

Abstract
Purpose
HPV-associated (HPV+) oropharyngeal squamous cell carcinomas (OPSCC) have different molecular and biological characteristics and clinical behavior compared to HPV-negative (HPV−) OPSCC. PIK3CA mutations are more common in HPV(+)OPSCC. To define molecular differences and tumor subsets, protein expression and phosphorylation were compared between HPV(+) and HPV(−) OPSCC and between tumors with and without PIK3CA mutations.
Experimental design
Expression of 137 total and phosphorylated proteins was evaluated by reverse phase protein array(RPPA) in 29 HPV(+) and 13 HPV(−)prospectively collected OPSCCs. 47 OPSCCs were tested for hotspot activating mutations in PIK3CA and AKT. Activation of PIK3CA downstream targets and sensitivity to pathway inhibitors were determined in HPV(+) head and neck cancer cells overexpressing wild-type or mutant PIK3CA.
Results
Analyses revealed forty-one differentially expressed proteins between HPV(+) and HPV(−) OPSCC categorized into functional groups: DNA repair, cell cycle, apoptosis, PI3K/AKT/mTOR, and receptor kinase pathways. All queried DNA repair proteins were significantly upregulated in HPV(+) samples. 8 of 33 HPV(+) and 0 of 14 HPV(−) tumors contained activating PIK3CA mutations. Despite all activating PIK3CA mutations occurring in HPV(+) samples, HPV(+) tumors had lower mean levels of activated AKT and downstream AKT target phosphorylation. Ectopic expression of mutant PIK3CA in HPV(+) cells increase dmTOR, but not AKT activity. HPV E6/E7 overexpression inhibited AKT phosphorylation in HPV-negative cells. Mutant PIK3CA overexpressing cells were more sensitive to a dual PI3K/mTOR inhibitor compared to an AKT inhibitor.
Conclusions
Protein expression analyses suggest that HPV(+) and HPV(−) OPSCC differentially activate DNA repair, cell cycle, apoptosis, PI3K/AKT/mTOR, and receptor kinase pathways. PIK3CA mutations are more common in HPV(+) OPSCC and are associated with activation of mTOR, but not AKT. These data suggest that inhibitors for mTOR may have activity against HPV(+) PIK3CA mutant oropharyngeal cancers.
Keywords: head and neck cancer, PIK3CA, AKT, HPV, RPPA, prognosis
Introduction
Head and neck squamous cell carcinoma (HNSCC) represents 3% of adult malignancies, and the incidence of human-papilloma virus (HPV)-related oropharyngeal squamous cell carcinoma (OPSCC) in the United States has dramatically increased over the last two decades (1, 2). Over the same timeframe, OPSCC patients have become younger, more affluent, and with less tobacco exposure, and therapy for advanced cancers has shifted from surgery followed by radiation to concomitant chemotherapy and radiation (3–5). Overall and disease-free survival for HPV-associated OPSCC is much better than HPV(−) OPSCC following treatment with concurrent platin-based chemotherapy and radiation; however, long-term treatment side effects are significant (6), which has driven interest in treatment de-escalation that will likely involve rational targeted therapeutics.
In addition to distinctions in etiology and patient demographics, significant molecular differences have been described that distinguish HPV(+) and HPV(−) OPSCC (7, 8). Because HPV oncoproteins E6 and E7 inactivate p53 and Rb respectively, HPV(+) OPSCC have few p53 mutations and highly express the Rb upstream regulator p16INK4a. Beyond differences related to p53 and Rb tumor suppressors, HPV(+) and HPV(−) tumors are easily distinguished by gene profiling and HPV(+) tumors are associated with fewer chromosomal abnormalities (9, 10). The majority of genes differently expressed in HPV(+) versus HPV(−) tumors function to regulate cell cycle, DNA replication and repair (11, 12). A proteomics study of HPV(+) and HPV(−) OPSCC using liquid chromatography and tandem mass spectroscopy (LC-MS/MS) also found marked differences in proteins primarily involved in metabolism (11). Unfortunately, identification of molecular differences between HPV(+) and HPV(−) OPSCC, has not led to improved therapies.
The PI3K/AKT/mTOR pathway is implicated in HNSCC tumorigenesis with next generation sequencing identifying activating pathway defects (8, 12–15) and indicating that the PI3K pathway is amongst the most frequently altered pathways in human head and neck cancer.
Here, expression of 137 total and phosphorylated proteins was evaluated by RPPA in 29 HPV-positive, 12 HPV-negative prospectively collected OPSCCs and one normal tonsil tissue collected from healthy patient. Comparison of HPV(+) and HPV(−) OPSCC revealed that nearly 1/3 of queried proteins (33 proteins and 8 phospho-proteins) were significantly differentially expressed between tumors with and without HPV, highlighting four major distinguishing pathways: 1) DNA repair, 2) cell cycle, 3) apoptosis, and 4) PI3K/AKT/mTOR.
