

Abstract

An improved synthesis of a phosphopeptidomimetic prodrug targeting the Src Homology 2 (SH2) domain of signal transducer and activator of transcription 6 (STAT6) is reported. In our convergent methodology, we employed a phosphotyrosine surrogate active ester harboring pivaloyloxymethyl groups, which efficiently coupled to tert-butylglycinyl proline diarylamide. Biological evaluation of 1 has not been reported. We show that it inhibits STAT6 phosphorylation in intact human bronchial epithelial cells, suggesting potential application in the treatment of asthma.
Keywords: Peptidomimetic inhibitor, SH2, STAT6, prodrug
Asthma is a complex inflammatory disease of the lungs characterized by mucus production, airway hyperresponsiveness (AHR), eosinophil recruitment, and T-Helper cell 2 (Th2) activation which results in immunoglobulin class switching to IgE (reviewed in refs (1−3)). Activated Th2 cells release cytokines, especially inerleukin-4 (IL-4) and interleukin-13 (IL-13), which then bind to receptors at the cell surface and recruit Janus kinases 1 and 3 (JAK1 and JAK3), and the tyrosine kinase 2 (Tyk2), leading to phosphorylation of IL-4Rα, the subunit common to both cytokine receptors. Cytoplasmic signal transducer and activator of transcription 6 (STAT6) is recruited to the phosphorylated receptor via its Src homology 2 (SH2 domain) and is phosphorylated on Tyr641 by the associated JAK kinases. Phosphorylated STAT6 (pSTAT6) dimerizes via reciprocal SH2 domain interactions, translocates to the nucleus, binds to specific DNA promoter sequences, and participates in the expression of genes leading to asthma and AHR. Elevated pSTAT6 levels have been found in the bronchial epithelium of asthma patients,4 and STAT6 knockout mice do not develop AHR or lung pathology associated with asthma.5 Therefore, inhibiting the activity of STAT6 is a potential modality for asthma treatment.
Of the steps in the IL4/13-JAK-STAT6 signaling cascade, blocking association of STAT6 with IL-4Rα by targeting the SH2 domain is an attractive strategy. A cell-penetrating phosphopeptide derived from Tyr631 of IL-4Rα, a docking site for STAT6, inhibited STAT6 phosphorylation stimulated by IL-4 in Ramos cells.6 Another cell-penetrating phosphopeptide, STAT-6-IP, derived from the phosphorylation site of STAT6, Tyr641, inhibited in vitro IL-4 and IL-13 expression from splenocytes from mice challenged with ovalbumin (OVA).7 Importantly, in vivo intranasal administration inhibited OVA-induced lung inflammation and mucus production, eosinophil migration, and AHR. Furthermore, intranasal administration of STAT-6-IP inhibited many features of allergic airway disease symptoms in a mouse asthma model induced by ragweed pollen.8 These reports provide proof of concept that steric block of the SH2 domain of STAT6 prevents recruitment to IL-4Rα, phosphorylation of Tyr641, and subsequent transcriptional activity leading to asthma symptoms.
In the early 2000s Tularik, Inc. (now part of Amgen, Inc.) published identical US and World patents on small molecule phosphopeptide mimetics targeting the SH2 domain of STAT6.9,10 Although extensive structure–affinity relationship studies were reported, the inventors described the synthesis of only one compound with the potential to inhibit STAT6 phosphorylation in intact cells (1, Figure 1). In this compound, phosphotyrosine was replaced with the conformationally constrained 4-phosphoryloxycinnamic acid unit, and the phosphate was replaced with the noncleavable phosphonodifluoromethyl group.11
Figure 1.

Structure of the Tularik inhibitor, 1.
