

Abstract
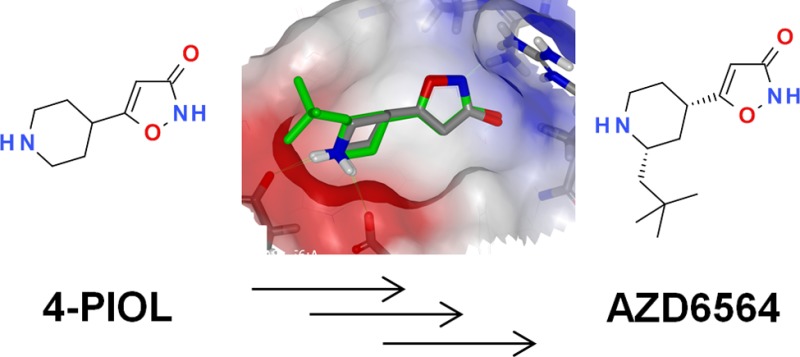
A class of novel oral fibrinolysis inhibitors has been discovered, which are lysine mimetics containing an isoxazolone as a carboxylic acid isostere. As evidenced by X-ray crystallography the inhibitors bind to the lysine binding site in plasmin thus preventing plasmin from binding to fibrin, hence blocking the protein–protein interaction. Optimization of the series, focusing on potency in human buffer and plasma clotlysis assays, permeability, and GABAa selectivity, led to the discovery of AZD6564 (19) displaying an in vitro human plasma clot lysis IC50 of 0.44 μM, no detectable activity against GABAa, and with DMPK properties leading to a predicted dose of 340 mg twice a day oral dosing in humans.
Keywords: Fibrinolysis, plasminogen, inhibitor, protein−protein interaction, isoxazolone, tranexamic acid
The physiological role of plasma coagulation is to prevent excessive blood loss from injured vessels, and the role of fibrinolysis is to dissolve fibrin clots after wound repair. Fibrinolysis is a cascade reaction with zymogene-to-enzyme conversion, feedback loops, and inhibitors.1 The key protein in fibrinolysis is plasminogen, which is transformed into plasmin by the plasminogen activators (tPA and uPA). The localized activation of plasminogen and action of plasmin on the fibrin surface is facilitated by binding of C-terminal lysine residues on fibrin to lysine binding sites (LBS) in the kringle domains in plasminogen or plasmin, respectively.2 Inhibition of fibrinolysis through blockade of LBS on plasminogen is an effective and safe treatment of bleeding conditions.3 Two LBS inhibitors that are used clinically, ε amino caproic acid (EACA) and tranexamic acid (TXA), elicit their effects by preventing the protein–protein interaction between plasminogen and fibrin (Figure 1).4
Figure 1.

ε-Amino caproic acid (EACA), tranexamic acid (TXA), and 4-PIOL.5
The main use of EACA and TXA are in the treatment of heavy menstrual bleeding, although reduction of blood loss during surgery and trauma are other application areas.4 In the recent CRASH-2 trial, it was demonstrated that TXA can be administered safely to a wide spectrum of patients with traumatic bleeding.6 EACA and TXA have also been shown to have an effect as add-on treatment of mild hemophilia and in von Willebrands disease.7
TXA is more potent than EACA, but typical TXA efficacious doses in humans are 1–1.5 g given 3–4 times per day. Despite attempts to reduce the dose by novel LBS inhibitors8 or prodrugs of TXA,9 no other LBS inhibitors have reached the market. Oral dosing of TXA is associated with dose-dependent gastrointestinal (GI) side effects, including nausea, vomiting, diarrhea, and cramping. This has limited the use and efficacious dosing in menorrhagia. There are also indications that treatment of menorrhagia in von Willebrands disease requires higher doses, and as a consequence, the dose-dependent side effects may become efficacy limiting.10 In an attempt to prolong the exposure of TXA, it has been formulated as a modified release tablet, Lysteda (1.3 g three times daily), and this has recently received approval by the FDA.11 Interestingly, GI side effects in the treatment groups were not more frequent than in the placebo control group, indicating that the local amount of TXA released in the GI lumen may be causative for the GI side effects.12
Finally, there are rare reports of seizures during treatment with TXA, probably caused by a weak GABAa antagonistic activity.12−14 It can be speculated that GI side effects associated with high dose TXA is also GABAa mediated. Thus, a novel oral fibrinolysis inhibitor, with the same mechanism of action as TXA but with more convenient dosing and higher selectivity over GABAa is hypothesized to be advantageous.
