

Abstract

Given the threat of drug resistance, there is an acute need for new classes of antimalarial agents that act via a unique mechanism of action relative to currently used drugs. We have identified a set of druglike compounds within the Tres Cantos Anti-Malarial Set (TCAMS) which likely act via inhibition of a Plasmodium aspartic protease. Structure–activity relationship analysis and optimization of these aminohydantoins demonstrate that these compounds are potent nanomolar inhibitors of the Plasmodium aspartic proteases PM-II and PM-IV and likely one or more other Plasmodium aspartic proteases. Incorporation of a bulky group, such as a cyclohexyl group, on the aminohydantion N-3 position gives enhanced antimalarial potency while reducing inhibition of human aspartic proteases such as BACE. We have identified compound 8p (CWHM-117) as a promising lead for optimization as an antimalarial drug with a low molecular weight, modest lipophilicity, oral bioavailability, and in vivo antimalarial activity in mice.
Keywords: Malaria, antimalarial, aminohydantoin, medicinal chemistry, aspartic protease inhibitors
Malaria is a devastating mosquito-borne infectious disease caused by a parasite of the genus Plasmodium, placing over one billion people at high risk for infection. According to the World Health Organization, there were an estimated 225 million cases of malaria in 2010 with 610,000–971,000 deaths.1 Especially hard hit is sub-Saharan Africa, where 80% of the deaths occur, mostly in children under the age of 5 years old. Although there are a number of drugs used to treat the disease, resistance to most of these drugs is widespread.2 The introduction of artemisinin and artemisinin combination therapies (ACTs) in 2005 has begun to reverse the trend. While this is a good sign, there have been reports of resistance to artemisinin in Southeast Asia.3 As a consequence, there is an urgent push for developing antimalarial therapies targeting novel modes of action. Drug discovery efforts in this area have been recently reviewed.4−6
Despite the urgent need, most antimalarial drugs in late stage clinical development do not target new mechanisms of action. Rather, many of these efforts have been focused on enhancing existing antimalarials, such as artemisinin. Recently, scientists at GlaxoSmithKline and elsewhere have published on the antimalarial activity of thousands of compounds.7,8 Within this data set, we have identified a class of aspartic protease inhibitors with the potential to be a good starting point for antimalarial drug discovery. This manuscript details our evaluation of this series of compounds as leads for optimization as antimalarial drugs with a novel mechanism of action as aspartic protease inhibitors.
P. falciparum, the most lethal to man of the Plasmodium species, has multiple aspartic proteases,9,10 including Plasmepsin V (PM-V), which has been demonstrated to be essential to the parasite’s survival due to its role in the export of hundreds of Plasmodium proteins to the host erythrocyte.11,12 Most of the early drug discovery efforts in this area have focused on inhibition of the plasmepsins of the digestive vacuole (PM-I, II, IV, and III or HAP), but these efforts have not been very successful due to the redundancy of the function of the digestive vacuole plasmepsins and other proteases. The function of the other plasmepsins (PM-VI, VII, VIII, IX, X) is unknown. Recently, the Plasmodium signal peptide peptidase (PfSPP), an aspartic protease member of a family of intramembrane cleaving proteases, has been demonstrated to be essential to parasite survival in both blood and liver stages.13,14
It is anticipated that inhibition of one or more aspartic proteases would be lethal to the parasite. However, there are currently few tools available to identify inhibitors of these more promising aspartic protease targets in an efficient manner. For example, the only known inhibitors of PM-V are pepstatin A and HIV protease inhibitors, such as lopinavir and ritonavir.11,12 These inhibitors are very weak inhibitors of PM-V (IC50 >20 μM) and, due to their large molecular weights and peptidomimetic character, make relatively poor starting points for a new antimalarial drug discovery program. Recently, optimization of the antimalarial activity of a hydroxylethylamine series was reported, presumably activating via an aspartic protease mechanism.15
Given the loose sequence homology between the aspartic protease β-secretase (BACE) and PM-V and the key role of the dual aspartic acid residues in the protease active site, we hypothesized that inhibitors of BACE may inhibit some members of the family of Plasmodium aspartic proteases as well. One of the scaffolds we selected was the aminohydantoin core common to BACE screening hit 1 and related optimized compounds such as 2 and 3.16,17 Compounds 2 and 3 are orally bioavailable, demonstrating the druglike nature of this chemotype. Furthermore, there are several X-ray crystal structures that demonstrate the key interactions between the protonated aminohydantoin core and the aspartic acid residues of the BACE active site (Figure 1).16 We used this aminohydantoin core to perform substructure searches on the more than 13,000 compounds (TCAMS) known to have antimalarial activity in Pf 3D7 infected red blood cells (RBC) published by GlaxoSmithKline.7 Our strategy is outlined in Figure 1.
