

Abstract
Human African Trypanosomiasis (HAT) is a severe, often fatal disease caused by the parasitic protist Trypanosoma brucei. The glycolytic pathway has been identified as the sole mechanism for ATP generation in the infective stage of these organisms, and several glycolytic enzymes, phosphofructokinase (PFK) in particular, have shown promise as potential drug targets. Herein, we describe the discovery of ML251, a novel nanomolar inhibitor of T. brucei PFK, and the structure–activity relationships within the series.
Keywords: Trypanosoma brucei, Trypanosoma cruzi, phosphofructokinase, inhibitors, glycolysis, high-throughput screening
Human African Trypanosomiasis (HAT), also known as sleeping sickness, is a disease caused by the parasitic protist, Trypanosoma brucei, and is endemic to sub-Saharan Africa. More than 60 million people are estimated to be at risk of HAT infection, due to the widespread infestation of the parasite’s tsetse fly vector, as well as the density of humans and cattle in the region, which serve as reservoirs for the causative parasites.1,2 HAT is almost always fatal if left untreated, and classic therapies used to treat it have historically suffered from a range of deleterious side effects, due in part to their nonselective mechanisms of action, and most approved therapies require either intravenous or intramuscular administration, further complicating treatment.3,4 Second-stage HAT, characterized by the infiltration of parasites into the central nervous system, remains a particular challenge because of the need for therapeutic molecules to cross the blood–brain barrier.5 Furthermore, growing resistance to these drugs has highlighted a need for the identification of novel therapies.6
The glycolytic enzyme phosphofructokinase (PFK) has recently been recognized as a potential drug target in T. brucei, as the infectious bloodstream stage of the parasite is solely dependent on the metabolism of glucose for ATP generation.7−10 PFK catalyzes an essentially irreversible reaction under physiological conditions in parasites and thus represents the first committed step of glycolysis.11 In addition, the necessity of glycolysis for parasite growth has been confirmed by siRNA-mediated knockdown; a 50% decrease in glycolytic flux was found to be sufficient to significantly decrease parasite viability, further suggesting that inhibition of glycolysis may be a viable strategy for HAT therapy.9 Currently, no inhibitors of Tb PFK have been described that demonstrate submicromolar potency or selectivity for the enzyme. Reported inhibitors include suramin, a classic antitrypanosomal drug in use since 1920 with noted promiscuous activity, inhibiting 80 assays among 370 tested in PubChem (IC50 ≤ 1 μM in 26 assays), as well as a furanose based analogue reported by Nowicki et al. with an IC50 of 23 μM (Figure 1).11,12
Figure 1.

(a) Chemical structures of Tb PFK inhibitors suramin, a glycoside analogue, and our qHTS hit (1). Compound 1 is an obvious derivative of the antibiotic sulfamethoxazole, which shows no inhibitory activity against PFK. (b) Synthetic route to 1, which was the general synthetic route to most analogues in Tables 1 and 2. (c) Synthetic route to 31, which was also used for analogues in Table 3
We report here the discovery and structure–activity relationship (SAR) of novel and potent inhibitors of T. brucei and T. cruzi PFK. A library of 330,683 compounds from the Molecular Libraries Small Molecule Repository (MLSMR, http://mli.nih.gov/mli/compound-repository/mlsmr-compounds/) was screened at 6 concentrations (spanning a concentration range from 57.5 μM to 3.7 nM) against recombinant Tb PFK for inhibitory activity.13 PFK activity was assessed by coupling PFK-mediated ADP production to a modified luciferase-based detection assay (ADP-Glo), providing a luminescent end point readout. A stepwise description of the 1536-well assay is shown in Supplementary Table 1. Complete screening and follow-up data have been made available in PubChem (PubChem BioAssay summary identifier 488768; http://pubchem.ncbi.nlm.nih.gov/assay/assay.cgi?aid=488768). Subsequent hit confirmation with an orthogonal ATP depletion-based assay (description in Supplementary Table 2; data not shown) led to the identification of the para-amidosulfonamide scaffold as one of the hit series, typified by the screening hit (1) with an IC50 of 1.1 μM (Figure 1). While 1 contains the same core as the antibiotic sulfamethoxazole, the latter shows no inhibitory activity against T. brucei PFK.
