

Abstract
Tubulin modulating agents such as the taxanes are among the most effective antimitotic cancer drugs, although resistance and toxicity present significant problems in their clinical use. However, most tubulin modulators are derived from complex natural products, which can make modification of their structure to address these problems difficult. Here, we report the discovery of new antimitotic compounds with simple structures that can be rapidly synthesized, through the phenotypic screening of a diverse compound library for the induction of mitotic arrest. We first identified a compound, which induced mitotic arrest in human cells at submicromolar concentrations. Its simple structure enabled rapid exploration of activity, defining a biphenylacetamide moiety required for activity, A family of analogues was synthesized, yielding optimized compounds that caused mitotic arrest and cell death in the low nanomolar range, comparable to clinically used antimitotic agents. These compounds can be synthesized in 1–3 steps and good yields. We show that one such compound targets tubulin, partially inhibiting colchicine but not vinblastine binding, suggesting that it acts allosterically to the known colchicine-binding site. Thus, our results exemplify the use of phenotypic screening to identify novel antimitotic compounds from diverse chemical libraries and characterize a family of biphenylacetamides (biphenabulins) that show promise for further development.
Keywords: Phenotypic screening, antimitotics, tubulin, colchicine, allosteric
Phenotypic screening involves testing small molecules in cellular or in vivo models directly, without any target bias.1 It has recently seen a resurgence, with the obervation that most first-in-class drugs with novel mechanisms of action originate from such approaches.2 Historically, the most successful drugs have been identified in this way. Phenotypic screening is particularly suitable when diverse compound collections are available. These are predicted to display a broader range of biological activity, which can be identified more efficiently when a range of targets can elicit the same phenotype when modulated. Diverse chemical libraries are often produced by diversity-oriented synthesis (DOS).3−6 This approach aims to build complex diverse small molecule libraries in an efficient manner. The combination of DOS with phenotypic screening has been successful in identifying antibacterial, antimalarial, and anticancer compounds, validating this approach.7−9
Our group has recently produced several distinct libraries using DOS and other more focused approaches,10−12 and their potential to modulate biological systems has not been evaluated. Recently, we have described an optimized phenotypic high content screen (HCS) to identify compounds that cause mitotic arrest. Causing mitotic arrest is a validated anticancer mechanism, and several mitotic inhibitors are approved clinically.13,14 All approved compounds target tubulin,15−17 and many unfortunately suffer from administration and/or resistance problems.18 Additionally, as current clinical compounds are mostly complex natural products or derivatives thereof, modification to improve properties can be limited to a few functional handles. Despite this, impressive progress has been made in the total synthesis and derivatization of tubulin-targeted antimitotics.19−21 New antimitotic compounds could offer an alternative to existing therapies, and several groups have dedicated significant efforts in this area, utilizing natural product inspired compound collections,22−24 as well as more traditional screening approaches.25−28 Our own efforts in this area resulted in the identification of dosabulin, a novel tubulin inhibitor from a DOS library of 35 compounds.29 In this work, we describe the evaluation of several compound libraries produced by DOS and other methods, for the induction of mitotic arrest. We identify one compound that causes significant mitotic arrest followed by cancer cell death. We obtain structure–activity relationships (SARs) to discover more potent analogues, and the mechanism of action is identified as perturbation of tubulin dynamics by binding to a site that causes partial displacement of colchicine from tubulin. These compounds, termed biphenabulins, are structurally simple and can be assembled in just 2–3 synthetic steps, providing a potential future alternative to currently used microtubule targeting agents (MTAs) and promising molecules for further optimization and biological evaluation.