Figure 1.

Medicinal chemistry strategy.
Chart 1. Example Aminohydantoin BACE Inhibitors with Common Core Colored Blue.
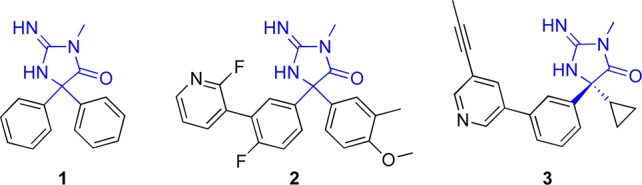
From the TCAMS collection, we identified 35 examples of aminohydantoins, with antimalarial activities ranging from 140 to 1500 nM, as exemplified by the racemate TCMDC-136879 (4), which had a published IC50 of 140 nM.7 We resynthesized compound 4 according to the procedure in Scheme 1, wherein diketone 5 was condensed with the corresponding thiourea 6 to give thiohydantoin 7. Thiohydantoin 7 was then converted to the aminohydantoin to give 4. Compound 4 was assayed for activity in the Pf 3D7 infected RBC assay.18 Compound 4 has a more modest Pf 3D7 IC50 of 2.76 μM in our hands but, interestingly, is also a potent inhibitor of PM-II (IC50 = 12 nM) and a modest inhibitor of the PM-V enzyme12 (IC50 = 977 nM). Given this data, we initiated a medicinal chemistry effort to prepare additional analogs to determine our ability to optimize for antimalarial potency, develop SAR, and understand the mechanism of action of this novel class of antimalarial compounds.
Scheme 1. General Synthesis of Aminohydantoins.

Reagents and conditions: (a) KOH, DMSO, heat. (b) X = S, t-BuOOH, NH4OH, MeOH.
Our initial efforts were to replace the flexible, lipophilic phenylbutyl side chain R1 and its chiral center. Analogs containing simple phenyl and p-methoxyphenyl were readily prepared from commercially available diketones (Scheme 1). The Pf 3D7 IC50 values for representative compounds are shown in Table 1. Compounds prepared in the R2/R3p-methoxyphenyl series were approximately 10-fold more potent than those in the unadorned phenyl series (e.g., 4 vs 8a, 8f vs 8g, and 8o vs 8p). Simple replacement of the phenylbutyl side chain with smaller alkyl groups, such as methyl, ethyl, and propyl, resulted in size-dependent losses in potency (8b–d). Large flexible groups, such as benzyl and phenethyl (8g–i), and secondary branched alkyls, such as isopropyl (8j), were tolerated but not tert-butyl (8k). Cyclic alkyl groups were identified to provide an optimal combination of potency and reduced lipophilicity (8n, 8p, 8q).
Table 1. SAR Analysis of N-R1 Substituents.
