

1. Introduction
In the field of drug discovery, some classes of proteins have been deemed undruggable. This typically means that no small molecule (≤800 Da) has been found that is capable of binding to a given site with sufficient potency (typically KD ≪ 10 μM) and that elicits a biological response. Protein–protein interactions (PPIs) have generally been deemed undruggable because high-throughput screens (HTS) for small-molecule inhibitors have often failed to identify viable hits. The central issue complicating PPIs is that the interacting surfaces are usually larger and flat (1500–3000 Å) compared to those of other targets that have been found to be druggable. This suggests that high-affinity (low-micromolar KD) compounds are unlikely to be found from a random screen; rather, compounds of more modest affinity are expected.1,2 In addition, HTS campaigns often rely exclusively on spectrophotometric, plate-based assays to test a large collection of small molecules. This approach is notoriously plagued by a large number of artifacts such as promiscuous aggregators, nonspecific binders, protein denaturing compounds, and redox compounds. There are also other artifacts due to liquid handling and compound instability.3−15 Under these conditions, weaker positive hits are often buried in the noise produced by frequent false positives.
It seems therefore intuitive that alternative approaches based on more robust biophysical methods for the detection of ligand binding are likely to be more successful than spectrophotometric HTS approaches, especially when targeting PPIs. Although a number of biophysical methods are available, protein-based NMR spectroscopy is the most robust and reliable method to study ligand binding.16−35 While such NMR assays are most often adopted for hit validation studies as part of a lead discovery campaign, their direct deployment for de novo drug discovery campaigns targeting PPIs is warranted by recent examples.36−51
In this paper we will briefly review the fundamental concepts of NMR-based approaches to drug discovery and then describe the use of these approaches to derive inhibitors of PPIs.
2. Targeting PPIs: A Case for Biophysical Approaches to Ligand Discovery
Most therapeutically relevant PPIs can be regarded as one protein functioning as a “receptor” and the other playing the role of its “ligand”. Typically, the ligand consists of a peptide region adopting an α-helical, a β-strand, or a loop conformation. Hence, PPIs can often be targeted by peptides mimicking these secondary structure elements. The most common strategy in this regard consists in chemical modification of the peptides aimed at stabilizing these secondary structures to increase affinity along with the half-life in biological media, cell permeability, and overall druglikeness.52−55
Rather than mimicking the entire peptide though, recent approaches have focused on identifying and targeting essential “hot spots” on the PPI interfaces.2,56 For example, a typical binding pocket for an α-helix is formed by a few adjacent subpockets that collectively form an elongated crevice that can accommodate side chains projecting out from one side of the α-helix. At the edge of the cavities, electrostatic interactions are formed with charged residues on the other side of the helix (Figure 1A). One can easily imagine that this arrangement of adjacent subpockets makes this type of PPI particularly amenable to fragment-based lead (or drug) discovery (FBLD or FBDD) strategies.57−64 In its original description,40,65 FBLD consists of identifying pairs of binding fragments that can occupy adjacent sites and then be linked chemically into more potent bidentate compounds. Small molecules designed to occupy the hot spots in these subpockets are expected to effectively displace the binding of the entire α-helix, even if the ligand does not occupy the entire protein surface.66 Protein NMR spectroscopy has been used for the identification, structural characterization, and design of such binders, as exemplified in the pioneering structure–activity relationship (SAR) by NMR approach40,65 described later in section 4.1. When applied to the PPI formed between the antiapoptotic Mcl-1 protein, a member of the Bcl-2 family of proteins, and its α-helical BH3-containing binding partners, SAR by NMR readily identified viable inhibitors (Figure 1A).48 When applied to other Bcl-2 family proteins, this approach led to the Abbott drug candidates ABT-73739 and ABT-19967,68 (see section 4.1), currently under clinical investigations for treating cancer. To date, these compounds are the first antagonists of PPIs to reach the clinic. Notably, HTS approaches against the same Bcl-2 targets by the same laboratories failed to produce viable hits.39
Figure 1.
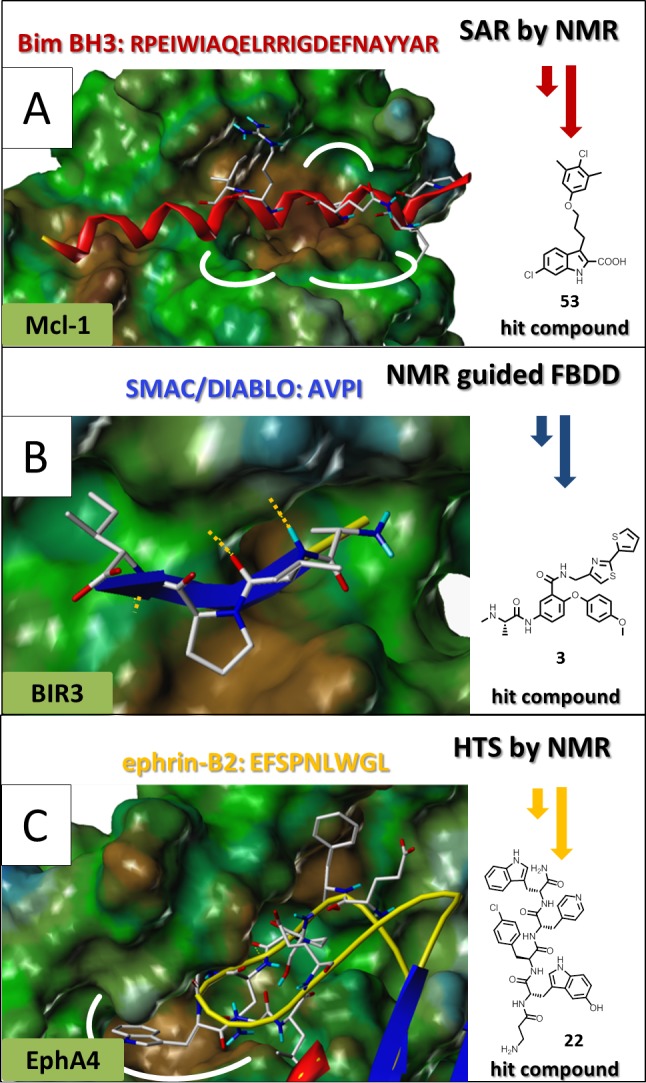
Representative PPIs mediated by an α-helix (A), an extended β-strand (B), or a loop region (C) and corresponding NMR approaches used to guide the discovery of hit compounds. Panel A displays the structure of Mcl-1 (surface representation) in complex with a BH3 peptide from Bim (red ribbon) (PDB ID 2NL9).166 The chemical structure of the SAR-by-NMR-derived compound inhibitor 53(48) is shown. Panel B displays the structure of the third BIR domain (BIR3) of XIAP (surface representation) in complex with a peptide of sequence AVPI (ribbon and stick representation) from Smac/DIABLO (PDB ID 1G73).167 The chemical structure of an AVPI mimetic derived from an NMR-fragment-based approach is also shown.42 Panel C displays a close-up view of the structure of EphA4 (surface representation) in complex with its ephrin-B2 ligand (stick and tube model) (PDB ID 3GXU).168 Obtained by the HTS by NMR approach, the chemical structure of a compound capable of displacing these interactions is also shown.45 Surface representations were obtained with MOLCAD169 as implemented in Sybyl-X 2.0 (Certara, NC). The surfaces are color coded according to lipophilic potential (brown, more lipophilic; green, more hydrophilic).
When β-strands mediate a protein–protein interface, the surface of the receptor is usually shallower than those involved in α-helix-mediated interactions (Figure 1B). The major anchoring contacts are often formed by intermolecular backbone hydrogen bonds that are supported by amino acid side chains occupying shallow subpockets. These backbone hydrogen bonds are critical as small molecules that mimic side chain interactions and are joined by a chemical linker typically do not bind with sufficiently high affinity to block PPIs. In these cases, FBDD combined with fragment linking is less likely to produce potent inhibitors. Rather, combinatorial strategies that start from either the natural peptide or the critical amino acid(s) (or their mimetics) are more likely to succeed. For example, an NMR-based approach was used to perform a stepwise replacement of binding events in the PPI between the AVPI natural tetrapeptide and the BIR3 domain of XIAP (Figure 1B).42 While several laboratories have approached this class of protein targets in recent years,69−76 scientists at Genentech have recently demonstrated that orally active compounds can be obtained starting with the natural tetrapeptide AVPI as a template followed by careful replacement of amino acid side chains to increase pharmacological properties and druglikeness.77 This example suggests that the optimal starting point for lead optimization for β-sheet ligands is to identify short peptide sequences and the critical residues essential for binding (see section 4.2).
Finally, the ligand in a PPI can be represented by peptides adopting a loop conformation (Figure 1C). Also, in this case, the surface area of these interactions is often much larger than those of readily druggable targets. This can be further exacerbated when multiple loops form the interacting surfaces, making the identification of small molecules against such targets very challenging. Nevertheless, the binding pockets for these flexible loops are often also dynamic and thus are more likely to accommodate small molecules either from a fragment-based approach or from a library of peptide mimetics. For example, an NMR-based screen of a combinatorial library of peptide mimetics identified compound 22 as a relatively potent and selective agent, stable in biological media and capable of antagonizing the interactions between EphA4 and ephrin ligands (Figure 1C; see section 4).45
In each of these instances, NMR-based assays were used for the unambiguous detection and characterization of PPI inhibitors throughout the lead discovery process, from initial hit identification to the hit-to-lead optimizations. In the remainder of this paper we will focus on reiterating the basic principles and technical aspects of NMR binding assays and their implementation in drug discovery campaigns targeting PPIs.
3. NMR Assays To Detect and Characterize Ligand Binding to Protein Targets
Key to the success of any lead discovery campaign is the ability to unambiguously detect and characterize the binding of test ligands to a given protein target. As mentioned above, this task can be particularly difficult when using indirect fluorimetric assays to identify inhibitors of PPIs. While these indirect assays are often sufficient to detect and characterize displacement by very potent ligands (KD < 1 μM), these methods are less reliable for detecting weaker binders. This is particularly problematic when these assays are used in HTS for the de novo identification of initial hits. Numerous factors are often evoked as the cause for the low success rate in such campaigns. The compound libraries used in HTS typically do not contain molecules that mimic peptides, and the large and often shallow surface binding area is not too receptive to small ligands. Perhaps an even greater factor is the prevalence of false positives in HTS campaigns, including promiscuous aggregators and other assay- or compound-dependent artifacts.3−15,78 Taken together, these considerations suggest that assays that can unambiguously detect the binding of weaker hits to a protein surface may provide a much needed alternative or complement to spectrophotometric assays, especially at early stages of hit discovery and optimizations.
NMR spectroscopy allows one to study the interaction of proteins and compounds in solution using sensitive and robust assays that are less prone to artifacts.16−35 By the term “NMR-based assays”, we broadly mean any study of the excitation and subsequent relaxation properties of nuclear spins in a strong external magnetic field within test molecules, observed in the presence and absence of binding partners. In drug discovery of PPI inhibitors, NMR can be used to study nuclei of 1H, 15N, 13C, 19F, or 31P.
Two general NMR-based assays can be envisioned. We classify protein-based assays as those in which the observed nuclei belong to the protein receptor. In these cases, the effect of a test ligand on observable nuclei is directly monitored by collecting NMR spectra of the protein receptor in the absence and presence of various concentrations of ligand(s). This typically is done by observing 1H, 15N, or 13C nuclei in isotopically enriched targets. Conversely, we classify ligand-based techniques as those in which the NMR spectra of test ligands are monitored in the absence and presence of the target protein. This typically is done by observing 1H or sometimes 19F nuclei of the ligand. The nature of the NMR spectrum of a molecule, whether it is the protein target or a test ligand, is greatly influenced by its chemical-physical characteristics and chemical environment. As the formation of a complex causes changes in these properties, binding events can be readily and unambiguously detected by NMR spectroscopy.
The ability of NMR to detect binding events is of general use. Given however the sensitivity of these methods, NMR-based assays have been particularly useful in finding inhibitors of PPIs. Several excellent reviews are available that describe in great detail the critical and technical aspects related to the use of NMR spectroscopy in drug discovery.16−35 In this review, we will reiterate only the most important aspects of these strategies with an emphasis on their utility in the identification of PPI inhibitors.
