

Abstract
Protein kinases are key regulators that govern complex cellular processes. Dysregulation of kinase signaling is associated in many human diseases, particularly cancers and developmental and metabolic disorders. Tyrosine kinase inhibitors have achieved great success in molecular targeted therapies for cancer and now is expanding to other therapeutic areas. The onset of drug resistance to prolonged TKI treatment brings new challenges in TKI drug development. The deep understanding of disease pathologies related to TKs and drug resistance mechanisms will generate new waves for seeking highly selective, potent, and safe TKIs.
Protein kinases are key regulators that govern complex cellular processes, including cell growth, differentiation, proliferation, and apoptosis. There are more than 518 distinct kinases encoded by ∼2% of all human genes, and among them, 90 proteins are tyrosine kinases.1 Since the first discovery of the transforming oncogene of the Rous sarcoma virus (v-Src) as a protein kinase in 1978, the dysregulation of kinase signaling has been recognized to underlie many human diseases, particularly cancers, and developmental and metabolic disorders, leading to the search for potent and selective protein kinase inhibitors for therapeutic interventions. Identification of the causative genetic lesion BCL-ABL in chronic myelogenous leukemia (CML) resulted in the breakthrough medicine imatinib, a tyrosine kinase ABL inhibitor for the treatment of CML in 2001. Targeting oncogenic driver mutations has been a proven therapeutic strategy to control tumor growth and disease progression. A number of tyrosine kinase inhibitors have achieved clinical success, including gefitinib (2003), erlotinib (2004), icotinib (2011), and afatinib (2013) targeting activating mutant EGFRs for nonsmall cell lung cancer (NSCLC); sorafenib (2005), sunitinib (2006), pazopanib (2009), and axitinib (2012) targeting VEGFRs for renal cell carcinoma; lapatinib (2007) targeting EGFR and ERBB2 for breast cancer; crizotinib (2011) targeting ALK for late stage lung cancer; ruxolitinib (2011) targeting JAK1/2 for myelofibrosis; vandetanib (2011) and cabozantinib (2012) targeting RET for metastatic medullary thyroid cancer; tofacitinib (2013) targeting JAK1/3 for rheumatoid arthritis; and ibrutinib (2013) targeting BTK for mantle cell lymphoma.
Although the overall response rate of these targeted therapies is impressive, the durability of the response is limited by the emergence of drug resistance. The clinical implementation of cancer genome sequencing is leading to a better understanding of the genetic basis of acquired drug resistance. The mechanism of drug resistance can be either intrinsic (altering the original target) or extrinsic (compensatory signaling through other pathways and pharmacokinetic factors that primarily reduce drug concentration in targeted cells). Common intrinsic resistance mechanisms to abrogate the effectiveness of kinase inhibitor drugs include target gene amplification, overexpression or epigenetic activation, and the development of secondary missense mutations.2 Selective pressure by drug treatment induces the clonal expansion of subsets of cancer cells with different genomic alterations that confer resistance.3 Drug-resistant point mutations often arise in protein regions involved in either drug interactions or in the transitions between active and inactive kinases. These mutations typically selectively weaken the binding affinity of the drug but not the ATP substrate with the targeted kinase. Therefore, drug-resistant mutations in different kinases share common “hotspots” for conserved resistance mechanisms.2 Gatekeeper mutants are the most frequent clinical drug-resistant mutants. Examples include ABLT315I in CML, PDGFRT674I/M in hypereosinophilic syndrome, EGFRT790M in nonsmall cell lung cancer, KITT670I in gastrointestinal stromal tumors, and ALKL1196M in NSCLC. Gatekeeper mutations mainly stabilize the active conformation leading to increased ATP binding affinity, catalytic power, and transforming potential between the active and inactive conformations. To overcome gatekeeper mutant resistance, additional interactions with inactive kinases need to be introduced to compensate for the increased transforming energy required in going from the disease-driven active conformation to the inactive conformation if the inhibitor is designed to target the inactive conformation. This may lead to increased molecular weight, higher lipophilicity, and poorer drug-like properties. New chemical entities targeting the mutant active conformation should be pursued to achive more efficient inhibition of mutant active kinases.