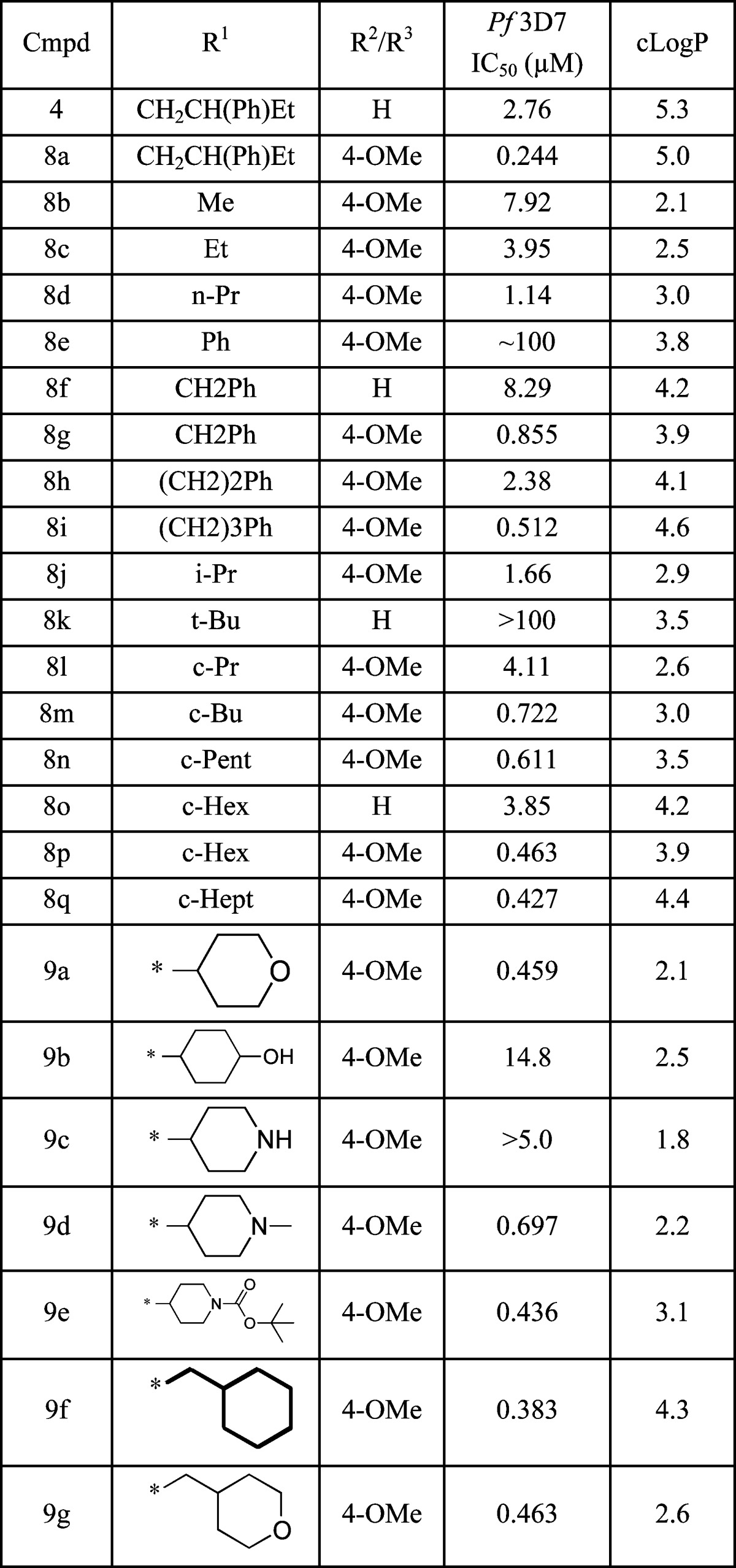
Cyclohexyl analog 8p was selected for further optimization studies, as derivitization of the cyclohexyl ring could be accomplished without adding additional stereocenters to the compounds (9a–g). Pyran derivative 9a has equivalent potency to cyclohexyl 8p while reducing the lipophilicy by 2 orders of magnitude. In contrast, the presence of an alcohol on the cyclohexyl ring (9b) was not tolerated. N-Alkylated and N-acylated piperidines (9d, 9e) are also suitable replacements for the cyclohexyl ring while the more polar NH piperidine was not tolerated (9c). Extension of the cyclohexyl (9f) and pyran (9g) rings by one carbon resulted in nearly identical potencies to those of the corresponding analogs 8p and 9a.
Having satisfactorily improved potency against the parasite by approximately 10-fold, we turned our attention toward evaluating the SAR of the aryl rings while keeping R1 constant as the cyclohexyl ring. While most of these substitutions elevated the lipophilicity of the compounds, our aim was to develop the SAR and to see if the SAR was consistent with inhibition of an aspartic protease active site (Table 2). More elaborate substitution patterns of the aryl rings in the R2 and R3 position required synthesis of diketone 5 by well-known methods (see Supporting Information).
