

Abstract
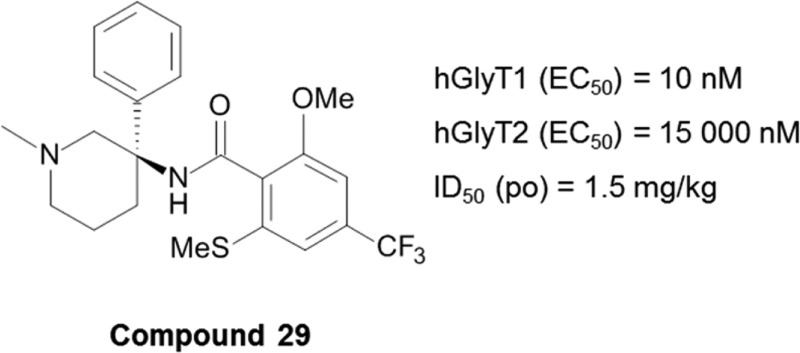
3-Amido-3-aryl-piperidines were discovered as a novel structural class of GlyT1 inhibitors. The structure–activity relationship, which was developed, led to the identification of highly potent compounds exhibiting excellent selectivity against the GlyT2 isoform, drug-like properties, and in vivo activity after oral administration.
Keywords: GlyT1 inhibitor, SAR, 3-amido-3-aryl-piperidines, Schizophrenia
Schizophrenia is a devastating and chronic mental illness that affects close to 1% of the population. Symptoms of schizophrenia, which typically arise at young age (adolescence or early adulthood), are categorized as positive, negative, or cognitive. Currently approved drugs are efficacious in the treatment of positive symptoms but do not address the negative and cognitive symptoms. Numerous lines of evidence suggest that hypofunction of glutamatergic transmission via N-methyl-d-aspartate (NMDA) receptors may represent the final common pathway leading to symptoms in schizophrenic patients.1 One approach to normalize the reduced NMDA receptor activity is to elevate the concentration of the coagonist glycine at its modulatory site on the receptor through blockade of the glycine transporter type 1 (GlyT1), which is colocalized with the NMDA receptor.2 In the past decade, ample efforts have been focused on the discovery and development of selective GlyT1 inhibitors.3 The first examples reported were sarcosine derivatives including 1(4) and 2(5) (Figure 1).
Figure 1.

Selection of published GlyT1 inhibitors.
More recently, nonsarcosine-based compounds6 have been described such as DCCCyB (3),7 GSK1018921 (4),8 and bitopertin (5),9,10 which all progressed to clinical studies (Figure 1). In a phase II proof of concept study, Bitopertin demonstrated a beneficial effect in patients with schizophrenia characterized with predominant negative symtoms.11 However, recently, Hoffmann-La Roche announced that two phase III studies of bitopertin did not meet their clinical end points.12 Four additional phase III studies with bitopertin are ongoing.
As part of our continued effort to discover and develop novel and selective GlyT1 inhibitors, we considered the pyrrolidino-ethyl-amide 4 as a potential starting point for further structural modification. In the low energy conformation obtained for compound 4 (Figure 2), the carbon (C1) adjacent to the pyrrolidine nitrogen and the benzylic carbon (C2) bearing the phenyl and amide substituents appeared as suitable attachment points for forming, via a linker, a novel cyclic system (Figure 2). We thus decided to investigate this cyclization approach further. As connecting elements between C1 and C2, a bond, a methylene, and an ethylene unit were evaluated (in red, Figure 2). To make our designed molecules chemically tractable, the gem dimethyl group and the pyrrolidine ring present in 4 were deleted. As a result, our approach led us to the generation of the 3,3′-substituted-N-methyl-azetidine 6, pyrrolidine 7, and piperidine 8 (Figure 2). In a glycine uptake inhibition assay performed in CHO cells transfected with hGlyT1, azetine 6 and pyrrolidine 7 were found inactive or poorly active. In contrast, and to our delight, piperidine 8 displayed a promising GlyT1 potency with an IC50 of 140 nM. The X-ray crystal structure of 8(13) (Figure 2) showed that the amide and phenyl substituents at the position 3 on the piperidine ring adopted, respectively, an axial and equatorial orientation. In addition, because of the presence of the two ortho groups on the benzamide side, the carbonyl function adopted a highly twisted orientation with a torsion angle of 78°. Overall, as depicted in Figure 2, the crystal structure of 8 aligned very well with the low energy conformation of 4, a result perfectly in line with the good GlyT1 potency measured for 8. A literature and patent search revealed that, although quite structurally simple, 3-amido-3-aryl-piperidines such as 8 had not been previously described. Thus, the discovery of 8 offered us the unique opportunity to develop a novel series of potent GlyT1 inhibitors having an exquisite intellectual proprietary status. In this letter, we report on our structure–activiy relationship (SAR) exploration effort in this series14 that culminated in the identification of potent, selective, and orally active GlyT1 inhibitors such as compound 29.