Full length plasminogen (Glu-plasminogen) may exist in a compact closed conformation and as an open extended confirmation.15 There are five kringle domains in plasmin(ogen), and each has an LBS. The amino acid sequence identity between the kringle domains are around 70%. The X-ray crystal structure of the full length plasminogen (pdb code: 4A5T) revealed that only kringle-1 (K1) is exposed on the surface of the closed complex, as such available for ligand binding. The respective LBS from K2, K4, and K5 are engaged in interdomain interactions contributing to the formation of the closed conformation, whereas the LBS on K3, being less exposed, does not engage any lysine from the neighboring domain. However, in the N-terminal truncated Lys-plasminogen, having an open conformation, all LBS should be available for binding. Full length Glu-plasminogen with the closed conformation has one strong affinity binding site for TXA (Kd = 1.1 μM, K1) and several low affinity binding sites (Kd = 750 μM), while elongated Lys-plasminogen has two high affinity binding sites (Kd = 2.2 μM, K1, and Kd = 36 μM, probably K4).16,17 TXA binds to plasmin(ogen) noncovalently by a charge interaction (pdb code: 1CEB). The carboxylic acid of TXA interacts with two arginines, ARG34 and ARG70, and the basic piperidine binds to two aspartate residues, ASP54 and ASP56, in the small and shallow pocket.18 Thus, a novel ligand would require both positive and negative charges, i.e., to be a zwitterion, to effectively bind to K1. Although other kringles may be important in the binding and activation of plasminogen and subsequent fibrinolysis, this report focuses on the K1 ligand–protein interactions.
In the search for new chemical leads as plasmin(ogen) inhibitors, we previously reported that 4-PIOL (Figure 1) was identified via a small-scale virtual screening campaign using an electrostatic similarity approach using TXA as a seed molecule (Figure 2).19 4-PIOL was also found to bind to the shallow but very polar pocket of the K1 domain of plasmin. This interaction occurs in a similar way to TXA, with ionic interactions to the basic nitrogens in the two aspartates (ASP54 and ASP56) and acidic interactions to the two arginine residues (ARG34 and ARG70).
Figure 2.
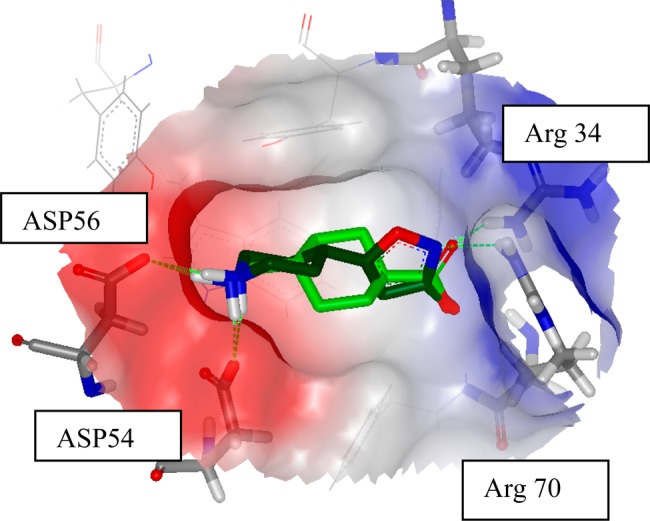
Overlay of TXA (light green) and 4-PIOL (dark green) in the binding site of plasmin(ogen).19,21 The position of 4-PIOL is generated by docking into the binding pocket using Glide from Schrödinger.
4-PIOL is a very interesting lead compound as it displays approximately 4-fold improvement of potency in both human buffer and plasma clot lysis assays compared with TXA (Table 1). However, 4-PIOL has also been reported to be a GABAa partial agonist.20 In an in-house GABAa binding assay, measuring displacement of radiolabeled muscimol from rat brain vesicles, the IC50 was 35 μM. This can be compared to the IC50 of TXA in the same assay of 1600 μM. Although 4-PIOL serves as a promising lead from a potency perspective, it has poor permeability properties as measured in a Caco-2 cell permeability assay (Papp A to B < 0.01 × 10–6 cm s–1) and development of a new oral pharmaceutical compound will require not only optimization of potency but also increased GABAa selectivity and increased permeability. These parameters became the important key factors in our optimization program described below.
Table 1. Initial SAR Development around 4-PIOL.
| compd | clotlysis IC50 (μM)a buffer/plasma | GABAa IC50 (μM)b | Caco-2 Papp A to B (10–6 cm s–1) |
|---|---|---|---|
| EACA | 105/40 | ||
| TXA | 12/3.1 | 1600 | |
| 4-PIOL | 2.8/0.8 | 35 | <0.01 |
| 1 | 100/100 | 1.0 | |
| 2 | 31/40 | 0.4 | |
| 3 | 5.7/2.0 | 51 | |
| 4 | 146/– | 0.4 | |
| 5c | 6.3/2.1 | >2000 | 0.6 |
| 6 | 12.2/– | 267 | 0.5 |
Values are means of three experiments.
Top concentration tested was 2000 μM.
Compound 5 is the racemic cis-isomer. – = not measured.
