

Abstract

Tuberculosis (TB), a disease caused by Mycobacterium tuberculosis (Mtb), is a global public health concern because of the emergence of various resistant strains. Benzothiazin-4-ones (BTZs), represented by BTZ043, are a promising new class of agents for the treatment of tuberculosis and have been shown to kill Mtb in vitro, ex vivo, and in mouse models of TB. Herein we report the design and syntheses of nitroaromatic sulfonamide, reverse-amide, and ester classes of anti-TB agents using a scaffold simplification strategy based on BTZ043. The presented work explores the effect of functional groups such as sulfonamides, reverse-amides, and esters that are attached to the nitroaromatic rings on their anti-TB activity. The in vitro activity of the compounds evaluated against the H37Rv strain of Mtb show that nitroaromatic sulfonamides and nitrobenzoic acid esters with two nitro substituents were most active and highlights the importance of the electronic character (electron deficient aromatic ring) of the nitroaromatic ring as a central theme in these types of nitroaromatic anti-TB agents.
Keywords: Mycobacterium tuberculosis, scaffold simplification strategy, BTZ043, nitroaromatics, DprE1
Tuberculosis (TB), is a disease caused by Mycobacterium tuberculosis (Mtb). Approximately 2 billion people are infected by TB worldwide, and it accounts for approximately 2 million deaths every year. TB is considered a global public health concern because of the emergence of multidrug resistant (MDR), extensively drug resistant (XDR), and, recently, totally drug resistant forms.1,2 Coinfection with human immunodeficiency virus (HIV) causes both TB and HIV to progress more rapidly. An estimated 1.3 million deaths occurred among HIV negative TB cases in 2009, whereas 0.4 million deaths occurred in HIV cases with TB coinfection the same year.1,3
Recently, TMC207 (bedaquiline) was the first new anti-TB agent to be approved in over 40 years.4 Still, the current anti-TB treatment regimen suffers from poor patient compliance due to the required simultaneous and long-term administration of multiple drugs, side effects, and treatment costs. This noncompliance also has contributed to the emergence of MDR and XDR strains. Therefore, there is a growing demand for new agents that are effective against TB.5,6
Recently, 1,3-benzothiazin-4-ones (BTZs) were discovered and found to be promising new agents for the treatment of tuberculosis.7,8 BTZs, especially BTZ043 (compound 1, Figure 1), have been shown to kill Mtb in vitro, ex vivo, and in mouse models of TB. The minimum inhibitory concentration (MIC) of BTZ043 was found to be 1 ng/mL against Mtb, which is extraordinary relative to existing drugs, such as isoniazid and ethambutol.7,8 More recently, additional structure–activity relationship (SAR) studies led to the development of pBTZ, which is reported to have efficacy in mouse and zebrafish models of tuberculosis.9
Figure 1.

Structures of a representative 1,3-benzothiazinone anti-TB agent, BTZ043, and other recently developed nitroaromatic amide anti-TB agents.
Mycobacteria possess a thick, impermeable hydrophobic cell wall. This cell wall includes peptidoglycan and d-arabinofuranose, containing arabinogalactan, arabinomannan polysaccharides, mannans, glucans, mycolic acids, lipids, glycolipids, poly glutamate-glutamine polymers, and proteins.9,10d-Arabinofuranose, a component of arabinogalactan and arabinomannan is biosynthesized in a process catalyzed by decaprenyl-phospho-ribose-2′-epimerase (DprE1).10,11 BTZ043 was shown to be a suicide inhibitor of DprE1, a key enzyme of the cell wall assembly for mycobacteria.7,9,13
A long-standing goal of our lab and many others is to develop effective syntheses of anti-TB agents. Herein we report design and syntheses of simplified anti-TB agents based on the molecular mode of action of BTZ043.
The SAR studies carried out by Makarov et al.7 and the detailed mechanistic studies carried out by Trefzer et al.9,13 and our group,12 indicate that the anti-TB activity of BTZs depends on the electron deficient nitroaromatic core; whereas substitution of the (S)-2-methyl-1,4-dioxa-8-azaspiro[4.5]decane group of 1 by either its enantiomer or by variety of functional groups, including substituted piperazines,7,14 is not as influential.