Using an optimized assay for identifying molecules that cause mitotic arrest,11,30 a library of over 400 compounds was screened. The library was composed of recently reported DOS efforts focusing on macrocycles,10−12 as well as more focused compound sets from methodology or medicinal chemistry efforts in our group (Supplementary Figure S1). The assay is undertaken in U2OS osteosarcoma cells and involves staining for phosphorylated histone H3 (pH3), a mitotic marker, following 20 h treatment with the test compounds. Cells are visualized with Hoechst and imaged with the Cellomics Arrayscan, a high content microscope. An automated algorithm then calculates the percentage of cells arrested in mitosis. The primary screen consisted of screening all compounds at a single concentration of 50 μM in triplicate.29
From the primary screen one compound (1), which caused mitotic arrest in 40–50% of all cells at 50 μM, was identified (Figure 1). This hit was reconfirmed and showed a clear dose-dependent effect, with an EC50 for mitotic arrest of 0.51 μM. This was particularly significant as the structure of the molecule was very simple and thus amenable to optimization. With this result in hand, we sought to investigate the mode of action of 1 and determine the SARs. To synthesize analogues, several similar strategies relying on amide and Suzuki couplings were employed (Scheme 1). Commercial carboxylic acids were subjected to amide coupling conditions with furfurylamine to explore right-hand side substitutions. All couplings proceeded in moderate to good yields (2a–f, 45–69%) using T3P and DIPEA in ethyl acetate. Using similar amide coupling conditions, 10 analogues were synthesized in moderate to excellent yield (3a–j, 32–98%) from biphenyl acetate and commercial amines. In most cases, column chromatography was not required to purify the final compounds. To further investigate SARs on the right-hand side of the molecule, a range of Suzuki couplings with commercially available boronic acids were performed with the para-bromoacetamide intermediates 4a and 4b. Finally, amide couplings on building block 6 delivered p-amido substituted analogues 7a–b in moderate yield.
Figure 1.
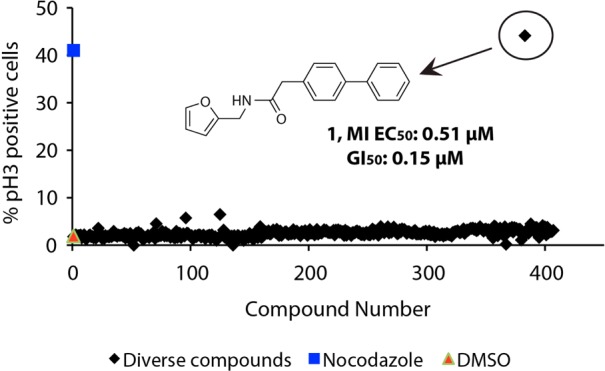
Data of the phenotypic screen for mitotic arrest. Each compound was screened at 50 μM in U2OS cells, which were stained with Hoechst and a phospho-histone H3 specific antibody. The structure and associated data for hit compound 1 are overlaid. Data shown as the mean of a single experiment conducted in triplicate; nocodazole was used at 200 nM.
Scheme 1. Synthesis of Antimitotic Biphenylacetamides and Analogues.

The importance of the biphenylacetate moiety was initially investigated. This was replaced with a series of aromatic and benzylic groups (Table 1, compounds 2a–f). Interestingly, only the naphthyl acetate group retained some antimitotic activity, albeit at higher concentrations than 1, while all other analogues were inactive. Having established that the biphenylacetate group was important for activity, we began exploring the amine substituents (Table 1, 3a–j). All compounds with the exception of the benzylamine-substituted 3a were less active than 1. Compound 3a exhibited a 2-fold increase in activity, which could be attributed to the increase in lipophilicity. Methylation on the benzylic carbon (3b and 3c) was not tolerated, with the (S)-enantiomer 3c being completely inactive, while the (R)-enantiomer was an order of magnitude less potent than 1. Methylation on the amide nitrogen was also not tolerated (3d), leading to a reduction in activity of almost 2 orders of magnitude. Substitution at the para-position led to a small reduction in activity (3e), while substitution with a bromide at the meta-position led to complete loss of activity (3f). Pyridines 3g and 3h also showed a significant reduction in activity, while substitution for a phenyl (3i) or aliphatic iso-propyl group (3j) completely abolished activity.
Table 1. Synthesis and Biological Evaluation of Biphenylacetamides with Differing Substituents on the Left- and Right-Hand Sides.