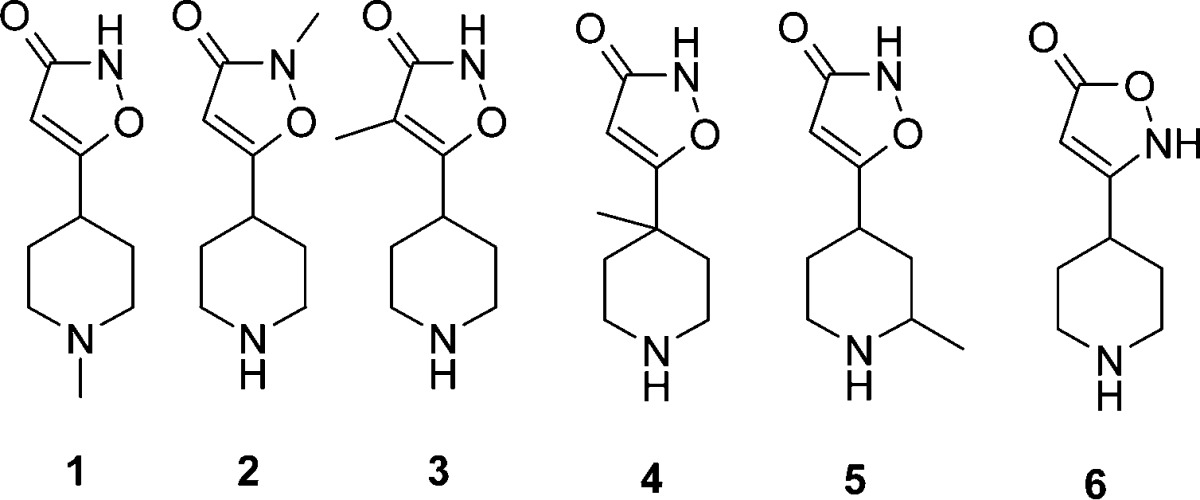
First, a methyl scanning strategy was performed to evaluate structure and activity/property relationships (Table 1, synthetic details for compounds 1–6 can be found in the Supporting Information).
N-Substitution of 4-PIOL either at the piperidine (1) or isoxazolone (2) resulted in a drastic decrease in potency suggesting a more optimal geometry and zwitterionic character for 4-PIOL as compared to 1 and 2. Further substitution on the isoxazolone ring, as in 3, was tolerated from a potency perspective, but no improvement on the GABAa selectivity could be seen. Methyl substitution in the 4-position of the piperidine ring (4) decreased potency. However, methyl substitution in the 2-position of the piperidine ring as in 5 gave a potency comparable with 4-PIOL, and most importantly, compound 5 dramatically improved the selectivity over GABAa with no detectable activity at a concentration of 2000 μM, hence revealing the 2-position as a site for further exploration. Finally, the regioisomeric isoxazolone 6 also lost potency compared with 4-PIOL.
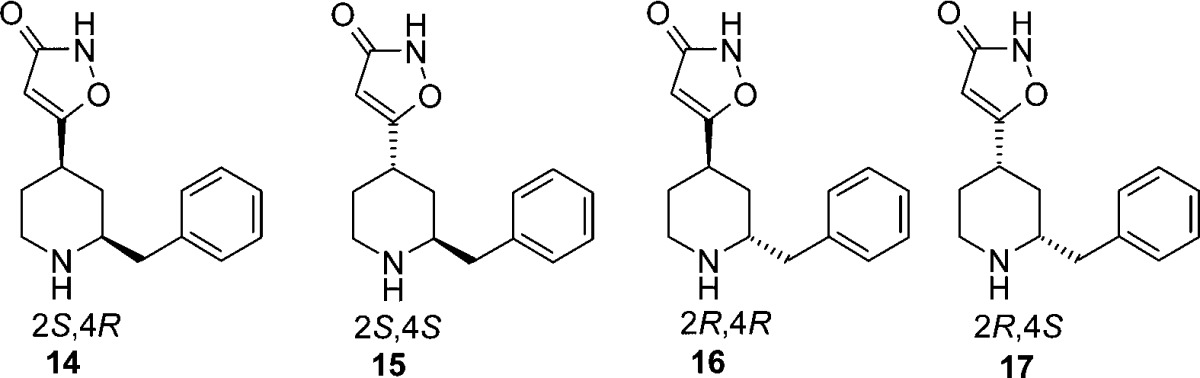
Encouraged by the improved GABAa selectivity and equivalent potency of 5 as compared with 4-PIOL, further substituents in the 2-position of the piperidine ring were investigated. In Scheme 1, the synthetic route to the benzyl substituted compounds 14–17 is depicted.
Scheme 1. General Synthesis for 14–17.