The resynthesis of 1 is shown in Figure 1b and followed a simple sequence of sulfonamide formation, acid catalyzed acetyl hydrolysis, and finally amide formation. Gratifyingly, the resynthesized version had an IC50 of 410 nM (Table 1). Though the original screening campaign was designed to identify inhibitors of PFK in T. brucei, the related organism, Trypanosoma cruzi, responsible for Chagas disease, possesses a similar reliance on ATP generation through glycolysis; the T. brucei and T. cruzi PFK isoforms display 77% overall sequence identity, with greater than 90% sequence identity within the active site.14 As such, the general synthetic strategy shown in Figure 1b was used to initiate synthesis, with all analogues being tested against both T. brucei and T. cruzi PFK isoforms. The SAR tracked very well with few exceptions between the species, though the discussion of SAR here will focus solely on T. brucei activity. The first changes attempted were to understand if we could modify the para-amidosulfonamide core (Table 1). The 1,4-disubstitution arrangement on the phenyl ring proved to be optimal as the meta-substituted analogue (2) suffered from greatly diminished activity. Attempts to change to saturated (3), alkyl chain (4), and heteroaryl (5) cores all resulted in less active analogues. This initial round of SAR concluded with efforts to understand the importance of the sulfonamide and amide linkers. Very tight SAR was observed with respect to both of these functionalities, as indicated in analogues 6–12.
Table 1. SAR Assessing the Core of PFK Inhibitors.
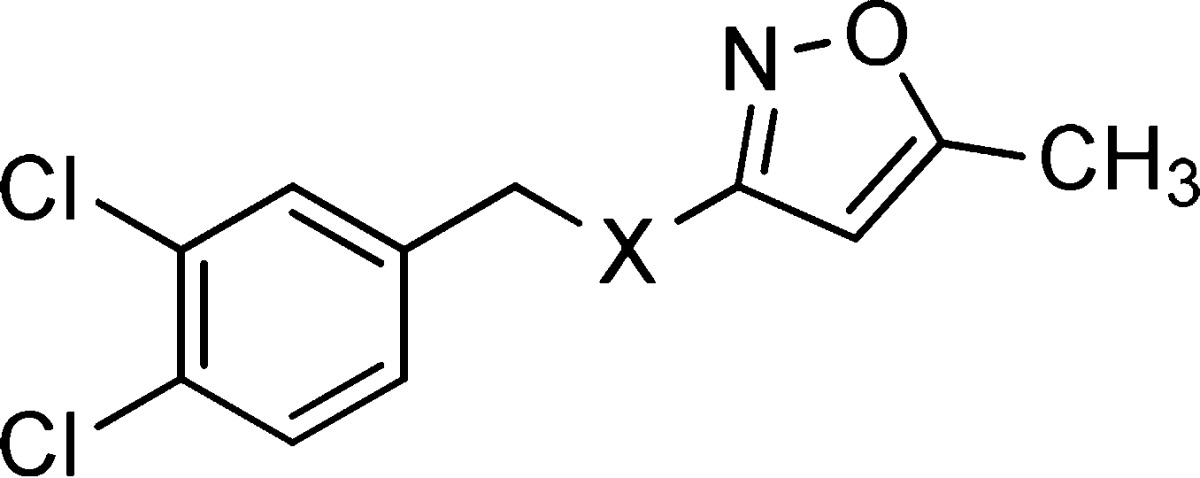
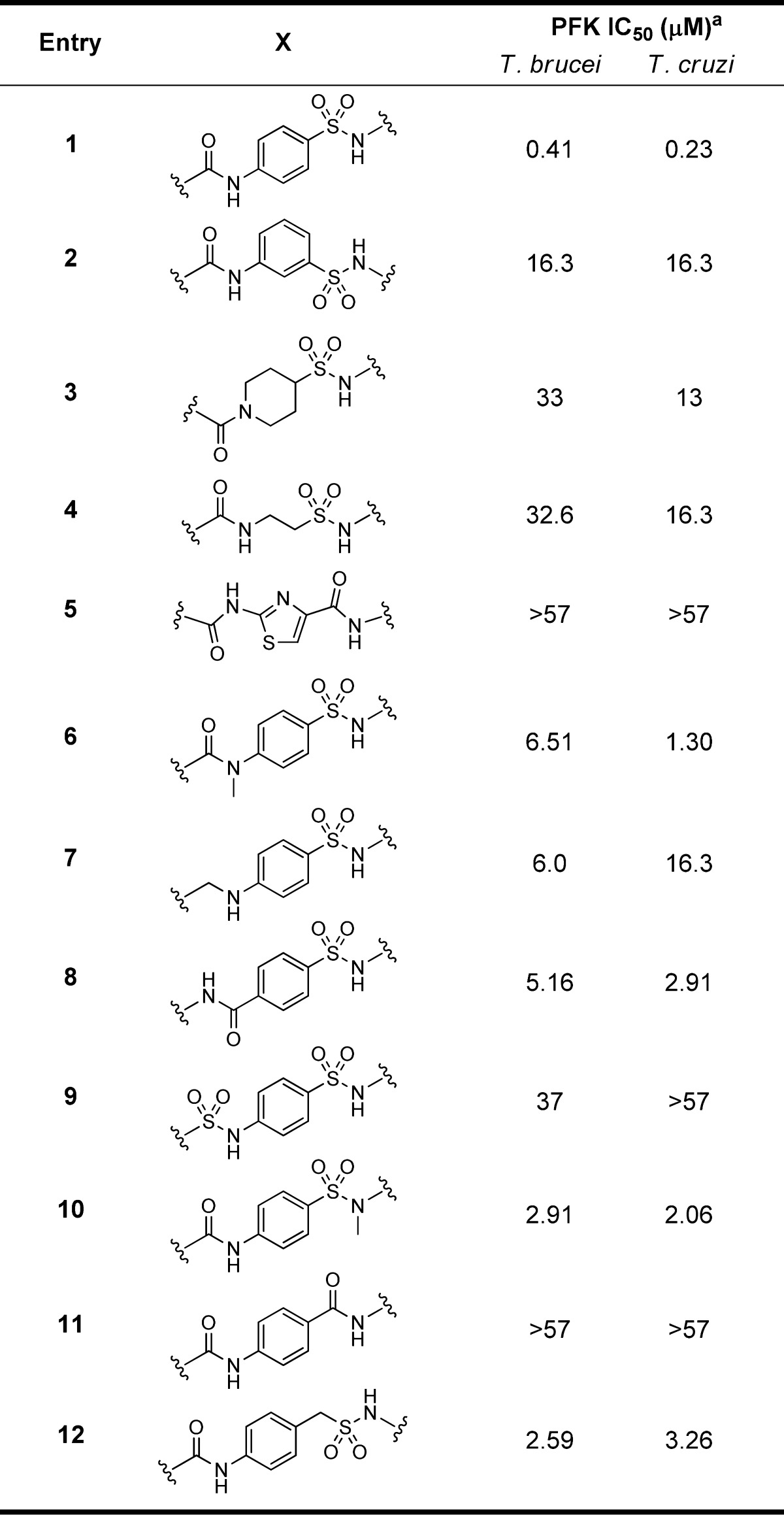
IC50 values represent the average of at least 3 separate experiments reported as the half maximal (50%) inhibitory concentration as determined in the ADP-Glo assay. All IC50s <57 μM have a maximum inhibition of >80% at 57 μM.
As a number of hits from the quantitative high-throughput screen (qHTS) campaign, as well as the furanose analogue reported by Nowicki, contained the 3,4-dichlorobenzyl motif present in 1, we wanted to test the role of these substituents for inhibitory activity (Table 2). While both single- and double-point deletions of the chloro groups led to less active compounds (13–15), we realized that chloro substitution at the 3- or 