Figure 2.
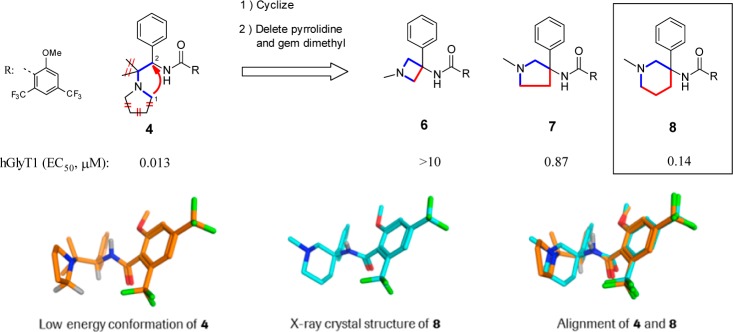
Design principles leading to the identification of active 3-amido-3-aryl-piperidine 8 using pyrrolidino-ethyl-amide 4 as a seed structure.
N-Methyl-3-amido-3-aryl-piperidine derivatives 8 and 18–43 were synthesized under standard amide coupling conditions from N-methyl-3-amino-3-aryl-piperidines 44 (Scheme 1). Derivatives 10–17 having different R1 groups on the piperidine nitrogen were obtained upon alkylation, reductive amination, acylation, or sulfonylation of the unsubstituted piperidine 9, itself prepared from N-Boc-protected-3-amino-3-phenyl-piperidine building block 45. N-Methyl-3-amino-3-aryl-piperidine intermediates 44a–d carrying the aromatic R3 substituents indicated in Scheme 2 were prepared from the 3-aryl-3-piperidin-ol derivatives 46a–d under modified Ritter reaction conditions15 in the presence of sodium azide and trifluoroacetic acid, followed by reduction of the obtained azido intermediates 47a–d with lithium aluminum hydride.
Scheme 1. Synthesis of 3-Amido-3-aryl-piperidines 8–43.

Reagents and conditions: (a) R2COCl, DIPEA, CH2Cl2, RT, 4–90%; (b) R2CO2H, HATU, DIPEA, DMF, RT, 18–68%; (c) 2- OMe,2′,4-(CF3)2-PhCOCl, DIPEA, CH2Cl2, RT, 86%; (d) HCl, dioxane, RT, 86%; (e) R1I, DIPEA, CH2Cl2, RT, 58%; (f) ketone or aldehyde, NaCNBH3, AcOH, MeOH, RT, 63–89%; (g) acyl- or sulfonyl-chloride, DIPEA, CH2Cl2, RT, 65–99%.
Scheme 2. Synthesis of N-Methyl-3-amino-3-aryl-piperidines 44a–d.

Reagents and conditions: (a) NaN3, TFA, H2O, RT, 87–100%; (b) LiAlH4, THF, RT, 32–63%.
However, this synthetic approach turned out to be not versatile enough. In particular, the azidation reaction performed on 3-aryl-3-piperidinols carrying electron rich aryl R3 groups such as 4-OMe-Ph failed. During the search of a more general access route, we discovered that N-methyl-3-amino-3-aryl-piperidine intermediates 44 could be efficiently prepared from easily accessible 4-aryl-4-nitro-butyric acid methyl esters 51 via a novel route involving as a key step a Mannich in situ lactamization reaction as depicted in Scheme 3. When performed in the presence of methylamine and formaldehyde, this sequence provided with good to excellent yields N-methyl-5-aryl-5-nitro-piperidin-2-one intermediates 52, which led to the target building blocks 44 after the reduction of the nitro and carbonyl functions. In addition, the synthetic access route we have established for 44 was successfully applied to the preparation of N-Boc-3-amino-3-phenyl-piperidine building block 45 using ammonium acetate and formaldehyde as reagents in the key Mannich in situ lactamization reaction (Scheme 3).
Scheme 3. Synthesis of N-Methyl-3-amino-piperidines 44f–o and N-Boc-3-amino-3-phenyl-piperidine 45.