Reagents and conditions: (a) (i) BnMgCl, THF, (ii) methyl chloroformate, (iii) HCl; (b) Pd/C, EtOAc; (c) TOSMIC, KtBuO; (d) conc. HCl; (e) (i) CDI, THF, (ii) MgCl2, potassium ethylmalonate, (iii) diastereoisomer separation; (f) (i) NaOH, MeOH, H2O, −20 °C, (ii) NH2OH, −20 °C, (iii) 6 N HCl, 80 °C; (g) (i) chiral HPLC, (ii) 33% HBr in HOAc.
The addition of benzyl magnesiumchloride to 4-methoxypyridine 7 via the in situ formed 1-acyl pyridinium salt gave, after acidic workup, the 2-substituted dihydropyridone 8 in a method analogous to the one pioneered by Comins et al.22 Hydrogenation over Pd/C gave the corresponding piperidone 9 that could be converted to the nitrile 10 by treatment with TOSMIC.23 Hydrolyis of the nitrile in the presence of the methylcarbamate could be achieved by conc. HCl to yield the carboxylic acid 11. The 3-isoxazolone moiety could be prepared in analogy to the method reported by Krogsgaard-Larsen et al.24 via formation of the β-keto-ester 12, and subsequent treatment of 12 with NaOH and then cyclization at −20 °C using hydroxylamine for the formation of 13. The cis- and trans-diastereomers were conveniently separated by chromatography on the stage of the β-keto-esters 12, whereas separation of the enantiomers by chiral HPLC was best undertaken with intermediates 13 before final removal of the methyl carbamate with HBr in acetic acid to give the benzyl substituted isoxazolone derivatives 14–17.
To understand the stereoisomeric significance of substitution in the 2-position of the piperidine ring, the four stereoisomers of the benzyl substituted piperidines (14–17) were studied in more detail (Table 2). It appears that an (S)-configuration at the 4-position is critical for potency as highlighted with 15 and 17, while the 2-position is more open for both configurations with 17 (2R,4S), having a cis-conformation, being somewhat more potent than the corresponding trans-analogue 15. Introduction of a larger group such as benzyl into the 2-position resulted in the desired selectivity over GABAa, which is in line with what was seen for methyl-substituted 5, but also gave an increased permeability as shown for 14–17.
Table 2. Stereoisomeric Evaluation of 2,4-Disubstituted Piperidines 14–17.
| compd | clotlysis IC50, (μM)a buffer/plasma | GABAa IC50 (μM)b | Caco-2 Papp A to B (10–6 cm s–1) |
|---|---|---|---|
| 14 (2S,4R) | 150/– | 1350 | 4.1 |
| 15 (2S,4S) | 2.9/0.58 | >2000 | 1.2 |
| 16 (2R,4R) | 169/– | >2000 | 1.0 |
| 17 (2R,4S) | 0.95/0.28 | >2000 | 1.9 |
Values are means of three experiments.
Top concentration tested was 2000 μM.
A crystal structure was obtained for 17 binding to K1 of plasminogen (pdb code: 4CIK; Figure 3).
Figure 3.
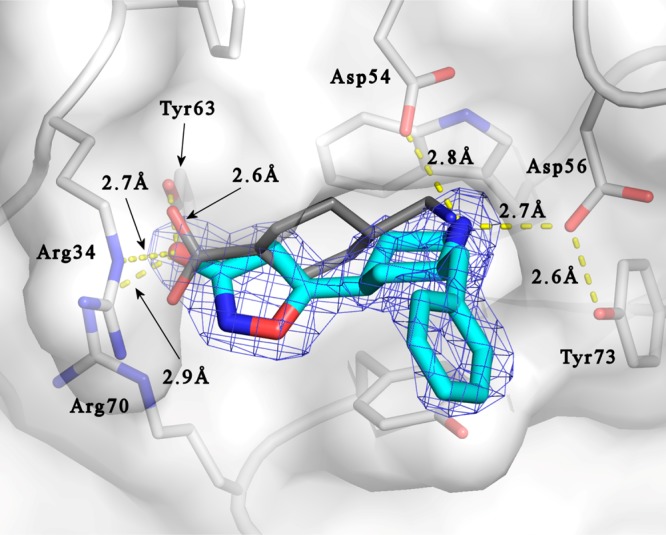
X-ray crystal structure of 17 (blue) binding to plasminogen (pdb code: 4CIK). The structure was overlaid with the kringle 1/TXA complex (pdb code: 1CEB). The position of TXA (gray) is shown for clarity.