However, the mechanistic details related to the reductive activation of BTZ043 to its nitroso intermediate are yet to be completely understood.9,12,13 It was hypothesized that the cofactor FADH2 was responsible for the reduction of the nitro group of BTZ043 to its nitroso intermediate, which in turn reacts with the cysteine at the active site of DprE1 to form a covalent semimercaptal adduct.
Our recent work further demonstrated that BTZ043 and other nitroaromatic compounds such as nitrobenzamides and nitroaromatic-BTZ hybrids undergo similar chemistry and metabolic transformations and thus may have common activating and metabolizing targets. In particular, our work suggests that these related nitroaromatic anti-TB agents can undergo cine addition reactions with either nucleophilic thiolates present in the active site of the enzyme (DprE1 in the case of BTZ043) or by a hydride from cofactor (FADH2). Activation initiated by cine addition to the nitroaromatic prodrugs initiates conversion to the corresponding target-active nitroso intermediates thereby causing the subsequent suicide inhibition of the enzyme (DprE1 in the case of BTZ043).12
Therefore, for the design and syntheses of BTZ043-based analogues, we retained the reactive nitroaromatic core and envisioned the simplification (defragmentation) of the BTZ043 scaffold based on electron withdrawing 1,3,5-substituents (namely, the trifluoromethyl, nitro, and carbonyl highlighted in purple in Figure 2) into three easily synthesized and cost-effective nitroaromatic analogues, namely, the sulfonamide based, the aniline based, and finally the benzyl ester based scaffolds shown in Figure 2.
Figure 2.

Design of anti-TB agents inspired by BTZ043. The molecular fragments of BTZ043 considered for the design are colored in purple.
The sulfonamide derivatives were designed as bioisosteres of the carbonyl fragment colored in purple (Figure 2), whereas the aniline derivatives are termed reverse-amides to reflect that they were designed to explore SAR by variation of previously reported substituted nitro benzamides.3 The sulfonamide and the reverse-amide based versions retained the dioxa-8-azaspiro[4.5]decane group from BTZ043 in order to keep their molecular surface areas similar to that of BTZ043, whereas the design of the ester based class of nitroaromatic agents was further simplified to facilitate syntheses while exploring the SAR.
This modification was inspired by recent X-ray crystallographic studies by Neres et al.15 on a covalently bound BTZ043-derived nitroso semimercaptal adduct with DprE1. This crystal structure revealed only a complementary weak hydrophobic interaction of the side chain leucine with the piperidine ring of BTZ043, whereas the dioxa-spirocycle is exposed without any noticeable interaction with DprE1.
In order to validate our scaffold simplification strategy by computational docking, a representative dinitro compound from each of the presented classes was docked into the crystal structure of DprE1 (PDB code 4F4Q)15 using Glide.16,17 Since compound 1 was crystallized as a covalent adduct with DprE1, it was necessary to optimize the docking protocol. This was achieved by docking the nitroso intermediate of 1 into DprE1 (see Supporting Information). Therefore, the S–N covalent bond of the semimercaptal adduct in the crystal structure was deleted, and the resulting ligand was converted into its nitroso intermediate (see Supporting Information for complete details) and docked. As can be seen from Figure 3A, the nitroso intermediate of BTZ043 was docked in the same binding pocket formed by the semimercaptal adduct.
Figure 3.