3.1. Protein-Based NMR Assays
Protein-based NMR assays detect ligand binding by observing changes in NMR nuclei in the target protein in response to test ligands. Because this is by far the most direct and reliable NMR-based assay, it is our opinion that these methods should be the preferred approach to either hit identification or hit validation when initial putative hits are selected by other methods.17,29,34,35 A ligand binding to the surface of a protein will almost always cause a change in the electron density that surrounds certain observable protein nuclei. These changes could result in a shielding or deshielding effect, either of which readily translates into significant changes in the resonance frequency (or chemical shift) of the observed nuclei. These general principles are the same by which NMR can be used to elucidate the chemical structure of a molecule or to determine the three-dimensional structure of a macromolecule.79−83 Indeed, one could observe the spectrum of a target protein and monitor binding of a test ligand by observing changes in the resonance frequency of the protein nuclei upon ligand titration.16,17,34 One significant challenge though is to distinguish the resonances of the target from those of the test ligands. This can be accomplished in several ways as discussed below.
The most sensitive and straightforward protein-based NMR binding assay measures signals in the aliphatic region of the protein’s spectrum (usually below 0.7 ppm, 1D 1H-aliph NMR spectrum) in the absence and presence of test ligands (Figure 2).16,42,44,64,84,85 This spectral region contains protein signals belonging to methyl protons that are likely proximal to aromatic side chains due to the three-dimensional structure of the protein. This causes their resonances to be shifted to the extreme right region of the spectrum, a region that is rarely (almost never) populated by signals from small peptides or small molecules (Figure 2). This means that it is very likely that there is no spectral overlap between ligand and protein resonances occurring in this region. In addition, methyl signals usually appear as intense, sharper peaks because methyl protons are chemically equivalent and less prone to rapid nuclear spin relaxation than any other proton in a protein. Depending on the molecular weight of the protein, it is possible to directly observe the 1D 1H-aliph NMR spectrum of a given protein in just a few minutes using a modern high-field NMR instrument (for example, operating at 600 MHz 1H frequency or above) with relatively low protein concentrations (about 1–10 μM depending on the protein MW). Experiments that use as little protein as possible not only have the obvious advantage of reducing costs but also increase the sensitivity by allowing for the identification of weaker interacting molecules, hence increasing the hit rate of a given screen. Under ideal conditions, a ligand that binds with off rates fast on the NMR time scale (as a rule of thumb, KD ranging from 1 μM to 1 mM or above) and particularly well-resolved resonances in the 1D 1H-aliph NMR region, it is possible to provide an estimate of the dissociation constant of the complex by measuring the chemical shift upon ligand titration.17,29
Figure 2.

Example of 1D 1H protein-based NMR assays. The 1D 1H NMR spectra of a protein collected in the absence (red) and presence (blue) of a ligand are reported. Protein spectral resonance regions of different proton nuclei within the protein are identified. 1H resonances of small molecules or peptides resonate usually in the region between 1 and 10 ppm. Therefore, two small spectral regions outside this range (insets) can be used to monitor protein NMR signals in the absence and presence of the test ligand. The spectra were collected with the protein vSrc-SH2 domain in complex with a pY-mimetic ligand.
Detection of ligand binding by 1H-aliph 1D NMR does have its limitations. Not all protein targets will have methyl resonances shifted below 0.7 ppm that are located in proximity to the binding site of the protein. In our experience though to date, most targets do have methyl resonances that are sensitive to ligand binding. Notably, ligand-induced conformational changes can also cause resonances of residues that are not in direct contact with the ligand to be perturbed; hence, the resolved methyl resonances do not necessarily need to be located in the site of binding to “report” a binding event. Spectral crowding in the 1H-aliph 1D NMR spectrum can, however, limit the detection of binding events, especially for proteins that are larger than 30 kDa. Nonetheless, we strongly recommend investigation of the potential utility of these simple 1D NMR protein spectra for any protein target. The use of a known ligand such as a reference peptide can be used to define the sensitivity of the method and to provide a reference with which to compare test ligands. Although simply a binary binding assay, the ease of implementation and sensitivity of this approach combined with the relatively low amounts of protein needed make this a powerful primary assay for ligand screening and for hit validation.
There is also useful information on the extreme left side of the protein 1D 1H spectrum. Trp side chain 1Hε resonances usually reside above 10 ppm (Figure 2), a region of the spectrum that is rarely populated by resonances from small molecules or peptides (with few exceptions). If one or more Trp residues are in the proximity of the binding site, this region can also be suitable for detecting ligand binding. One disadvantage though of working with Trp side chains is that they are more prone to exchange broadening due to relaxation unlike the methyl groups. As a consequence, detecting such signals usually requires longer measurement times and/or higher protein concentration, as only a single proton is being measured instead of three as in the case of methyl groups. Finally, spectral crowding and line broadening, both of which get worse as proteins get larger, also limit the utility of this approach. In general, these assays are only applicable for proteins of <30 kDa, although exceptions to this limit may occur.
For larger proteins, ligand binding is more frequently measured using protein targets that are labeled uniformly or selectively with NMR-observable nuclei such as 15N and 13C. Isotopically enriched recombinant proteins are typically produced in Escherichia coli using appropriately labeled media. Although a variety of labeled rich media are available, minimal medium containing 15NH4Cl as the sole source of nitrogen and/or 13C-glucose as the sole source of carbon is typically sufficient to produce a uniformly labeled recombinant protein. Either [1H, 15N] or 2D [1H, 13C] NMR spectra can be used to analyze the chemical shift perturbations caused by a test ligand upon titration. When combined with sequence-specific resonance assignments, chemical shift perturbations induced by a given test ligand can be mapped on the three-dimensional structure of the target to roughly identify the site of binding.
For uniformly 15N-labeled proteins of small to medium size (up to 30 kDa) at concentrations of 20–50 μM, collection of 2D SOFAST-HMQC (HMQC = heteronuclear multiple-quantum correlation) spectra86,87 can be accomplished within 30 min using a modern high-field instrument equipped with a cryogenic probe (Figure 3A). For larger proteins, deuteration and transverse relaxation optimized spectroscopy (TROSY)-type correlation spectra88 may overcome line-broadening effects.
Figure 3.
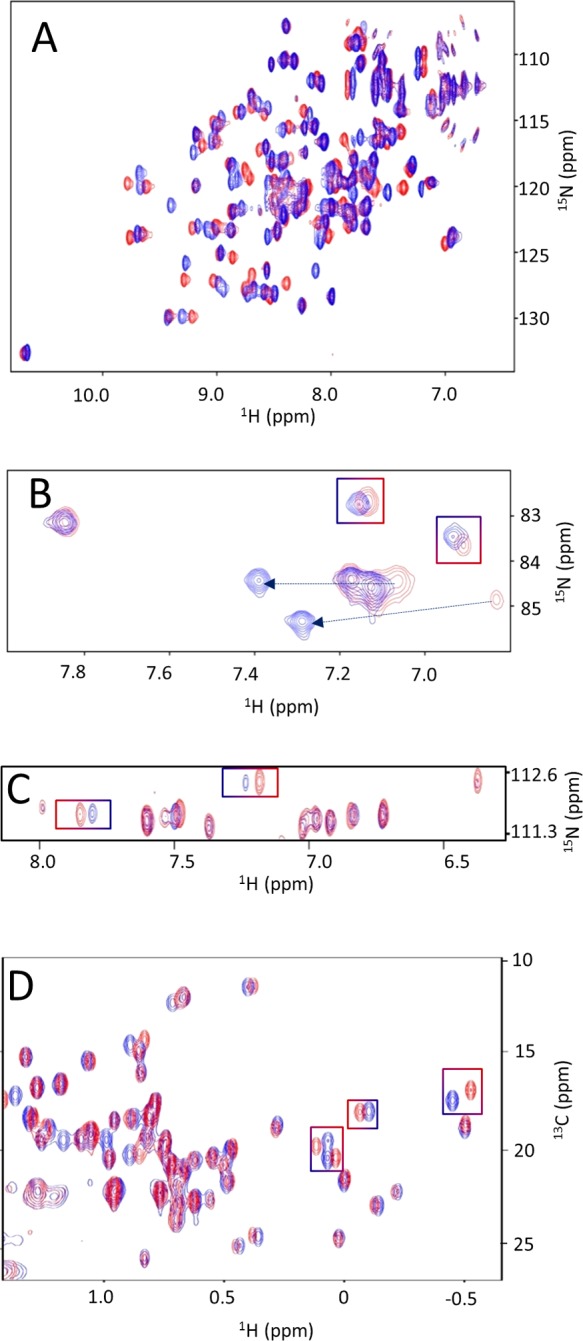
Examples of 2D protein-based NMR assays. Overlays of 2D heteronuclear NMR spectra of the vSrc-SH2 domain collected in the absence (red) and presence (blue) of a ligand. Panels A and B show [1H, 15N] SOFAST-HMQC spectra collected for backbone 1HN, 15N (A) or Arg 1Hε, 15Nε side chains (B). Panel C shows the spectra obtained with a modified [1H, 15N] HSQC (heteronuclear single-quantum coherence) experiment that selects for the 1Hδ, 15Nδ and 1Hε, 15Nε side chains of Asn and Gln, respectively. Panel D shows the [1H, 13C] HSQC spectra in the aliphatic region of the protein. Selected chemical shift perturbations in well-resolved regions of each spectrum are highlighted.
While the majority of screening campaigns can reliably use 1HN, 15N backbone resonances (observing the region of ∼100–130 ppm in the 15N dimension) to detect and characterize ligand binding, [1H, 15N] correlation spectra based on the side chains of Trp, Arg, Asn, Gln, and His can also be used. Trp side chain 1Hε, 15Nε resonances are usually well resolved in the [1H, 15N] correlation spectra resonating around 10 and 130 ppm in the 1H and 15N dimensions, respectively. In addition, selective 15N excitation pulses can be used to select Arg 1Hε, 15Nε resonances as they appear in a distinct spectral region (∼80 ppm in the 15N dimension) (Figure 3B). Similarly, correlation spectra of His side chains 1Hε/δ, 15Nε/δ (∼160–200 ppm in the 15N dimension) can provide information on different protonation states of the imidazole rings, particularly useful especially in cases when these residues are present in the binding sites. Asn and Gln side chains resonate around 110 ppm in the 15N dimension and in principle overlap with backbone resonances. However, these side chains can be isolated using a simple modification of the magnetization transfer step in a [1H, 15N] correlation experiment (Figure 3C) that selects for the −NH2 groups over other −NH groups. Other 1H, 15N resonances of Lys and Arg side chains are generally less visible in the NMR spectra due to their rapid exchange with water.
Uniformly 13C-labeled protein targets can also be used for side chain specific binding studies using 2D [1H, 13C] correlation NMR spectra collected in the presence and absence of test ligand(s). Typical spectral regions that can be well resolved include again the aliphatic region of the spectrum (∼10–30 ppm in the 13C dimension) (Figure 3D) and the aromatic region (∼100–130 ppm in the 13C dimension when observing resonances of the side chains of Tyr, Trp, Phe, and His). Spectral crowding for larger protein targets (>30 kDa) can be resolved with selective labeling of amino acids. This can easily be attained by supplementing the bacterial growth medium with an excess of the desired labeled amino acids (usually about 100–200 mg/L of culture) just prior to the induction of protein expression. While some metabolic scrambling occurs, successful incorporation of different amino acids has been reported also by using labeled precursors or inhibitors of enzymes involved in the biosynthesis of a given amino acid.89−93 This approach can result in simplified 2D [1H, 13C] correlation spectra, making it easier to directly observe perturbations induced by test ligands on a particular residue or set of residues. Combined with deuteration and the 2D [1H, 1H] NOESY (nuclear Overhauser effect spectroscopy) type of experiments16,84 (or by directly using 3D 13C-resolved [1H, 1H] NOESY), these protein samples can be used to indirectly measure intermolecular distances useful to dock ligands into the binding site of the target.16,84
The ability to unambiguously detect ligand binding while simultaneously providing information about the site and mode of binding makes NMR-based protein methods particularly powerful for hit identification and optimizations, especially for the design of PPI antagonists as will be discussed later in this paper (section 4).