4-position contributed significantly to the potency as the phenyl analogue was 5- and 20-fold less potent than the 4- and 3-chloro analogues, respectively, for T. brucei. Using this information, we next synthesized a few analogues with p-halo or -methyl substituents and found that the p-halo analogues (17–19) had similar activity among themselves and slightly diminished activity compared to the dichloro analogue. The p-methyl analogue (20) showed a ∼8-fold loss in potency compared to 18 highlighting the importance of these halogens at the 4-position. Further manipulations of these 3,4-substituents led to significantly lower or a complete loss of activity (21–24). Finally, removal, extension, and substitution of the benzylic methylene group all resulted in reduced potencies (25–27).
Table 2. SAR Assessing the Benzyl Group of PFK Inhibitors.
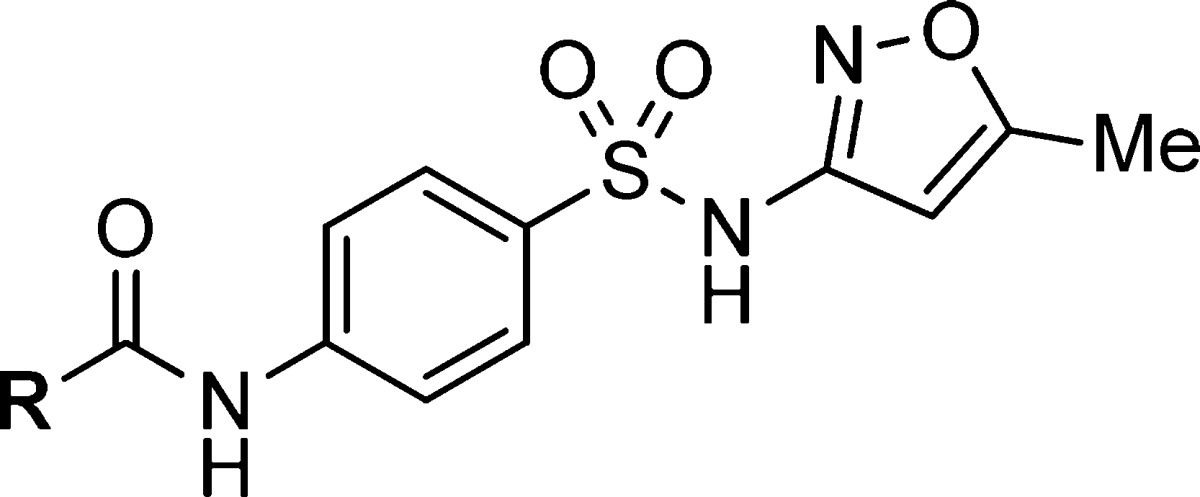
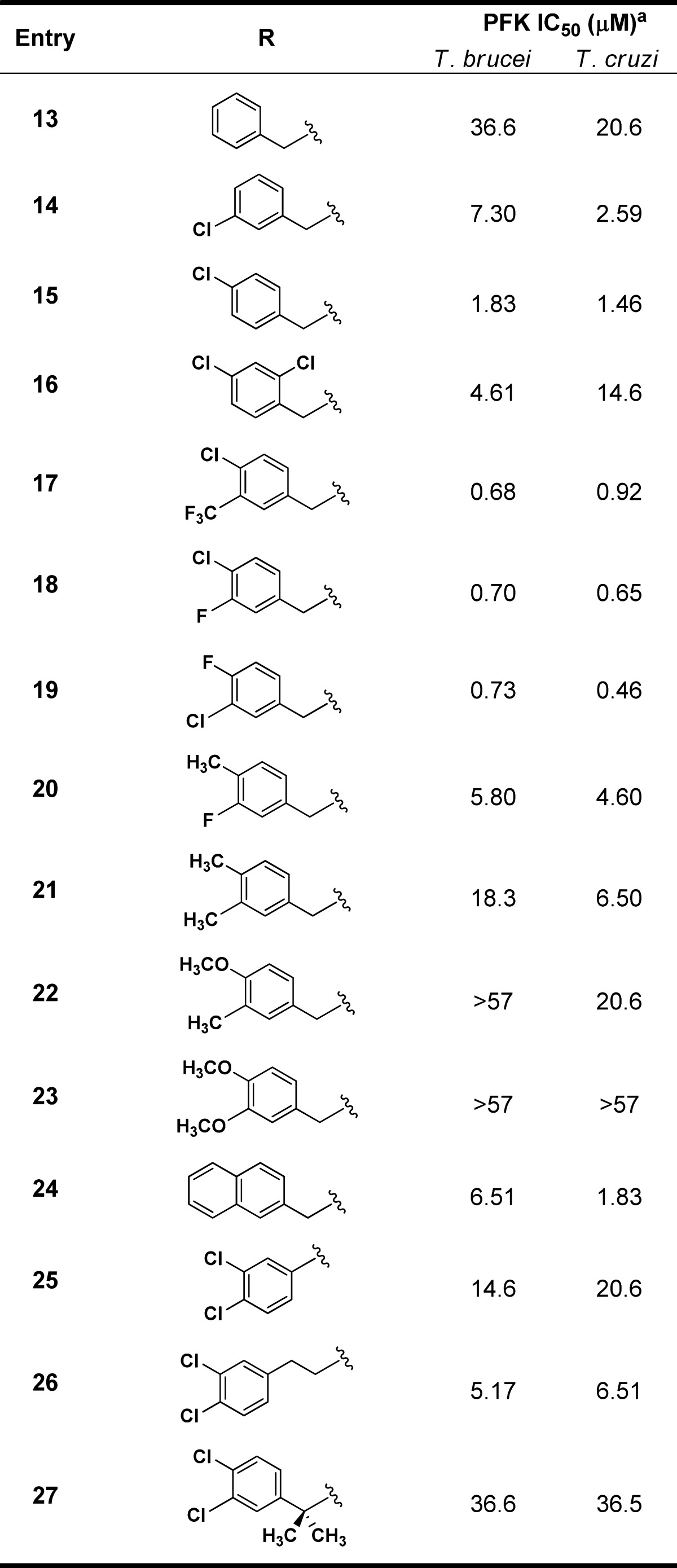
IC50 values represent the average of at least 3 separate experiments reported as the half maximal (50%) inhibitory concentration as determined in the ADP-Glo assay. All IC50s <57 μM have a maximum inhibition of >80% at 57 μM.