The structure shows that 17 binds in the same binding pocket as TXA, interacting with the same key amino acids. The isoxazolone carbonyl overlays well with the carboxylic acid moiety of TXA and is interacting with Arg34, Tyr63, and Arg70. The piperidine nitrogen of 17 takes a similar position as the amino group of TXA (about 1 Å apart between the respective nitrogen atoms), both are within H-bond distance to Asp54 (2.8 Å) and Asp56 (2.7 Å). Evidently, the benzyl substituent does not make any specific interactions with the K1 kringle, which is in line with the observed small difference in potency between the two stereoisomeric compounds 15 and 17. Nevertheless, as indicated by the lower GABAa affinity for 2-substituted 4-PIOL analogues as 14–17, it suggests that the benzyl groups populate an unfavorable region in the GABAa binding site. Similar substitution effects on the GABAa affinity has been observed for a related structural series indicating the generality of this finding.25
In order to assess the scope of the 2-substituted 4-PIOL derivatives, we further evaluated a range of analogues focusing on the 2R,4S piperidine configuration (Table 3, synthetic details for compounds 18–25 can be found in the Supporting Information). Introduction of larger aliphatic groups in the 2-position resulted in an increase in potency compared with methyl-substituted 5 as seen for the isobutyl 18 and neopentyl 19 derivatives. Further elongation with an additional methylene linker (20) gave a slight decrease in potency indicating a limitation in length and volume of the side chain. Introduction of an aliphatic ring as in 21 reduced potency. In contrast to 20, elongation of the benzyl substituent to a phenethyl group was tolerated as shown for 22 displaying excellent potency as well as GABAa selectivity and permeability. Fluorine atoms in the phenyl ring (23–25) were also tolerated, but no significant improvement in the desired properties was achieved.
Table 3. Optimization of the Substituent in the 2-Position of the Piperidine.
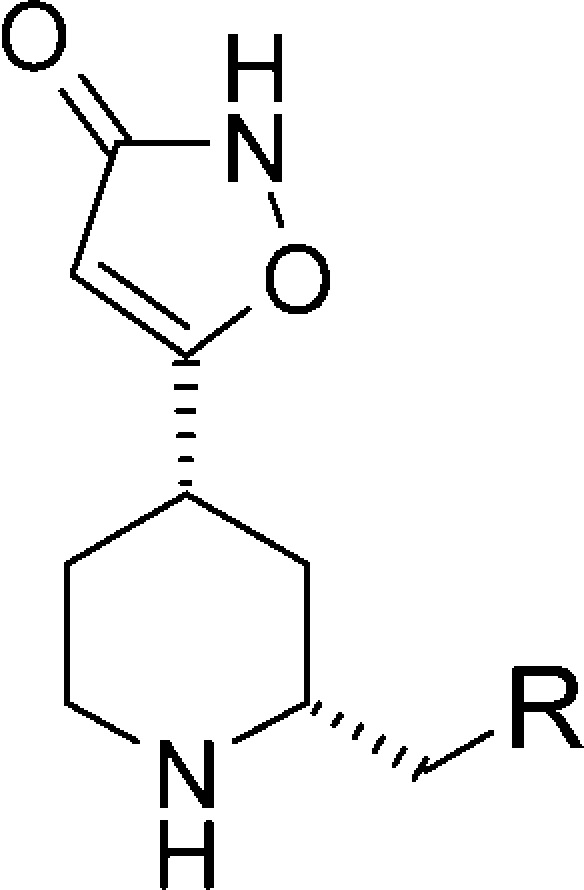
| compd | R | clotlysis IC50 (μM)a plasma | GABAa IC50 (μM)b | Caco-2 A to B (10–6 cm s–1) |
|---|---|---|---|---|
| 17 | Ph | 0.37 | >2000 | 1.9 |
| 18 | iPr | 1.02 | >2000 | 1.0 |
| 19 | tBu | 0.44 | >2000 | 2.6 |
| 20 | –CH2C(CH3)3 | 0.77 | >2000 | 4.5 |
| 21 | –C6H11– | 1.06 | >2000 | 6.2 |
| 22 | –CH2Ph | 0.39 | >2000 | 2.6 |
| 23 | 2-F–Ph | 1.26 | >2000 | 1.8 |
| 24 | 3-F–Ph | 0.36 | >2000 | 3.1 |
| 25 | 4-F–Ph | 0.45 | 1090 | 3.7 |
Values are means of three experiments.
Top concentration tested was 2000 μM.
The analogues in Table 3 all show a narrow span in potency. Despite varying the side chain from aliphatic to aromatic, varying chain length, and introducing fluorine atoms, the potency varies less than 5-fold. This is in agreement with the conclusions from the crystal structure of 4-PIOL and 17 (Figure 3) that the substituent in the 2-position does not make any specific interactions to the protein. However, some steric limitations could be seen, for example, 20. The small variation noted could possibly be attributed to differences in solvation and entropic effects.
Substitution in the 2-position of the piperidine ring led to more permeable compounds compared to 4-PIOL (Table 3). Introduction of the 2-substituents led to an increase in lipophilicity, which has been shown to increase membrane permeability, especially in the low lipophilicity range of these derivatives.26 Systematic modification on the substitution pattern and stereochemistry revealed how the zwitterionic nature of the compounds contribute to the permeability profile. This is seen by stereoisomers 14 and 15 in Table 2, where the experimentally determined pKa difference between the basic and acidic centers falls from 4.9 for compound 15 to 4.4 for compound 14, while Papp increases from 1.2 to 4.1 × 10–6 cm s–1, respectively. For compounds with similar lipophilicity, the compound with a lower difference between the acid and base pKa values had the higher Papp value. Although not evident with classical molecular descriptors related to permeability such as logD or number of H-bond donors, which are identical for the stereoisomers, the permeability is also influenced by the pKa difference between the acid and base within the molecule, and permeability increases with reducing zwitterionic character. This effect has also been showed for other molecular series.27 For more details on the relationship between pKa and permeability for several compounds in this article, please see the Supporting Information.