3.2. Ligand-Based NMR Assays
Just as the binding of a test compound can influence the NMR spectra of the target protein, the spectra of the ligand can be perturbed by binding to a receptor. A key chemical-physical property that affects the NMR spectrum of a biomolecule is its rotational correlation time. Small magnetic fields induced by neighboring spins fluctuate with the rotational correlation time of a molecule. In slowly rotating macromolecules, these small fluctuating fields cause fast nuclear spin relaxation. Fast nuclear spin relaxation times manifest in NMR spectra as line broadening. Small molecules though have a much lower spin density and rotate much faster in solution than macromolecules. As such, they usually relax more slowly and consequently present NMR spectra with much sharper resonance lines than proteins. However, if a small-molecule ligand binds to a macromolecule, it assumes the overall slow rotational correlation time characteristic of the macromolecule, resulting in the NMR signals broadening. This phenomenon is still appreciable even if the ligand binds transiently to a macromolecule. For ligands that bind with fast off rates with respect to the NMR relaxation time scale (again, as a rule of thumb for ligands with KD ranging from 1 μM to 1 mM and above), this phenomenon is still appreciable even if the ligand is in excess compared to the protein.94−97 This means that the NMR relaxation effects on ligand resonances, even if the ligand only binds weakly to its target, can be observed even when the protein is present at very low concentrations compared to the ligand. Hence, ligand-based NMR binding and displacement assays are typically based on observing changes in the NMR spectra of the ligands (at 100 μM to 1 mM concentrations) induced by low concentrations of protein (typically present at 1–10 μM concentration).
The simplest implementation of these relaxation experiments is the T1ρ experiment,98 in which the magnetization is “locked” perpendicular to the static magnetic field for a certain relaxation time during which transverse nuclear spin relaxation takes place. The duration of the relaxation delay is set to be sufficiently long to cause nuclear spin relaxation to occur to macromolecules (usually between 100 and 400 ms), but not too long that signals from unbound small molecules or peptides remain unperturbed. If a small molecule is bound to the protein, it will behave like the macromolecule, and its signal will also be largely attenuated during the relaxation time (Figure 4A). One advantage of this approach is the ability to test several ligands (in general between 10 and 50) at once.
Figure 4.
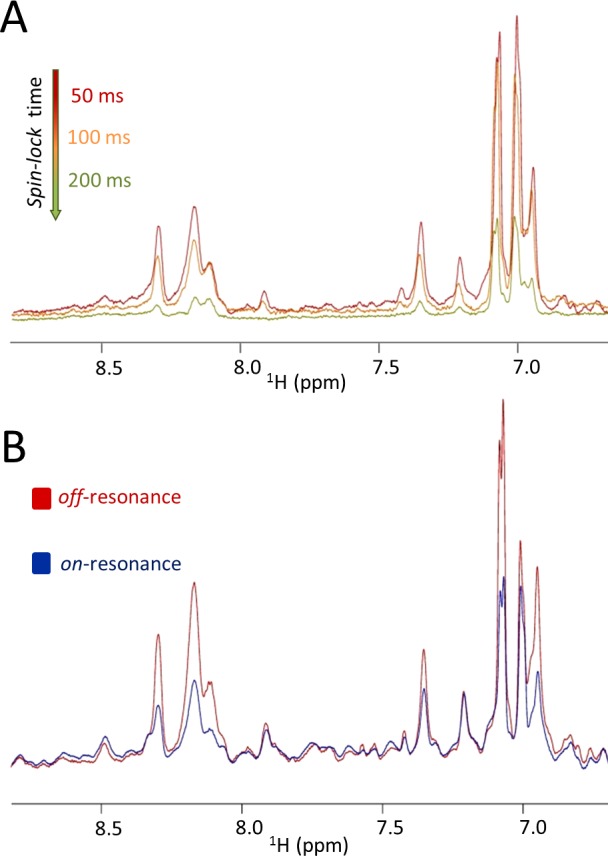
Examples of 1D 1H ligand-based NMR binding assays. In both panels, 1D 1H NMR spectra of a test ligand are collected in the presence of a substoichiometric amount of target (vSrc-SH2). Panel A displays data from the T1ρ experiments collected at the indicated relaxation times. Panel B displays data from the WaterLOGSY experiment.
A second popular ligand-based approach is the saturation transfer difference (STD) experiment.99,100 The protein target is irradiated at selected radio frequencies resulting in a resonance signal in the aliphatic region of its 1H spectrum. In a second reference experiment, the irradiation is placed well outside the spectral region of the protein. A difference spectrum between the 1H-aliph saturated and reference spectra is then created. The signal intensity of test ligands is largely attenuated for binders, while it remains unaltered for nonbinders. Therefore, in a mixture of test ligands (usually up to 50 compounds), only those that bind will present resonances in the difference spectrum. The technique though is only effective if the active site of the protein contains sufficient methyl resonances in the aliphatic regions. An alternative to the STD is the WaterLOGSY experiment (Figure 4B).101,102 Instead of saturating the aliphatic region of the protein spectrum, the entire protein surface is indirectly irradiated by saturating water molecules, which invariantly will be present at the binding site. In both experiments, however, an NMR phenomenon known as spin diffusion will ensure that the saturation is spread from the site of irradiation to the entire macromolecule and from there to the bound ligand.
Similarly, 2D [1H, 1H] NOESY experiments103 are based on the transfer of magnetization between proton nuclei that are within 5 Å of one another. This effect though is larger for protons that are within a slowly tumbling molecule (such as a typical protein) and negligible for protons that are within quickly tumbling molecules (such as a small molecule). The detection of intramolecular NOEs in 2D [1H, 1H] NOESY spectra of a test ligand exposed to its macromolecular target is thus indirect evidence of binding (see section 4).
Ligand-based methods require far less protein than protein-based methods and are not limited by the size of a protein. In fact, larger proteins will result in larger ligand relaxation transfer effects, making binders easier to detect. However, one has to remember that these are indirect effects and thus do not provide direct observations of binding events like in protein-based NMR methods. Transient aggregation and/or nonspecific binding to the protein is often the cause of false positives. Also, quantification of binding, while in principle possible,99 is not as reliably attainable as in protein-based approaches. Finally, no information about the binding site is obtained by these general methods. To address some of these issues, one approach is to include known binders in displacement assays.104−107 In one clever application, a reference ligand is labeled with a paramagnetic moiety, a molecule with an unpaired electron that causes fast nuclear spin relaxation to adjacent nuclei. The relaxation enhancement induced by the unpaired spin is used to detect ligands that bind in proximity to the paramagnetic reference ligand (see section 4.1). Another interesting ligand-based NMR application uses an immobilized target.108 This approach appears even more sensitive than the STD in detecting weak binding fragments.109
As ligand-based approaches are technically easier and still result in highly sensitive observations, they have been more widely adopted than protein-based NMR methods. On the basis of our experience though, we firmly believe that protein-based NMR experiments should always be the method of choice when addressing challenging targets such those involving PPIs. Ligand-based NMR experiments, somewhat similar to fluorimetric assays and other binding methods such as surface plasmon resonance (SPR),110−114 do not provide the same level of reliability and unambiguity as protein-based NMR experiments. This is especially valuable when targeting PPIs. Hence, we recommend that others not be charmed by the apparent shortcut that ligand-based NMR experiments provide, as these methods are not equivalent in their type of information and reliability to protein-based experiments.
4. NMR Approaches To Guide the Design and Optimization of PPI Antagonists
As should be clear from the previous section, protein- and ligand-based NMR assays can be employed in drug discovery campaigns as tools for screening, to guide hit optimizations, or, more simply, for hit validation when combined with other screening techniques. However, a common and important feature of the NMR-based binding assays described above is that these methods are very sensitive to weak binding events and therefore very useful for fragment-based drug design. FBDD is an emerging modular approach to drug design aimed at deriving high-affinity ligands starting by identifying weakly interacting small molecular fragments (<300 Da). Because protein binding sites are modular, this approach is likely to be particularly suitable for designing inhibitors to PPIs. While different biophysical methods have been used for fragment-based drug discovery—including most often SPR or isothermal titration calorimetry (ITC)110−114 and various fragment optimization approaches, including SAR and structure-based design aided by computational115,116 or X-ray117−125 studies—the pioneering work known as SAR by NMR65 described in section 4.1 is arguably the father of the current field.
In addition to FBDD, NMR can also be used to directly screen larger compound libraries. Typical primary screens using protein- or ligand-based NMR assays consist of mixtures of 10–50 compounds per test sample. Combined with automated sampling, this approach can be used in medium-throughput screening campaigns for on the order of several thousand compounds. When testing positional scanning combinatorial libraries, mixtures containing thousands of test ligands can be screened, allowing one to test >100000 compounds in a given campaign. This latter approach is particularly efficient at identifying peptides and peptide mimetics from large libraries of tri- or tetrapeptides against a given protein target (see section 4.2)
When protein-based NMR methods are deployed, whether for a fragment screen or for a screen of a larger compound library, these approaches present a unique advantage over a typical HTS campaign. Specifically, NMR strategies enable the identification of hit compounds and also generate initial hypotheses about the binding site and binding mode. While these seem to be obvious advantages for any drug discovery program, these factors become even more crucial when tackling a challenging target such as PPIs. The following sections will illustrate two general NMR-based strategies for drug discovery of PPI antagonists.
4.1. NMR-Guided Fragment-Based Drug Discovery
Recognizing the power of protein-based NMR assays in unambiguously detecting even weakly interacting molecules, a group at Abbott Laboratories led by Dr. Fesik came up with a simple and powerful drug discovery strategy: SAR by NMR.65 In this approach (Figure 5), a library of fragments, typically a few thousand small compounds (<300 Da), is screened against a protein target using 2D [1H, 15N] correlation spectra. Hit molecules are identified, and their dissociation constant is measured by NMR titration. The most interesting hits are mapped on the three-dimensional structure of the protein by examining changes in backbone 1HN, 15N chemical shifts upon complex formation. Selected hits are then used in a second screening campaign in which a second library of fragments (a few hundred) is screened in the presence of a saturating concentration of the first hit. The goal of this screen is to identify second-site binders, compounds that occupy a site adjacent to that occupied by the first ligand. Finally, these two potentially weak binders are chemically linked without perturbing their original binding poses with the hope of generating a high-affinity bidentate molecule. This process is guided by the structure of the ternary complex between the target and the two fragments (Figure 5).
Figure 5.
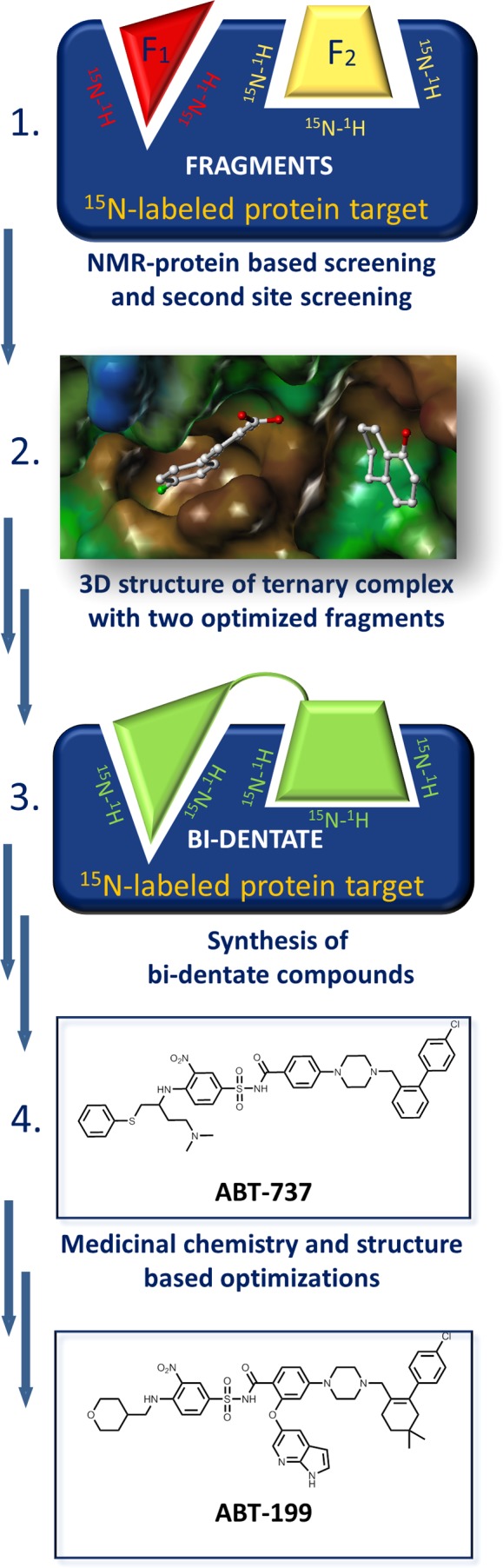
Schematic illustration of the SAR by NMR approach as applied to Bcl-xL and Bcl-2. (1) Starting from the 15N-labeled target, a pair of fragments were identified that bound to adjacent regions of the protein surface. (2) The three-dimensional structure of the ternary complex (PDB ID 1YSG) with these two small fragments was used to guide (3) the design of bidentate compounds, resulting in the first clinical candidate (4) ABT-737. Additional structural studies with optimized bidentate compounds led to the design of a further clinical candidate, ABT-199, with improved pharmacological properties and selectivity for Bcl-2.