Reagents and conditions: (a) methyl acrylate, amberlyst A-21, dioxane, RT, 41–85%; (b) cat. Pd2dba3, cat. di-tert-butyl-(2′-methyl-biphenyl-2-yl)-phosphane, Cs2CO3, DME, reflux, 10–90%; (c) MeNH2 (41% in water), CH2O (36% in water), dioxane, RT then 65 °C, 49–94%; (d) zinc, HCl, dioxane, RT, 13–86%; (e) LiAlH4, THF, RT, 71–86%; (f) Lawesson’s reagent, toluene, 80 °C, 77–100%; (g) NaBH4, MeOH, RT, 49–76%; (h) Ra–Ni, H2 (1 atm.), THF, 0 °C, 87–100%; (i) NH4OAc, CH2O (36% in water), EtOH, reflux, 84%; (j) Boc2O, Et3N, CH2Cl2, RT, 70%.
Our first effort aimed at establishing the SAR at the piperidine nitrogen (Table 1). Starting from 8, deletion of the methyl group (compound 9) led to a 5-fold reduction in GlyT1 activity. A drop of activity was equally observed upon replacing the methyl group with larger alkyl (10, 11) or cycloalkyl groups (12, 13). Moreover, N-benzylation (14) as well as N-acylation and sulfonylation (15–17) practically abolished the GlyT1 potency suggesting that a certain level of basicity of the piperidine nitrogen is required for GlyT1 activity. Overall, our exploration at this position revealed a highly restricted SAR with the methyl group being the only well tolerated substituent.
Table 1. In Vitro Inhibitory Activity of 9–17 at GlyT1a.
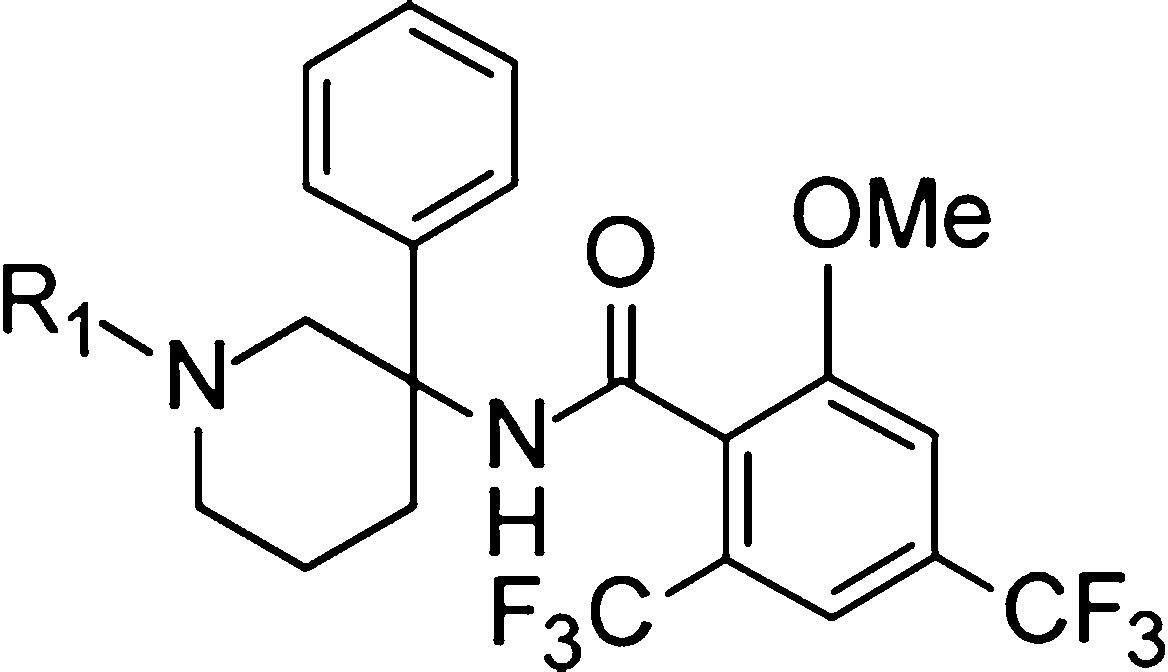
| compd | R1 | GlyT1 EC50 (μM)b |
|---|---|---|
| 9 | H | 0.66 |
| 10 | Et | 0.23 |
| 11 | i-Pr | 0.23 |
| 12 | c-C5H9 | 0.27 |
| 13 | CH2-c-Pr | 0.38 |
| 14 | CH2–Ph | 6.7 |
| 15 | C(O)–Me | >10 |
| 16 | C(O)–Ph | >10 |
| 17 | SO2Me | >10 |
EC50 values are the average of at least two independent experiments.