A prodrug strategy was employed in which the negative charges of the phosphonate were capped with carboxyesterase-labile pivaloyloxymethyl (POM)12 groups to allow passage into cells. The theory was that, on entering cells, esterases will cleave the POM groups, thereby freeing the phosphonate to bind to the phosphotyrosine binding pocket on the SH2 domain of STAT6. In the patents the synthesis of 1 was inefficient, and the last step did not have yield data nor was the final compound characterized by NMR, although the molecular weight was confirmed by mass spectrometry. More importantly, no data on the biological activity of this compound was reported in patent or peer-reviewed literature. Given the potential impact of inhibiting STAT6 activity by targeting the SH2 domain, our laboratory developed an improved synthesis of 1 and found it to be a relatively potent inhibitor of STAT6 phosphorylation in IL-4 and IL-13 stimulated immortalized bronchial epithelial cells, Beas-2B. Our improved synthesis scheme and biological evaluation of 1 are reported here (see also Supporting Information).
The Tularik researchers used a convergent scheme in which the phosphotyrosine surrogate, 4′-phosphonodifluoromethylcinnamate (2), was coupled to a modified dipeptide 3 followed by installation of the POM groups (Scheme 1). Coupling of 2 and 3 was carried out in the presence of HBTU, HOBt, and DIEA to give intermediate 4 in 11% yield. The POM groups were installed by converting the phosphonate oxygens to silver salts and treating with iodomethyl pivalate to get the desired STAT6 inhibitor 1. As mentioned, no yield was given for this step.
Scheme 1. Published Synthesis of 1.

Reagents and conditions: (a) HBTU, HOBt, DIPEA, DMF, overnight, rt, 11%; (b) (i) NaOH, (ii) AgNO3; (iii) iodomethyl pivalate, toluene.
Our laboratory reported a convergent method that was used to synthesize phosphatase-stable, cell-permeable peptidomimetic prodrugs targeted to the SH2 domain of STAT3.15 The key to our synthesis is the use of the fully protected phosphonodifluoromethylcinnamate, 9 (Scheme 2B). Blocking the phosphonate oxygens with POM groups prior to coupling to 3 prevents unproductive depletion of coupling agent due to the free phosphonate group in 2. Further, POM groups are typically installed on the silver salts of phosphonates in toluene, which is a poor solvent for peptides and peptidic compounds. In addition to POM protection, the carboxyl group of 9 is derivatized as the pentachlorophenyl (Pcp) ester, a known activator of carboxyl groups. The Pcp ester possesses sufficient stability to serve as a protecting group for the POM installation as we described.14,15 Our overall yields for the synthesis of the protected phosphotyrosine surrogate 13 were 30–40%14,15 in contrast to 13% for the overall synthesis of 2.9,10 This strategy resulted in marked improvements in the synthesis of 1.
Scheme 2. Improved Synthesis of STAT6 Inhibitor, 1.

Reagents and conditions: (a) 4-iodoaniline, EDC, DCM, rt, 4 h, 88%; (b) Ph3 Bi, Cu(OAc)2, TEA, DCM, overnight, rt, 48%; (c) (i) TFA/DCM (1:1), 0.5 h, rt; (ii) Fmoc-Tle-OH, EDC, TEA, DCM, 6 h, 78%; (d) 20% piperidine/DMF, 47%; (e) 3, NMM, DMAP (cat), DMF, rt 2 h, 66%.
For the synthesis of 3, we followed almost the same procedure as that in the patent with some modifications. Boc-Pro-OH (5) was coupled with 4-iodo-aniline in the presence of EDC to get 6. The yield of the triphenylbismuth-mediated replacement of the amide proton14 of 6 to give 7 was 48%. Yields of coupling of sterically hindered Fmoc-tert-buytlglycine to the deprotected proline amide were higher with HBTU than with EDC. The Fmoc-protected dipeptide was obtained in 78% yield after flash chromatography. The Fmoc group was removed with piperidine, and 3 was obtained in 47% yield after reverse-phase HPLC purification. One of the key aspects of our method is high yield coupling of the bis-POM protected pTyr surrogate to the dipeptide, depicted in Scheme 2. The building block 9 was coupled to the dipeptide 3 in the presence of N-methylmorpholine (NMM) and catalytic DMAP to give 1 in 66% yield after purification by reverse-phase HPLC.