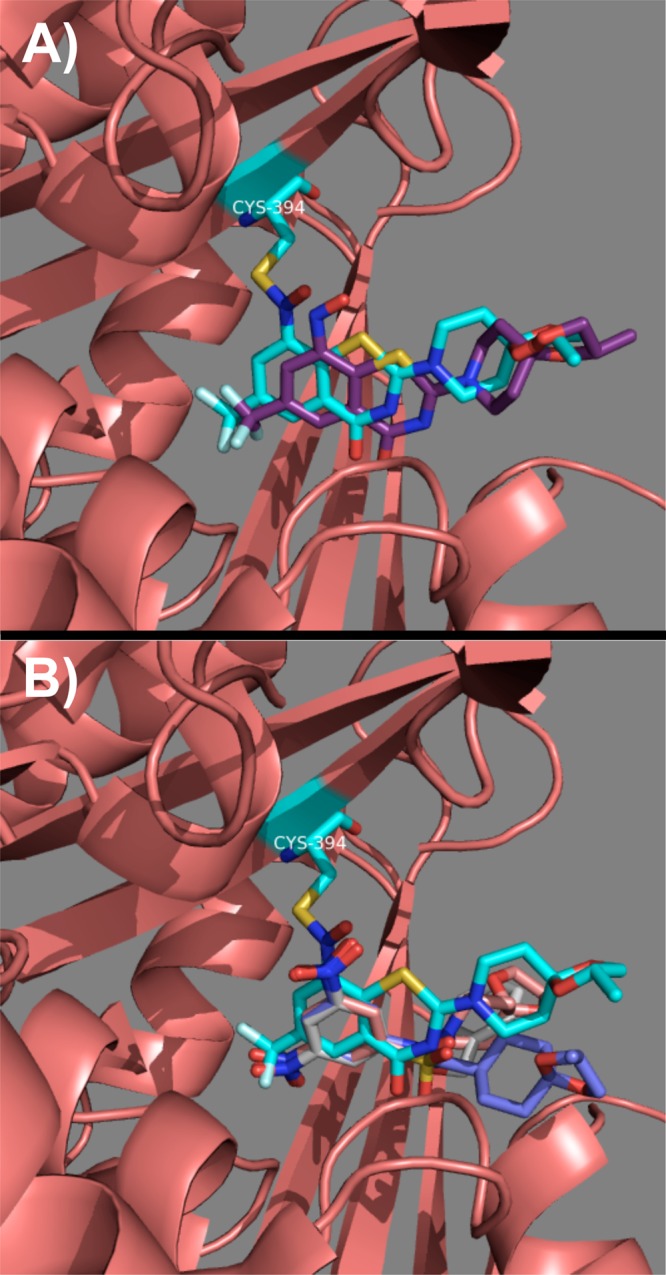
(A) Overlay of the docking nitroso intermediate of BTZ043 (carbons are purple) on the crystallized semimercaptal adduct (carbons are cyan). (B) Overlay of the docked structures of compounds 9 (carbons are pink), 19 (carbons are blue), and 24 (carbons are colored white) on the crystallized semimercaptal adduct (carbons are cyan).
Next, we repeated the docking with compounds 9, 19, and 24 to explore and compare their binding with that of semimercaptal adduct in DprE1. Our docking study indicated that the dinitroaromatic portion present in all these compounds binds essentially in the same pocket as that of 1 (Figure 3B). Thus, this observed docking pattern of sulfonamides, reverse amide, and nitrobenzoic acid ester class of compounds further justified our design strategy of these classes using a scaffold simplification strategy.
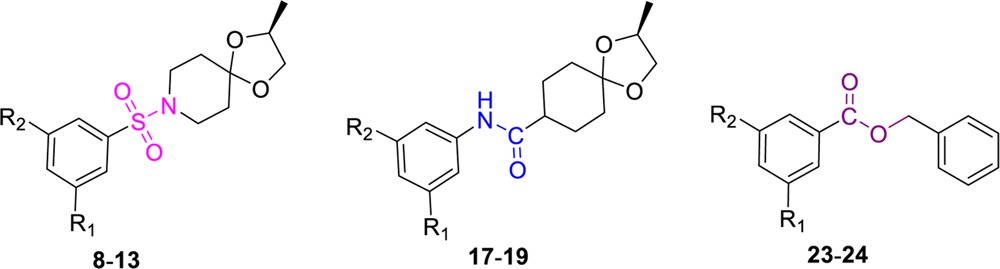
As shown in Scheme 1, preparation of the first sulfonamide subset of compounds (8–10) only required reactions between appropriately substituted aryl sulfonyl chlorides with (S)-2-methyl-1,4-dioxa-8-azaspiro [4.5]decane (7) in the presence of triethylamine. The aryl sulfonyl chloride precursors were obtained from commercial sources or prepared by diazotization of the corresponding 1,3-disubstituted anilines with sodium nitrite followed by reaction with sulfur dioxide gas in the presence of Cu(I)Cl.18 The phenyl-substituted analogue 11 was synthesized in 69% yield by the Suzuki coupling of 10 with phenyl boronic acid in the presence of CsF.19
Scheme 1. Synthesis of BTZ043 Inspired Sulfonamide Based Nitroaromatics.