[3H]-glycine uptake inhibition assay in cells transfected with hGlyT1.10
Next, keeping the preferred N-methyl group in place, the SAR evaluation at the benzamide region was conducted (Table 2). Starting from 8, selective deletion of the ortho, ortho′, and para substituents (compounds 18 to 21) resulted in a significant drop of GlyT1 potency suggesting that substitution at all three positions on the benzamide moiety is important for reaching a good level of potency. In particular, the lack of activity observed for the p-CF3 monosubstituted compound 21, devoid of any ortho groups suggested that for GlyT1 activity the amide carbonyl group should adopt a twisted oriention as seen in the X-ray structure of our initial compound 8. Next, starting from inactive compound 21 and keeping the p-CF3 group in place, we explored various substituents at the ortho position (compounds 22–26). No improvement of activity was observed with polar groups in place such as nitrile (22) as well as with small substituents like fluorine (23). With larger lipophilic groups such as methyl (24) or bromine (25), the activity improved by more than 30-fold. Further exploration in that direction resulted finally in the identification of the thiomethyl group as one of the best substituents at the ortho position providing a compound displaying a low nanomolar activity (26, 67 nM). Our initial SAR studies (vide supra) suggested that a further gain in activity could be predicted upon introducing a second ortho group on 26. Pleasingly, by adding a methoxy group (27), a very potent compound was generated demonstrating an EC50 of 24 nM. This compound existing as a racemic mixture was subsequently separated by chiral HPLC to provide the two enantiomers 28 ((S)-enantiomer) and 29 ((R)-enantiomer). The R-configurated isomer 29(16) showed the best GlyT1 potency, reaching an EC50 as low as 10 nM at the hGlyT1 (5 nM at the mouse GlyT1).
Table 2. In Vitro Inhibitory Activity of 18–30 at GlyT1a.
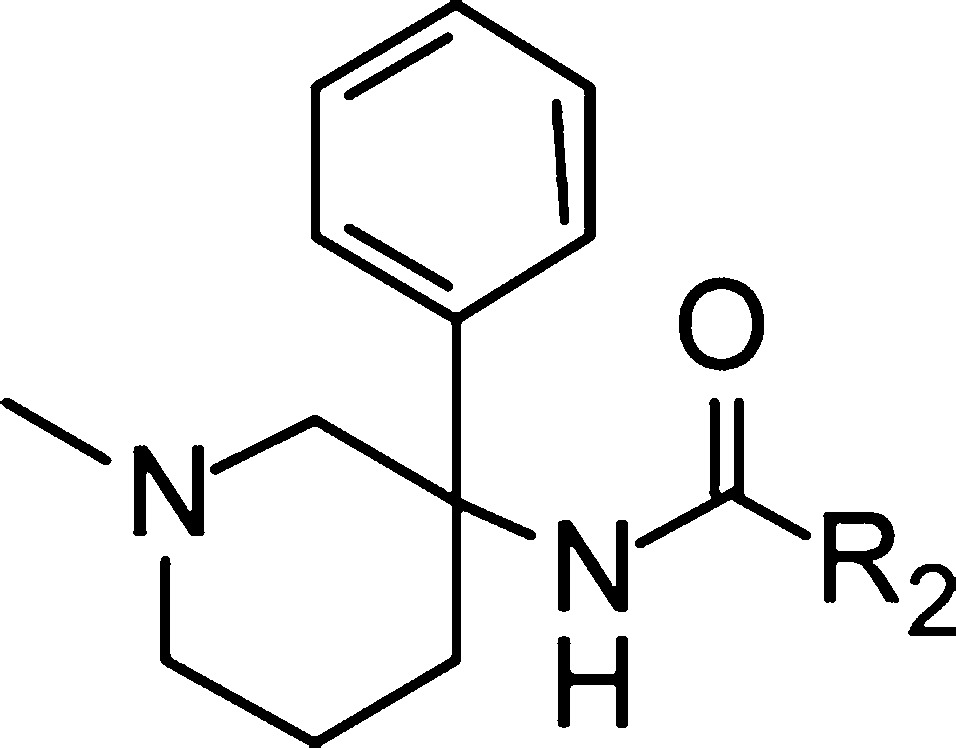
| compd | R2 | GlyT1 EC50 (μM)b |
|---|---|---|
| 18 | 2-OMe, 6-CF3–Ph | 1.5 |
| 19 | 2-OMe, 4-CF3–Ph | 4.8 |
| 20 | 2-CF3, 4-CF3–Ph | 0.54 |
| 21 | 4-CF3–Ph | >10 |
| 22 | 2-CN, 4-CF3–Ph | >10 |
| 23 | 2-F, 4-CF3–Ph | 9.9 |
| 24 | 2-Me, 4-CF3–Ph | 0.32 |
| 25 | 2-Br, 4-CF3–Ph | 0.30 |
| 26 | 2-SMe, 4-CF3–Ph | 0.067 |
| 27 | 2-SMe, 2′-OMe, 4-CF3–Ph | 0.024 |
| 28 | S-enantiomer of 27 | 0.18 |
| 29 | R-enantiomer of 27 | 0.010 |
| 30 | 2,4-Cl2–Ph | 0.67 |
EC50 values are the average of at least two independent experiments.