Because the biological activity has not been reported, prodrug 1 was evaluated for its ability to inhibit STAT6 Tyr641 phosphorylation in intact Beas-2B cells. Compound 1 was added to cells from a DMSO stock solution. Cells were preincubated with 1 for 2 h and were then stimulated with IL-4 or IL-13 for 1 h. Levels of phospho-STAT6(Y641) and total STAT6 proteins were estimated by Western blotting of the cell lysates (Figure 2A). Prodrug 1 inhibited pSTAT6 phosphorylation at 1–10 μM in both IL-4 and IL-13 stimulated Beas-2B cells. This result suggests that 1 enters cells, the POM groups are cleaved, and the resulting phosphonate binds to the SH2 domain of STAT6 thereby preventing recruitment to the cytokine receptors and subsequent phosphorylation by JAKs. The free phosphate version of 1 was reported to inhibit the binding of a fluorescently tagged phosphopeptide at <1 μM using a solid phase competition assay.9 Prodrug 1 at concentrations up to 10 μM was found not to be toxic to Beas-2B cells in a 72 h MTS assay (Figure 2C). The duration of pSTAT6 inhibition was assayed by treating Beas-2B cells with a single dose of 1 (10 μM). Cells were incubated for predefined time intervals and were stimulated with IL-4 for 1 h prior to cell lysis. Western blot analysis showed that nearly complete inhibition of phospho-STAT6(Y641) occurred at 2 h and was maintained up to 48 h (Figure 2B). The reduction in pSTAT6 was not due to toxicity as the MTS assay showed not effect on proliferation.
Figure 2.
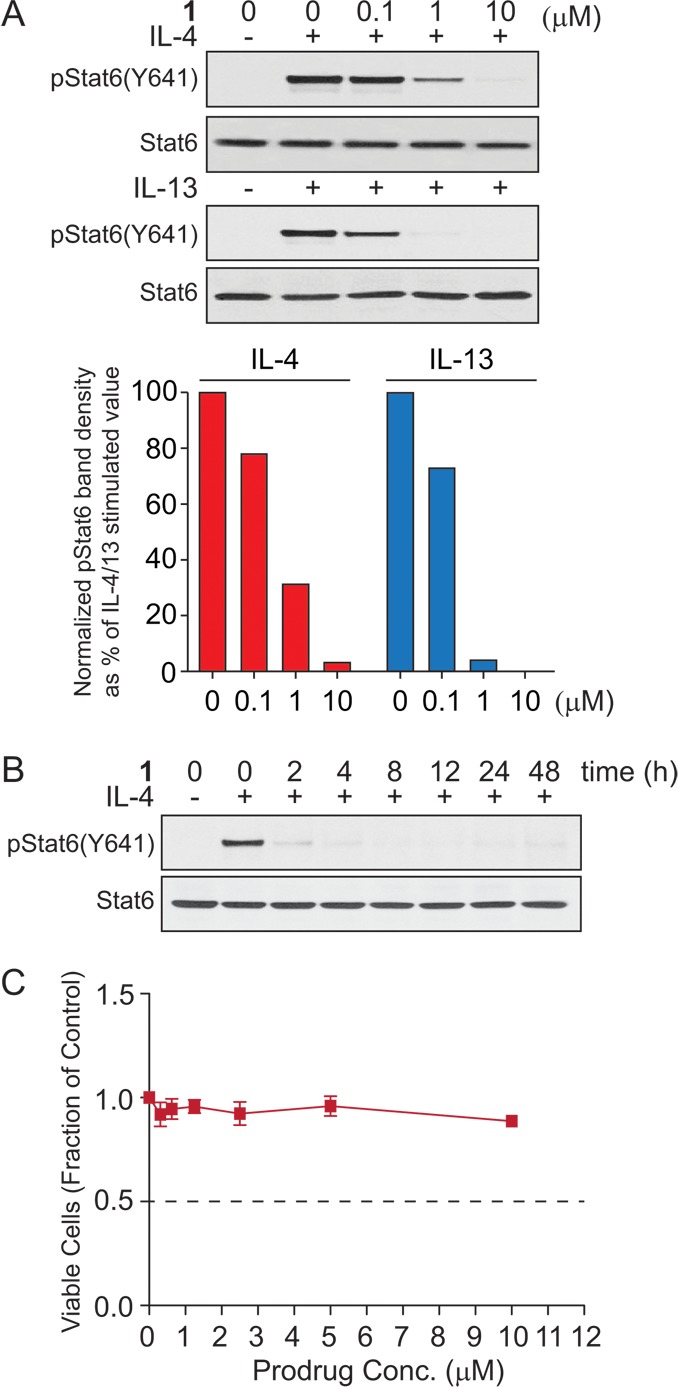
Biological evaluation of 1 in Beas-2B cells. (A) Western blotting showing the inhibitory effect of 1 on STAT6(Y641) phosphorylation in Beas-2B cells stimulated with IL-4 or IL-13 and relative quantification of protein-band intensites by densitometric analysis. (B) Time course of pSTAT6(Y641) inhibition of 1 in IL-4 stimulated Beas-2B cells. (C). Effect of 1 on Beas-2B cell proliferation after 72 h as determined by MTS assay.