Reagents and conditions: (a) (i) NaNO2, HCl, CH3COOH; (ii) SO2, CH3COOH; (b) 7, Et3N, DCM, rt; (c) phenyl boronic acid, Pd(PPh3)4, CsF, 1,4-dioxane, reflux, 69%; (d) pyridine-N-oxide, PtBu3·HBF4, Pd(OAc)2, K2CO3, toluene, 110 °C, 38%; (e) iron dust, 0.1 M CH3COOH.
The effect on the anti-TB activity by the replacement of the nitro groups of aryl sulfonamides by pyridyl or pyridyl-N-oxides was also investigated. The pyridyl-N-oxide 12 was synthesized in 38% yield from 10 by utilizing the coupling procedure described by Fagnou et al.20,21 The pyridine analogue 13 was then synthesized by the reduction of compound 12 with Fe/CH3COOH.
The in vitro activity of compounds 8–10 and 11–13 in two different mycobacterial growth media (7H12 and GAS)22−24 are summarized in Table 1. As can be seen from Table 1, only compounds 8 and 9 showed significant activity against the H37Rv strain of Mtb, while others were less active. The SAR studies further highlighted the importance of the nitro group for the anti-TB activity of these compounds. It is probable that the sulfonamide class, in general, is still not as electron deficient as the 1,3-benzothiazinones. Therefore, the nitroaromatic ring present in 8 is still not as electron deficient as in dinitro-substituted compound 9, which is consistent with the difference in anti-TB activity (see Table 2). Additionally, compound 9 did not show any toxicity in the VERO assay, whereas compound 8 was found to be toxic (see Supporting Information for VERO and LORA activities). Both compounds 8 and 9 were only weakly active against nonreplicating Mtb (LORA).
Table 1. In Vitro Activity of the Analogues Inspired by BTZ043 against the H37Rv Strain of Mtb in 7H12 and GAS Media (MIC in μM)a.
| R1 | R2 | MABA: 7H12 (μM) | MABA: GAS (μM) | |
|---|---|---|---|---|
| 8 | CF3 | NO2 | 6.99 | 10.2 |
| 9 | NO2 | NO2 | 1.53 | 2.05 |
| 10 | CF3 | Br | >10 | nd |
| 11 | CF3 | Ph | >64 | 17.4 |
| 12 | CF3 | pyridine-N-oxide | >64 | >64 |
| 13 | CF3 | pyridine | >50 | 49.3 |
| 17 | CF3 | H | >50 | 17.9 |
| 18 | CF3 | NO2 | 23.0 | 18.4 |
| 19 | NO2 | NO2 | 47.9 | 22.6 |
| 23 | CF3 | NO2 | >50 | 2.03 |
| 24 | NO2 | NO2 | 13.03 | 0.27 |
| BTZ043 | <0.02 | <0.02 |
MABA, microplate alamar blue assay; GAS, glycerol-alanine-salts medium; 7H12, 7H9 medium plus casitone, palmitic acid, albumin, and catalase.
Table 2. Mulliken Charges on the Indicated Positions Calculated by AM1 Methods for Representative Dinitroaromatic Scaffoldsa.

| Mulliken charges | 9 | 19 | 24 |
|---|---|---|---|
| 1 | 0.24 | 0.17 | 0.22 |
| 2 | 0.26 | 0.10 | 0.22 |
| 3 | 0.26 | 0.11 | 0.22 |
The Mulliken charges are shown only for the numbered atoms for comparison purposes.