We previously found that HPV(+) OPSCC more frequently contained activating mutations of PIK3CA (16). Here, PIK3CA mutations were also found more frequently in HPV-associated tumors and these mutations were correlated with protein expression and patient outcome. Unexpectedly, we found that in HPV(+) OPSCC, PIK3CA mutations were associated with a non-significant tendency for improved patient survival. In addition, mutant PIK3CA in HPV-associated tumors correlated with activation of downstream mTOR, but not Akt pathway. To confirm these results and provide therapeutic insight, we tested the ability of mutant PIK3CA to activate downstream targets in HPV-positive and HPV-negative head and neck cancer cells lines expressing the HPV oncogenes E6 and E7. We also examined response of HPV(+) cells expressing wild-type or mutant PIK3CA to AKT inhibition or combined PI3K/mTOR inhibition.
Materials and Methods
Tumor Selection
Forty-nine patients with OPSCC(HPV-positive = 33 patients; HPV-negative = 16 patients), for which clinical data was available were consented and enrolled in this study. Tumors were collected upon biopsy or excision and frozen in liquid nitrogen for further analyses.
Reverse phase protein array (RPPA)
Tumor was available from 42 of 49 patients in the study. RPPA procedures for antibody staining and signaling quantification were performed as described (17). Briefly, five 2-fold serial dilutions of cellular lysates from macrodissected tumors, containing greater than 70% cancer cells, were performed. Serially diluted lysates were arrayed on nitrocellulose-coated slides (Grace Biolab) using Aushon 2470 Arrayer (Aushion BioSystems). Diluted samples were robotically printed on multiple slides that included positive and negative controls.
Slides were probed with primary antibodies (one per slide) followed by a biotin-conjugated secondary antibody. Only antibodies with a Pearson correlation coefficient between RPPA and Western blotting of greater than 0.7 were used in the RPPA study.
Signals were amplified using a Dakocytomation-catalyzed system (Dako) and visualized by DAB colorimetric reaction. Slides were scanned, analyzed, and quantified using customized software Microvigene (VigeneTech Inc.) to generate spot intensity.
HPV and PIK3CA mutation status
HPV status of each patient sample was determined by PCR amplification of L1, E6, and E7 genes, as well as analysis of E6, E7, and p16 expression by qRT-PCR. PIK3CA mutation status from primary patient tumors was determined by direct sequencing of PCR amplified exons 1, 9, and 20, as previously described (18).
Cell lines and expression vectors
The HPV(+) cell line UMSCC-47 was cultured in DMEM with nonessential amino acids, HPV(−) cell lines, SCC61 and UNC7, were cultures in Dulbecco’s modified Eagle’s (DMEM)/F12 medium supplemented with 0.4 μg/mL hydrocortizon; for all cell lines medium was supplemented with 10% FBS (Invitrogen), 50 μg/mL penicillin, and 50 μg/mL streptomycin (Invitrogen). Cells were cultured in a humidified atmosphere of 5% CO2 at 37°C.
Wild-type p110α subunit of PIK3CA and E545K mutant of PIK3CA were purchased from Addgene and UMSCC-47 cells were transfected with indicated plasmids using lipofectamine reagent (Invitrogen). Stable transfectants were selected in the presence of 250 ug/ml G418 (American Bioanalytical) for 2 weeks, and overexpression of PIK3CA was verified by immunoblotting.
LXSN and HPV16 E6/E7-containing LXSN replication-incompetent MLV retroviral constructs were obtained from Dr. John H. Lee.
RNA extraction and quantitative real-time reverse transcription PCR
Total RNA was extracted by Roche RNA extracting kit and cDNA was synthesized using iScript cDNA Synthesis Kit (Bio-Rad) according to the manufacturer’s instructions. qRT-PCR was done using iQ SYBR Green Supermix (Bio-Rad) on an iCycleriQ (Bio-Rad). Primers for HPV-16 E6 were as follows: forward: CTCTGAATTCGCCACCATGCACCAAAAGAGAACTGCA, reverse: CCCTCGAGGTATCTCCATGCATGATTACA. A housekeeping gene, GUSB, was amplified in parallel and used as an endogenous control to quantify relative gene expression.
Immunoblotting
Cells were collected by trypsinization and lysed in RIPA lysis buffer (Sigma)for 10 minutes on ice with addition of protease (Roche) and phosphatase (Sigma) inhibitors. Insoluble material was removed by centrifugation at 14,000 rpm for 15 minutes at 4°C. Proteins were separated in 4–12% NUPAG gels (Invitrogen) and transferred to PVDF membrane. Membranes were blocked with 3% BSA in PBS and incubated with antibodies against total S6, pS6 S240/244, pAKT S473 (Cell Signaling), and PCNA (Sigma). After incubation with primary antibodies, membranes were washed, incubated with secondary DyLight anti-mouse and anti-rabbit antibodies (Thermo Scientific), and signals was visualized using Bio-Rad imager.
Survival assay
Cells were plated in 6 well plates (100,000 cells/well) in duplicates and treated with increasing concentration of BEZ-235 (Selleckem)or MK-2206 (Selleckem). After 8 days of treatment, viable cells, based on trypan blue exclusion, were counted.