Table 2. SAR Analysis of Aryl Substituents.
| cmpd | R1 | R2 | R3 | Pf 3D7 IC50 (μM) | cLogP |
|---|---|---|---|---|---|
| 10a | cHex | 3-OMe | 3-OMe | 0.768 | 3.9 |
| (±)-10b | cHex | 4-OMe | H | 0.533 | 4.1 |
| (±)-10c | cHex | 3-OMe | H | 0.917 | 4.1 |
| 10d | cHex | 4-OCF3 | 4-OCF3 | 2.20 | 7.1 |
| 10e | cHex | 4-OEt | 4-OEt | 0.466 | 4.6 |
| 10f | cHex | 4-Me | 4-Me | 1.26 | 5.3 |
| 10g | cHex | 3-Me | 3-Me | 0.533 | 5.3 |
| 10h | cHex | 4-Cl | 4-Cl | 1.07 | 5.5 |
| 10i | cHex | 3-Cl | 3-Cl | 1.06 | 5.5 |
| (±)-10j | cHex | 3-Ph | 4-OMe | 1.49 | 5.7 |
| (±)-10k | cHex | 3-(3-pyr) | 4-OMe | 0.595 | 4.5 |
| (±)-10l | cHex | 3-(3-pyr) | 3-OMe | 0.787 | 4.5 |
| (±)-10m | cHex | 3-(3-pyr) | H | 0.75 | 4.7 |
| (±)-10n | cHex | 3-(2-pyr) | 4-OMe | 1.81 | 4.9 |
| (±)-10o | cHex | 3-(4-pyr) | 3-OMe | 0.318 | 4.5 |
| 10p | cHex | 3-(3-pyr) | 3-(3-pyr) | 0.725 | 5.1 |
| (±)-10q | cHex | 3-OPh | H | 0.424 | 5.7 |
| (±)-10r | cHex | 3-OBn | H | 0.641 | 5.8 |
| (±)-11 | Me | 3-(3-pyr) | H | 6.88 | 5.8 |
Moving the methoxy groups to the 3-position results in only a 2-fold loss of potency (10a vs 8p) while deletion of the second methoxy group had a negligible effect on potency (10b–c). Replacement of methoxy with triflouromethoxy (10d) had a detrimental effect on potency while the ethoxy (10e) analog was equipotent. The more lipophilic methyl and chloro derivatives were tolerated in the 4- and 3-positions but did not enhance potency (10f–i).
Many advanced aminohydantoin inhibitors of BACE (e.g., 2 and 3) exhibit an aryl group in the 3-position of the aromatic ring. This substitution pattern allows the biaryl group of the inhibitor to occupy both the S1 and the S3 binding pockets simultaneously (Supporting Information; Figure S1). Since this general aspartic protease active site topography might be present in our Plasmodium target, we prepared derivatives such as 10j–p (Table 2). Phenyl derivative 10j resulted in only a modest 3- to 4-fold loss of potency. As has been observed with BACE,17 replacement of the phenyl ring with a 3-pyridyl ring (10k) results in improved potency relative to phenyl. Interestingly, the methoxy group on the second aryl ring has a negligible effect on potency (10l, 10m). 4-Pyridyl substitution (10o) had a similar effect to that of 3-pyridyl while 2-pyridyl (10n) was more similar to phenyl. Phenyl and benzyl ethers 10q and 10r demonstrated a relatively tolerant substitution pattern at this position.
Given the consistency of the SAR with an aspartic protease binding site, we evaluated a subset of these antimalarial compounds for inhibition of Plasmodium aspartic proteases PM-II and PM-IV (Supporting Information; Table S2). Most of the aminohydantoins evaluated are potent inhibitors of both PM-II and PM-IV, some in the single digit nanomolar range. Compounds with weaker 3D7 potency tended to have weaker PM-II and PM-IV activity (9b and 9c). We assayed ∼80 analogs in our collection for PM-II activity and plotted this potency versus 3D7 potency (Supporting Information; Figure S2). While there is a loose correlation between PM-II potency and 3D7 potency, there is a 30–100 fold shift in potency. Furthermore, it has been previously demonstrated that inhibition of Plasmepsins I–IV is insufficient to kill the parasite.19−21 These data suggest that the observed PM-II and PM-IV activity may be indicative of inhibition of one or more other, albeit unidentified, Plasmodium aspartic proteases. Additionally, the more potent compounds (e.g., 8p, 10a, 10m) appear to be inactive against PM-V (data not shown).
To further demonstrate the likelihood of these compounds inhibiting an aspartic protease binding site, we also prepared two simple analogs of compound 8p wherein the imino nitrogen atom was replaced with oxygen (7a) and with sulfur (7b), both of which are unable to form the requisite hydrogen bonding network to the catalytic aspartic acid residues (see Figure 1). These minor changes resulted in complete loss of 3D7, PM-II, and PM-IV potency, consistent with an aspartic protease mechanism of action (Supporting Information Table S2).