Syntheses of the anilines or reverse-amide compounds (17–19, Scheme 2) required prior ketalization of commercially available 4-oxo-ethyl-cyclohexane carboxylate (14) with (S)-2-methyl-2,2-propandiol in the presence of catalytic pTsOH to obtain 15. Compound 15 was then saponified to afford the free carboxylic acid, which was coupled with m,m-disubstituted electron deficient anilines by reaction of the acid chloride of 15 in the presence of triethylamine to obtain compounds 17–19. On the basis of the earlier results with the sulfonamide class of analogues, other substitutions such as phenyl, pyridyl, or pyridyl-N-oxide for this class of analogues were not explored. The in vitro activities of compounds 17–19 in two different mycobacterial growth media (7H12 and GAS) are also included in Table 1.
Scheme 2. Syntheses of Reverse-Amide Nitroaromatics.

Reagent and conditions: (a) pTsOH, benzene, reflux; (b) LiOH, THF/water; (c) (i) oxalyl chloride, DMF, CH2Cl2, 4 h; (ii) Et3N, CH2Cl2, 12 h.
The change in the electronic character of the nitroaromatic ring brought by the reverse-amide functionality became evident by in vitro activities. The nitro aromatic ring in this class of compounds (17–19) is not as electron deficient as in the sulfonamides, and hence, this class of compounds is virtually devoid of any appreciable activity (see Table 2 for the Mulliken charges calculated for 19). Notably, unlike the previous class the dinitro compound (19) was found to be less active than the mononitro one (18).
The nitrobenzoic acid esters 23–24 (Scheme 3) were synthesized in 80–90% yields by coupling of the appropriately substituted nitrobenzoic acids, 20–21 with the various commercially available benzyl alcohol, 22 in the presence of triethylamine and a catalytic amount of DMAP. Both m-dinitro benzoic acids and m-trifluoromethyl-m-nitro benzoic acid esters were synthesized.
Scheme 3. Synthesis of Benzyl Esters of Nitrobenzoic Acids.

Reagent and conditions: (a) oxalyl chloride, cat. DMF; (b) Et3N, CH2Cl2, DMAP, 80–90%.
Overall, the SAR pattern displayed by this class of compounds was similar to both sulfonamides and reverse-amides (Table 1). Briefly, the m-dinitrobenzoate esters displayed more potent activity than the corresponding m-trifluoromethyl-m-nitro benzoates.
Surprisingly, this class of compounds displayed much weaker activity in the 7H12 media and displayed notable activity in the GAS media (Table 1).
In conclusion, from Table 1, it is clear that simple sulfonamides and nitrobenzoic acid ester analogues with dinitro substituents (compounds 9 and 24) were more active though not at the level of BTZ043 itself. The reverse-amide functionality drastically affected the in vitro anti-TB activity of the studied nitroaromatic scaffold. Table 2 shows the Mulliken charges25 calculated using AM1 method26 on dinitroaromatic 9, 19, and 24. It is evident from Table 2 that the nitroaromatic scaffold present in 9 is more electron deficient than 24, which in turn is more electron deficient than 19. As shown in Figure 3, the docking pattern for 9, 19, and 24 with DprE1 is similar; however, there is a marked difference in the electronic nature of the nitroaromatic rings brought about by the introduction of the sulfonamide, reverse-amide, and ester functionality. This difference is reflected in their anti-TB activity. Therefore, this work highlights the importance of the electronic character (electron deficient aromatic ring) of the nitroaromatic ring as a central theme in these nitroaromatic anti-TB agents.
Acknowledgments
We thank the University of Notre Dame, especially the Mass Spectrometry and Proteomics Facility (Bill Boggess, Michelle Joyce, and Nonka Sevova), which is supported by Grant CHE-0741793 from the National Science Foundation (NSF).