The fragment-linking approach of SAR by NMR seems particularly well suited to targeting PPIs whose binding sites are often formed by distinct, adjacent subpockets. An example of using this strategy to disrupt the antiapoptotic protein Bcl-xL binding to the α-helical BH3 peptide is shown in Figure 5. This approach gave rise to several drug candidates, including ABT-73739 and ABT-199 (Figure 5),68 both of which are currently under clinical investigation. Following the initial report, other fragment-based screening methods have been developed, including ligand-based approaches. In one adaptation, binding fragments that occupy adjacent pockets in the target can be identified on the basis of ligand-to-ligand NOEs (interligand NOEs, or ILOEs).126−128 In principle, this provides the same information as SAR by NMR, albeit indirectly (Figure 6). These fragments can again be chemically linked to derive a more potent bidentate compound. As shown in Figure 6, an Mcl-1 antagonist has been identified using this approach.43
Figure 6.
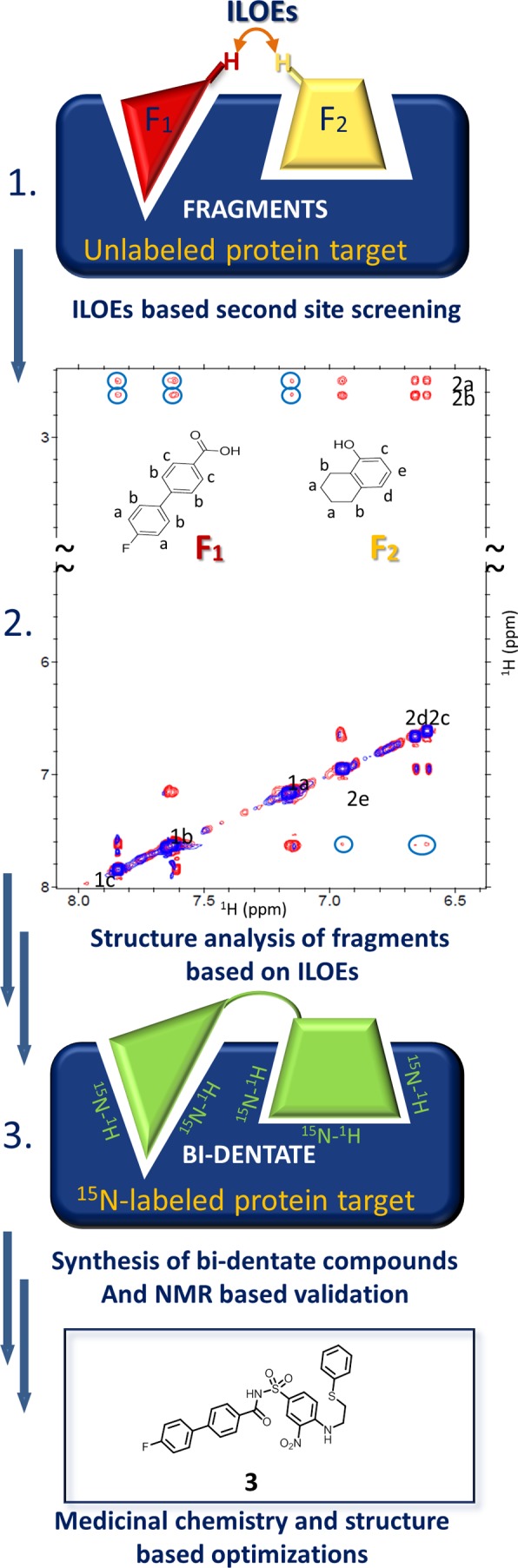
Schematic illustration of the NMR approach using ligand–ligand NOEs (ILOEs) as applied to Bcl-xL and Mcl-1. Starting from unlabeled targets, pairs of fragments (1), similar to those identified by the SAR by NMR approach, were directly identified by protein-mediated ligand-to-ligand NOEs (ILOEs) collected in the presence of a substoichiometric amount of target (2). Analysis of the ILOEs (3) guided the synthesis of bidentate molecules, namely, compound 3 from Rega et al.43 Adapted from ref (43). Copyright 2011 American Chemical Society.
Another ligand-based NMR method seeks to identify second-site binders using a first ligand labeled with a paramagnetic group. A commonly used spin label for this application is TEMPO (2,3,4,6-tetramethylpiperidine-1-oxyl).129,130 The unpaired electron of the paramagnetic moiety causes rapid NMR nuclear spin relaxation to nuclei that are within a few angstroms. Hence, second-site binders can be identified by selecting those ligands whose NMR signals are attenuated by the presence of the target and the first paramagnetic ligand.84,130−133 This approach has been used to identify inhibitors of protein kinases or phosphatases.132,134 In the example shown in Figure 7, a furanylsalicylic acid moiety was used as a phosphotyrosine mimic135 and chemically linked to TEMPO. Screening for second-site binders resulted in bidentate compounds that specifically target the given phosphatase, YopH from Yersinia pestis.132 In this case, specificity is largely driven by the second binder as the subpockets adjacent to the common pY binding site are not very well conserved among phosphates. Second-site binders were identified during the NMR-based screen, guiding the synthesis of bidentate compounds with increased affinity and selectivity (Figure 7).49 Proper compound linking is critical; hence, structural information on the ternary complex is particularly important to attain bidentate compounds with increased affinity. Nevertheless, the fragment-linking approach, almost invariably based on the NMR strategies described above, has resulted in bidentate compounds with dramatically increased affinity relative to the individual fragments. These approaches have successfully identified PPI antagonists where other methods have failed.
Figure 7.
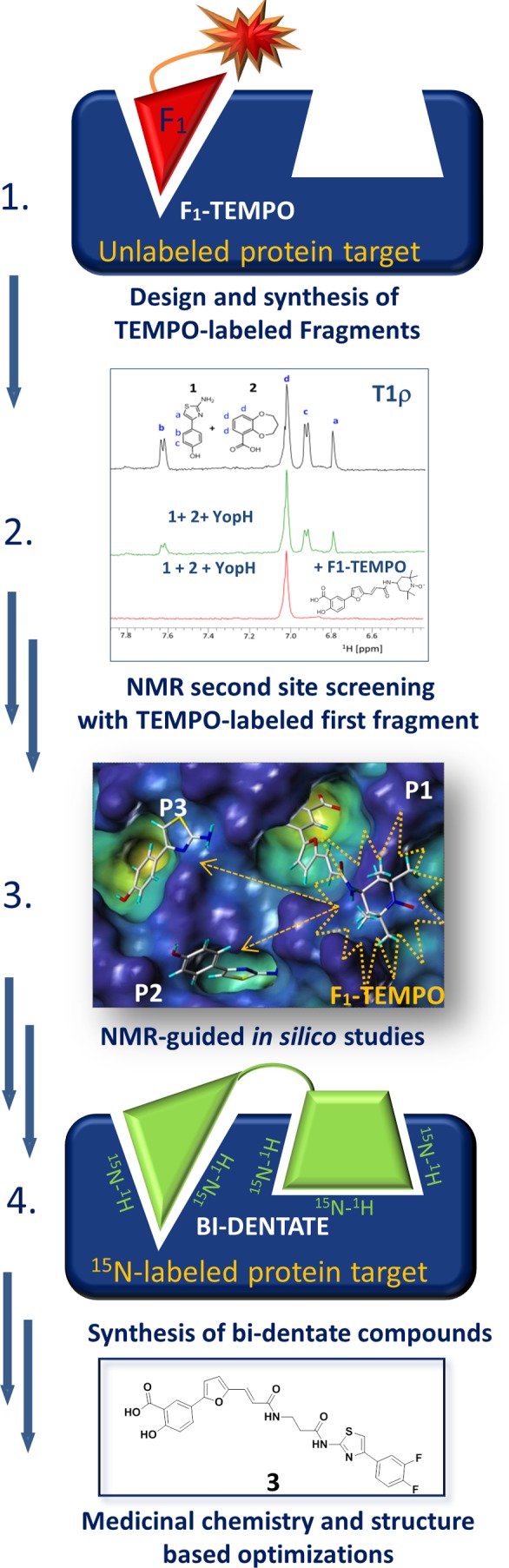
Schematic illustration of the paramagnetic enhancement approach for detection of second-site binders by using a spin-labeled first ligand (F1-TEMPO). In the example reported against the phosphotyrosine (pY) phosphatase YopH, a furanylsalicilate pY mimetic is used to design and synthesize a reference molecule (1) (F1-TEMPO).132 Subsequently, (2) using 1D 1H T1ρ NMR experiments, the binding of a test ligand is detected by a decrease of signal intensity in the presence of a substoichiometric amount of protein target and the F1-TEMPO compound. In the application reported here, analysis of the F1-TEMPO-mediated relaxation (3) enhancement second-site ligands was used to guide (4) the synthesis of bidentate molecules, namely, compound 3 from Leone et al.49 Further NMR-based validations and structural studies can be used to guide hit to lead optimization studies. Adapted with permission from ref (49). Copyright 2010 John Wiley & Sons A/S.
FBDD can also be used in the absence of information on a second-site binder. Stepwise iterative optimizations are performed on the initially weakly interacting scaffold. This fragment-growing approach represents a simpler and currently more common strategy than the previously described fragment-linking approach. After the identification of an initial hit, higher affinity compounds are sought first by testing commercially available molecules containing the hit fragment. In addition, structure-based refinements can be performed using computational models or experimentally derived by X-ray diffraction136−143 or NMR spectroscopy.16,64,65,84,127,144−146
4.2. NMR-Based Screening of Larger Compound Collections
The compounds in the fragment libraries used above are building blocks that can result in a hit compound, after either fragment-linking or fragment-growing optimizations. Hence, these compounds do not generally obey the traditional Lipinski rule-of-five147,148 but belong to a fragmentlike rule-of-three.149 Often, medicinal chemists get directly “inspired” by the initially discovered fragment hits and build libraries of analogues that may lead to more potent compounds. For example, an NMR-based screen, using a combination of STD and [1H, 15N] correlation experiments, resulted in the identification of PDK1 allosteric inhibitors targeting a docking site for kinases, known as the PIF pocket (Table 1).150 There are several recent examples of lead compounds and even drug candidates that target PPIs which were aided by NMR-based approaches, some of which are reported in Table 1 and in Figures 1 and 5–8.36−49,151−156 As mentioned in section 2, we believe that the FBDD approach may be best suited to targeting PPIs when the interaction is mediated by an α-helix. This is because α-helices produce a set of discrete interactions of spatially adjacent substructures that can be mimicked by small molecular fragments that are chemically linked. For PPIs that are mediated by the formation of an intermolecular β-strand, the situation is different, with most pivotal interactions being specific hydrogen bonds between the proteins augmented by some side chain interactions that are located in usually shallower subpockets. Likewise, PPIs mediated by loops may prove particularly difficult when more than one nonadjacent loop is part of the intermolecular interaction. Nonetheless, in both cases, only a few anchoring amino acids are critical for binding. Hence, libraries of compounds composed of mimetics of such anchoring amino acids can lead potentially to effective PPI antagonists. For example, the orally active XIAP antagonist GDC-015277 closely mimics the structure of the natural antagonist tetrapeptide motif AϕPϕ (where ϕ represents hydrophobic residues).
Table 1. Examples of PPIs Antagonists Discovered Using NMR Methodsa.