A number of more potent ABL kinase inhibitors with a broad spectrum of activities toward wild type and mutant ABL kinases have been developed and achieved clinical success in primary and imatinib-refractory CML patients, including dasatinib (2006), nilotinib (2007), bosutinib (2012), and ponatinib (2012) (Chart 1). Imatinib, nilotinib, and ponatinib stabilize ABL kinase in DFG-out inactive conformation. Nilotinib introduced an additional trifluoromethyl group to improve potency against both wild and mutant ABLs except ABLT315I mutant. Ponatinib uses acetylene group to replace the pyrimidinylamino linker leading to a favored interaction with the mutant hydrophobic gatekeeper residue I315 (Figure 1) and is the only ABL inhibitor active against the ABLT315I mutant although it induces significantly high cardiovascular events. Both dasatinib and nilotinib occupy space near the gatekeeper region that interferes with the preferred active conformation of the ABLT315I mutant leading to significantly less activity against the gatekeeper mutant. Future development of new generation of ABL inhibitors targeting the active conformation may provide more choices for CML patients after relapse from existing TKI treatments.
Chart 1. ABL Tyrosine Kinase Inhibitorsa.
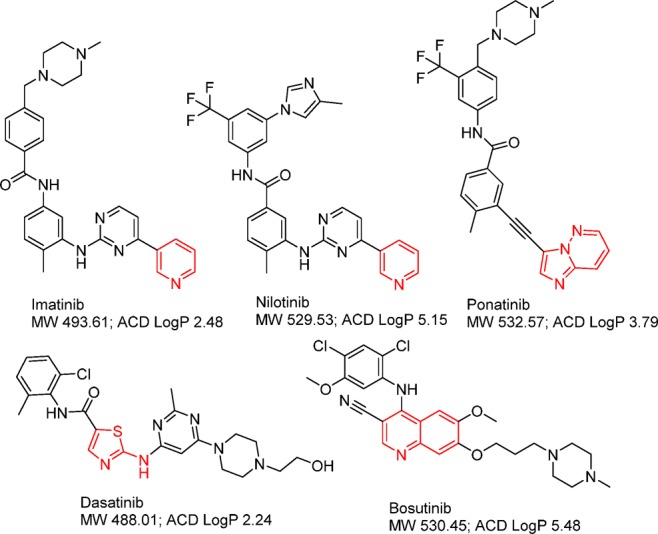
a The group in red functions as a kinase hinge binder.
Figure 1.
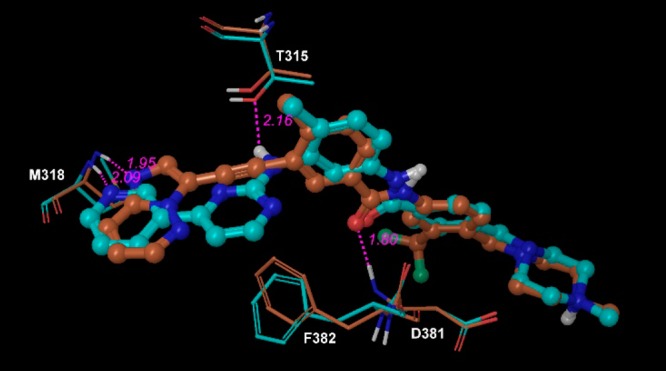
Imatinib (cyan) overlapping with ponatinib (brown) in ABL proteins (PDB IDs: 2hyy and 3oxz).
The success of imatinib and the second and third generation ABL tyrosine kinase inhibitors paves the road to the development of new targeted kinase inhibitors and the effective management of TKI resistance. Crizotinib (PF-02341066), a receptor tyrosine kinase drug targeting MET/ALK/ROS1 was granted fast track approval in 2011 for late-stage NSCLC patients who expresses the abnormal EML4-ALK fusion gene. Similar to imatinib, crizotinib patients invariably develop resistance after a period of treatment. The resistance mechanisms include ALK gene amplification, secondary ALK mutations including gatekeeper mutant EML4-ALKL1196M, and aberrant activation of other kinases including c-KIT and EGFR.4 A new generation of ALK inhibitors has been developed and demonstrates marked clinical efficacy for both crizotinib-naïve and -refractory ALK+ NSCLC patients. The front runners LDK378 and alectinib (Chart 2), compounds with more potent inhibition and broader activities against both wild and mutant ALK proteins, received FDA breakthrough designation in 2013, just two years after crizotinib’s FDA fast-track approval. Crizotinib, LDK378, and alectinib represent three distinct classes of ALK inhibitors and have significantly different interactions with the ALK protein at the gatekeeper region, leading to different sensitivity to the L1196M gatekeeper mutant. LDK378 is 3–6-fold more potent than crizotinib in antiproliferative cell assays corresponding to a response rate of about 60% and median progression-free survival of 8.3 months in ALK-positive patients who had relapsed on crizotinib.5 Interestingly, both imatinib and crizotinib, as first generation kinase inhibitors, have relatively weaker cellular activities (>100 nM). The more potent second generation inhibitors are almost as effective as the first generation with regards to response rate and duration of response in refractory patients. Sequential treatment following a weaker kinase inhibitor with a more potent and broad-spectrum second generation kinase inhibitor can significantly improve patient’s life expectancy.