It is also evident that while these compounds are potent inhibitors of Plasmodium PM-II and PM-IV, they are poor inhibitors of human aspartic proteases BACE, CatD, and CatE (Supporting Information Table S2). Lead compound cyclohexyl 8p is >1000-fold selective for PM-II over BACE and CatD and ∼300-fold over CatE. Compounds containing the S3-occupying aryl ring, such as 10m, also retained greater than 1000-fold selectivity over BACE and CatD and ∼200-fold over CatE. Furthermore, comparator compound 11 with a methyl group in place of the cyclohexyl substituent in the R1 position demonstrates the importance of the cyclohexyl group (or a secondary alkyl group) in enhancing antimalarial 3D7 and PM-II potency while reducing potency against BACE (11 vs 10m). Compounds with larger but more flexible R1 side chains (4, 9f, and 9g) were found to be less selective.
In order to evaluate this series for its potential as oral antimalarial agents, compound 8p and closely related 9a, 9f, and 9g were further profiled (Supporting Information, Table S1). All four compounds retained nearly equivalent potency against 3D7 at 48 h and against the chloroquine- and pyrimethamine-resistant Dd2 strain, indicating the novel mechanism of action and the potential utility of this class of antimalarials in targeting drug resistant parasites.
The compounds were also profiled against a number of human enzymes and cell lines (Supporting Information Table S1). The compounds were found to have limited activity in a HepG2 cytotoxicity assay and against most CYP enzymes evaluated, with the exception of CYP3A4. Compound 8p showed good metabolic stability in mouse, rat, and human liver microsomes (MLM, RLM, and HLM, respectively) and is orally bioavailable in rats with a half-life of 2.9 h.
Given the overall properties of compound 8p, we elected to characterize compound 8p and related compounds in the in vivo P. chabaudi murine Peter’s 4-day suppressive test (Table 3).18 Parasitemia levels were determined 24 h after the last treatment. Compound 8p gave a strong effect (89% inhibition) at 100 mpk, while closely related 9a, 9f, and 9g were efficacious but to a lesser extent than 8p. These results may be explained in part by the apparent shorter half-lives of these compounds relative to 8p. At 24 h postdose, only compound 8p has greater than 1 μM concentration in the plasma. In a separate experiment with 8p, a clear dose–response relationship was obtained, with complete elimination of parasitemia achieved at 300 mpk (Figure 2). In this experiment, all of the mice survived at least 10 days post-treatment with 8p. 100% suppression of parasitemia was achieved in 10 of 12 animals in the 150 and 300 mpk dose groups.
Table 3. Oral in Vivo Efficacy of Lead Compounds (P. chabaudi ASS-Infected Murine Four-Day Suppressive Test).
| plasma [cmpd]d (ng/mL) |
|||||
|---|---|---|---|---|---|
| cmpd | dose (mpk)b | %Inh of parasitemiac | 1 h | 6 h | 24 h |
| CQ | 4.5 | 99.99 | nda | nd | nd |
| 8p | 100 | 89 | 2790 | 3393 | 575 |
| 9a | 110 | 63 | 4140e | nd | 29 |
| 9f | 100 | 73 | 2517 | 2187 | 7 |
| 9g | 100 | 41 | 10363 | 3027 | 123 |
nd = not determined.
Four hours after infection, compounds were dosed orally once daily for four days.
Determined on Day 4 postinoculation.
Compound concentration in the plasma determined 1, 6, and 24 h post last dose on Day 3.
Determined at 30 min post dose.
Figure 2.

Dose–response of compound 8p and CQ in the P. chabaudi ASCQ-infected murine four-day suppressive test. Four hours after infection, compound 8p was dosed orally once daily for four days. Parasitemia was determined on Day 4 postinoculation. The vehicle group was within error of the four lowest doses of compound 8p.
In conclusion, the SAR analysis of the aminohydantoin class of antimalarial compounds presented here demonstrates that these compounds are potent nanomolar inhibitors of the Plasmodium aspartic proteases PM-II and PM-IV and likely one or more other Plasmodium aspartic acid proteases. Incorporation of a bulky group, such as a cyclohexyl group, on the aminohydantion N-3 position gives enhanced antimalarial potency while reducing inhibition of human aspartic proteases such as BACE. Compound 8p (CWHM-117) presents a promising lead for optimization as an antimalarial drug with a low molecular weight, modest lipophilicity, oral bioavailability, and in vivo antimalarial activity in mice. Efforts are underway to further improve the whole cell potency in 3D7 and Dd2 strains and potency in in vivo models of malaria.