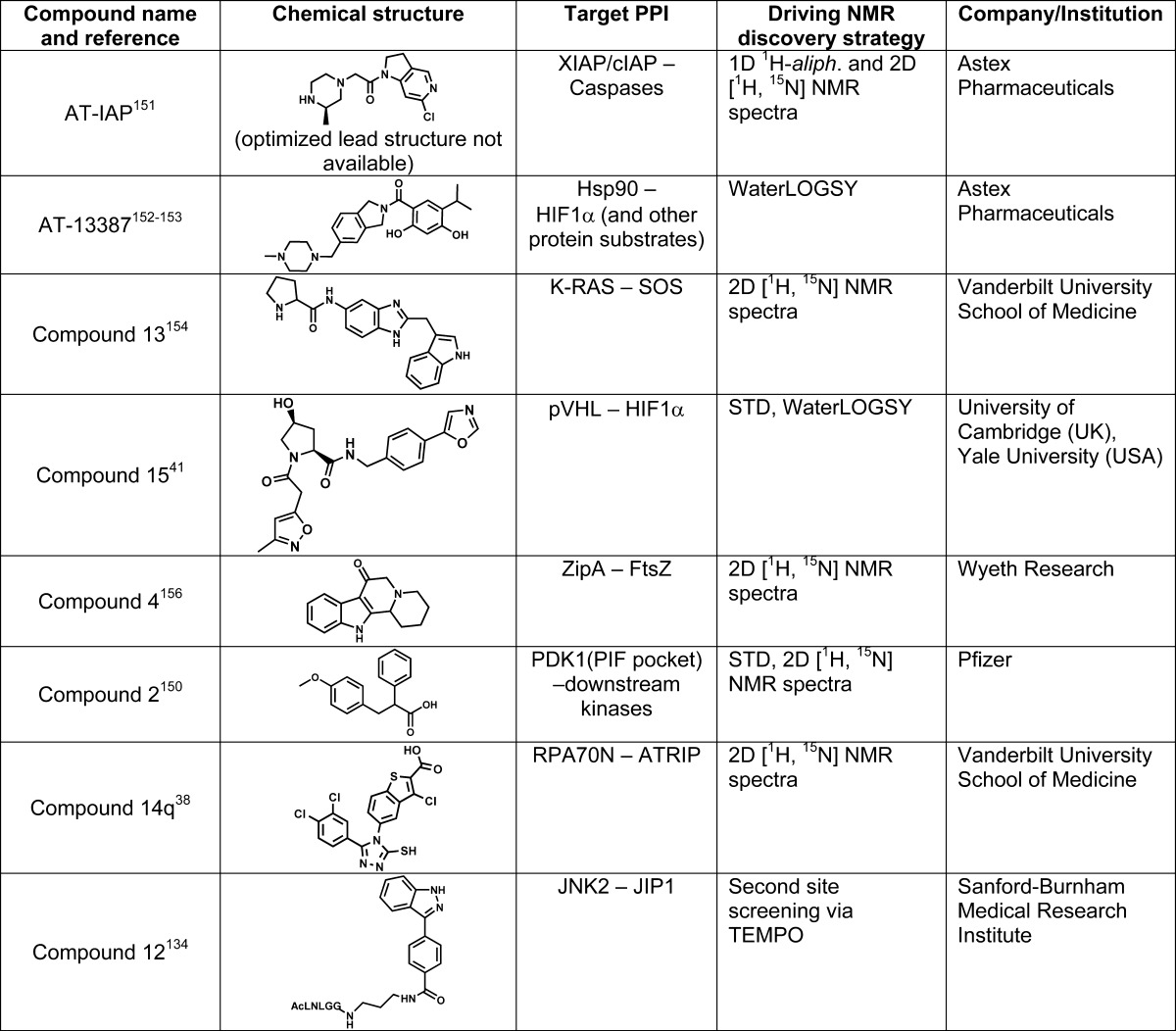
For each ligand, the NMR methods involved for the discovery and characterization of the ligands are indicated.
Figure 8.
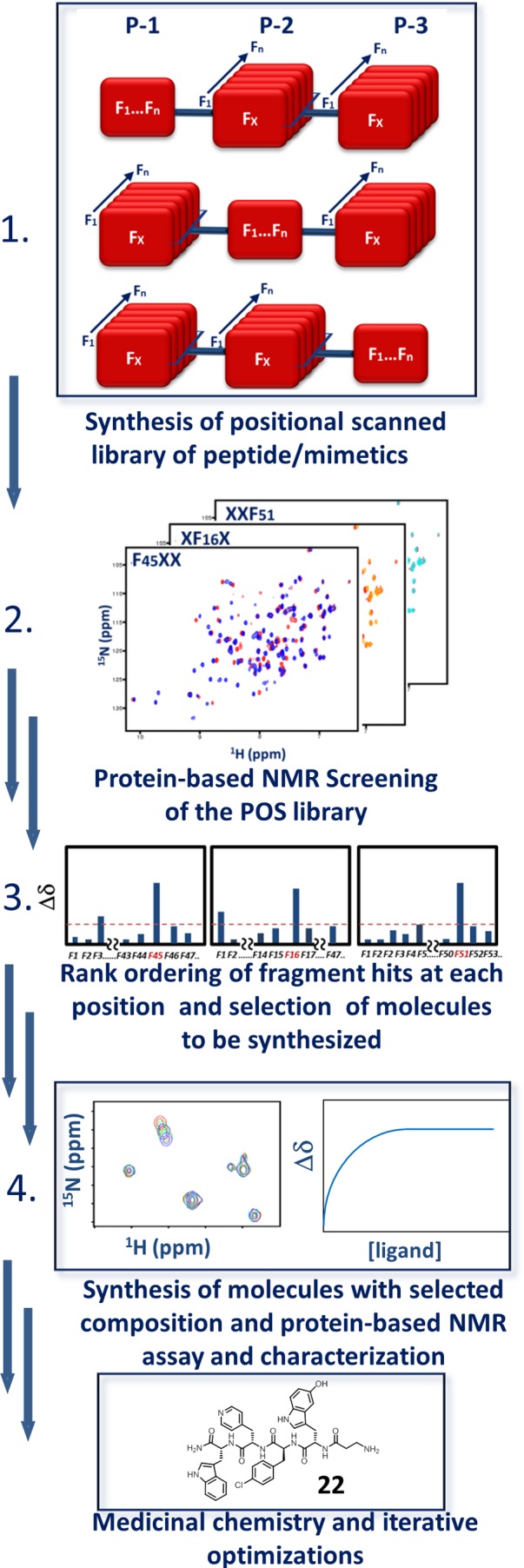
Schematic illustration of HTS by NMR of combinatorial libraries. (1) The synthesis of the library is performed in a combinatorial position scanning fashion. In the example, a three-position combinatorial library is prepared with n fragments (F1, ..., Fn). This entails the synthesis of 3n mixtures, each containing n2 molecules. Following testing of such mixtures using protein-based NMR assays (2), preferential binding fragments at each position are deduced (3). Individual compounds with proper combinations of fragments are synthesized and tested using protein-based NMR experiments (4) for follow-up hit to lead optimizations. In the example reported, antagonists of EphA4–ephrin-B4 are identified from an initial HTS by NMR screen of a positional scanned library of ∼200000 compounds, resulting in compound 22 from Wu et al.45 Adapted with permission from ref (45). Copyright 2013 Cell Press.
These examples suggest that, as mentioned in the Introduction, short peptides or, better, peptide mimetics represent ideal starting points for hit to lead optimizations of PPI antagonists.
A recent approach we have developed is based on combining positional scanning combinatorial libraries of short tri- or tetrapeptides with protein-based NMR screening. Starting with libraries of well over 100000 possible compounds, this approach can select and characterize those that bind selectively and efficiently to a given protein target. This strategy is called HTS by NMR to emphasize the high throughput relative to other fragment screening campaigns. It has proven particularly useful in identifying short peptide sequences (such as AVPI binding to BIR3) and initial lead compounds using libraries consisting of non-natural amino acids. In our recent example, we screened a library consisting of 58 natural and non-natural amino acids linked in tripeptoids to identify an inhibitor of EphA4–ephrin-B4 PPIs. The compounds in the library had an average molecular weight of about 500 Da. The library was assembled in 174 positional scanning mixtures (58 + 58 + 58), each of which contained 3364 compounds (1 × 58 × 58). In combinatorial chemistry, positional scanning157−160 implies that each mixture is built systematically with one given element fixed at one position while the other positions comprise all combinations (Figure 8). Screening the 174 mixtures is easily accomplished using protein-based NMR methods. 1D 1H-aliph was used as the primary screening method44 to identify preferred amino acids at each position.45 Following synthesis of each selected tripeptoid (usually between 5 and 10 possible tripeptoids), 2D [1H, 15N] and/or [1H, 13C] correlation NMR experiments are used to characterize the binding of these potential ligands. Hence, this relatively simple strategy can sample all possible tripeptoids from a potential pool of nearly 200000 compounds using sensitive and unambiguous protein-based NMR binding assays. When used against a variety of targets, the dissociation constants of the initial hits range from 5 to 300 μM. Further optimization can follow different routes, but most are based on the traditional SAR in which individual “scaffolds” (each of the non-natural amino acids) are iteratively optimized. When applied to the EphA4–ephrin-B4 PPIs, this approach led to the discovery of a compound with high binding affinity and high selectivity relative to other Eph members. In addition, the compound was very resistant to proteases present in biological fluids, thereby conferring high stability in plasma.45 The method can be further extended by synthesizing and testing nonpeptide libraries arranged in the same positional scanning format.161 Compared to SAR by NMR, this approach has the advantage that, once the library has been prepared, it can be used for several targets. Also, the resulting scaffolds are already preassembled on a common backbone and thus will not require extensive medicinal chemistry expertise to obtain properly linked compounds. This latter consideration may prove particularly useful in initiating a PPI targeting project when dedicated medicinal chemistry resources are not readily available.
In another variation of the method, libraries of compounds can be produced with one anchoring fragment (or amino acid) fixed while other positions are randomized. For example, in searching for possible SH2 domain antagonists, positional scanned libraries of tri- or tetrapeptides composed of non-natural amino acids can be synthesized, all containing phosho-Tyr (or a pY mimetic) at a given position. The pY will provide the anchoring residue, and an NMR screen can be used to identify the most suitable side chains in neighboring positions. Considering the versatility of the positional scanning libraries and the unambiguous detection provided by protein-based NMR experiments, we are confident that this approach may find general and widespread utility in the identification of inhibitors of PPIs.
Like any protein-based NMR method, these screening approaches are not biased to a particular site of the protein surface. This will allow in principle the identification of additional hot spots on the surface of the given target, recognizing other potential interaction sites and/or allosteric sites and their inhibitors.
5. Conclusions and Perspectives
One of the most appealing aspects of using NMR in drug discovery is that challenging targets can be addressed, particularly when using protein-based methods. Given the sensitivity and unambiguous binding data, artifacts that plague nearly every other binding assay can be avoided. The SAR by NMR approach not only pioneered the entire field of current FBDD, but also produced the very first inhibitors of PPIs that reached the clinic. Unfortunately, setting up an NMR protein-based screening laboratory requires a variety of expertise, including NMR spectroscopy, molecular biology, protein chemistry, and, of course, medicinal chemistry. These specialties are often not centralized in a typical industrial setting but rather are compartmentalized in different groups. This is unfortunate as we believe that the power of protein-based NMR approaches has been underutilized. Most current FBDD campaigns rely on less sensitive and less informative approaches, such as ligand-based NMR techniques or other methods such as SPR. These methods are generally easier to implement than protein-based NMR assays and have the advantages of being less limited by the size of the macromolecule and not requiring isotope labeling. Nonetheless, we believe that protein-based NMR approaches are the most suited to identify PPI antagonists. The key advantages are the availability early in the discovery process of structural information on the mode of binding and the reliability of robust and unambiguous binding data. Hence, we hope and encourage that those interested in deriving antagonists of therapeutically viable PPIs will consider first those targets that are amenable to protein NMR spectroscopy. With the resurgence of peptide mimetics as therapeutics, we also envision that protein-based NMR will be used in HTS campaigns. Direct NMR-based screening of large libraries of peptide mimetics as in HTS by NMR may provide viable hit compounds for more immediate hit-to-lead optimizations. Because the synthetic chemistry methods for the production of the positional scanned libraries are well established160,162−165 and amenable to outsourcing from specialized peptide-synthesis companies, we are hopeful that these approaches will become widely used in both industry and academic research.
Acknowledgments
This review is not meant to be a comprehensive account of all possible NMR experiments that have thus far been proposed for applications in ligand binding and drug discovery but represents the authors’ personal and general perspectives of the use of NMR spectroscopy in the context of its applicability to the discovery, design, and optimization of antagonists of protein–protein interactions. Financial support to M.P. was obtained in part by NIH Grant CA168517. We thank Dr. Jason Kreisberg (Contract Scientific Writer) for careful proofreading and editing of the manuscript.
Biographies

Elisa Barile obtained an M.S. in Pharmaceutical Chemistry from the University of Naples (Naples, Italy) and a Ph.D. in Applied Chemistry and Biochemistry with a focus on natural product chemistry and molecular pharmacology. Soon after her doctoral studies, she joined the Sanford-Burnham Medical Research Institute in 2008, where she currently continues to work as a scientist in Dr. Pellecchia’s laboratory. Her research interest is in molecular recognition, medicinal chemistry, and pharmacology applied to targets in oncology and infectious diseases. To date, Dr. Barile has published over 30 manuscripts in these areas.