Compounds in this series, including 17, 19, and 22, showed very high metabolic stability in human hepatocytes (<1 μL/min/106 cells) and liver microsomes (<5 μL/min/mg), as well as displaying no notable inhibition of CYP450s and no inhibition of hERG. Biotransformation experiments in human hepatocytes supported the observed low metabolism. The only observed metabolism of these compounds, if any could be seen, was trace amounts of glucuronidation on the isoxazolone ring.
Compounds 17, 19, and 22 were selected for further exploration in vivo based on the balanced potency, permeability, and GABAa selectivity properties. The pharmacokinetic profiles of 17, 19, and 22 were studied in rat and dog after i.v. and oral dosing (Table 4).
Table 4. Pharmacokinetic Profiles of Selected Compounds.
| compd | CLtot/CLrenal rat (mL/min/kg)a | F% rata | CLtot/CLrenal dog (mL/min/kg)b | F% dogb |
|---|---|---|---|---|
| 17 | 46/11 | 33 | 2.9/0.2 | 73 |
| 19 | 13/6 | 39 | 1.1/0.8 | 53 |
| 22 | 24/6 | 19 | 1.8/0.12 | 59 |
Doses used: i.v. 5 μmol/kg and oral 20 μmol/kg.
Doses used: i.v. 3 μmol/kg and oral 6 μmol/kg.
Compound 17 had higher clearance in rat and dog compared with 19 and 22. Compound 19 demonstrated good bioavailability of 39% and 53% in rat and dog, respectively, whereas compound 22 showed 19% and 59%, respectively. Investigation of the elimination pathways revealed that between 25% (17 and 22) and 45% (19) of the dose was eliminated renally in the rat and that between 7% (17) and 70% (19) was eliminated renally in the dog. To confirm reduction in bleeding in vivo, compounds 17, 19, and 22 were tested in a rat bleeding model (please refer to Supporting Information for details). It could be demonstrated that inhibition of fibrinolysis measured as the prolongation of the human plasma clot lysis time in vitro translated to a shortening of tPA-prolonged bleeding time in vivo (Table 5). For example, compound 19 reduced bleeding times from an incision in the tail by 50% at a 19- and 12-fold lower dose and plasma concentration, respectively, as compared to TXA. The corresponding values for 17 and 22 were 25- and 11-fold reductions in dose and 24- and 10-fold decrease in plasma concentration, respectively.
Table 5. In Vivo Fibrinolysis Inhibition EC50 and ED50 Values in a Rat Bleeding Modela.
| compd | rat in vivo EC50 (μM) | rat in vivo ED50 (μmol/kg·min) |
|---|---|---|
| TXA | 19.2 | 15.9 |
| 17 | 0.79 | 0.65 |
| 19 | 1.62 | 0.84 |
| 22 | 1.92 | 1.44 |
Compounds were infused at four different doses in 6 individual animals per dose. The ED50 and EC50 were calculated as the infused dose and plasma concentration giving 50% reduction in bleeding time, respectively.
From clinical experience, it is known that a total TXA plasma concentration of at least 30 μM is necessary to achieve the desired clinical effect suggesting that a 95% receptor occupancy is required.28 For compound 19, the plasma and blood clot lysis IC95 estimates fall in the range of 1.5 and 6.6 μM. These results are used as potency estimates in the dose prediction in man. If, as observed in rat and dog studies, hepatic metabolism and glomerular filtration are the routes of elimination the total human clearance is estimated to be 17.5 L h–1, a composite of metabolism and renal filtration. Considering the 95% coverage of LBS and the absorption and clearance properties of 19 gives a human predicted dose estimate of twice daily dosing of 340 mg.29
In summary, we have described the discovery of a novel series of fibrinolysis inhibitors acting via interference of a protein–protein interaction. On the basis of the improved potency over TXA, excellent GABAa selectivity, no inhibition of CYP450 isozymes or hERG30,31 (the human ether-à-go-go-related gene), good pharmacokinetic properties with both hepatic and renal routes of elimination, and a much improved predicted daily dose to man, compound 19 was selected as the drug candidate AZD6564 for development. Further profiling and testing will be the subject of future publications.
Acknowledgments
Maria Thorstensson and Ahlke Hayen are acknowledged for early lead generation work in this project. Ola Fjellström is acknowledged for interesting discussions around the article topic.