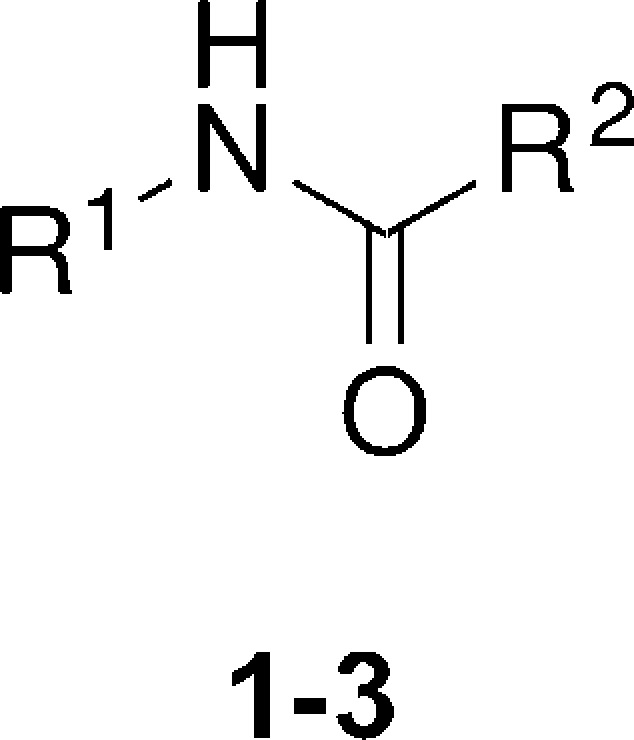
| Code | -R1 | -R2 | mitotic index (MI) EC50 (μM)a |
|---|---|---|---|
| 1 | 2-furfuryl | 4-phenylbenzyl | 0.51 |
| 2a | 2-furfuryl | 2- naphthylmethyl | 12.9 |
| 2b | 2-furfuryl | 2-phenylbenzene | >40 |
| 2c | 2-furfuryl | 4-benzoylphenyl | >40 |
| 2d | 2-furfuryl | 2-benzofuran | >40 |
| 2e | 2-furfuryl | 3-quinoline | >40 |
| 2f | 2-furfuryl | biphenyl | >40 |
| 3a | benzyl | 4-phenylbenzyl | 0.25 |
| 3b | (R)-N-(1-phenyl ethyl) | 4-phenylbenzyl | 4.83 |
| 3c | (S)-N-(1-phenyl ethyl) | 4-phenylbenzyl | >40 |
| 3d | N-(Me)-N-(Bn) | 4-phenylbenzyl | 29.9 |
| 3e | p-methoxybenzyl | 4-phenylbenzyl | 1.90 |
| 3f | m-bromobenzyl | 4-phenylbenzyl | >40 |
| 3g | 4-pyridyl | 4-phenylbenzyl | 16.6 |
| 3h | 2-pyridyl | 4-phenylbenzyl | 9.70 |
| 3i | phenyl | 4-phenylbenzyl | >40 |
| 3j | iso-butyl | 4-phenylbenzyl | >40 |
MI EC50 data is the mean of at least two independent experiments conducted in triplicate.
It must be noted that because the SAR was obtained from a phenotypic screen, factors such as cell permeability and metabolic stability may affect activity. To address this issue, physiochemical properties were calculated for all molecules synthesized in this series using Schrodinger’s QikProp application (Supplementary Table 1).31 Their cell permeability, clogP, and solubility were not predicted to vary greatly between the series, suggesting that phenotypic SAR may be a viable surrogate in the absence of on-target activity. In summary, even small substitutions in the left-hand portion of the molecules had a very big impact on activity. This suggests that it is not possible to obtain large increases in activity from changes in this region.
To investigate SAR on the right-hand side of the molecule, the para-bromoacetamide intermediates 4a and 4b, were synthesized in one step using standard amide coupling conditions. Following this, a range of Suzuki couplings with commercially available boronic acids were performed to explore different substitution patterns at this position. The substrates selected were predominantly aromatics with substituents of differing sizes and electronic properties attached to different positions. Twelve final compounds (5a–l) were screened for their ability to induce mitotic arrest in U2OS cells (Table 2).
Table 2. Effect of Substitution on the Right-Hand Side Aryl Ring on the Induction of Mitotic Arrest and Growth Inhibition in U2OS Cellsa.