[3H]-glycine uptake inhibition assay in cells transfected with hGlyT1.10
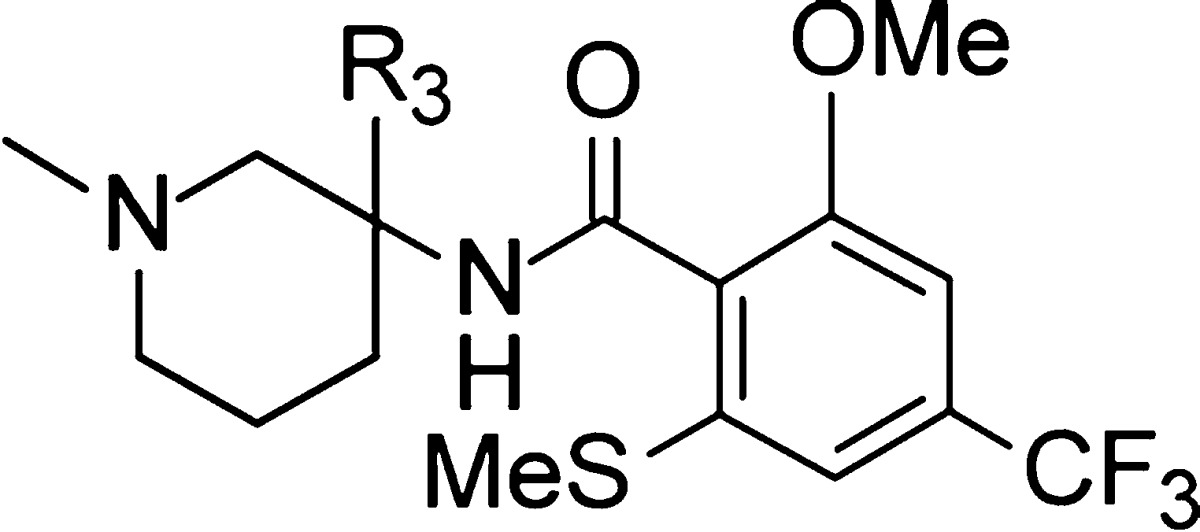
Finally, the SAR around the aryl group at position 3 on the piperidine ring was explored (Table 3).
Table 3. In Vitro Inhibitory Activity of 31–43 at GlyT1a.
| compd | R3 | GlyT1 EC50 (μM)b |
|---|---|---|
| 31 | 4-F–Ph | 0.026 |
| 32 | 4-Cl–Ph | 0.028 |
| 33 | 4-Me–Ph | 0.23 |
| 34 | 4-OMe–Ph | 0.17 |
| 35 | 3-F–Ph | 0.048 |
| 36 | 3-Cl–Ph | 0.025 |
| 37 | 3-OMe–Ph | 0.110 |
| 38 | pyridin-3-yl | 0.054 |
| 39 | pyridin-4-yl | 0.060 |
| 40 | 5-F-pyridin-2-yl | 0.180 |
| 41 | pyrazinyl-2-yl | 1.670 |
| 42 | pyrimidin-2-yl | >10 |
| 43 | pyrimidin-4-yl | 1.7 |
EC50 values are the average of at least two independent experiments.
[3H]-glycine uptake inhibition assay in cells transfected with hGlyT1.10
This study was performed keeping the optimized 2-thiomethyl-2-methoxy-4-trifluoromethyl-phenyl group in place at the benzamide moiety. A very similar SAR was seen at the para and meta position on the aryl moiety. Indeed, at both positions, introduction of EWGs like fluorine or chlorine (compounds 31–32 and 35–36) was well tolerated (EC50 = 23–48 nM), whereas with EDGs such as methoxy group (34, 37) the potency dropped significantly (EC50 = 110–170 nM). Moreover, a 6-membered-ring-heteroaryl scan revealed that the meta and para positions were the best centers to insert a nitrogen atom as exemplified with compounds 38 (54 nM) and 39 (60 nM). All the synthetized heteroaryl derivatives having a nitrogen atom in ortho (40–43) displayed much reduced potencies.