Glossary
ABBREVIATIONS
- DMF
N,N-dimethyl formamide
- DprE1
decaprenylphosphoryl-β-d-ribose 2′ oxidase
- MDR
multidrug resistant
- M. tuberculosis
Mycobacterium tuberculosis
- XDR
extensively drug resistant
- TB
tuberculosis
Supporting Information Available
Complete experimental details along with the characterizations of the synthesized compound. This material is available free of charge via the Internet at http://pubs.acs.org.
This research was supported in part by grant 2R01AI054193 from the National Institutes of Health (NIH).
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Global Tuberculosis Report 2013. http://www.who.int/tb/publications/global_report/en/.
- Udwadia Z. F.; Amale R. A.; Ajbani K. K.; Rodrigues C. Totally drug-resistant tuberculosis in India. Clin. Infect. Dis. 2011, 54, 579–581. [DOI] [PubMed] [Google Scholar]
- Brodin P.; Christophe T.; No Z.; Kim J.; Genovesio A.; Fenistein D. P. C.; Jeon H.; Ewann F. A.; Kang S.; Lee S.; Seo M. J.; Park E.; Contreras Dominguez M.; Nam J. Y.; Kim E. H. Patent Appl. WO 2010003533, Jan 14, 2010.
- Rouan M.-C.; Lounis N.; Gevers T.; Dillen L.; Gilissen R.; Raoof A.; Andries K. Pharmacokinetics and pharmacodynamics of TMC207 and its N-desmethyl metabolite in a murine model of tuberculosis. Antimicrob. Agents Chemother. 2012, 56, 1444–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zumla A.; Nahid P.; Cole S. T. Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug Discovery 2013, 12, 388–404. [DOI] [PubMed] [Google Scholar]
- Rivers E. C.; Mancera R. L. New anti-tuberculosis drugs with novel mechanisms of action. Curr. Med. Chem. 2008, 15, 1956–1967. [DOI] [PubMed] [Google Scholar]
- Makarov V.; Manina G.; Mikusova K.; Möllmann U.; Ryabova O.; Saint-Joanis B.; Dhar N.; Pasca M. R.; Buroni S.; Lucarelli A. P.; Milano A.; De Rossi E.; Belanova M.; Bobovska A.; Dianiskova P.; Kordulakova J.; Sala C.; Fullam E.; Schneider P.; McKinney J. D.; Brodin P.; Christophe T.; Waddell S.; Butcher P.; Albrethsen J.; Rosenkrands I.; Brosch R.; Nandi V.; Bharath S.; Gaonkar S.; Shandil R. K.; Balasubramanian V.; Balganesh T.; Tyagi S.; Grosset J.; Riccardi G.; Cole S. T. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science 2009, 324, 801–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov V.; Lechartier B.; Zhang M.; Neres J.; van der Sar A. M.; Raadsen S. A.; Hartkoorn R. C.; Ryabova O. B.; Vocat A.; Decosterd L. A.; Widmer N.; Buclin T.; Bitter W.; Andries K.; Pojer F.; Dyson P. J.; Cole S. T. Towards a new combination therapy for tuberculosis with next generation benzothiazinones. EMBO Mol. Med. 2014, 10.1002/emmm.201303575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trefzer C.; Rengifo-Gonzalez M.; Hinner M. J.; Schneider P.; Makarov V.; Cole S. T.; Johnsson K. Benzothiazinones: prodrugs that covalently modify the decaprenylphosphoryl-beta-d-ribose 2′-epimerase DprE1 of Mycobacterium tuberculosis. J. Am. Chem. Soc. 2010, 132, 13663–13665. [DOI] [PubMed] [Google Scholar]
- Barry C. E.; Crick D. C.; McNeil M. R. Targeting the formation of the cell wall core of M. tuberculosis. Infect. Disord. Drug Targets 2007, 7, 182–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolucka B. A. Biosynthesis of d-arabinose in mycobacteria: a novel bacterial pathway with implications for antimycobacterial therapy. FEBS J. 2008, 275, 2691–711. [DOI] [PubMed] [Google Scholar]