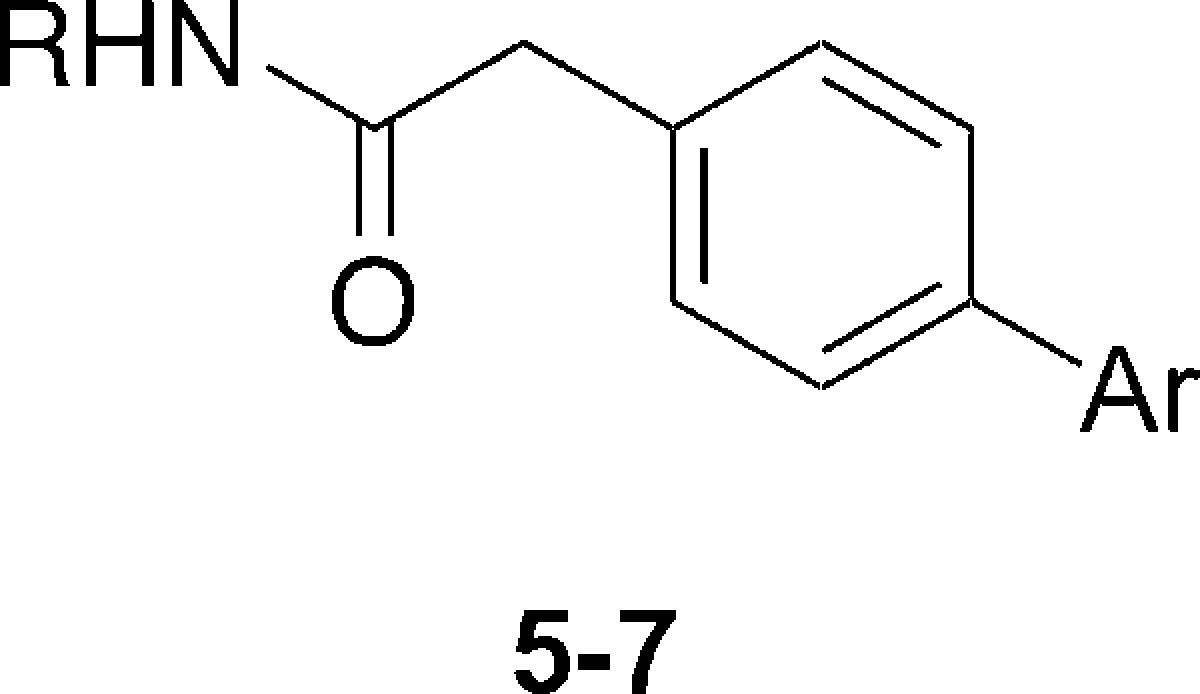
| code | R | Ar | mitotic index (MI) EC50 (μM) | growth inhibition GI50 (μM) |
|---|---|---|---|---|
| 1 | 2-furfuryl | phenyl | 0.51 | 0.15 |
| 5a | 2-furfuryl | 2-acetylphenyl | 12.9 | N/D |
| 5b | 2-furfuryl | 3-acetamido phenyl | 0.490 | 0.120 |
| 5c | 2-furfuryl | 3-fluoro-4-methyl phenyl | 0.340 | 0.100 |
| 5d | 2-furfuryl | 3,4,5-trimethoxyl phenyl | >40 | N/D |
| 5e | 2-furfuryl | 3,5-dimethyl-4-methoxy phenyl | 1.17 | 0.603 |
| 5f | 2-furfuryl | 4-trifluoromethoxy phenyl | N/D | 0.120 |
| 5g | 2-furfuryl | 4-acetyl phenyl | 0.076 | 0.024 |
| 5h | 2-furfuryl | 4-benzoate methyl ester | 0.073 | 0.024 |
| 5i | 2-furfuryl | 4-cyanophenyl | 0.526 | 0.111 |
| 5j | 2-furfuryl | benzodioxole | 0.144 | 0.031 |
| 5k | benzyl | 4-acetyl phenyl | 0.028 | 0.011 |
| 5l | benzyl | 4-benzoate methyl ester | 0.081 | 0.027 |
| 6 | 2-furfuryl | 4-benzoic acid | >20 | N/D |
| 7a | 2-furfuryl | 4-(methyl carbamoyl) phenyl | 0.051 | 0.021 |
| 7b | 2-furfuryl | 4-(isobutyl carbamoyl) phenyl | 0.101 | 0.046 |
MI EC50 and GI50 values are the mean of at least two independent experiments conducted in triplicate. N/D = not determined.
A compound with an ortho-substituent (5a) was considerably less active than the original hit, while the activity of those containing a meta-substituent varied depending on the other substituents present. Compounds 5b and 5c both displayed similar activity to 1, whereas the trimethoxy-substituted 5d was completely inactive. Reducing the size and polarity of the substituents flanking the para-methoxy group with two methyl groups restored an active compound (5e), though this was still 2-fold less potent than 1. The trifluoromethoxy analogue (5f) displayed potent cytotoxicity; however, mitotic arrest was not measurable. This suggests that 5f is acting by a different mechanism. The trifluoromethoxy functional group is potentially reactive and thus was not pursued further. When a para-substituent bearing a carbonyl was introduced, a 7-fold increase in activity was observed. Both the ketone 5g and ester 5h displayed very high levels of activity in the mitotic index assay. The carbonyl functionality was essential for maximal levels of activity, as the para-nitrile 5i and benzodioxole 5j both displayed lower acitivities.