Maurizio Pellecchia holds an M.S. in Organic Chemistry and a Ph.D. in Pharmaceutical Chemistry from the University of Naples, (Naples, Italy), followed by postdoctoral studies at the ETH (Zurich, Switzerland) and at the University of Michigan (Ann Arbor, MI). After spending a few years at a biotechnology company, Dr. Pellecchia joined the Sanford-Burnham Medical Research Institute in 2002 as Associate Professor and was subsequently promoted to Professor in 2007. Dr. Pellecchia’s research focuses on the characterization of intermolecular interactions in the development of small-molecule inhibitors of protein targets involved in cell signaling, virulence factors, and host–pathogen interactions and has published to date over 150 publications in these areas. The resulting compounds are then used as molecular probes to provide further understanding of the mechanism of action of their respective targets. The overall goal of the laboratory is to successfully bring together basic sciences involving modern NMR spectroscopy techniques, computer modeling, and medicinal chemistry to elucidate the molecular basis of disease and to develop novel therapeutic compounds. An area in which Dr. Pellecchia remains particularly interested is the development of novel strategies to aid the design and synthesis of novel ligands against protein–protein interactions.
The authors declare the following competing financial interest(s): Dr. Pellecchia is co-founder of AnCoreX Therapeutics, which uses several of the proposed strategies for ligand discovery.
Funding Statement
National Institutes of Health, United States
References
- Wells J. A.; McClendon C. L. Nature 2007, 450, 1001. [DOI] [PubMed] [Google Scholar]
- Arkin M. R.; Wells J. A. Nat. Rev. Drug Discovery 2004, 3, 301. [DOI] [PubMed] [Google Scholar]
- Bocker A.; Bonneau P. R.; Edwards P. J. J. Biomol. Screening 2011, 16, 765. [DOI] [PubMed] [Google Scholar]
- Coan K. E.; Maltby D. A.; Burlingame A. L.; Shoichet B. K. J. Med. Chem. 2009, 52, 2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. J. Med. Chem. 2010, 53, 2719. [DOI] [PubMed] [Google Scholar]
- Rishton G. M. Drug Discovery Today 2003, 8, 86. [DOI] [PubMed] [Google Scholar]
- Hann M. M.; Oprea T. I. Curr. Opin. Chem. Biol. 2004, 8, 255. [DOI] [PubMed] [Google Scholar]
- Coan K. E.; Shoichet B. K. Mol. Biosyst. 2007, 3, 208. [DOI] [PubMed] [Google Scholar]
- Feng B. Y.; Shelat A.; Doman T. N.; Guy R. K.; Shoichet B. K. Nat. Chem. Biol. 2005, 1, 146. [DOI] [PubMed] [Google Scholar]
- Feng B. Y.; Shoichet B. K. Nat. Protoc. 2006, 1, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng B. Y.; Shoichet B. K. J. Med. Chem. 2006, 49, 2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng B. Y.; Simeonov A.; Jadhav A.; Babaoglu K.; Inglese J.; Shoichet B. K.; Austin C. P. J. Med. Chem. 2007, 50, 2385. [DOI] [PubMed] [Google Scholar]
- McGovern S. L.; Helfand B. T.; Feng B.; Shoichet B. K. J. Med. Chem. 2003, 46, 4265. [DOI] [PubMed] [Google Scholar]
- Seidler J.; McGovern S. L.; Doman T. N.; Shoichet B. K. J. Med. Chem. 2003, 46, 4477. [DOI] [PubMed] [Google Scholar]
- Shoichet B. K. J. Med. Chem. 2006, 49, 7274. [DOI] [PubMed] [Google Scholar]
- Pellecchia M.; Sem D. S.; Wuthrich K. Nat. Rev. Drug Discovery 2002, 1, 211. [DOI] [PubMed] [Google Scholar]
- Pellecchia M. Chem. Biol. 2005, 12, 961. [DOI] [PubMed] [Google Scholar]
- Stockman B. J.; Dalvit C. Prog. Nucl. Magn. Reson. Spectrosc. 2002, 41, 187. [Google Scholar]
- Lepre C. A.; Peng J.; Fejzo J.; Abdul-Manan N.; Pocas J.; Jacobs M.; Xie X.; Moore J. M. Comb. Chem. High Throughput Screening 2002, 5, 583. [DOI] [PubMed] [Google Scholar]
- Wyss D. F.; McCoy M. A.; Senior M. M. Curr. Opin Drug Discovery Dev. 2002, 5, 630. [PubMed] [Google Scholar]
- Jahnke W.; Widmer H. Cell. Mol. Life Sci. 2004, 61, 580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huth J. R.; Sun C.; Sauer D. R.; Hajduk P. J. Methods Enzymol. 2005, 394, 549. [DOI] [PubMed] [Google Scholar]
- Klages J.; Coles M.; Kessler H. In NMR-Based Screening in Exploiting Chemical Diversity for Drug Discovery; Bartlett P. A., Etzeroth M., Eds.; RSC Publishing: London, 2006; Vol. 12. [Google Scholar]
- Coles M.; Heller M.; Kessler H. Drug Discovery Today 2003, 8, 803. [DOI] [PubMed] [Google Scholar]
- Kessler H.; Klages J. In Comprehensive Medicinal Chemistry II; Triggle D. J., Taylor J. B., Eds.; Elsevier: Oxford, U.K., 2006; Vol. 3. [Google Scholar]
- Luy B.; Frank A.; Kessler H. In Molecular Drug Properties: Measurement and Prediction; Mannhold R., Ed.; Wiley-VCH: Weinheim, Germany, 2008. [Google Scholar]
- Takeuchi K.; Wagner G. Curr. Opin. Struct. Biol. 2006, 16, 109. [DOI] [PubMed] [Google Scholar]
- Zartler E. R.; Shapiro M. J. Curr. Pharm. Des. 2006, 12, 3963. [DOI] [PubMed] [Google Scholar]
- Pellecchia M.; Becattini B.; Crowell K. J.; Fattorusso R.; Forino M.; Fragai M.; Jung D.; Mustelin T.; Tautz L. Expert Opin. Ther. Targets 2004, 8, 597. [DOI] [PubMed] [Google Scholar]
- Fry D. C.; Emerson S. D. Drug Des. Discovery 2000, 17, 13. [PubMed] [Google Scholar]
- Salvatella X.; Giralt E. Chem. Soc. Rev. 2003, 32, 365. [DOI] [PubMed] [Google Scholar]
- Betz M.; Saxena K.; Schwalbe H. Curr. Opin. Chem. Biol. 2006, 10, 219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klages J.; Coles M.; Kessler H.. NMR-Based Screening; RSC Publishing: London, 2006. [Google Scholar]
- Pellecchia M.; Bertini I.; Cowburn D.; Dalvit C.; Giralt E.; Jahnke W.; James T. L.; Homans S. W.; Kessler H.; Luchinat C.; Meyer B.; Oschkinat H.; Peng J.; Schwalbe H.; Siegal G. Nat. Rev. Drug Discovery 2008, 7, 738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harner M. J.; Frank A. O.; Fesik S. W. J. Biomol. NMR 2013, 56, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias D. M.; Van Molle I.; Baud M. G.; Galdeano C.; Geraldes C. F.; Ciulli A. ACS Med. Chem. Lett. 2014, 5, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson F. M.; Fedorov O.; Chaikuad A.; Philpott M.; Muniz J. R.; Felletar I.; von Delft F.; Heightman T.; Knapp S.; Abell C.; Ciulli A. J. Med. Chem. 2013, 56, 10183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patrone J. D.; Kennedy J. P.; Frank A. O.; Feldkamp M. D.; Vangamudi B.; Pelz N. F.; Rossanese O. W.; Waterson A. G.; Chazin W. J.; Fesik S. W. ACS Med. Chem. Lett. 2013, 4, 601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltersdorf T.; Elmore S. W.; Shoemaker A. R.; Armstrong R. C.; Augeri D. J.; Belli B. A.; Bruncko M.; Deckwerth T. L.; Dinges J.; Hajduk P. J.; Joseph M. K.; Kitada S.; Korsmeyer S. J.; Kunzer A. R.; Letai A.; Li C.; Mitten M. J.; Nettesheim D. G.; Ng S.; Nimmer P. M.; O’Connor J. M.; Oleksijew A.; Petros A. M.; Reed J. C.; Shen W.; Tahir S. K.; Thompson C. B.; Tomaselli K. J.; Wang B.; Wendt M. D.; Zhang H.; Fesik S. W.; Rosenberg S. H. Nature 2005, 435, 677. [DOI] [PubMed] [Google Scholar]
- Hajduk P. J.; Sheppard G.; Nettesheim D. G.; Olejniczak E. T.; Shuker S. B.; Meadows R. P.; Steinman D. H.; Carrera G. M.; Marcotte P. A.; Severin J.; Walter K.; Smith H.; Gubbins E.; Simmer R.; Holzman T. F.; Morgan D. W.; Davidsen S. K.; Summers J. B.; Fesik S. W. J. Am. Chem. Soc. 1997, 119, 5818. [Google Scholar]
- Buckley D. L.; Gustafson J. L.; Van Molle I.; Roth A. G.; Tae H. S.; Gareiss P. C.; Jorgensen W. L.; Ciulli A.; Crews C. M. Angew. Chem., Int. Ed. 2012, 51, 11463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J. W.; Zhang Z.; Wu B.; Cellitti J. F.; Zhang X.; Dahl R.; Shiau C. W.; Welsh K.; Emdadi A.; Stebbins J. L.; Reed J. C.; Pellecchia M. J. Med. Chem. 2008, 51, 7111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rega M. F.; Wu B.; Wei J.; Zhang Z.; Cellitti J. F.; Pellecchia M. J. Med. Chem. 2011, 54, 6000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stebbins J. L.; Zhang Z.; Chen J.; Wu B.; Emdadi A.; Williams M. E.; Cashman J.; Pellecchia M. J. Med. Chem. 2007, 50, 6607. [DOI] [PubMed] [Google Scholar]
- Wu B.; Zhang Z.; Noberini R.; Barile E.; Giulianotti M.; Pinilla C.; Houghten R. A.; Pasquale E. B.; Pellecchia M. Chem. Biol. 2013, 20, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krajewski M.; Rothweiler U.; D’Silva L.; Majumdar S.; Klein C.; Holak T. A. J. Med. Chem. 2007, 50, 4382. [DOI] [PubMed] [Google Scholar]
- Petros A. M.; Dinges J.; Augeri D. J.; Baumeister S. A.; Betebenner D. A.; Bures M. G.; Elmore S. W.; Hajduk P. J.; Joseph M. K.; Landis S. K.; Nettesheim D. G.; Rosenberg S. H.; Shen W.; Thomas S.; Wang X.; Zanze I.; Zhang H.; Fesik S. W. J. Med. Chem. 2006, 49, 656. [DOI] [PubMed] [Google Scholar]
- Friberg A.; Vigil D.; Zhao B.; Daniels R. N.; Burke J. P.; Garcia-Barrantes P. M.; Camper D.; Chauder B. A.; Lee T.; Olejniczak E. T.; Fesik S. W. J. Med. Chem. 2013, 56, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone M.; Barile E.; Vazquez J.; Mei A.; Guiney D.; Dahl R.; Pellecchia M. Chem. Biol. Drug Des. 2010, 76, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raimundo B. C.; Oslob J. D.; Braisted A. C.; Hyde J.; McDowell R. S.; Randal M.; Waal N. D.; Wilkinson J.; Yu C. H.; Arkin M. R. J. Med. Chem. 2004, 47, 3111. [DOI] [PubMed] [Google Scholar]