In conclusion, we have improved on the synthesis of a novel STAT6 inhibitor and show that it potently inhibits the phosphorylation of STAT6 in human bronchial epithelial cells stimulated with either IL-4 or IL-13. Key to the synthesis is having the phophonate oxygens protected prior to coupling to the amine. Furthermore, preactivation of the cinnamate building block as pentachlorophenyl ester improved coupling yields compared to the published method. As mentioned, this class of inhibitor has not been evaluated and shown to inhibit STAT6 to date. Given the importance of this transcription factor in asthma and allergic lung disease, our promising results suggest that targeting the SH2 domain with phosphatase-stable, cell-permeable phosphopeptide mimetic prodrugs is a potential treatment modality for this disease.
Acknowledgments
Beas-2B cells were a kind gift from Walter Hittelman.
Glossary
Abbreviations
- DMAP
4-dimethylaminopyridine
- EDC
N-ethyl-N′-(3-dimethylaminopropyl)carbodiimide hydrochloride
- HBTU
O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- NMM
N-methylmorpholine
- NMP
N-methylpyrrolidone
- POM
pivaloyloxymethyl
Supporting Information Available
Procedures for the synthesis, NMR, and biological characterization of 1. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
P.M.: Department of Genomic Medicine, The University of Texas M. D. Anderson Cancer Center, 1901 East Road, Houston, Texas 77054.
Author Contributions
P.M. and P.K.M. contributed equally. The manuscript was written through contributions of all authors. Dr. Pietro Morlacchi performed biological evaluation of compound 1 and contributed to the preparation of the manuscript. Dr. Pijus Mandal synthesized compound 1 and contributed to preparation of the manuscript. Dr. John S. McMurray supervised the work and contributed to preparation of the manuscript.
We are grateful to the American Asthma Foundation for financial support of this work (Grant # 11-0360). We also acknowledge the Cancer Center Support Grant (P30 CA16672-38) for support of the NMR laboratory and the Translational Chemistry Core Facility who provided mass spectrometry.
The authors declare no competing financial interest.