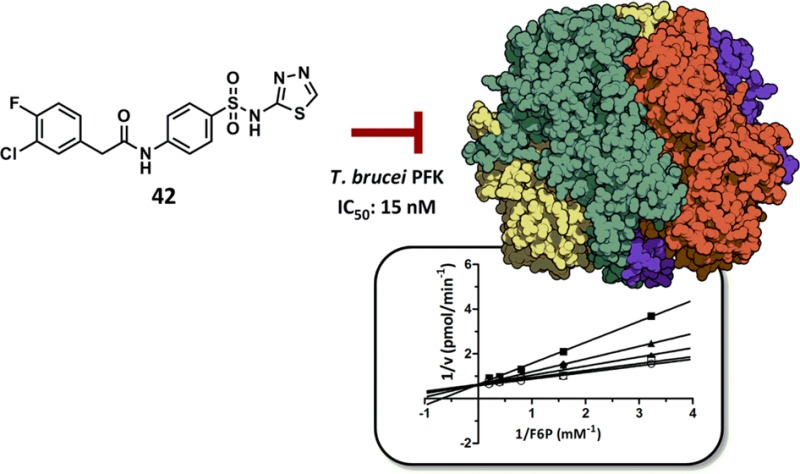
Realizing that the 3,4-dihalo substitution pattern was key for activity, this motif was carried on to an additional round of analogues aimed at investigating the SAR around the isoxazole moiety using the chemistry shown in Figure 1c (Table 3). Interestingly, sulfonic acid analogue 28 had submicromolar activity while the more hydrophobic phenyl analogue (29) lost considerable activity. The 5-des-methyl analogue 30 (ML251) showed a slight improvement in potency compared to the hit 1 and led us to hypothesize that analogues with hydrophilic and/or hydrogen bond donors/acceptors at this position may lead to improved activity. Therefore, we synthesized analogues 31–42 containing various heterocycles and substituted phenyl groups. While pyrazole analogues 31 and 32 had appreciable activity, a significant jump in potency against PFKs from both species was observed for the carboxylic acids 33 and 35. Interestingly, the parent ester (34) had no inhibitory activity. Pyrimidine, phenol, and aniline containing analogues 36–38 all showed diminished activity, leading us to investigate small heterocyclic analogues. Switching from the isoxazole to a thiazole resulted in an improvement in activity with analogues 39 and 40 having 79 and 24 nM potency, respectively, for T. brucei and 41 and 120 nM for T. cruzi, respectively. The thiadiazole analogue (41) saw only a very slight decrease in activity against both T. brucei and T. cruzi compared to 39. Interestingly, the thiadiazole analogue with the 4-chloro-3-fluoro substitution pattern (42) gave the most potent analogue against T. brucei at (IC50 = 15 nM), while 39 remained the most potent inhibitor of T. cruzi (IC50 = 41 nM).
Table 3. SAR of the Heterocycle.
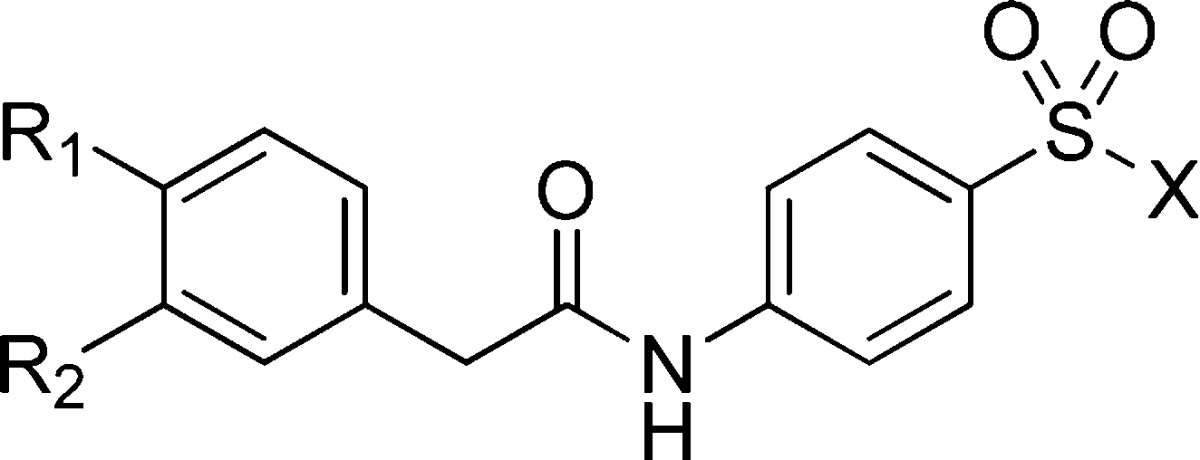
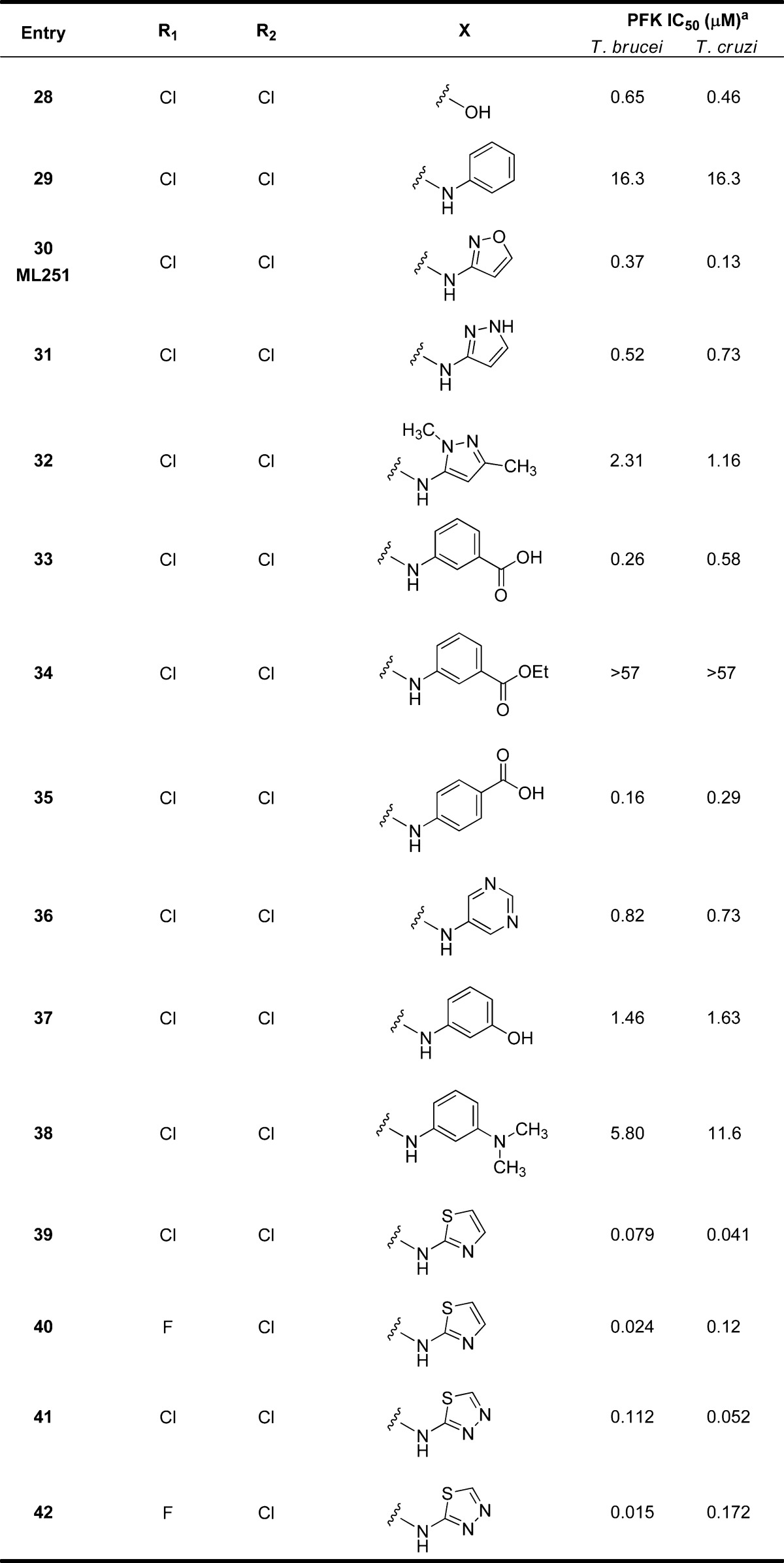
IC50 values represent the average of at least 3 separate experiments reported as the half maximal (50%) inhibitory concentration as determined in the ADP-Glo assay. All IC50s <57 μM have a maximum inhibition of >80% at 57 μM.