Statistical analyses
The Kaplan–Meier method was used to generate recurrence-free survival and overall survival curves, and log-rank test analysis was used to compare HPV-positive and HPV-negative patient groups. Other statistical analyses were done using Fisher exact and χ2 for trend tests.
Predictive linear modeling was used to identify differentially expressed proteins.
Results
Protein expression profiles distinguish HPV(+) and HPV(−) OPSCC
Patient characteristics, including gender, age at diagnosis, tumor and nodal staging, treatment and smoking status are summarized in Table 1. Research-consented patients were treated according to institutional norms and treatment was not influenced by participation in this study. The prospectively collected cohort consisted of 39 males (~80%) and 10 females (~20%), all with oropharyngeal squamous cell carcinoma that had not been previously treated. Although males were the majority for both HPV(+) and HPV(−) OPSCC, a greater percentage of patients with HPV(+) tumors were male compared to patients with HPV(−) tumors (88% vs. 62%, p=0.048, Table 1). As expected, patients with HPV-associated tumors were younger than patients with HPV(−) tumors (median age 56 vs. 68.5, p=0.003) and fewer were smokers (51% vs. 94%, p=0.009). As a group, patients with HPV(+) tumors have overall survival advantages as compared to patients with HPV(−) OPSCC (Figure 2, p<0.0001).
Table 1.
Patient Characteristics
| Characteristics | Total | HPV-positive | HPV-negative | p-value | mut-PI3KCA | wt-PI3KCA | p-value |
|---|---|---|---|---|---|---|---|
| N of cases | 49 | 33 | 16 | 8 | 39 | ||
|
| |||||||
| Sex | |||||||
| Male | 39 (79.6%) | 29 (87.9%) | 10(62.5%) | 0.048 | 7 (87.5%) | 30 (76.9%) | 0.45 |
| Female | 10 (20.4%) | 4 (12.1%) | 6 (37.5%) | 1 (12.5%) | 9 (23.1%) | ||
|
| |||||||
| Age at Dx | |||||||
| Median | 59 | 56 | 68.5 | 0.003 | 57 | 52 | 0.098 |
| Range | 35–80 | 35–74 | 44–80 | 35–60 | 38–80 | ||
| Mean | 59 | 55 | 64.6 | 49.4 | 60.2 | ||
|
| |||||||
| Staging * | |||||||
| T1–T2 | 29 (65.9%) | 21 (67.7%) | 9 (56.3%) | 4 (66.7%) | 23 (65.7%) | ||
| T3–T4 | 15 (34.1%) | 10 (32.3%) | 7 (43.7%) | 0.32 | 2 (33.3%) | 12 (34.3%) | 0.67 |
| N0 | 6 (13.6%) | 3 (9.7%) | 3 (18.8%) | 0 (0%) | 6 (17.1%) | ||
| N1–3 | 38 (86.4%) | 28 (90.3%) | 13 (81.2%) | 0.41 | 6 (100%) | 29 (82.9%) | 0.5 |
| I–II | 4 (9.1%) | 2 (6.5%) | 2 (12.5%) | 0 (0%) | 4 (11.4%) | ||
| III–IV | 40 (90.9%) | 29 (93.5%) | 14 (87.5%) | 0.42 | 6 (100%) | 31 (88.6%) | 0.52 |
|
| |||||||
| Smoking history | |||||||
| Smoker | 32 (65.3%) | 17 (51.51%) | 15 (93.75%) | 0 (0%) | 26 (70.3%) | ||
| Non-smoker | 14 (28.6%) | 13 (39.39%) | 1 (6.25%) | 0.009 | 6 (85.7%) | 8 (21.6%) | 0.0008 |
| N/A** | 3 (6.1%) | 3 (9.1%) | 0 (0%) | 1 (14.3%) | 3 (8.1%) | ||
|
| |||||||
| Treatment | |||||||
| RT-Chemo | 9 (18.4%) | 5 (15.2%) | 4 (25.0%) | 4 (10.3%) | 1 (12.5%) | ||
| Surgery-RT-Chemo | 4 (8.2%) | 4 (12.1%) | 0 (0%) | 2 (5.1%) | 2 (25.0%) | ||
| Chemo-RT-Chemo | 11 (22.5%) | 8 (24.2%) | 3 (18.7%) | 7 (17.8%) | 3 (37.5%) | ||
| Chemo-RT-Chemo-Surgery | 1 (2.0%) | 1 (3.0%) | 0 (0%) | 0 (0%) | 0 (0%) | ||
| Surgery | 4 (8.2%) | 1 (3.0%) | 3 (18.7%) | 4 (10.3%) | 0 (0%) | ||
| Surgery-RT | 6 (12.2%) | 5 (15.2%) | 1 (6.3%) | 6 (15.4%) | 0 (0%) | ||
| Chemo-RT | 3 (6.1%) | 0 (0%) | 3 (18.7%) | 6 (15.4%) | 0 (0%) | ||
| Chemo | 1 (2.0%) | 0 (0%) | 1 (6.3%) | 1 (2.6%) | 0 (0%) | ||
| N/A** | 10 (20.4%) | 9 (27.3%) | 1 (6.3%) | 9 (23.1%) | 2 (25.0%) | ||
Four Patients had unknown TNM staging status
No information available for smoking status (3/49)
Figure 2. PIK3CA mutations correlate with improved survival.
