperidines as HDM2 inhibitors. Bioorg. Med. Chem. Lett. 2014, 24, 1026–1030. [DOI] [PubMed] [Google Scholar]
- Ma Y.; Lahue B.; Shipps G.; Wang Y.; Bogen S.; Voss M.; Nair L.; Tian Y.; Doll R.; Guo Z.; Strickland C.; Zhang R.; McCoy M.; Pan W.; Siegel E.; Gibeau C. Preparation of substituted piperidines that increase p53 activity and the uses thereof. U.S. Pat. Appl. Publ. US2008004287, 2008.
- Brubaker A.; Colley M. Synthesis and pharmacological evaluation of some 6-substituted 7-methyl-1,4-dioxa-7-azaspiro[4.5]decanes as potential dopamine agonists. J. Med. Chem. 1986, 29, 1528. [DOI] [PubMed] [Google Scholar]
- Cvetovich R. J.; Chung J. Y. L.; Kress M. H.; Amato J. S.; Matty L.; Weingarten M. D.; Tsay F.-R.; Li Z.; Zhou G. An efficient synthesis of a dual PPAR α/γ agonist and the formation of a sterically congested α-aryloxyisobutyric acid via a Bargellini reaction. J. Org. Chem. 2005, 70, 8560–8563. [DOI] [PubMed] [Google Scholar]
- Rychnovsky S. D.; Beauchamp T.; Vaidyanathan R.; Kwan T. Synthesis of chiral nitroxides and an unusual racemization reaction. J. Org. Chem. 1998, 63, 6363–6374. [DOI] [PubMed] [Google Scholar]
- Zhang R.; Mayhood T.; Lipari P.; Wang Y.; Durkin J.; Syto R.; Gesell J.; McNemar C.; Windsor W. Fluorescence polarization assay and inhibitor design for MDM2/p53 interaction. Anal. Biochem. 2004, 331, 138–146. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.