Supplementary Material
References
- Kuperman D. A.; Schleimer R. P. Interleukin-4, interleukin-13, signal transducer and activator of transcription factor 6, and allergic asthma. Curr. Mol. Med. 2008, 8, 384–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasaian M. T.; Miller D. K. IL-13 as a therapeutic target for respiratory disease. Biochem. Pharmacol. 2008, 76, 147–155. [DOI] [PubMed] [Google Scholar]
- Popescu F. D. New asthma drugs acting on gene expression. J. Cell. Mol. Med. 2003, 7, 475–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullings R. E.; Wilson S. J.; Puddicombe S. M.; Lordan J. L.; Bucchieri F.; Djukanovic R.; Howarth P. H.; Harper S.; Holgate S. T.; Davies D. E. Signal transducer and activator of transcription 6 (STAT-6) expression and function in asthmatic bronchial epithelium. J. Allergy Clin. Immunol. 2001, 108, 832–838. [DOI] [PubMed] [Google Scholar]
- Darcan-Nicolaisen Y.; Meinicke H.; Fels G.; Hegend O.; Haberland A.; Kuhl A.; Loddenkemper C.; Witzenrath M.; Kube S.; Henke W.; Hamelmann E. Small interfering RNA against transcription factor STAT6 inhibits allergic airway inflammation and hyperreactivity in mice. J. Immunol. 2009, 182, 7501–7508. [DOI] [PubMed] [Google Scholar]
- Stolzenberger S.; Haake M.; Duschl A. Specific inhibition of interleukin-4-dependent Stat6 activation by an intracellularly delivered peptide. Eur. J. Biochem. 2001, 268, 4809–4814. [DOI] [PubMed] [Google Scholar]
- McCusker C. T.; Wang Y.; Shan J.; Kinyanjui M. W.; Villeneuve A.; Michael H.; Fixman E. D. Inhibition of Experimental Allergic Airways Disease by Local Application of a Cell-Penetrating Dominant-Negative STAT-6 Peptide. J. Immunol. 2007, 179, 2556–2564. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Li Y.; Shan J.; Fixman E.; McCusker C. Effective treatment of experimental ragweed-induced asthma with STAT-6-IP, a topically delivered cell-penetrating peptide. Clin. Exp. Allergy 2011, 41, 1622–1630. [DOI] [PubMed] [Google Scholar]
- McKinney J.; Raimundo B. C.; Cushing T. D.; Yoshimura H.; Ohuchi Y.; Hiratate A.; Fukushima H. Preparation of peptides as inhibitors of STAT function. U.S. Patent US 6426331 B1, 2002.
- McKinney J.; Raimundo B. C.; Cushing T. D.; Yoshimura H.; Ohuchi Y.; Hiratate A.; Fukushima H.; Xu F.; Peto C. STAT4 and STAT6 binding dipeptide derivatives. World Patent WO 2001083517 20000503, 2001.
- Burke T. R. Jr.; Smyth M. S.; Otaka A.; Nomizu M.; Roller P. P.; Wolf G.; Case R.; Shoelson S. E. Nonhydrolyzable phosphotyrosyl mimetics for the preparation of phosphatase-resistant SH2 domain inhibitors. Biochemistry 1994, 33, 6490–6494. [DOI] [PubMed] [Google Scholar]
- Farquhar D.; Khan S.; Srivastva D. N.; Saunders P. P. Synthesis and antitumor evaluation of bis[(pivaloyloxy)methyl] 2′-deoxy-5-fluorouridine 5′-monophosphate (FdUMP): a strategy to introduce nucleotides into cells. J. Med. Chem. 1994, 37, 3902–3209. [DOI] [PubMed] [Google Scholar]
- Mandal P. K.; Liao W. S.-L.; McMurray J. S. Synthesis of phosphatase-stable, cell-permeable peptidomimetic prodrugs that target the SH2 domain of Stat3. Org. Lett. 2009, 11, 3394–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal P. K.; Gao F.; Lu Z.; Ren Z.; Ramesh R.; Birtwistle J. S.; Kaluarachchi K. K.; Chen X.; Bast R. C. Jr.; Liao W. S.; McMurray J. S. Potent and Selective Phosphopeptide Mimetic Prodrugs Targeted to the Src Homology 2 (SH2) Domain of Signal Transducer and Activator of Transcription 3. J. Med. Chem. 2011, 54, 3549–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.