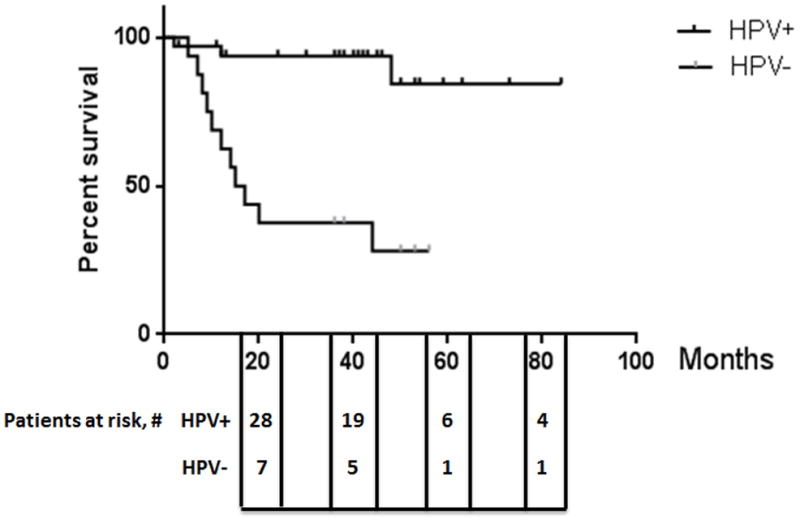
Kaplan–Meier curves showing overall survival of patients with HPV-positive versus HPV-negative OPSCC.
Recent gene expression profiling of HPV(+) and HPV(−) cancers has not clearly defined biological alterations that explain marked differences in the behavior and response. Since protein expression does not always mirror mRNA expression and protein modifications are not captured by RNA analyses, protein and phospho-protein levels were measured by RPPA on 42 primary oropharyngeal squamous carcinomas to further define biological differences based on HPV status and to gain mechanistic insight.
Predictive linear modeling of the raw RPPA data revealed that 41 total or phospho-proteins were significantly differentially expressed between HPV(+) and HPV(−) OPSCC (Figure 1A and Table 2, p<0.05). This remarkable portion of queried proteins that were differentially expressed (30%) highlights the magnitude of molecular differences between HPV(+) and HPV(−) tumors. Described functions of differentially expressed proteins identified distinguishing pathways: 1) cell cycle, 2) DNA repair, 3) apoptosis,4) receptor, kinase, signaling and 5) PI3K/AKT/mTOR (Table 2).
Figure 1. Reverse Phase Protein Array data.

(A) 41 proteins are significantly (p<0.05) differentially expressed in HPV(+) as compared to HPV(−) OPSCC. HPV(+) tumors are labeled in orange, HPV(−) tumors are labeled in yellow. (B) 30 proteins are significantly (p<0.05) differentially expressed in HPV(+) PIK3CA mutants as compared to HPV(+) PIK3CA wild type OPSCC. PIK3CA mutant tumors are labeled in red; HPV(+), PIK3CA wild type tumors are labeled in black.
Table 2.
Proteins differentially expressed in HPV(+) vs. HPV(−) OPSCC
| Pathways | Direction of change HPV(+) vs. HPV(−) | p-value | Notes |
|---|---|---|---|
| Proteins | |||
|
| |||
| DNA repair | |||
| ATR pS428 |
|
7.64 E-05 | Kinase responsible unknown |
| ATRIP | 0.0020 | ||
| BRCA2 | 0.0028 | ||
| CHK2 | 0.0003 | ||
| KU80 | 0.0382 | ||
| MSH2 | 0.0006 | ||
| PARP | 0.0082 | ||
| PCNA | 0.0007 | ||
| XRCC1 | 0.0119 | ||
| ATM | 0.0253 | ||
| MGMT | 0.0196 | ||
| ATM pS1981 |
|
0.0124 | Auto-phosphorylation site |
| Cell cycle | |||
| Cyclin.B1 |
|
0.0243 | |
| Cyclin.E1 | 0.0048 | ||
| Stathmin | 0.0010 | ||
| Rb |
|
0.0321 | |
| Rb pS807 | 0.0033 | Cdk4/Cdk6 target | |
| Cyclin D1 | 8.74 E-05 | ||
| Apoptosis | |||
| BCL2 |
|
0.0002 | |
| BIM | 2.77 E-07 | ||
| Cl. Caspase.7 | 0.0105 | ||
| JNK2 | 0.0196 | ||
| PI3K/AKT/mTOR | |||
| Raf pS259 |
|
0.0402 | Downstream AKT target |
| GSK3a/b pS21/S9 | 0.0499 | Downstream AKT target | |
| TSC2_pT1462 |
|
0.0217 | Downstream AKT target |
| GSK3a/b | 0.0155 | ||
| TSC2 |
|
0.0037 | |
| Receptors/kinases/signaling | |||
| EGFR |
|
0.0120 | EGFR |
| IRS1 | 0.0083 | Insulin/Insulin-like receptor | |
| IRS2 | 0.0185 | Insulin/Insulin-like receptor | |
| STAT3 | 0.0036 | EGFR, cytokines | |
| Caveolin1 | 8.15 E-05 | Insulin/Insulin-like receptor | |
| AMPKα | 0.0097 | Multiple input, metabolism | |
| MEK1/2 pS217 | 0.0152 | Ras signaling, multiple inputs | |
| PKCα | 0.0004 | Cell adhesion/junctions, cell cycle | |
| Others | |||
| Src |
|
0.0023 | Src |
| c.Myc pT58 | 0.0408 | Myc | |
| ATF2 | 0.0311 | Transcriptional factor, JNK target | |
| MCP1 |
|
0.0227 | Immune response |
| PTCH | 0.0466 | Wnt/Hedgehog/NOTCH | |
| Cox2 | 0.0184 | Metabolism/Prostaglandins | |
 - Inhibitory phosphorylation
- Inhibitory phosphorylation
 - Activating phosphorylation
- Activating phosphorylation