From our SAR and optimization studies, compound 29 emerged as the most potent congener from our newly discovered piperidine series. Further profiling revealed that 29 had excellent selectivity against GlyT2 (15 μM) as well as a fairly good selectivity profile in a CEREP screen performed against a panel of 80 targets including transmembrane and soluble receptors, ion channels, and monoamine transporters.17 Moreover, as indicated in Table 4, 29 displayed good physicochemical properties with a moderate lipophilicity, a good aqueous solubility and membrane permeability. The metabolic stability of 29 in mouse and human liver microsomes was found to be intermediate with a measured intrinsic clearance of 54 and 19 μL/min/mg protein, respectively. Not surprisingly, in vitro metabolism studies on 29 revealed that the corresponding N-des-methyl piperidine derivative was a major metabolite after incubation in human hepatocytes. In agreement with our SAR results (vide supra), this compound showed only weak GlyT1 activity (EC50 = 0.45 μM). Gratifyingly, 29 showed low inhibitory activity at the hERG potassium channel (IC20 = 5.4 μM), a result that may be attributed to the relatively low basicity of its piperidine nitrogen (pKa = 7.72). For comparison, pyrrolidino-ethyl-amide seed structure 4, which displayed a higher basicity (pKa = 9.12), showed a significantly higher hERG activity (IC20 = 0.7 μM). Moreover, 29 demonstrated a low level of inhibition at the major drug metabolizing CYP450 enzymes (IC50 > 16 μM) and was inactive in the genotoxicity assays (Ames and MNT tests). Finally, 29 was not a substrate for the human and mouse P-gp efflux systems.
Table 4. In Vitro Properties of 29.
| parameter | value |
|---|---|
| log Da | 3.12 |
| solubilityb (μg/mL) | 147 |
| pampa pec (10–6 cm/s) | 7.4 |
| pKad | 7.72 |
| human, mouse CLint (μL/min/mg protein) | 19, 54 |
| hERG IC20e (μM) | 5.4 |
| CYP450 3A4, 2D6, 2C9, 2C19 IC50 (μM) | >50, 16, >50, 46 |
| human, mouse P-gp ERf | 0.9, 1.5 |
Determined in 1-octanol/phosphate buffer, pH = 7.4.
Measured in a lyophilization solubility assay (LYSA) at pH = 6.5.18
Pe: PAMPA permeation constant through artificial membranes.19
Determined by capillary electrophoresis.
Patch clamp assay.
ER = efflux ratio in LLC-PK1 cells stably expressing human MDR1 and mouse MDR1A.
In vivo pharmacokinetic studies in mouse (Table 5) revealed, in addition, that 29 had an attractive profile, with, in particular, a complete oral bioavailability despite a medium systemic clearance and an excellent brain penetration (brain/plasma = 4.3), a result in agreement with its low P-gp efflux ratio (1.5) and large volume of distribution. Interestingly, 29 demonstrated a much superior brain penetration profile over seed compound 4 for which a P-gp efflux ratio of 3.9 and a brain/plasma ratio of only 0.4 was measured in the mouse.
Table 5. Pharmacokinetics Properties of 29 in Mouse.
| parameter | valuea |
|---|---|
| iv dose (1.7 mg/kg) | |
| CL (mL/min/kg) | 54 |
| Vss (L/kg) | 8 |
| po dose (7.7 mg/kg) | |
| bioavailability F (%) | 100 |
| t1/2 (h) | 4.2 |
| brain/plasma (at 1.5 h) | 4.3 |
| PPB (% unbound) | 1.7 |
Values are the average of two independent experiments.
These favorable data led us to evaluate the oral effect of 29 in the mouse L-687,414-induced hyperlocomotion assay, a novel and straightforward functional screening model,20 that allows the detection of the in vivo activity of GlyT1 inhibitors (Figure 3). Pleasingly, 29 displayed a robust pharmacodynamic activity after oral administration and reached an ID50 (dose producing 50% inhibition of L-687,414-induced hyperlocomotion) of 1.5 mg/kg po. Noteworthy, the in vivo efficacy of 29 was reached at a very low plasma concentration (8 ng/mL), a result that can be explained by its high GlyT1 potency and its excellent brain penetration in the mouse.
Figure 3.

Dose-dependent inhibition of L-687,414-induced hyper-locomotion by 29 in mice. The data represent mean horizontal activity counts per group recorded over a 60 min time period; error bars indicate SEM (n = 8 per group). ##, p < 0.01 versus vehicle (Veh) alone; ***, p < 0.001 versus L-687,414 alone.
In addition, the effect of 29 on the extracellular level of glycine in rat striatum in a microdialysis study was evaluated (Table 6). We were pleased to observe that, at an oral dose of 10 mg/kg, 29 produced a 1.7-fold glycine increase over basal levels.
Table 6. Effect of 29 on the Extracellular Glycine Levels in Rat Striatum at 10 mg/kg poa and Its Plasma and Brain Exposures.
| max. fold incr. of glycine | fold incr. of glycine at 3 h | plasmab (ng/mL) | brainb (ng/mL) | brainb GlyT1 EC50 |
|---|---|---|---|---|
| 1.7 | 1.5 | 713 | 60 | 13 |
n = 6.