- Frank A. O.; Feldkamp M. D.; Kennedy J. P.; Waterson A. G.; Pelz N. F.; Patrone J. D.; Vangamudi B.; Camper D. V.; Rossanese O. W.; Chazin W. J.; Fesik S. W. J. Med. Chem. 2013, 56, 9242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henchey L. K.; Jochim A. L.; Arora P. S. Curr. Opin. Chem. Biol. 2008, 12, 692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzarito V.; Long K.; Murphy N. S.; Wilson A. J. Nat. Chem. 2013, 5, 161. [DOI] [PubMed] [Google Scholar]
- Mahalakshmi R.; Balaram P. Methods Mol. Biol. 2006, 340, 71. [DOI] [PubMed] [Google Scholar]
- Glenn M. P.; Fairlie D. P. Mini-Rev. Med. Chem. 2002, 2, 433. [DOI] [PubMed] [Google Scholar]
- Arkin M. R.; Randal M.; DeLano W. L.; Hyde J.; Luong T. N.; Oslob J. D.; Raphael D. R.; Taylor L.; Wang J.; McDowell R. S.; Wells J. A.; Braisted A. C. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valkov E.; Sharpe T.; Marsh M.; Greive S.; Hyvonen M. Top. Curr. Chem. 2012, 317, 145. [DOI] [PubMed] [Google Scholar]
- Erlanson D. A. Top. Curr. Chem. 2012, 317, 1. [DOI] [PubMed] [Google Scholar]
- Zartler E. R.; Shapiro M. J. Curr. Opin. Chem. Biol. 2005, 9, 366. [DOI] [PubMed] [Google Scholar]
- Erlanson D. A.; McDowell R. S.; O’Brien T. J. Med. Chem. 2004, 47, 3463. [DOI] [PubMed] [Google Scholar]
- Talamas F. X.; Ao-Ieong G.; Brameld K. A.; Chin E.; de Vicente J.; Dunn J. P.; Ghate M.; Giannetti A. M.; Harris S. F.; Labadie S. S.; Leveque V.; Li J.; Lui A. S.; McCaleb K. L.; Najera I.; Schoenfeld R. C.; Wang B.; Wong A. J. Med. Chem. 2013, 56, 3115. [DOI] [PubMed] [Google Scholar]
- Fischer M.; Hubbard R. E. Mol. Interventions 2009, 9, 22. [DOI] [PubMed] [Google Scholar]
- Rees D. C.; Congreve M.; Murray C. W.; Carr R. Nat. Rev. Drug Discovery 2004, 3, 660. [DOI] [PubMed] [Google Scholar]
- Fragment-Based Approaches in Drug Discovery; Jahnke W., Erlanson D. A., Eds.; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Shuker S. B.; Hajduk P. J.; Meadows R. P.; Fesik S. W. Science 1996, 274, 1531. [DOI] [PubMed] [Google Scholar]
- Pellecchia M. J. Med. Chem. 2013, 56, 13. [DOI] [PubMed] [Google Scholar]
- Vandenberg C. J.; Cory S. Blood 2013, 121, 2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souers A. J.; Leverson J. D.; Boghaert E. R.; Ackler S. L.; Catron N. D.; Chen J.; Dayton B. D.; Ding H.; Enschede S. H.; Fairbrother W. J.; Huang D. C.; Hymowitz S. G.; Jin S.; Khaw S. L.; Kovar P. J.; Lam L. T.; Lee J.; Maecker H. L.; Marsh K. C.; Mason K. D.; Mitten M. J.; Nimmer P. M.; Oleksijew A.; Park C. H.; Park C. M.; Phillips D. C.; Roberts A. W.; Sampath D.; Seymour J. F.; Smith M. L.; Sullivan G. M.; Tahir S. K.; Tse C.; Wendt M. D.; Xiao Y.; Xue J. C.; Zhang H.; Humerickhouse R. A.; Rosenberg S. H.; Elmore S. W. Nat. Med. 2013, 19, 202. [DOI] [PubMed] [Google Scholar]
- Finlay D.; Vamos M.; Gonzalez-Lopez M.; Ardecky R. J.; Ganji S. R.; Yuan H.; Su Y.; Cooley T. R.; Hauser C. T.; Welsh K.; Reed J. C.; Cosford N. D.; Vuori K. Mol. Cancer Ther. 2014, 13, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardecky R. J.; Welsh K.; Finlay D.; Lee P. S.; Gonzalez-Lopez M.; Ganji S. R.; Ravanan P.; Mace P. D.; Riedl S. J.; Vuori K.; Reed J. C.; Cosford N. D. Bioorg. Med. Chem. Lett. 2013, 23, 4253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y.; Sun H.; Lu J.; Liu L.; Cai Q.; Shen R.; Yang C. Y.; Yi H.; Wang S. J. Med. Chem. 2012, 55, 106. [DOI] [PubMed] [Google Scholar]
- Sun H.; Lu J.; Liu L.; Yi H.; Qiu S.; Yang C. Y.; Deschamps J. R.; Wang S. J. Med. Chem. 2010, 53, 6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H.; Stuckey J. A.; Nikolovska-Coleska Z.; Qin D.; Meagher J. L.; Qiu S.; Lu J.; Yang C. Y.; Saito N. G.; Wang S. J. Med. Chem. 2008, 51, 7169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H.; Nikolovska-Coleska Z.; Lu J.; Meagher J. L.; Yang C. Y.; Qiu S.; Tomita Y.; Ueda Y.; Jiang S.; Krajewski K.; Roller P. P.; Stuckey J. A.; Wang S. J. Am. Chem. Soc. 2007, 129, 15279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C. M.; Sun C.; Olejniczak E. T.; Wilson A. E.; Meadows R. P.; Betz S. F.; Elmore S. W.; Fesik S. W. Bioorg. Med. Chem. Lett. 2005, 15, 771. [DOI] [PubMed] [Google Scholar]
- Oost T. K.; Sun C.; Armstrong R. C.; Al-Assaad A. S.; Betz S. F.; Deckwerth T. L.; Ding H.; Elmore S. W.; Meadows R. P.; Olejniczak E. T.; Oleksijew A.; Oltersdorf T.; Rosenberg S. H.; Shoemaker A. R.; Tomaselli K. J.; Zou H.; Fesik S. W. J. Med. Chem. 2004, 47, 4417. [DOI] [PubMed] [Google Scholar]
- Flygare J. A.; Beresini M.; Budha N.; Chan H.; Chan I. T.; Cheeti S.; Cohen F.; Deshayes K.; Doerner K.; Eckhardt S. G.; Elliott L. O.; Feng B.; Franklin M. C.; Reisner S. F.; Gazzard L.; Halladay J.; Hymowitz S. G.; La H.; LoRusso P.; Maurer B.; Murray L.; Plise E.; Quan C.; Stephan J. P.; Young S. G.; Tom J.; Tsui V.; Um J.; Varfolomeev E.; Vucic D.; Wagner A. J.; Wallweber H. J.; Wang L.; Ware J.; Wen Z.; Wong H.; Wong J. M.; Wong M.; Wong S.; Yu R.; Zobel K.; Fairbrother W. J. J. Med. Chem. 2012, 55, 4101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorpp K.; Rothenaigner I.; Salmina E.; Reinshagen J.; Low T.; Brenke J. K.; Gopalakrishnan J.; Tetko I. V.; Gul S.; Hadian K. J. Biomol. Screening 2014, 10.1177/1087057113516861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billeter M.; Wagner G.; Wuthrich K. J. Biomol. NMR 2008, 42, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wider G.; Wuthrich K. Curr. Opin. Struct. Biol. 1999, 9, 594. [DOI] [PubMed] [Google Scholar]
- Wuthrich K. Nat. Struct. Biol. 1998, 5Suppl.492. [DOI] [PubMed] [Google Scholar]
- Wuthrich K. J. Biol. Chem. 1990, 265, 22059. [PubMed] [Google Scholar]
- Wuthrich K. Angew. Chem., Int. Ed. 2003, 42, 3340. [DOI] [PubMed] [Google Scholar]
- Pellecchia M.; Meininger D.; Dong Q.; Chang E.; Jack R.; Sem D. S. J. Biomol. NMR 2002, 22, 165. [DOI] [PubMed] [Google Scholar]
- Wu B.; Rega M. F.; Wei J.; Yuan H.; Dahl R.; Zhang Z.; Pellecchia M. Chem. Biol. Drug Des. 2009, 73, 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal M.; Schanda P.; Brutscher B.; Frydman L. J. Am. Chem. Soc. 2007, 129, 1372. [DOI] [PubMed] [Google Scholar]
- Schanda P.; Kupce E.; Brutscher B. J. Biomol. NMR 2005, 33, 199. [DOI] [PubMed] [Google Scholar]
- Pervushin K.; Riek R.; Wider G.; Wuthrich K. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Religa T. L.; Kay L. E. J. Biomol. NMR 2010, 47, 163. [DOI] [PubMed] [Google Scholar]
- Tugarinov V.; Kanelis V.; Kay L. E. Nat. Protoc. 2006, 1, 749. [DOI] [PubMed] [Google Scholar]
- Goto N. K.; Gardner K. H.; Mueller G. A.; Willis R. C.; Kay L. E. J. Biomol. NMR 1999, 13, 369. [DOI] [PubMed] [Google Scholar]
- Rosen M. K.; Gardner K. H.; Willis R. C.; Parris W. E.; Pawson T.; Kay L. E. J. Mol. Biol. 1996, 263, 627. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Mias R. A.; Pellecchia M. J. Am. Chem. Soc. 2003, 125, 2892. [DOI] [PubMed] [Google Scholar]
- Gronenborn A. M.; Clore G. M. Biochem. Pharmacol. 1990, 40, 115. [DOI] [PubMed] [Google Scholar]
- Meyer E. F. Jr.; Clore G. M.; Gronenborn A. M.; Hansen H. A. Biochemistry 1988, 27, 725. [DOI] [PubMed] [Google Scholar]
- Sykes B. D. Curr. Opin. Biotechnol. 1993, 4, 392. [DOI] [PubMed] [Google Scholar]
- Leone M.; Freeze H. H.; Chan C. S.; Pellecchia M. Curr. Drug Discovery Technol. 2006, 3, 91. [DOI] [PubMed] [Google Scholar]
- Hajduk P. J.; Olejniczak E. T.; Fesik S. W. J. Am. Chem. Soc. 1997, 119, 12257. [Google Scholar]
- Mayer M.; Meyer B. Angew. Chem., Int. Ed. 1999, 38, 1784. [DOI] [PubMed] [Google Scholar]
- Mayer M.; Meyer B. J. Am. Chem. Soc. 2001, 123, 6108. [DOI] [PubMed] [Google Scholar]
- Dalvit C.; Pevarello P.; Tato M.; Veronesi M.; Vulpetti A.; Sundstrom M. J. Biomol. NMR 2000, 18, 65. [DOI] [PubMed] [Google Scholar]
- Dalvit C.; Fogliatto G.; Stewart A.; Veronesi M.; Stockman B. J. Biomol. NMR 2001, 21, 349. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Ernst R. R.; Wuthrich K. Biochem. Biophys. Res. Commun. 1980, 95, 1. [DOI] [PubMed] [Google Scholar]
- Wang Y. S.; Liu D.; Wyss D. F. Magn. Reson. Chem. 2004, 42, 485. [DOI] [PubMed] [Google Scholar]
- Dalvit C.; Flocco M.; Veronesi M.; Stockman B. J. Comb. Chem. High Throughput Screening 2002, 5, 605. [DOI] [PubMed] [Google Scholar]
- Dalvit C.; Flocco M.; Knapp S.; Mostardini M.; Perego R.; Stockman B. J.; Veronesi M.; Varasi M. J. Am. Chem. Soc. 2002, 124, 7702. [DOI] [PubMed] [Google Scholar]
- Dalvit C.; Fasolini M.; Flocco M.; Knapp S.; Pevarello P.; Veronesi M. J. Med. Chem. 2002, 45, 2610. [DOI] [PubMed] [Google Scholar]
- Vanwetswinkel S.; Heetebrij R. J.; van Duynhoven J.; Hollander J. G.; Filippov D. V.; Hajduk P. J.; Siegal G. Chem. Biol. 2005, 12, 207. [DOI] [PubMed] [Google Scholar]
- Kobayashi M.; Retra K.; Figaroa F.; Hollander J. G.; Ab E.; Heetebrij R. J.; Irth H.; Siegal G. J. Biomol. Screening 2010, 15, 978. [DOI] [PubMed] [Google Scholar]
- Giannetti A. M. Methods Enzymol. 2011, 493, 169. [DOI] [PubMed] [Google Scholar]
- Frostell A.; Vinterback L.; Sjobom H. Methods Mol. Biol. 2013, 1008, 139. [DOI] [PubMed] [Google Scholar]
- Elinder M.; Geitmann M.; Gossas T.; Kallblad P.; Winquist J.; Nordstrom H.; Hamalainen M.; Danielson U. H. J. Biomol. Screening 2011, 16, 15. [DOI] [PubMed] [Google Scholar]
- Danielson U. H. Curr. Top. Med. Chem. 2009, 9, 1725. [DOI] [PubMed] [Google Scholar]