Unsupervised hierarchical clustering using differentially expressed proteins based on HPV status of 42 tumors and one normal tonsillar mucosa revealed two major groups(designated Group I and Group II, separated by solid line, Figure 1A). Group I contained the majority of tumors and was composed of two major subgroups (designated Group IA and Group IB, separated by the dashed line, Figure 1A). Seven of 13 HPV negative tumors, as well as the normal mucosa, clustered exclusive of HPV(+) tumors into Group II. Within Group I, which included all HPV(+) and 6 HPV(−) tumors, Subgroup IA consisted, nearly exclusively, of HPV(+) tumors (20 HPV(+) tumors and 1 HPV(−) tumor). In contrast, a mix of HPV(+) and HPV(−) tumors comprised Subgroup IB(9 HPV(+) and 5 HPV(−)).
Using liquid chromatography -mass spectrometry, we previously reported that proteins involved in DNA replication, many of which were linked by transcription factor enrichment analysis to E2F activity, were differentially expressed in HPV(+) and HPV(−) OPSCC (11). Validating the important role of HPV oncoproteins in dysregulation of cell cycle progression, RPPA analyses revealed that HPV(−) OPSCC expressed higher levels of cyclin D1 and corresponding higher levels of phospho-Rb, whereas HPV(+) tumors expressed lower levels of Rb, as expected from E7 activity to destabilize Rb (19), and higher levels of S and G2/M cyclins E1 and B1, respectively (Figure 1A, Table 2).
In addition to expected distinctions in cell cycle dysregulation between HPV(+) and HPV(−) tumors, RPPA analyses identified markedly altered expression patterns for proteins involved in DNA repair (Figure 1A, Table 2). In fact, proteins involved in DNA repair represented the largest functional category of differentially expressed proteins between HPV(+) and HPV(−) tumors (12 of 41 differentially expressed proteins, 29%) with all differentially expressed total proteins being up-regulated in HPV(+) as compared to HPV(−) tumors (Figure 1A and Table 2). Interestingly, although total ATM was relatively increased in HPV-associated tumors, ATM phospho-S1981 expression was decreased. In addition, activated Chk1 was also relatively, but non-significantly, increased in HPV(+) OPSCC (CHK1 pS345, p = 0.0790, Supplementary Table 1).
As is true for abrogation of cell cycle control, genes involved in apoptosis were also dysregulated by HPV oncogenes. RPPA analyses revealed that HPV(+) and HPV(−) tumors differentially express both pro- and anti-apoptotic proteins (Figure 1A and Table 2). HPV(+) tumors expressed relatively more cleaved caspase7 and showed a non-significant increase in cleaved caspase3 (p = 0.1009, Supplemental Table 1). Using cleavage of caspase 3 and 7 as markers of apoptosis, these data suggest that apoptosis is increased in untreated HPV(+) tumors.
EGFR is the most frequently activated receptor tyrosine kinase in HNSCC, being amplified in a subset and highly expressed in the majority. RPPA analyses revealed that HPV(+) OPSCC expressed lower levels of EGFR (Figure 1A and Table 2) and, although not significant, had decreased expression of activated EGFR (EGFR pY1173, p = 0.05, and EGFR pY992, p = 0.07, Supplemental Table 1). These data support described inverse correlation between HPV-positivity and EGFR expression (20). Downstream targets of EGFR activity were also expressed at relatively lower levels in HPV(+) compared to HPV(−) tumors, including STAT3 and activated MEK1/2 (phospho-S217). Although not significantly different, MKK2 total levels were downregulated in HPV(+) tumors (p = 0.0619, Supplemental Table 1). We found significant decrease of proteins critical for insulin receptor and insulin like growth factor receptor signaling in HPV(+) tumors including IRS1, IRS2, and caveolin (Figure 1A and Table 2). Together, these data suggest that HPV(+) OPSCC may be less reliant on EGFR and insulin/insulin-like growth factor signaling compared to HPV(−)OPSCC.