Glossary
ABBREVIATIONS
- TXA
tranexamic acid
- 4-PIOL
5-(4-piperidyl)-3-isoxazolone
- EACA
epsilon aminocaproic acid
- tPA
tissue-plasminogen activator
- PLG
plasminogen
- LBS
lysine binding site
- DMPK
drug metabolism and pharmacokinetics
- GABA
γ-aminobutyric acid
- TBSCl
tert-butyl-dimethyl silyl chloride
- DMF
dimethyl formamide
- DCM
dichloromethane
- THF
tetrahydrofuran
- TBAF
tert-butyl ammonium fluoride
- CDI
carbonyldiimidazole
- HPLC
high performance liquid chromatography
Supporting Information Available
Synthetic procedures and experimental details (1–25). This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
⊥ (L.C.) E-mail: leifengcheng88@gmail.com.
Author Present Address
# (P.S.) GPPS (Global Product and Portfolio Strategy) RiA IMED, Astrazeneca Mölndal, SE-43183 Mölndal, Sweden.
Author Present Address
○ (E.E.) Medicinal Chemistry RIA iMED, Astrazeneca Mölndal, SE-43183 Mölndal, Sweden.
Author Present Address
∇ (W.L.) DMPK RIA iMED, Astrazeneca Mölndal, SE-43183 Mölndal, Sweden.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Project leader L.C.; Lead author D.P.; Medicinal chemists P.S., D.P., B.O., M.K., S.P., M.J., A.P., and T.F.; Computational chemists E.E. and J.B.; Bioscientists A.T., G.W., and D.G.; DMPK C.H., W.L., and L.L.; X-ray Y.X.
The authors declare no competing financial interest.
Supplementary Material
References
- Medcalf R. L.Plasminogen Activator Inhibitor. Part II. In Methods in Enzymology; Whisstock J., Bird P., Eds.; Academic Press: New York, 2007. [Google Scholar]
- Castellino F. J.; Ploplis V. A. Structure and function of the plasminogen/plasmin system. Thromb. Haemost. 2005, 93, 647–654. [DOI] [PubMed] [Google Scholar]
- McCormack P. L. Tranexamic acid: a review of its use in the treatment of hyperfibrinolysis. Drugs 2012, 72, 585–617. [DOI] [PubMed] [Google Scholar]
- Okamoto S.; Okamoto U. Amino-methyl-cyclohexane-carboxylic acid. A new potent inhibitor of fibrinolysis. Keio J. Med. 1962, 11, 105–115. [Google Scholar]
- Roberts I.; Perel P.; Prieto-Merino D.; Shakur H.; Coats T.; Hunt B. J.; Lecky F.; Brohi K.; Willett K. Effect of tranexamic acid on mortality in patients with traumatic bleeding. Brit. Med. J. 2012, 345, 5839–5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochschwander S. M.; Laursen R. A. The lysine binding sites of human plasminogen. J. Biol. Chem. 1981, 256, 11172–11176. [PubMed] [Google Scholar]
- Dunn C. J.; Goa K. L. Tranexamic acid: a review of its use in surgery and other indications. Drugs 1999, 57, 1005–1032. [DOI] [PubMed] [Google Scholar]
- Baumgarten W.Synthetic Antifibrinolytic Agents. In Hematologic Reviews; Ambrus J. L., Ed.; Marcel Dekker: New York, 2002. [Google Scholar]
- Edlund M.; Andersson K.; Rybo G.; Lindoff C.; Astedt B.; von Schoultz B. Reduction of menstrual blood loss in women suffering from idiopathic menorrhagia with a novel antifibrinolytic drug (Kabi 2161). Br. J. Obstet. Gynaecol. 1995, 102, 913–917. [DOI] [PubMed] [Google Scholar]
- Mohri H. High dose of tranexamic acid for treatment of severe menorrhagia in patients with von Willebrand disease. J. Thromb. Thrombolysis 2002, 14, 255–257. [DOI] [PubMed] [Google Scholar]
- Lukes A. S.; Kouides P. A.; Moore K. A. Tranexamic acid: a novel oral formulation for the treatment of heavy menstrual bleeding. Womens Health 2011, 7, 151–158. [DOI] [PubMed] [Google Scholar]
- van der Maarl D. L.; Hilkens P.; Bosch F. The epileptogenic effect of tranexamic acid. J. Neurol. 1999, 246, 843. [DOI] [PubMed] [Google Scholar]
- Koster A.; Börgermann J.; Zittermann A.; Lueth J. U.; Gillis-Januszewski T; Schirmer U. Moderate dosage of tranexamic acid during cardiac surgery with cardiopulmonary bypass and convulsive seizures: incidence and clinical outcome. Brit. J. Anaesth. 2013, 110, 34–40. [DOI] [PubMed] [Google Scholar]
- Furtmüller R.; Schlag M. G.; Berger M.; Hopf R.; Huck S.; Sieghart W.; Redl H. Tranexamic acid, a widely used antifibrinolytic agent, causes convulsions by a gamma-aminobutyric acid(A) receptor antagonistic effect. J. Pharmacol. Exp. Ther. 2002, 301, 168–173. [DOI] [PubMed] [Google Scholar]