Chart 2. ALK Tyrosine Kinase Inhibitorsa.
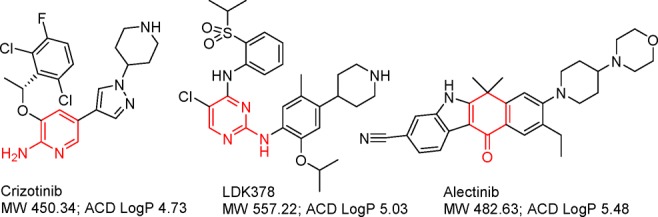
a The group in red functions as a kinase hinge binder.
The development of the next generation of EGFR inhibitors for gefitinib or erlotinib-refractory NSCLC patients illustrates the importance of capturing the disease-driven kinase conformation. The four approved EGFR inhibitors (Chart 3) share the same binding mode, stabilizing EGFR in the inactive conformation. Although afatinib provides much more potent inhibition against EGFR through covalent-binding at the ATP binding site, the drug-resistant T790M mutant prefers the active conformation. Therefore, the T790M mutant EGFR is in the wrong conformation for afatinib to bind effectively first to permit the covalent interaction to occur. The new generation of irreversible EGFR inhibitors AZD-9291 and CO-1686 (Chart 3) uses a pyrimidine scaffold, eliminating the structure elements close to the gatekeeper, and increasing interactions with the G-loop. Both AZD-9291 and CO-1686 have demonstrated promising clinical efficacies in EGFR TKI-resistant patient population in phase I clinical studies. Reversible EGFR inhibitors targeting the active conformation of EGFRT790M mutant will certainly diversify the collection of EGFR inhibitors to benefit the refractory patients from EGFR TKI treatments.
Chart 3. EGFR Tyrosine Kinase Inhibitorsa.
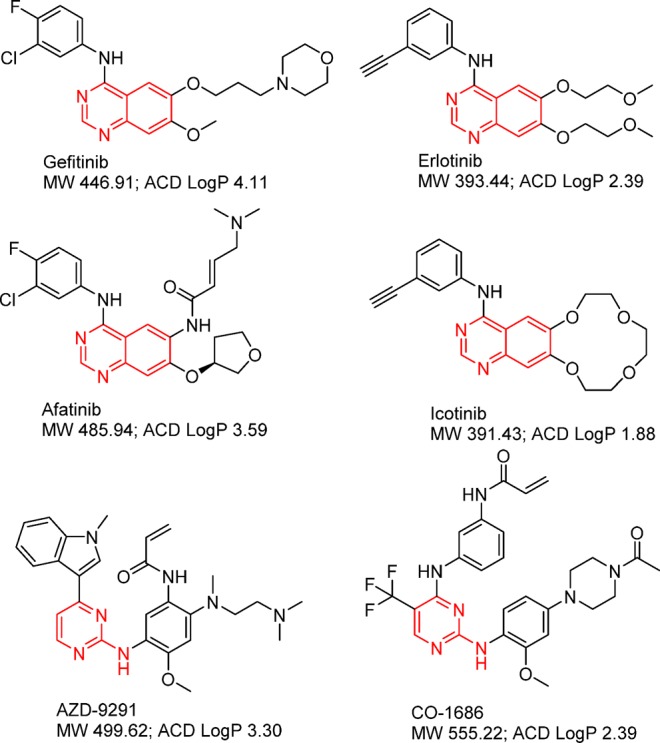
a The group in red functions as a kinase hinge binder.