Having established the substituents that dictated activity in the left and right portions of the molecule, these were combined to observe whether an additive effect existed. For the left-hand side, the benzyl substituent was selected, while for the right-hand side the para-acetyl (5k) and para-methylester (5l) groups were chosen. Both compounds showed an approximate 2-fold increase in activity compared to the furan containing analogues 5g and 5h, consistent with the increase observed from 1 to 3a. Compound 5k was particularly active at inducing mitotic arrest and growth inhibition, with values in the low nanomolar range (Table 2 and Supplementary Figure S2). However, this compound was poorly soluble even in organic solvents at concentrations higher than 1 μM, which was predicted to cause problems at later stages of biological evaluation. At this stage it was also observed that the activity of 5l decreased over time. This could be attributed to a slow hydrolysis of the ester upon storage of the compound in DMSO. To overcome these problems, analogues with improved stability and solubility were sought. It was hypothesized that an analogue with an amide substituent at the para-position could improve stability. Returning to the furan or an alternative heterocycle on the left-hand side was predicted to improve solubility. The precursor for such compounds (6) was synthesized by Suzuki coupling and subjected to amide coupling conditions using a very small and a sterically hindered amine to assess what size substituent could be tolerated at this position (Table 2).
Both compounds synthesized retained very good levels of antimitotic and growth inhibitory activity, with 7a being the most active (Table 2 and Supporting Information Figure S2). This suggests that a larger amide substituent is less tolerated than a simple methyl amide, though the difference in activity was not large. The precursor containing the carboxylic acid (6) was completely inactive, which may be the result of a predicted lack of cell permeability (Supplementary Table S1).
Having obtained compounds with low nanomolar activity for mitotic arrest and growth inhibition, the identification of their targets was the next goal. Confocal microscopy was used for in-depth analysis of the phenotype resulting from compound treatment. The DNA dye 4′,6-diamidino-2-phenylindole (DAPI) and an α-tubulin specific antibody were selected to image mitotic arrest. Compounds 5k and 7a were selected for analysis, as these were the most potent and stable compounds identified. Images of the interphase cells showed a disrupted tubulin network, which suggested that tubulin itself could be the target of these molecules (Figure 2a).
Figure 2.

Target identification of the biphenylacetamide antimitotic compounds. (a) Confocal microscopy images of U2OS cells stained with DAPI (blue) and an α-tubulin specific antibody (green). Cells treated with 5k and biphenabulin (7a) showed a significantly disrupted tubulin network, suggesting that this may be their molecular target; scale bar, 18 μm. (b) Representative tubulin polymerization assay with compounds from the biphenylacetamide series. Tubulin (3 mg/mL) was incubated with test compounds and GTP at 37 °C for 1 h. All active compounds from the mitotic index assay were also inhibitors of tubulin polymerization, whereas 3f, the inactive analogue, was not. All compounds were tested at 10 μM. (c) A fluorescence polarization (FP) assay using FL-BODIPY-vinblastine (2 μM) as a tracer and tubulin (2 μM) showed that biphenabulin did not interfere with vinblastine binding to tubulin; data shown as mean ± s.e.m. for an experiment conducted in triplicate (ex/em wavelengths 485/520 nm). (d) Fluorescence intensity (FI) assay data of biphenabulin inhibiting tubulin at the colchicine site. Colchicine (3 μM), tubulin (3 μM), and biphenabulin were incubated for 1 h at 37 °C. While a dose-dependent decrease in FI was observed for this compound, it did not reach the same level of inhibition as the positive control nocodazole. This could suggest that biphenabulin is a tubulin inhibitor that acts near or allosteric to the colchicine site; data shown as mean ± s.e.m. for an experiment conducted in triplicate (ex/em wavelengths 365/435 nm).