Acknowledgments
The authors would like to thank Eva Istvan for supplying the PM-II and PM-IV DNA constructs, and Felix Calderon, Elena Fernandez Alvaro, Lluis Ballell-Pages, Esther Pilar Fernandez-Velando, Inigo Angulo-Barturen, and Santiago Ferrer-Bazaga for helpful discussions.
Glossary
Abbreviations
- ACT
artemisinin combination therapy
- PM
plasmepsin
- HAP
histo-aspartic peptidase
- SPP
signal peptide peptidase
- HIV
human immunodeficiency virus
- SAR
structure–activity relationships
- BACE
β-site APP cleaving enzyme 1
- TCAMS
Tres Cantos Antimalarial Set
- RBC
red blood cells
- CatD
cathepsin D
- CatE
cathepsin E
- MLM
mouse liver microsomes
- RLM
rat liver microsomes
- HLM
human liver microsomes
- PK
pharmacokinetics
- PPB
plasma protein binding
- CYP
cytochrome P450
- CQ
chloroquine
- mpk
milligrams per kilogram
Supporting Information Available
Supporting tables, experimental procedures, characterization of compounds, and assay methods. This material is available free of charge via the Internet at http://pubs.acs.org.
Research reported in this publication at Saint Louis University was supported by Saint Louis University and the National Institute Of Allergy And Infectious Diseases of the National Institutes of Health under Award Number R01AI106498. Research reported in this publication at the Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, was supported by Bureau of Science and Information Technology of Guangzhou Municipality, Grant Number 2009Z1-E841, and Natural Science Foundation of China (NSFC). D.E.G. is supported by the National Institutes of Health under Award Number AI047798. The content is solely the responsibility of the authors and does not necessarily represent the official views of Saint Louis University, the Guangzhou Institutes of Biomedicine and Health, Chinese Academy of Sciences, the Natural Science Foundation of China, or the National Institutes of Health.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- World Malaria Report 2012; World Health Organization: 2012.
- Hyde J. E. Drug-resistant malaria. Trends Parasitol. 2005, 21, 494–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dondorp A. M.; Yeung S.; White L.; Nguon C.; Day N. P.; Socheat D.; von Seidlein L. Artemisinin resistance: current status and scenarios for containment. Nat. Rev. Microbiol. 2010, 8, 272–80. [DOI] [PubMed] [Google Scholar]
- Burrows J. N.; Chibale K.; Wells T. N. C. The State of the Art in Anti-malarial Drug Discovery and Development. Curr. Top. Med. Chem. 2011, 11, 1226–1254. [DOI] [PubMed] [Google Scholar]
- Burrows J. N.; Burlot E.; Campo B.; Cherbuin S.; Jeanneret S.; Leroy D.; Spangenberg T.; Waterson D.; Wells T. N.; Willis P. Antimalarial drug discovery—the path towards eradication. Parasitology 2013, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biamonte M. A.; Wanner J.; Le Roch K. G. Recent advances in malaria drug discovery. Bioorg. Med. Chem. Lett. 2013, 23, 2829–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamo F. J.; Sanz L. M.; Vidal J.; de Cozar C.; Alvarez E.; Lavandera J. L.; Vanderwall D. E.; Green D. V.; Kumar V.; Hasan S.; Brown J. R.; Peishoff C. E.; Cardon L. R.; Garcia-Bustos J. F. Thousands of chemical starting points for antimalarial lead identification. Nature 2010, 465, 305–10. [DOI] [PubMed] [Google Scholar]
- Calderón F.; Barros D.; Bueno J. M.; Coterón J. M.; Fernández E.; Gamo F. J.; Lavandera J. L.; León M. L.; Macdonald S. J. F.; Mallo A.; Manzano P.; Porras E.; Fiandor J. M.; Castro J. An Invitation to Open Innovation in Malaria Drug Discovery: 47 Quality Starting Points from the TCAMS. ACS Med. Chem. Lett. 2011, 2, 741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coombs G. H.; Goldberg D. E.; Klemba M.; Berry C.; Kay J.; Mottram J. C. Aspartic proteases of Plasmodium falciparum and other parasitic protozoa as drug targets. Trends Parasitol. 2001, 17, 532–7. [DOI] [PubMed] [Google Scholar]