- Tiwari R.; Moraski G. C.; Krchnak V.; Miller P. A.; Colon-Martinez M.; Herrero E.; Oliver A. G.; Miller M. J. Thiolates chemically induce redox activation of BTZ043 and related potent nitroaromatic anti-tuberculosis agents. J. Am. Chem. Soc. 2013, 135, 3539–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trefzer C.; Skovierova H.; Buroni S.; Bobovska A.; Nenci S.; Molteni E.; Pojer F.; Pasca M. R.; Makarov V.; Cole S. T.; Riccardi G.; Mikusova K.; Johnsson K. Benzothiazinones are suicide inhibitors of mycobacterial decaprenylphosphoryl-beta-d-ribofuranose 2′-oxidase DprE1. J. Am. Chem. Soc. 2012, 134, 912–915. [DOI] [PubMed] [Google Scholar]
- Gao C.; Ye T.-H.; Wang N.-Y.; Zeng X.-X.; Zhang L.-D.; Xiong Y.; You X.-Y.; Xia Y.; Xu Y.; Peng C.-T.; Zuo W.-Q.; Wei Y.; Yu L.-T. Synthesis and structure–activity relationships evaluation of benzothiazinone derivatives as potential anti-tubercular agents. Bioorg. Med. Chem. Lett. 2013, 23, 4919–4922. [DOI] [PubMed] [Google Scholar]
- Neres J.; Pojer F.; Molteni E.; Chiarelli L. R.; Dhar N.; Boy-Rottger S.; Buroni S.; Fullam E.; Degiacomi G.; Lucarelli A. P.; Read R. J.; Zanoni G.; Edmondson D. E.; De Rossi E.; Pasca M. R.; McKinney J. D.; Dyson P. J.; Riccardi G.; Mattevi A.; Cole S. T.; Binda C. Structural basis for benzothiazinone-mediated killing of Mycobacterium tuberculosis. Sci. Transl. Med. 2012, 4, 150ra121–150ra121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: A new approach for rapid, accurate docking and scoring. 1. method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [DOI] [PubMed] [Google Scholar]
- Hoffman R. V.Organic Syntheses; Wiley & Sons: New York, 1990; Vol. 7, p 508. [Google Scholar]
- Wright S. W.; Hageman D. L.; McClure L. D. Fluoride-mediated boronic acid coupling reactions. J. Org. Chem. 1994, 59, 6095–6097. [Google Scholar]
- Campeau L.-C.; Rousseaux S.; Fagnou K. A solution to the 2-pyridyl organometallic cross-coupling problem: Regioselective catalytic direct arylation of pyridine N-oxides. J. Am. Chem. Soc. 2005, 127, 18020–18021. [DOI] [PubMed] [Google Scholar]
- Sun H.-Y.; Gorelsky S. I.; Stuart D. R.; Campeau L.-C.; Fagnou K. Mechanistic analysis of azine N-oxide direct arylation: Evidence for a critical role of acetate in the Pd(OAc)2 precatalyst. J. Org. Chem. 2010, 75, 8180–8189. [DOI] [PubMed] [Google Scholar]
- Cho S. H.; Warit S.; Wan B.; Hwang C. H.; Pauli G. F.; Franzblau S. G. Low-oxygen-recovery assay for high-throughput screening of compounds against nonreplicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2007, 51, 1380–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins L.; Franzblau S. G. Microplate alamar blue assay versus BACTEC 460 system for high-throughput screening of compounds against Mycobacterium tuberculosis and Mycobacterium avium. Antimicrob. Agents Chemother. 1997, 41, 1004–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Voss J. J.; Rutter K.; Schroeder B. G.; Su H.; Zhu Y.; Barry C. E. III. The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc. Natl. Acad. Sci. U.S.A. 2000, 97, 1252–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulliken R. S. Electronic population analysis on LCAO-MO [linear combination of atomic orbital-molecular orbital] molecular wave functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar]
- Dewar M. J. S.; Zoebisch E. G.; Healy E. F.; Stewart J. J. P. Development and use of quantum mechanical molecular models. 76. AM1: A new general purpose quantum mechanical molecular model. J. Am. Chem. Soc. 1985, 107, 3902–3909. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.