Having identified a potential target of the series, several compounds were selected for testing in a tubulin polymerization assay.22 Four compounds with differing potencies in the cell-based mitotic index assay were selected to give a representative spread in activity (Figure 2b). All compounds that were active in the cell-based mitotic index assay were also inhibitors of tubulin polymerization, whereas the inactive compound 3f showed no activity in this assay. Interestingly, the order of activity in the mitotic index assay was also reflected in the tubulin polymerization assay, with 7a being the most potent at reducing polymerization followed by 1 and then 5i (Supplementary Table S2). Compound 7a was therefore termed biphenabulin, for biphenyl tubulin inhibitor. Nocodazole was significantly more potent at reducing the tubulin polymerization rate than the biphenylacetamides despite having similar potencies in the MI assay (EC50 = 50 nM for nocodazole and 30–70 nM for the biphenylacetamides). This could suggest a differing mechanism of inhibition of polymerization for the biphenylacetamide compounds. To investigate their potential binding site(s), biphenabulin was selected for further analysis as it displayed good antimitotic activity, solubility, and stability. Several binding sites on tubulin have been reported for inhibitors of tubulin polymerization of which the vinca alkaloid and the colchicine ones are the best studied.32,33 For other small molecules such as noscapine, the binding site has not yet been identified.23 The Vinca alkaloid and colchicine sites were selected for analysis, as we had recently optimized assays to study their engagement.29 Biphenabulin was initially tested in a fluorescence polarization assay monitoring the interaction of vinblastine with tubulin (Figure 2c). The data clearly showed that this compound was not able to inhibit the binding of fluorescent vinblastine to tubulin, while the positive control (unlabeled vinblastine) was.
Following this, biphenabulin was also assayed for its ability to inhibit tubulin at the colchicine site, using a fluorescence intensity (FI) assay previously described.29 A dose-dependent decrease in colchicine FI was seen with biphenabulin, although the decrease did not occur to the same extent as the positive control nocodazole (Figure 2d). This corresponded to a maximal inhibition of approximately 45% for biphenabulin, while nocodazole reached 85–90% inhibition. We would expect a molecule that bound in the same site as colchicine to completely displace it and thus reduce fluorescence, which could suggest that biphenabulin is a tubulin inhibitor that acts allosterically to the colchicine site, resulting in a reduction of the ability of tubulin to bind colchicine.
In summary, a high content screen of a small diverse library for induction of mitotic arrest was implemented. One compound was identified that arrested cells in mitosis at submicromolar concentration. Expedient synthesis of analogues resulted in the rapid exploration of SAR. After exploration of the SAR and having obtained compounds with low nanomolar activity for mitotic arrest and growth inhibition, the identification of their targets was the next goal.
Using confocal microscopy, a disrupted tubulin network was observed, and the mechanism of action was confirmed as tubulin depolymerization. One of the most active compounds, termed biphenabulin, was shown not to interfere with vinblastine binding to tubulin, but to partially inhibit colchicine binding. This suggests that it may be binding allosterically to the colchicine site, as direct inhibitors of the colchicine site such as nocodazole cause complete displacement of colchicine from tubulin. The mode of action is very similar to dosabulin, which we recently discovered. However, biphenabulin is 2 orders of magnitude more potent and is synthesized in fewer (3 compared to 7) steps. Additionally, only 27 analogues were required before molecules with a cellular potency in the low nanomolar range (similar to clinically used drugs such as vinblastine) were identified. Many anticancer drugs that target tubulin display formulation and resistance problems; therefore, new, simple modulators of tubulin dynamics amenable to rapid modification would provide promising alternatives. We hope that the molecules presented herein will serve as a starting point or inspiration for future efforts in this area.
Glossary
ABBREVIATIONS
- DOS
diversity-oriented synthesis
- HCS
high content screening
- SAR
structure–activity relationship
Supporting Information Available
Synthetic procedures and compound characterization as well as biological assay protocols. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written by L.L with contributions from all authors. All authors have given approval to the final version of the manuscript.
We thank Cancer Research U.K. (to L.L., A.R.V., and D.R.S.) and the U.K. Medical Research Council (to J.S., A.E., G.J.M., and A.R.V.) for support.
The authors declare no competing financial interest.