We previously reported that PIK3CA mutations occur more frequently in HPV-associated vs. HPV(−) HNSCC and suggested that HPV(+) tumors express more PIK3CA compared to HNSCC lacking HPV (16, 21). RPPA analyses confirm differential dysregulation of the PI3K/AKT/mTOR pathway (Figure 1A and Table 2). Direct substrates of Akt (phospho-Raf; phospho-GSK3α/β and phospho-TSC2) were expressed at relatively lower levels in HPV(+) OPSCC (Figure 1A and Table 2), which was surprising, since AKT is a major downstream target of PI3K. The PI3K pathway also alters insulin signaling and protein metabolism through regulation of GSK3 and TSC/mTOR with downstream effects on protein translation. GSK3α/β and TSC2 are phosphorylated and inhibited by activated AKT, increasing translation initiation through relief of inhibition of eIF2B. Likewise, inhibition of TSC2 results in mTOR activation and increased translation initiation through relief of inhibition of eIF4E. HPV(−) tumors had increased expression of phospho-TSC2 and phospho-GSK3α/β as well as increased expression of phospho-AKT (AKT pT308, p = 0.0841, Supplemental Table 1), all of which indicate that HPV(−) OPSCC tumors have activated AKT signaling compared to HPV(+) tumors.
LKB1/AMPK is a master regulator of glucose uptake and fatty acid synthesis in response to the AMP:ATP ratio, and AMPK activation is linked to enhanced insulin sensitivity (22). It is notable that AMPK alpha (AMPKα) was more highly expressed in HPV(−) compared to HPV(+) OPSCC. Although not significant, LKB1 expression trended in the same direction (p = 0.1031, Supplemental Table 1). Insulin receptor (IR) and insulin-like growth factor receptor (IGF-1R) signaling are major regulators of glucose metabolism. Calveolin mediates IR and IGF-1R signaling (23)and protects IRS-1 from proteasomal degradation (24). RPPA analyses revealed that HPV(+) OPSCC expressed lower levels of caveolin, and perhaps as a consequence, lower levels of IRS-1 and IRS-2.
PIK3CA mutations are more frequent in HPV(+) OPSCC
Activation of PI3K pathway is frequent in cancer occurring through mutation of PIK3CA, AKT, PTEN, or the PIK3CA regulatory subunit, p85, and is often associated with resistance to therapy and poor prognosis in human cancer (25, 26). Given the importance of the PI3K/AKT/mTOR pathway and differential expression of pathway proteins between HPV(+) and HPV(−) OPSCC, PIK3CA hotspot (exons 1, 9, 20) and AKT1 hotspot (E17K)mutations were determined from 47 of the original 49 OPSCC, for which DNA was available. No AKT1 mutations were identified. PIK3CA nucleic acid sequences that would cause amino acid substitutions were identified from 9 of 47 tumors analyzed. Eight tumors had activating missense point mutations resulting in amino acid substitutions E545K or E542K and one sample contained a described non-functional mutation Q60K (27) and was further grouped together with wild-type PIK3CA tumors. One of the 8 OPSCC with activating PIK3CA mutation contained 2 nucleotide alterations, an activating E545K mutation and a yet-to-be described mutation, P104L. Importantly, all PIK3CA mutations were observed in HPV(+) OPSCC (Table 1). Although the cohort was too small to draw conclusions related to survival, there was no indication that PIK3CA mutations were associated with worse survival in patients with HPV(+) tumors. In fact, all deaths in the HPV(+) group occurred in patients whose tumors did not have PIK3CA mutations (Supplementary Fig. S1).
Mutant PIK3CA tumors have a distinct protein expression profile within HPV(+) OPSCC
Protein expression was compared between HPV(+)tumors with and without PIK3CA mutations. Seven of the 8 PIK3CA mutant OPSCC had RPPA analyses performed, and predictive linear modeling identified 30 differentially expressed proteins between PIK3CA mutant versus wild-type tumors.(Table 3, p<0.05). Unsupervised hierarchical clustering using the 30 differentially expressed proteins clustered 6 of 7 PIK3CA mutant tumors into one of 2 major groups (Group II, Figure 1B). Described functions of differentially expressed proteins highlighted several cancer pathways including: 1) PI3K/AKT/ mTOR, 2) insulin signaling, and 3) receptor/kinases signaling (Table 3 and Figure 1B).
Table 3.