Measured at 3 h postdosing.
After 3 h, at which time the extracellular fluid sampling was stopped, the PD effect was slightly diminished (1.5-fold). Determination of the plasma and brain exposures at 3 h postdosing revealed a much lower brain penetration (B/P: 0.08) for 29 in this rat experiment compared to its penetration measured in the mouse (B/P: 4.3) (vide supra). In spite of this, 27 achieved a total brain exposure above its GlyT1 EC50 (13-fold), sufficient to produce the effect on glycine levels observed at the 3 h time point.21
Noteworthy, in a dedicated mechanistic study performed in the rat, we observed that the brain/plasma ratio increased by 4-fold after coadministration of 29 with the P-gp inhibitor tariquidar22 indicating that 29 is a rat P-gp substrate. This result is in sharp contrast with the low P-gp activity of 29 measured in the mouse (vide supra). The marked difference in brain penetration observed in the mouse and in the rat may thus be attributed to the differentiated P-gp activity profile of 29 in these two rodent species. Compound 29, which is not a substrate of the human P-gp, is thus predicted to display in human an excellent brain penetration, quite similar to the one measured in the mouse.
In summary, we report here on the discovery of 3-amido-3-aryl-piperidines, a novel structural class of GlyT1 inhibitors designed using the previously reported pyrrolidino-ethyl-amide 4 as a seed structure. From the initial compound 8, the exploration of the SAR at the three exit vectors has led to the identification of potent and selective compounds such as 29, which demonstrated drug-like properties, a promising in vitro safety profile and in vivo efficacy in rodents after oral administration.
Acknowledgments
We would like to thank André Alker, Theo Stoll, Christian Schnider, and Daniel Zimmerli for their dedicated technical assistance.
Glossary
ABBREVIATIONS
- CYP
cytochrome P
- dba
dibenzylideneacetone
- DIPEA
N,N-diisopropylethylamine
- EWG
electron withdrawing group
- EDG
electron donating group
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyri-dinium 3-oxid hexafluorophosphate
- hERG
human ether-à-go-go-related gene
- MDR
multidrug resistance protein
- TFA
trifluoro acetic acid
Supporting Information Available
Experimental details for the synthesis of compounds 8–43 and intermediates. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Kantrowitz J. T.; Javitt D. C. N-Methyl-d-aspartate (NMDA) receptor dysfunction or dysregulation: The final common pathway on the road to schizophrenia?. Brain Res. Bull. 2010, 83, 108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chue P. Glycine reuptake inhibition as a new therapeutic approach in schizophrenia: focus on the glycine transporter 1 (GlyT1). Curr. Pharm. Des. 2013, 19, 1311–1320. [DOI] [PubMed] [Google Scholar]
- Hashimoto K. Glycine transport inhibitors for the treatment of schizophrenia. Open Med. Chem. J. 2010, 4, 10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson B. N.; Bell S. C.; De Vivo M.; Kowalski L. R.; Lechner S. M.; Ognyanov V. I.; Tham C.-S.; Tsai C.; Jia J.; Ashton D.; Klitenick M. A. ALX 5407: a potent, selective inhibitor of the hGlyT1 glycine transporter. Mol. Pharmacol. 2001, 60, 1414–1420. [DOI] [PubMed] [Google Scholar]
- Brown A.; Carlyle I.; Clark J.; Hamilton W.; Gibson S.; McGarry G.; McEachen S.; Rae D.; Thorn S.; Walker G. Discovery and SAR of Org 24598: a selective glycine uptake inhibitor. Bioorg. Med. Chem. Lett. 2001, 11, 2007–2009. [DOI] [PubMed] [Google Scholar]
- Wolkenberg S. E.; Sur C. Recent progress in the discovery of non-sarcosine based GlyT1 inhibitors. Curr. Top. Med. Chem. 2010, 10, 170–186. [DOI] [PubMed] [Google Scholar]
- Blackaby W. P.; Lewis R. T.; Thomson J. L.; Jennings A. S. R.; Goodacre S. C.; Street L. J.; MacLeod A. M.; Pike A.; Wood S.; Thomas S.; Brown T. A.; Smith A.; Pillai G.; Almond S.; Guscott M. R.; Burns H. D.; Eng W.; Ryan C.; Cook J.; Hamill T. G. Identification of an orally bioavailable, potent, and selective inhibitor of GlyT1. ACS Med. Chem. Lett. 2010, 7, 350–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffini P.; James A. D.; Roberts A. D.; Pellegatti M. Metabolites in safety testing: issues and approaches to the safety evaluation of human metabolites in a drug that is extensively metabolized. J. Drug Metabolism. Toxicol. 2010, 1, 1000102. [Google Scholar]