Supplementary Material
References
- O’Connor C. J.; Laraia L.; Spring D. R. Chemical genetics. Chem. Soc. Rev. 2011, 40, 4332–4345. [DOI] [PubMed] [Google Scholar]
- Swinney D. C.; Anthony J. How were new medicines discovered?. Nat. Rev. Drug. Discovery 2011, 10, 507–519. [DOI] [PubMed] [Google Scholar]
- Schreiber S. L. Target-oriented and diversity-oriented organic synthesis in drug discovery. Science 2000, 287, 1964–1969. [DOI] [PubMed] [Google Scholar]
- Galloway W. R. J. D.; Isidro-Llobet A.; Spring D. R. Diversity-oriented synthesis as a tool for the discovery of novel biologically active small molecules. Nat. Commun. 2010, 1, 80. [DOI] [PubMed] [Google Scholar]
- O’Connor C. J.; Beckmann H. S. G.; Spring D. R. Diversity-oriented synthesis: producing chemical tools for dissecting biology. Chem. Soc. Rev. 2012, 41, 4444–4456. [DOI] [PubMed] [Google Scholar]
- Tan D. S. Diversity-oriented synthesis: exploring the intersections between chemistry and biology. Nat. Chem. Biol. 2005, 1, 74–84. [DOI] [PubMed] [Google Scholar]
- Thomas G. L.; Spandl R. J.; Glansdorp F. G.; Welch M.; Bender A.; Cockfield J.; Lindsay J. A.; Bryant C.; Brown D. F. J.; Loiseleur O.; Rudyk H.; Ladlow M.; Spring D. R. Anti-MRSA agent discovery using diversity-oriented synthesis. Angew. Chem., Int. Ed. 2008, 47, 2808–2812. [DOI] [PubMed] [Google Scholar]
- Heidebrecht R. W.; Mulrooney C.; Austin C. P.; Barker R. H.; Beaudoin J. A.; Cheng K. C.-C.; Comer E.; Dandapani S.; Dick J.; Duvall J. R.; Ekland E. H.; Fidock D. A.; Fitzgerald M. E.; Foley M.; Guha R.; Hinkson P.; Kramer M.; Lukens A. K.; Masi D.; Marcaurelle L. A.; Su X.-Z.; Thomas C. J.; Weiwer M.; Wiegand R. C.; Wirth D.; Xia M.; Yuan J.; Zhao J.; Palmer M.; Munoz B.; Schreiber S. Diversity-oriented synthesis yields a novel lead for the treatment of malaria. ACS Med. Chem. Lett. 2012, 3, 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S.; Nam H. J.; Park J.; Beak S. H.; Park S. B. Development of a benzopyran-containing androgen receptor antagonist to treat antiandrogen-resistant prostate cancer. ChemMedChem 2010, 5, 529–533. [DOI] [PubMed] [Google Scholar]
- Isidro-Llobet A.; Murillo T.; Bello P.; Cilibrizzi A.; Hodgkinson J. T.; Galloway W. R. J. D.; Bender A.; Welch M.; Spring D. R. Diversity-oriented synthesis of macrocyclic peptidomimetics. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 6793–6798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell K. M. G.; Beckmann H. S. G.; Laraia L.; Horsley H. T.; Bender A.; Venkitaraman A. R.; Spring D. R. A two-directional strategy for the diversity-oriented synthesis of macrocyclic scaffolds. Org. Biomol. Chem. 2012, 10, 7545–7551. [DOI] [PubMed] [Google Scholar]
- Beckmann H. S. G.; Nie F.; Hagerman C. E.; Johansson H.; Tan Y. S.; Wilcke D.; Spring D. R. A strategy for the diversity-oriented synthesis of macrocyclic scaffolds using multidimensional coupling. Nat. Chem. 2013, 5, 861–867. [DOI] [PubMed] [Google Scholar]
- Manchado E.; Guillamot M.; Malumbres M. Killing cells by targeting mitosis. Cell Death Differ. 2012, 19, 369–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan K. S.; Koh C. G.; Li H. Y. Mitosis-targeted anti-cancer therapies: where they stand. Cell Death Dis. 2012, 3, e411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumontet C.; Jordan M. A. Microtubule-binding agents: a dynamic field of cancer therapeutics. Nat. Rev. Drug Discovery 2010, 9, 790–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan M. A.; Wilson L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [DOI] [PubMed] [Google Scholar]
- Perez E. A. Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol. Cancer Ther. 2009, 8, 2086–2095. [DOI] [PubMed] [Google Scholar]