To identify the mechanism of action of this para-amidosulfonamide chemotype, competition studies were performed by titrating individual substrates and the potent thiadiazole analogue, 42, using a coupled biochemical PFK assay. Lineweaver–Burk transformations of the initial velocities suggest that 42 engages in competition for binding with fructose 6-phosphate (F6P) (Figure 2a), while demonstrating mixed inhibition with respect to ATP (Figure 2b). Determination of inhibition constants for 42 against PFK for F6P showed a Ki of 52 nM (Supplemental Figure 1a), while ATP (under saturating F6P levels) further decreases the affinity of 42 by 4.5-fold (Ki′ = 240 nM, Supplemental Figure 1b). Indeed, previous studies have shown that T. brucei PFK undergoes a conformation transition upon ATP binding, inducing a significant change in the active site, which may contribute to the reduced affinity of these inhibitors in the presence of ATP.10 In this view, our study is consistent with the inhibitors binding to the free enzyme and inhibiting activity through direct competition with F6P. The commonality of the 3,4-dichlorobenzyl motif in our inhibitors with the furanose analogues reported by Nowicki also supports interaction with the sugar pocket and our enzyme.
Figure 2.

Assessment of mechanism of action of para-amidosulfonamide analogue 42. Lineweaver–Burk transformations of initial velocity are shown for (a) F6P and (b) ATP, demonstrating competitive and mixed modes of inhibition, respectively. The second (untitrated) substrates were held constant at saturating concentrations in both studies (0.5 mM ATP and 2 mM F6P, respectively). Concentration of 42 is shown at 0 (○), 1.5 (□), 12.5 (◆), 50 (▲), and 100 (■) nM. Secondary plots to derive inhibition constants for 42 are shown for (a) F6P (Ki) and (b) ATP (Ki′). (c) Selectivity of ML251 (circles) and 42 (triangles) against T. brucei (black) and T. cruzi (white) PFK isoforms. (d) Toxicity of 1, ML251, and 42 against in vitro T. brucei cultures (solid bars) and MRC-5 human lung cell line (dashed bars).