Proteins differentially expressed in PI3KCA mutant vs. wild-type tumors within HPV(+) OPSCC
| Pathways | Direction of change PIK3CA mutant. vs. wild-type | p-value | Notes |
|---|---|---|---|
| Proteins | |||
|
| |||
| PI3K/AKT/mTOR | |||
| S6 pS240 |
|
0.0081 | mTOR, S6K1, PDK target |
| S6 | 0.0090 | ||
| GSK3a/b | 0.0257 | ||
| TSC2 | 0.0242 | ||
| p85 PI3K |
|
0.0390 | |
| Insulin signaling | |||
| IGFBP2 |
|
0.0309 | |
| IRS1 pS307 | 0.0037 | JNK2, IR, IGF-1R, S6K1, mTOR target | |
| IGF-1R pY1135 |
|
0.0434 | Auto-phosphorylation site |
| Receptors/kinases/signaling | |||
| ACC pS79 |
|
0.0028 | |
| AMPKa pT172 | 0.0245 | AMPK target | |
| EGFR pY1173 |
|
0.0217 | LKB1, CaMKKβ target |
| STAT3 pY705 | 0.0078 | Auto-phosphorylation site | |
| ER pS118 | 0.0195 | EGFR, p38MAPK, other target | |
| ERK2 | 0.0031 | ERK1/2, CDK7 target | |
| c-kit | 0.0156 | ||
| Cell cycle | |||
| Cyclin.B1 |
|
0.0233 | |
| P27 |
|
0.0460 | |
| Apoptosis | |||
| 14.3.3. Zeta |
|
0.0233 | |
| PKCa |
|
0.0113 | |
| PKCa pS657 | 0.0057 | ||
| JNK2 | 0.0304 | Potential PDK target | |
| DNA repair | 0.0058 | ||
| ATR pS428 |
|
0.0164 | |
| PCNA | 0.0167 | ||
| ATM |
|
0.0030 | |
| MRE11 | |||
| Wnt/Hedgehog/ Notch | 0.0008 | ||
| Beta-catenin |
|
0.0044 | |
| E-cadherin | |||
| Others | 0.0052 | Transcriptional factor | |
| ATF1 |
|
0.0045 | Transcriptional factor |
| GATA3 | 0.0045 | Telomere maintenance | |
| Telomerase |
|
||
- Inhibitory phosphorylation
- Activating phosphorylation
EGFR, insulin receptor, and insulin-like growth factor receptor signaling are notable activators of PI3K (28, 29). HPV-associated HNSCCs are thought to rely less than HPV(−) tumors on receptor tyrosine kinase signaling as indicated by the absence of amplification events at loci encoding EGFR and FGFR1 in HPV(+) tumors, whereas amplification was observed in 13% and 10% of HPV(−) tumors respectively ((30) and TCGA data). Within the HPV(+) subset of OPSCC, tumors containing activating mutations of PIK3CA had decreased activation of EGFR as indicated by lower levels of phosphorylation (Y1173) (Figure 1B and Table 3). Likewise, tumors with PIK3CA mutations had decreased activating phosphorylation of IGF1R (Y1135) and increased inhibitory phosphorylation of IRS1 (S307), as well as increased expression of the IGF1R inhibitory protein, IGFBP2. Interestingly, two proteins from Wnt/Hedgehog/Notch pathway, namely E-cadherin and β-catenin, were expressed at higher levels in PIK3CA mutant as compared to wild-type tumors (Figure 1B and Table 3).
PIK3CA mutations are associated with activation of mTOR, but not AKT signaling pathway
It is widely held that AKT is the critical mediator of PI3K activation. Surprisingly, neither total nor phospho-AKT (S308 or S473)were increased in tumors with mutant PIK3CA; however, an indirect target of mTOR, S6, carried increased phosphorylation in PIK3CA mutant group (Table 3, Figure 3A and B). We compared phosphorylation of mTOR targets in HPV(+) tumors with and without PIK3CA activating mutations. In PIK3CA mutant tumors, mTOR targets S6 and IRS1 (S240 and S307, respectively) had statistically increased phosphorylation. In addition, direct mTOR sites of STAT3(S727, p=0.0862)(31), 4EBP1 (T37, p=0.0637) and an additional site in S6 (S235, p=0.0684)had relatively increased phosphorylation in PIK3CA mutant tumors (Figure 3B). We did not find significant differences in PTEN protein expression in mutant versus wild-type HPV(+) OPSCC (Figure 3C right), nor in HPV(+) as compared to HPV(−)tumors (Figure 3C left).
Figure 3. Downstream mTOR pathway, but not AKT pathway, is activated in HPV(+) PIK3CA mutant head and neck tumors and cells.

Scatter plots of proteins and phospho-proteins expression showing AKT (A) and mTOR (B) activation in HPV(+) PIK3CA wild type and mutant tumors as assessed by RPPA. (C) PTEN expression in in HPV(+) versus HPV(−) tumors (left), and in PIK3CA mutant versus wild type tumors within HPV(+) group (50)as assessed by RPPA. (D)Immunoblotting of HPV(+) UMSCC-47 cells stably transfected with either wild type or E545K mutant PIK3CA vectors with total S6, pS6, and pAKT antibodies. PCNA antibody was used as a loading control. (E) HPV-negative SCC61 and UNC7 cells were transiently transfected with empty vector or construct expressing HPV16 E6/E7; (left) pAKT (S473) levels were detected in immunoblotting. GPDH was used as a loading control. (50) Expression of E6 in SCC61 and UNC cells transfected with E6/E7 expression vector (SCC61 E6/E7, UNC7 E6/E7) compared with cells harboring empty vector (SCC61 and UNC7) was confirmed by qRT-PCR.