- Pinard E.; Alanine A.; Alberati D.; Bender M.; Borroni E.; Bourdeaux P.; Brom V.; Burner S.; Fischer H.; Hainzl D.; Halm R.; Hauser N.; Jolidon S.; Lengyel J.; Marty H. P.; Meyer T.; Moreau J. L.; Mory R.; Narquizian R.; Nettekoven M.; Norcross R. D.; Puellmann B.; Schmid P.; Schmitt S.; Stalder H.; Wermuth R.; Wettstein J. G.; Zimmerli D. Selective GlyT1 inhibitors: discovery of [4-(3-fluoro-5-trifluoromethylpyridin-2-yl)piperazin-1-yl][5-methanesulfonyl-2-((S)-2,2,2-trifluoro-1-methylethoxy)phenyl]methanone (RG1678), a promising novel medicine to treat Schizophrenia. J. Med. Chem. 2010, 53, 4603–4614. [DOI] [PubMed] [Google Scholar]
- Alberati D.; Moreau J. L.; Lengyel J.; Hauser N.; Mory R.; Borroni E.; Pinard E.; Knoflach F.; Schlotterbeck G.; Hainzl D.; Wettstein J. G. Glycine reuptake inhibitor RG1678: A pharmacologic characterization of an investigational agent for the treatment of schizophrenia. Neuropharmacology 2012, 62, 1152–1161. [DOI] [PubMed] [Google Scholar]
- Umbricht D.; Martin-Facklam M.; Pizzagalli F. E.; Youssef E.; Yoo K.; Dorflinger E.; Bausch A.; Arrowsmith R.; Alberati D.; Santarelli L. Glycine transporter type 1 (GLYT1) inhibition RG1678: Results of the proof-of-concept study for the treatment of negative symptoms in schizophrenia. Schiz. Bull. 2011, 371324. [Google Scholar]
- Investor update: January, 21, 2014. www.roche.com.
- The X-ray crystal structure of 8 was determined on its (+)-(1S,4R)-camphor-10-sulfonic acid salt.
- Kolczewski S.; Pinard E.; Stalder H. WO Patent 2010086251, 2010.
- Balderman D.; Kalir A. Selective reduction of azides. Improved preparation of α,α-disubstituted benzylamines. Synthesis 1978, 1, 24–26. [Google Scholar]
- The absolute configuration was determined by X-ray crystallography of the (+)-(1S,4R)-camphor-10-sulfonic acid salt of 29.
- The panel consisted in targets included in the standard CEREP high throughput profile screen. The targets that were inhibited by more than 50% at the concentration of 10 μM: H2R, MC4R, NK1, NK2, BZDR (peripheral), Na Ch (site 2), and SSTR were followed up in dose–response experiments. In these studies, a selectivity of at least 110-fold vs GlyT1 was measured.
- Solubility was measured in a lyophilization solubility assay: Compound initially in DMSO solution is lyophilized then dissolved in 0.05 M phosphate buffer (pH 6.5), stirred for one hour, and shaken for two hours. After one night, the solution is filtered and the filtrate analyzed by direct UV measurement or by HPLC-UV.
- Kansy M.; Senner F.; Gubernator K. Physicochemical high throughput screening: parallel artificial membrane permeation assay in the description of passive absorption processes. J. Med. Chem. 1998, 41, 1007–1010. [DOI] [PubMed] [Google Scholar]
- Alberati D.; Moreau J.-L.; Mory R.; Pinard E.; Wettstein J. G. Pharmacological evaluation of a novel assay for detecting glycine transporter 1 inhibitors and their antipsychotic potential. Pharmacol., Biochem. Behav. 2010, 97, 185–191. [DOI] [PubMed] [Google Scholar]
- Similar PD effects were observed at similar brain/GlyT1 EC50 multiples with our previously published structurally distinct series of GlyT1 inhibitors:Pinard E.; Alberati D.; Bender M.; Borroni E.; Brom V.; Burner S.; Fischer H.; Hainzl D.; Halm R.; Hauser N. Discovery of benzoylisoindolines as a novel class of potent, selective and orally active GlyT1 inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 6960–6965. [DOI] [PubMed] [Google Scholar]
- Bankstahl J. P.; Kuntner C.; Abrahim A.; Karch R.; Stanek J.; Wanek T.; Wadsak W.; Kletter K.; Mueller M.; Loescher W. Tariquidar-induced P-glycoprotein inhibition at the rat blood–brain barrier studied with (R)-11C-verapamil and PET. J. Nucl. Med. 2008, 49, 1328–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.