- Meyers M. J.; Goldberg D. E. Recent advances in plasmepsin medicinal chemistry and implications for future antimalarial drug discovery efforts. Curr. Top. Med. Chem. 2012, 12, 445–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boddey J. A.; Hodder A. N.; Gunther S.; Gilson P. R.; Patsiouras H.; Kapp E. A.; Pearce J. A.; de Koning-Ward T. F.; Simpson R. J.; Crabb B. S.; Cowman A. F. An aspartyl protease directs malaria effector proteins to the host cell. Nature 2010, 463, 627–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo I.; Babbitt S.; Muralidharan V.; Butler T.; Oksman A.; Goldberg D. E. Plasmepsin V licenses Plasmodium proteins for export into the host erythrocyte. Nature 2010, 463, 632–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Chen H.; Bahamontes-Rosa N.; Kun J. F.; Traore B.; Crompton P. D.; Chishti A. H. Plasmodium falciparum signal peptide peptidase is a promising drug target against blood stage malaria. Biochem. Biophys. Res. Commun. 2009, 380, 454–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbut M. B.; Patel B. A.; Yeung B. K.; McNamara C. W.; Bright A. T.; Ballard J.; Supek F.; Golde T. E.; Winzeler E. A.; Diagana T. T.; Greenbaum D. C. Targeting the ERAD pathway via inhibition of signal peptide peptidase for antiparasitic therapeutic design. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 21486–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciana C. L.; Siegrist R.; Aissaoui H.; Marx L.; Racine S.; Meyer S.; Binkert C.; de Kanter R.; Fischli C.; Wittlin S.; Boss C. Novel in vivo active anti-malarials based on a hydroxy-ethyl-amine scaffold. Bioorg. Med. Chem. Lett. 2013, 23, 658–62. [DOI] [PubMed] [Google Scholar]
- Malamas M. S.; Erdei J.; Gunawan I.; Turner J.; Hu Y.; Wagner E.; Fan K.; Chopra R.; Olland A.; Bard J.; Jacobsen S.; Magolda R. L.; Pangalos M.; Robichaud A. J. Design and synthesis of 5,5′-disubstituted aminohydantoins as potent and selective human beta-secretase (BACE1) inhibitors. J. Med. Chem. 2010, 53, 1146–58. [DOI] [PubMed] [Google Scholar]
- Cumming J. N.; Smith E. M.; Wang L.; Misiaszek J.; Durkin J.; Pan J.; Iserloh U.; Wu Y.; Zhu Z.; Strickland C.; Voigt J.; Chen X.; Kennedy M. E.; Kuvelkar R.; Hyde L. A.; Cox K.; Favreau L.; Czarniecki M. F.; Greenlee W. J.; McKittrick B. A.; Parker E. M.; Stamford A. W. Structure based design of iminohydantoin BACE1 inhibitors: identification of an orally available, centrally active BACE1 inhibitor. Bioorg. Med. Chem. Lett. 2012, 22, 2444–9. [DOI] [PubMed] [Google Scholar]
- Li X.; He Z.; Chen L.; Li Y.; Li Q.; Zhao S.; Tao Z.; Hu W.; Qin L.; Chen X. Synergy of the antiretroviral protease inhibitor indinavir and chloroquine against malaria parasites in vitro and in vivo. Parasitol. Res. 2011, 109, 1519–24. [DOI] [PubMed] [Google Scholar]
- Liu J.; Gluzman I. Y.; Drew M. E.; Goldberg D. E. The role of Plasmodium falciparum food vacuole plasmepsins. J. Biol. Chem. 2005, 280, 1432–7. [DOI] [PubMed] [Google Scholar]
- Omara-Opyene A. L.; Moura P. A.; Sulsona C. R.; Bonilla J. A.; Yowell C. A.; Fujioka H.; Fidock D. A.; Dame J. B. Genetic disruption of the Plasmodium falciparum digestive vacuole plasmepsins demonstrates their functional redundancy. J. Biol. Chem. 2004, 279, 54088–96. [DOI] [PubMed] [Google Scholar]
- Moura P. A.; Dame J. B.; Fidock D. A. Role of Plasmodium falciparum digestive vacuole plasmepsins in the specificity and antimalarial mode of action of cysteine and aspartic protease inhibitors. Antimicrob. Agents Chemother. 2009, 53, 4968–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.