- Torres F. E.; Recht M. I.; Coyle J. E.; Bruce R. H.; Williams G. Curr. Opin. Struct. Biol. 2010, 20, 598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Shoichet B. K. Nat. Chem. Biol. 2009, 5, 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark M.; Meshkat S.; Talbot G. T.; Carnevali P.; Wiseman J. S. J. Chem. Inf. Model. 2009, 49, 1901. [DOI] [PubMed] [Google Scholar]
- Recht M. I.; Sridhar V.; Badger J.; Hernandez L.; Chie-Leon B.; Nienaber V.; Torres F. E. J. Biomol. Screening 2012, 17, 469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig M.; Ruf A.; Huber W. Top. Curr. Chem. 2012, 317, 115. [DOI] [PubMed] [Google Scholar]
- Wyss D. F.; Wang Y. S.; Eaton H. L.; Strickland C.; Voigt J. H.; Zhu Z.; Stamford A. W. Top. Curr. Chem. 2012, 317, 83. [DOI] [PubMed] [Google Scholar]
- Drinkwater N.; Vu H.; Lovell K. M.; Criscione K. R.; Collins B. M.; Prisinzano T. E.; Poulsen S. A.; McLeish M. J.; Grunewald G. L.; Martin J. L. Biochem. J. 2010, 431, 51. [DOI] [PubMed] [Google Scholar]
- Wang Y. S.; Strickland C.; Voigt J. H.; Kennedy M. E.; Beyer B. M.; Senior M. M.; Smith E. M.; Nechuta T. L.; Madison V. S.; Czarniecki M.; McKittrick B. A.; Stamford A. W.; Parker E. M.; Hunter J. C.; Greenlee W. J.; Wyss D. F. J. Med. Chem. 2010, 53, 942. [DOI] [PubMed] [Google Scholar]
- Jhoti H.; Cleasby A.; Verdonk M.; Williams G. Curr. Opin. Chem. Biol. 2007, 11, 485. [DOI] [PubMed] [Google Scholar]
- Gill A.; Cleasby A.; Jhoti H. ChemBioChem 2005, 6, 506. [DOI] [PubMed] [Google Scholar]
- Hartshorn M. J.; Murray C. W.; Cleasby A.; Frederickson M.; Tickle I. J.; Jhoti H. J. Med. Chem. 2005, 48, 403. [DOI] [PubMed] [Google Scholar]
- Lesuisse D.; Lange G.; Deprez P.; Benard D.; Schoot B.; Delettre G.; Marquette J. P.; Broto P.; Jean-Baptiste V.; Bichet P.; Sarubbi E.; Mandine E. J. Med. Chem. 2002, 45, 2379. [DOI] [PubMed] [Google Scholar]
- Becattini B.; Culmsee C.; Leone M.; Zhai D.; Zhang X.; Crowell K. J.; Rega M. F.; Landshamer S.; Reed J. C.; Plesnila N.; Pellecchia M. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 12602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becattini B.; Pellecchia M. Chemistry 2006, 12, 2658. [DOI] [PubMed] [Google Scholar]
- Becattini B.; Sareth S.; Zhai D.; Crowell K. J.; Leone M.; Reed J. C.; Pellecchia M. Chem. Biol. 2004, 11, 1107. [DOI] [PubMed] [Google Scholar]
- Jahnke W.; Rudisser S.; Zurini M. J. Am. Chem. Soc. 2001, 123, 3149. [DOI] [PubMed] [Google Scholar]
- Jahnke W. ChemBioChem 2002, 3, 167. [DOI] [PubMed] [Google Scholar]
- Jahnke W.; Lawrence B.; Perez L.; Paris G.; Strauss A.; Fendrich G.; Nalin C. M. J. Am. Chem. Soc. 2000, 122, 7394. [Google Scholar]
- Vazquez J.; Tautz L.; Ryan J. J.; Vuori K.; Mustelin T.; Pellecchia M. J. Med. Chem. 2007, 50, 2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertini I.; Fragai M.; Lee Y. M.; Luchinat C.; Terni B. Angew. Chem., Int. Ed. 2004, 43, 2254. [DOI] [PubMed] [Google Scholar]
- Vazquez J.; De S. K.; Chen L. H.; Riel-Mehan M.; Emdadi A.; Cellitti J.; Stebbins J. L.; Rega M. F.; Pellecchia M. J. Med. Chem. 2008, 51, 3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tautz L.; Bruckner S.; Sareth S.; Alonso A.; Bogetz J.; Bottini N.; Pellecchia M.; Mustelin T. J. Biol. Chem. 2005, 280, 9400. [DOI] [PubMed] [Google Scholar]
- Hartshorn M. J.; Murray C. W.; Cleasby A.; Frederickson M.; Tickle I. J.; Jhoti H. J. Med. Chem. 2005, 48, 403. [DOI] [PubMed] [Google Scholar]
- Gill A.; Cleasby A.; Jhoti H. ChemBioChem 2005, 6, 506. [DOI] [PubMed] [Google Scholar]
- Rees D. C.; Congreve M.; Murray C. W.; Carr R. Nat. Rev. Drug Discovery 2004, 3, 660. [DOI] [PubMed] [Google Scholar]
- Allen K. N.; Lavie A.; Petsko G. A.; Ringe D. Biochemistry 1995, 34, 3742. [DOI] [PubMed] [Google Scholar]
- Rutenber E.; Fauman E. B.; Keenan R. J.; Fong S.; Furth P. S.; Ortiz de Montellano P. R.; Meng E.; Kuntz I. D.; DeCamp D. L.; Salto R. J. Biol. Chem. 1993, 268, 15343. [PubMed] [Google Scholar]
- Rutenber E. E.; McPhee F.; Kaplan A. P.; Gallion S. L.; Hogan J. C. Jr.; Craik C. S.; Stroud R. M. Bioorg. Med. Chem. 1996, 4, 1545. [DOI] [PubMed] [Google Scholar]
- Shoichet B. K.; Stroud R. M.; Santi D. V.; Kuntz I. D.; Perry K. M. Science 1993, 259, 1445. [DOI] [PubMed] [Google Scholar]
- Verlinde C. L. M. J.; Pijning T.; Kalk K. H.; van Calenbergh S.; Aershot A. V.; Herdewijn P.; Callens M.; Michels P.; Opperdoes F. R.; Wierenga R. K.; Hol W. G. J. In Perspectives in Medicinal Chemistry; Testa P., Kyburz E., Fuhrer W., Giger R., Eds.; Verlag Helvetica Chimica Acta: Basel, Switzerland, 1993. [Google Scholar]
- Hajduk P. J.; Greer J. Nat. Rev. Drug Discovery 2007, 6, 211. [DOI] [PubMed] [Google Scholar]
- Chen J.; Zhang Z.; Stebbins J. L.; Zhang X.; Hoffman R.; Moore A.; Pellecchia M. ACS Chem. Biol. 2007, 2, 329. [DOI] [PubMed] [Google Scholar]
- Schade M.; Oschkinat H. Curr. Opin. Drug Discovery Dev. 2005, 8, 365. [PubMed] [Google Scholar]
- Lipinski C. A. Drug Discovery Today 2003, 8, 12. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Adv. Drug Delivery Rev. 2001, 46, 3. [DOI] [PubMed] [Google Scholar]
- Congreve M.; Carr R.; Murray C.; Jhoti H. Drug Discovery Today 2003, 8, 876. [DOI] [PubMed] [Google Scholar]
- Stockman B. J.; Kothe M.; Kohls D.; Weibley L.; Connolly B. J.; Sheils A. L.; Cao Q.; Cheng A. C.; Yang L.; Kamath A. V.; Ding Y. H.; Charlton M. E. Chem. Biol. Drug Des. 2009, 73, 179. [DOI] [PubMed] [Google Scholar]
- Chessari G.; Maria A.; Buck I.; Chiarparin E.; Coyle J.; Day J.; Frederickson M.; Griffiths-Jones C.; Hearn K.; Howard S.; Heightman T.; Hillmann P.; Iqbal A.; Johnson C. N.; Lewis J.; Martins V.; Munck J.; Reader M.; Page L.; Hopkins A.; Millemaggi A.; Richardson C.; Saxty G.; Smyth T.; Tamanini E.; Thompson N.; Ward G.; Williams G.; Williams P.; Wilsher N.; Woolford A. AT-IAP, a Dual cIAP1 and XIAP Antagonist with Oral Antitumor Activity in Melanoma Models. Presented at the American Association for Cancer Research Annual Meeting, Washington, DC, Apr 6–10,2013; Poster 2944.
- Woodhead A. J.; Angove H.; Carr M. G.; Chessari G.; Congreve M.; Coyle J. E.; Cosme J.; Graham B.; Day P. J.; Downham R.; Fazal L.; Feltell R.; Figueroa E.; Frederickson M.; Lewis J.; McMenamin R.; Murray C. W.; O’Brien M. A.; Parra L.; Patel S.; Phillips T.; Rees D. C.; Rich S.; Smith D. M.; Trewartha G.; Vinkovic M.; Williams B.; Woolford A. J. J. Med. Chem. 2010, 53, 5956. [DOI] [PubMed] [Google Scholar]
- Murray C. W.; Carr M. G.; Callaghan O.; Chessari G.; Congreve M.; Cowan S.; Coyle J. E.; Downham R.; Figueroa E.; Frederickson M.; Graham B.; McMenamin R.; O’Brien M. A.; Patel S.; Phillips T. R.; Williams G.; Woodhead A. J.; Woolford A. J. J. Med. Chem. 2010, 53, 5942. [DOI] [PubMed] [Google Scholar]
- Sun Q.; Burke J. P.; Phan J.; Burns M. C.; Olejniczak E. T.; Waterson A. G.; Lee T.; Rossanese O. W.; Fesik S. W. Angew. Chem., Int. Ed. 2012, 51, 6140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley D. L.; Van Molle I.; Gareiss P. C.; Tae H. S.; Michel J.; Noblin D. J.; Jorgensen W. L.; Ciulli A.; Crews C. M. J. Am. Chem. Soc. 2012, 134, 4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsao D. H.; Sutherland A. G.; Jennings L. D.; Li Y.; Rush T. S. 3rd; Alvarez J. C.; Ding W.; Dushin E. G.; Dushin R. G.; Haney S. A.; Kenny C. H.; Malakian A. K.; Nilakantan R.; Mosyak L. Bioorg. Med. Chem. 2006, 14, 7953. [DOI] [PubMed] [Google Scholar]
- Dooley C. T.; Houghten R. A. Methods Mol. Biol. 1998, 87, 13. [DOI] [PubMed] [Google Scholar]
- Dooley C. T.; Houghten R. A. Life Sci. 1993, 52, 1509. [DOI] [PubMed] [Google Scholar]
- Houghten R. A.; Ostresh J. M.; Pratt S. M. NIDA Res. Monogr. 1991, 112, 239. [PubMed] [Google Scholar]
- Pinilla C.; Appel J. R.; Blanc P.; Houghten R. A. Biotechniques 1992, 13, 901. [PubMed] [Google Scholar]
- Houghten R. A.; Pinilla C.; Giulianotti M. A.; Appel J. R.; Dooley C. T.; Nefzi A.; Ostresh J. M.; Yu Y.; Maggiora G. M.; Medina-Franco J. L.; Brunner D.; Schneider J. J. Comb. Chem. 2008, 10, 3. [DOI] [PubMed] [Google Scholar]
- Houghten R. A.; Appel J. R.; Blondelle S. E.; Cuervo J. H.; Dooley C. T.; Pinilla C. Biotechniques 1992, 13, 412. [PubMed] [Google Scholar]
- Houghten R. A.; Pinilla C.; Appel J. R.; Blondelle S. E.; Dooley C. T.; Eichler J.; Nefzi A.; Ostresh J. M. J. Med. Chem. 1999, 42, 3743. [DOI] [PubMed] [Google Scholar]
- Houghten R. A.; Pinilla C.; Blondelle S. E.; Appel J. R.; Dooley C. T.; Cuervo J. H. Nature 1991, 354, 84. [DOI] [PubMed] [Google Scholar]
- Pinilla C.; Appel J.; Blondelle S.; Dooley C.; Dorner B.; Eichler J.; Ostresh J.; Houghten R. A. Biopolymers 1995, 37, 221. [DOI] [PubMed] [Google Scholar]
- Czabotar P. E.; Lee E. F.; van Delft M. F.; Day C. L.; Smith B. J.; Huang D. C.; Fairlie W. D.; Hinds M. G.; Colman P. M. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 6217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G.; Chai J.; Suber T. L.; Wu J. W.; Du C.; Wang X.; Shi Y. Nature 2000, 408, 1008. [DOI] [PubMed] [Google Scholar]
- Qin H.; Noberini R.; Huan X.; Shi J.; Pasquale E. B.; Song J. J. Biol. Chem. 2010, 285, 644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschner M.; Henn C.; Vollhardt H.; Reiling S.; Brickmann J. J. Mol. Graphics 1994, 12, 98. [DOI] [PubMed] [Google Scholar]