- Kavallaris M. Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer 2010, 10, 194–204. [DOI] [PubMed] [Google Scholar]
- Smith A. B.; Freeze B. S.; Xian M.; Hirose T. Total synthesis of (+)-discodermolide: a highly convergent fourth-generation approach. Org. Lett. 2005, 7, 1825–1828. [DOI] [PubMed] [Google Scholar]
- Ishikawa H.; Colby D. A.; Boger D. L. Direct coupling of catharanthine and vindoline to provide vinblastine: total synthesis of (+)-and ent-(−)-vinblastine. J. Am. Chem. Soc. 2008, 130, 420–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganem B.; Franke R. R. Paclitaxel from primary taxanes: Perspective on creative invention in organozirconium chemistry. J. Org. Chem. 2007, 72, 3981–3987. [DOI] [PubMed] [Google Scholar]
- Voigt T.; Gerding-Reimers C.; Ngoc Tran T. T.; Bergmann S.; Lachance H.; Schölermann B.; Brockmeyer A.; Janning P.; Ziegler S.; Waldmann H. A natural product inspired tetrahydropyran collection yields mitosis modulators that synergistically target CSE1L and tubulin. Angew. Chem., Int. Ed. 2013, 52, 410–414. [DOI] [PubMed] [Google Scholar]
- Zimmermann T. J.; Roy S.; Martinez N. E.; Ziegler S.; Hedberg C.; Waldmann H. Biology-oriented synthesis of a tetrahydroisoquinoline-based compound collection targeting microtubule polymerization. ChemBioChem 2013, 14, 295–300. [DOI] [PubMed] [Google Scholar]
- Antonchick A. P.; Gerding-Reimers C.; Catarinella M.; Schuermann M.; Preut H.; Ziegler S.; Rauh D.; Waldmann H. Highly enantioselective synthesis and cellular evaluation of spirooxindoles inspired by natural products. Nat. Chem. 2010, 2, 735–740. [DOI] [PubMed] [Google Scholar]
- Yang W. S.; Shimada K.; Delva D.; Patel M.; Ode E.; Skouta R.; Stockwell B. R. Identification of simple compounds with microtubule-binding activity that inhibit cancer cell growth with high potency. ACS Med. Chem. Lett. 2011, 3, 35–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tcherniuk S.; Deshayes S.; Sarli V.; Divita G.; Abrieu A. UA62784 is a cytotoxic inhibitor of microtubules, not CENP-E. Chem. Biol. 2011, 18, 631–641. [DOI] [PubMed] [Google Scholar]
- Yi X.; Zhong B.; Smith K. M.; Geldenhuys W. J.; Feng Y.; Pink J. J.; Dowlati A.; Xu Y.; Zhou A.; Su B. Identification of a class of novel tubulin inhibitors. J. Med. Chem. 2012, 55, 3425–3435. [DOI] [PubMed] [Google Scholar]
- Chen J.; Ahn S.; Wang J.; Lu Y.; Dalton J. T.; Miller D. D.; Li W. Discovery of novel 2-aryl-4-benzoyl-imidazole (ABI-III) analogues targeting tubulin polymerization as antiproliferative agents. J. Med. Chem. 2012, 55, 7285–7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibbeson B. M.; Laraia L.; Alza E.; O’Connor C. J.; Tan Y. S.; Davies H. M. L.; McKenzie G. J.; Venkitaraman A. R.; Spring D. R. Diversity-oriented synthesis as a tool for identifying new modulators of mitosis. Nat. Commun. 2014, 5, 3155. [DOI] [PubMed] [Google Scholar]
- Zheng S.; Aves S. J.; Laraia L.; Galloway W. R. J. D.; Pike K. G.; Wu W.; Spring D. R. A concise total synthesis of deoxyschizandrin and exploration of its antiproliferative effects and those of structurally related derivatives. Chem.—Eur. J. 2012, 18, 3193–3198. [DOI] [PubMed] [Google Scholar]
- QikProp, version 3.8; Schrödinger, LLC.: New York, 2013. [Google Scholar]
- Gigant B.; Wang C.; Ravelli R. B. G.; Roussi F.; Steinmetz M. O.; Curmi P. A.; Sobel A.; Knossow M. Structural basis for the regulation of tubulin by vinblastine. Nature 2005, 435, 519–522. [DOI] [PubMed] [Google Scholar]
- Ravelli R. B. G.; Gigant B.; Curmi P. A.; Jourdain I.; Lachkar S.; Sobel A.; Knossow M. Insight into tubulin regulation from a complex with colchicine and a stathmin-like domain. Nature 2004, 428, 198–202. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.