Typified by 30 and 42, comparable inhibitory activity was seen in both T. brucei and T. cruzi PFK isoforms (Figure 2c), and this trend was anticipated as these isoforms display significant sequence identity (vide supra).11 Furthermore, many analogues displayed increased potencies against the T. cruzi isoform compared to T. brucei, suggesting that this series may find further utility outside of T. brucei alone. Encouragingly, 42 was tested at 1 μM against the rabbit isoform of PFK and showed no significant inhibition (Supplemental Figure 2). These data suggest that a significant window of selectivity exists for this chemotype, and importantly, a mammalian isoform of the enzyme does not appear sensitive to inhibition. This is of particular importance for potential HAT therapies, as cross-species promiscuity and polypharmacology continue to plague many frontline treatments currently used to treat trypanosomiasis (e.g., suramin).
Further validation of this chemotype was performed by evaluating toxicity in T. brucei (strain Lister 427) bloodstream-form cultures in vitro. This subspecies, while not human-infective, is highly related to the HAT causative subspecies T. b. gambiense and T. b. rhodesiense and is therefore commonly used as a model in laboratory settings. Activity of a subset of para-amidosulfonamide analogues was determined (Figure 2d), and modest dose-dependent toxicity was seen with both 1 (ED50 = 15.18 μg/mL/26.8 μM) and 30 (ED50 = 7.24 μg/mL/16.3 μM). The most potent analogue in the PFK enzymatic assay, 42, did not show any appreciable parasite toxicity, though it is unclear whether the lack of activity is due to poor compound permeability or decreased potency/binding against the target in the parasite. These compounds were additionally tested for toxicity against the MRC-5 human lung cell line, a commonly used surrogate for human host toxicity. No appreciable toxicity was observed at any concentration of compound (ED50 > 20 μg/mL/46 μM), suggesting that these compounds do not exert any off-target or PFK-related effects in a human cell environment. It should be noted that the overall reduction in potency relative to the primary assay was anticipated, as multiple studies have shown translation from enzymatic to cultured parasite assays is often accompanied by a significant loss of activity.15,16
We also assessed these select analogues via in vitro ADME assays to evaluate aqueous solubility, stability in mouse, rat and human liver microsomes, and plasma stability (Table 4). Aside from a liability in rat microsomes for analogue 1 (t1/2 = 9.7 min), none of the analogues showed major issues with all having very good aqueous solubility.
Table 4. Select in Vitro ADME Properties for 1, ML251, and 42.
| aqueous kinetic solubility | liver microsomal
stability (t1/2 in min.) |
plasma stability (% remaining after 2 h) |
||||
|---|---|---|---|---|---|---|
| compd | μg/mL | mouse | rat | human | mouse | human |
| 1 | 23.3 | 408 | 9.7 | N/D | N/D | N/D |
| ML251 | >81.0 | 231 | >30 | 330 | >95 | >95 |
| 42 | >81.0 | N/D | >30 | N/D | >95 | >95 |
The para-amidosulfonamide chemotype, exemplified by 30 and 42, represent the first small molecules to possess submicromolar inhibitory activity against T. brucei and T. cruzi PFK. Furthermore, 1 and 30 had micromolar activity in cultured parasite growth assays. This, coupled with our mechanistic and selectivity data, provides the first evidence that specific inhibition of T. brucei PFK by a small molecule may be realized. Lastly, members of this series have demonstrated encouraging properties in a panel of in vitro ADME assessment assays. As such, they represent useful tools for advancing our understanding the role that PFK and glycolysis play in trypanosome infection.
Acknowledgments
We thank Sam Michael and Michael Balcom for assistance with robotic HTS and William Leister, Chris LeClair, Danielle van Leer, James Bougie, Heather Baker, Paul Shinn, and Tom Daniel for help with analytical chemistry and compound management. DF1020DE3 was received as a generous gift from Simon H. Chang (Louisiana State University, Baton Rouge, LA). We are grateful to Edinburgh Protein Production Facility (EPPF) and to staff at the synchrotron facility at Diamond, U.K. The crystal structure of tetrameric Tb PFK (PDB# 3F5M) in the graphical Table of Contents was generated using QuteMol open source molecular visualization software (http://qutemol.sourceforge.net).
Glossary
ABBREVIATIONS
- PFK
phosphofructokinase
- HAT
Human African Trypanosomiasis
- T. brucei
Trypanosoma brucei:
- T. cruzi
Trypanosoma cruzi
- MLSMR
Molecular Libraries Small Molecule Repository
- SAR
structureactivity relationship
- qHTS
quantitative high-throughput screen
- F6P
fructose-6-phosphate
- ADME
absorption, distribution, metabolism and excretion
Supporting Information Available
Supplemental Tables 1 and 2 and Figures 1 and 2, experimental procedures for the synthesis and characterization of the compounds, the in vitro activity assay, and the in vivo antibacterial assay. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
(D.S.A.) Lead Finding Platform, Center for Proteomic Chemistry, Novartis Institutes for BioMedical Research, Inc., Cambridge, Massachusetts 02139, United States.