In the past 20 years, protein kinases have been emerging as major cancer drug targets, and more than 50% of current cancer drug discovery programs are focused on protein kinase inhibitors.6 Many cancers seem to depend heavily on the specific activation of one or a few TKs. Designing TKIs targeting the mutant TKs is expected to selectively impact tumor cells and result in better safety profiles to patients. The intratumor heterogeneity of cancer and the adaptation of tumors to drugs will remain challenges in the development of targeted therapies as well as to the clinical management of advanced cancers. The combination of different TKIs or TKIs with other therapeutic agents should result in potent anticancer effects. As molecular mechanisms of resistance begin to be elucidated, new strategies to overcome or prevent the development of resistance are beginning to emerge. Kinase-directed drug resistance necessitates strategies to develop multiple inhibitors that target different topographies of the kinase active site. This has been an effective strategy employed in protease design for managing retroviral infection and may be a future path for transforming cancer into chronic disease.
Protein kinases play important roles not only in cancer but also in many other diseases. The survey by Lahiry et al. found 50 kinases that underlie 67 distinct single-gene clinical entities, and approximately half of these disease-associated kinases were TKs.7 Mutations in members of the kinase gene family underlie a broad range of disease phenotypes, including neurologic disorders, skeletal and craniosynostosis disorders, hematological and vascular disorders, immunological disorders, endocrine and metabolic disorders, and multiorgan disorders. The approval of tofacitinib, the first TKI for the treatment of inflammatory diseases, extends the small-molecule TKI for an indication outside oncology. Although there are 30 KIs entering into market and several hundred KIs in various stages of clinical development, the coverage of kinase targets and the structural diversity of KIs are limited. Deep understanding of protein kinase functions and their dysregulation in human diseases could lead to the identification of new kinase targets for unmet medicinal needs. Analyses of current kinase inhibitors indicate that the structural diversity, kinase selectivity, and toxicity profiles remain to be improved. The extension of TKIs in non-oncology fields and combination strategies for cancer therapies require the development of high quality TKIs. These certainly warrant further structural biology and medicinal chemistry efforts for discovering highly potent, selective, and safe TKIs.
Acknowledgments
The author is grateful to Drs. Ted Boritzki and Yishan Li for their critical reading, editing, and suggestions.
Views expressed in this editorial are those of the author and not necessarily the views of the ACS.
The authors declare no competing financial interest.
References
- Manning G.; Whyte D. B.; Martinez R.; Hunter T.; Sudarsanam S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [DOI] [PubMed] [Google Scholar]
- Barouch-Bentov R.; Sauer K. Mechanisms of drug resistance in kinases. Expert Opin. Invest. Drugs. 2011, 20, 153–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespan E.; Zucca E.; Maga G. Overcoming the drug resistance problem with second-generation tyrosine kinase inhibitors: from enzymology to structural models. Curr. Med. Chem. 2011, 18, 2836–2847. [DOI] [PubMed] [Google Scholar]
- Katayama R.; Shaw A. T.; Khan T. M.; Mino-Kenudson M.; Solomon B. J.; Halmos B.; Jessop N. A.; Wain J. C.; Yeo A. T.; Benes C.; Drew L.; Saeh J. C.; Crosby K.; Sequist L. V.; Iafrate A. J.; Engelman J. A. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci. Transl. Med. 2012, 4, 120ra17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw A. T.; Mehra R.; Kim D.-W.; Felip E.; Chow L. Q. M.; Camidge D. R.; Tan D. S.-W.; Vansteenkiste J. F.; Sharma S.; De Pas T.; Wolf J.; Katayama R.; Lau Y.-Y. Y.; Goldwasser M.; Boral A.; Engelman J. A. Clinical activity of the ALK inhibitor LDK378 in advanced, ALK-positive NSCLC. J. Clin. Oncol. 2013, 31, 8010. [Google Scholar]
- Cohen P.; Alessi D. R. Kinase drug discovery: what’s next in the field?. ACS Chem. Biol. 2013, 8, 96–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiry P.; Torkamani A.; Schork N. J.; Hegele R. A. Kinase mutations in human disease: interpreting genotype-phenotype relationships. Nat. Rev. Genet. 2010, 11, 60–74. [DOI] [PubMed] [Google Scholar]