- Kalavrouziotis D.; Voisine P.; Mohammadi S.; Dionne S.; Dagenais F. High-dose tranexamic acid is an independent predictor of early seizure after cardiopulmonary bypass. Ann. Thorac. Surg. 2012, 93, 148–155. [DOI] [PubMed] [Google Scholar]
- Xue Y.; Bodin C.; Olsson K. Crystal structure of the native plasminogen reveals an activation-resistant compact conformation. J. Thromb. Haemost. 2012, 10, 1385–1396. [DOI] [PubMed] [Google Scholar]
- Kim P. Y.; Tieu L. D.; Stafford A. R.; Fredenburgh J. C.; Weitz J. I. A high affinity interaction of plasminogen with fibrin is not essential for efficient activation by tissue-type plasminogen activator. J. Biol. Chem. 2012, 287, 4652–4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markus G.; Priore R. L.; Wissler F. C. The binding of tranexamic acid to native (Glu) and modified (Lys) human plasminogen and its effect on conformation. J. Biol. Chem. 1979, 254, 1211–1216. [PubMed] [Google Scholar]
- Boström J.; Grant A. J.; Fjellström O.; Thelin A.; Gustafsson D. Potent fibrinolysis inhibitor discovered by shape and electrostatic complementary to the drug tranexamic acid. J. Med. Chem. 2013, 56, 3273–3280. [DOI] [PubMed] [Google Scholar]
- Byberg J. R.; Labouta I. M.; Falch E.; Hjeds H.; Krogsgaard-larsen P.; Curtis D. R.; Gynther B. D. Synthesis and biological activity of a GABAa agonist which has no effect on benzodiazepine binding and structurally related glycine antagonists. Drug Des. Delivery 1987, 1, 261–274. [PubMed] [Google Scholar]
- Mathews I. I.; Vanderhoff-Hanaver P.; Castellino F. J.; Tulinsky A. Crystal structures of the recombinant kringle 1 domain of human plasminogen in complexes with the ligands ε-aminocaproic acid and trans-4-(aminomethyl)cyclohexane-1-carboxylic acid. Biochemistry 1996, 35, 2567–2576. [DOI] [PubMed] [Google Scholar]
- Comins D. L.; Brown J. D. Addition of Grignard reagents to 1-acyl-4-methoxypyridinium salts. An approach to the synthesis of quinolizidinones. Tetrahedron Lett. 1986, 27, 4549–4552. [Google Scholar]
- Oldenziel O. H.; Van Leusen D.; Van Leusen A. M. Chemistry of sulfonylmethyl isocyanides. 13. A general one-step training & development plan delivered synthesis of nitriles from ketones using tosylmethyl isocyanide. Introduction of a one-carbon unit. J. Org. Chem. 1977, 42, 3114–3118. [Google Scholar]
- Førlund B.; Tagmose L.; Liljefors T.; Stensbol T. B.; Engblom C.; Kristiansen U.; Krogsgaard-Larsen P. A novel class of potent 3-isoxazolol GABA(A) antagonists: design, synthesis, and pharmacology. J. Med. Chem. 2000, 43, 4930–4933. [DOI] [PubMed] [Google Scholar]
- Boström J. Manuscript in preparation.
- Waring M. J. Lipophilicity in drug discovery. Expert Opin. Drug. Discovery 2010, 5, 235–248. [DOI] [PubMed] [Google Scholar]
- Over B.; McCarren P.; Artursson P.; Foley M.; Giordanetto F.; Grönberg G.; Hilgendorf C.; Lee M. D.; Matsson P.; Muncipinto G.; Pellisson M., Perry M.; Svensson R.; Duvall R.; Kihlberg J.. Impact of Stereospecific Intramolecular Hydrogen-Bonding on Cell Permeability. J. Med Chem. Manuscript submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LYSTEDA. http://dailymed.nlm.nih.gov/dailymed/lookup.cfm?setid=84a65305-65d7-e7fd-66f3-dda8d8f920b1.
- Human dose was predicted
from the equation
where Cuss is the unbound plasma concentration, Clu is the unbound clearance, τ is the dosing interval, and F is the bioavailability. Inserting a Cuss of 4 μM, Clu of 50 L h–1, F of 0.65, and τ of 12 h gives a predicted dose of 340 mg BID.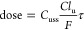
- IC50 for inhibition of CYP450 (3A4, 2D6, 1A2, 2C9, 2C19, and 2C8) enzymes > 20 mM. IC50 for inhibition of the hERG channel > 1000 mM.
- Sanguinetti M. C.; Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature 2006, 440, 463–469. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.