Author Present Address
(M.J.W.) Dow AgroSciences LLC, Crop-Protection Discovery Group, Bldg. 306/E2/980, 9330 Zionsville Road, Indianapolis, Indiana 46268, United States.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Author Contributions
∥ These authors (K.R.B. and M.J.W.) contributed equally to this work.
This research was supported in part by Award No. R03MH092153-01 from the Molecular Libraries Initiative of the NIH Roadmap for Medical Research (grant U54MH084681).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Verlinde C. L.; Hannaert V.; Blonski C.; Willson M.; Perie J. J.; Fothergill-Gilmore L. A.; Opperdoes F. R.; Gelb M. H.; Hol W. G.; Michels P. A. Glycolysis as a target for the design of new anti-trypanosome drugs. Drug Resist. Updates 2001, 4150–65. [DOI] [PubMed] [Google Scholar]
- Welburn S. C.; Maudlin I. Priorities for the elimination of sleeping sickness. Adv. Parasitol. 2012, 79, 299–337. [DOI] [PubMed] [Google Scholar]
- Barrett M. P.; Burchmore R. J.; Stich A.; Lazzari J. O.; Frasch A. C.; Cazzulo J. J.; Krishna S. The trypanosomiases. Lancet 2003, 36293941469–1480. [DOI] [PubMed] [Google Scholar]
- Burri C. Chemotherapy against human African trypanosomiasis: is there a road to success?. Parasitology 2010, 137141987–1994. [DOI] [PubMed] [Google Scholar]
- Sanderson L.; Dogruel M.; Rodgers J.; Bradley B.; Thomas S. A. The blood–brain barrier significantly limits eflornithine entry into Trypanosoma brucei brucei infected mouse brain. J. Neurochem. 2008, 10741136–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs R. T.; Nare B.; Phillips M. A. State of the art in African trypanosome drug discovery. Curr. Top. Med. Chem. 2011, 11101255–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunaway G. A. A review of animal phosphofructokinase isozymes with an emphasis on their physiological role. Mol. Cell. Biochem. 1983, 52175–91. [DOI] [PubMed] [Google Scholar]
- Opperdoes F. R. Compartmentation of carbohydrate metabolism in trypanosomes. Annu. Rev. Microbiol. 1987, 41, 127–151. [DOI] [PubMed] [Google Scholar]
- Albert M. A.; Haanstra J. R.; Hannaert V.; Van Roy J.; Opperdoes F. R.; Bakker B. M.; Michels P. A. Experimental and in silico analyses of glycolytic flux control in bloodstream form Trypanosoma brucei. J. Biol. Chem. 2005, 2803128306–28315. [DOI] [PubMed] [Google Scholar]
- McNae I. W.; Martinez-Oyanedel J.; Keillor J. W.; Michels P. A.; Fothergill-Gilmore L. A.; Walkinshaw M. D. The crystal structure of ATP-bound phosphofructokinase from Trypanosoma brucei reveals conformational transitions different from those of other phosphofructokinases. J. Mol. Biol. 2009, 38551519–1533. [DOI] [PubMed] [Google Scholar]
- Nowicki M. W.; Tulloch L. B.; Worralll L.; McNae I. W.; Hannaert V.; Michels P. A.; Fothergill-Gilmore L. A.; Walkinshaw M. D.; Turner N. J. Design, synthesis and trypanocidal activity of lead compounds based on inhibitors of parasite glycolysis. Bioorg. Med. Chem. 2008, 1695050–5061. [DOI] [PubMed] [Google Scholar]
- Sharma B. Modulation of phosphofructokinase (PFK) from Setaria cervi, a bovine filarial parasite, by different effectors and its interaction with some antifilarials. Parasites Vectors 2011, 4, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglese J.; Auld D. S.; Jadhav A.; Johnson R. L.; Simeonov A.; Yasgar A.; Zheng W.; Austin C. P. Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc. Natl. Acad. Sci. U.S.A. 2006, 1033111473–11478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez E.; Lander N.; Ramirez J. L. Molecular and biochemical characterisation of Trypanosoma cruzi phosphofructokinase. Mem Inst Oswaldo Cruz 2009, 1045745–748. [DOI] [PubMed] [Google Scholar]
- Frearson J. A.; Wyatt P. G.; Gilbert I. H.; Fairlamb A. H. Target assessment for antiparasitic drug discovery. Trends Parasitol 2007, 2312589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson S.; Jones D. C.; Shanks E. J.; Frearson J. A.; Gilbert I. H.; Wyatt P. G.; Fairlamb A. H. Synthesis and evaluation of 1-(1-(benzo[b]thiophen-2-yl)cyclohexyl)piperidine (BTCP) analogues as inhibitors of trypanothione reductase. ChemMedChem 2009, 481341–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.