Because no HPV(+) HNSCC cell line endogenously expresses a PIK3CA mutation (15), effects of HPV on PIK3CA signaling was explored using HPV(+) HNSCC cell lines, UMSCC47 and SCC090, engineered to stably express wild-type or mutant (E545K) PIK3CA. Consistent with RPPA data (Figure 1B and Table 3), expression of mutant PIK3CA in HPV(+) UMSCC47 cells resulted in phosphorylation of S6, but no increase in phospho-AKT was observed (Figure 3D). In line with these results, we found elevated phosphorylation of mTOR downstream targets, S6 and S6K, but no AKT phosphorylation in another HPV(+) head and neck cancer cell line SCC090 stably expressing E545K PI3KCA. (Supplementary Fig. S2). Overexpression of wild-type PIK3CA also increased phosphorylation of mTOR targets, but not Akt in HPV(+) cells (Figure 3D and Supplementary Figure 3).
To begin exploring mechanisms through which HPV may alter PIK3CA signaling, the major HPV oncogenes, E6 and E7, were expressed in HPV-negative head and neck cancer cell lines, SCC61 and UNC7 (Figure 3E). UNC7 expresses wild-type PIK3CA, whereas SCC61 harbors an endogenous mutation of PIK3CA (E545K; data not shown). Remarkably, transient transfection of HPV16 E6/E7 greatly diminished AKT phosphorylation in these cells regardless of PIK3CA gene mutation status (Figure 3E). These results suggest that expression of HPV oncogenes E6/E7 inhibits the ability of wild-type or mutant PIK3CA to activate AKT.
HPV(+)cells expressing mutant PIK3CA are more sensitive to PI3K/mTOR inhibition vs. AKT inhibition
Given that the mTOR pathway, but not Akt, was activated in HPV(+) tumors expressing mutant PIK3CA, and that E6/E7 expression decreased Akt phosphorylation in HPV(+) cells lines, we suspected that mTOR may be a better target for these tumors. To begin exploring these possibilities, survival of UMSCC47 cells with ectopic expression of wild-type or mutant PIK3CA, as well as parental cells, was determined following treatment with a dual PI3K/mTOR inhibitor (BEZ-235) or a pan-Akt inhibitor (MK-2206). Expression of mutant and wild-type PIK3CA sensitized HPV(+) cells to the dual PI3K/mTOR inhibition with expression of mutant PIK3CA resulting in more than 2-fold sensitization and expression of wild-type PIK3CA in modest sensitization (1.22-fold vs parental cells)(Supplementary Figure 1). As opposed to treatment with the PI3K/mTOR inhibitor, treatment with the pan-Akt inhibitor, MK-2206 resulted in no sensitization of HPV(+) cells expressing wild-type PIK3CA (1.0-fold) and only modest increased sensitivity in cells expressing mutant PIK3CA mutant (1.5-fold) (Supplementary Fig. S3). These data align with the absence of AKT activation upon expression of either wild-type or mutant PIK3CA in UMSCC47 (Figure 3D) and suggest that PIK3CA/mTOR may be a better target in HPV(+) tumors.
Discussion
Identification of dysregulated signaling pathways have led to rational targeted therapies for many cancer types. Although HPV(+) OPSCC is associated with improved survival, the molecular and signaling underpinnings of improved response, as well as rational targeted therapies for these tumors have yet to emerge. To better understand alterations in signaling pathways in HPV(+) and HPV(−) cancers, RPPA that allows for high-throughput analysis of expression and activation of multiple proteins involved in cancer progression, was performed on a prospectively collected cohort of 42 OPSCC patients.
Because HPV oncoproteins inhibit p53 and Rb, dysregulation of proteins involved in cell cycle progression in the HPV(+) group was anticipated. Indeed, expression of cyclin D1 was diminished in HPV-associated tumors, while expression of cyclin E1 and cyclin B1 was increased (Figure 1A and Table 2). Retinoblastomais an essential regulator of progression from the G1 phase of the cell cycle into S phase, and loss of Rb function as a result of HPV E7 expression likely contributes to altered expression of cyclins and unscheduled S phase entry, which can result in DNA damage including double strand breaks at stalled replication forks (32). Replication stress combined with the presence of viral DNA trigger DNA damage response likely contributing to increased expression of DNA repair proteins observed in HPV(+) OPSCC (Figure 1A, Table 2). Indeed, in HPV(+) versus HPV(−) OPSCC, overexpression of many cell cycle and DNA damage response proteins were found, including PARP-1, BRCA2, PCNA and XRCC1. Consistent with our data, upregulation of XRCC1, as well as PCNA, mRNA has been reported in HPV(+) OPSCC (12), and elevated expression of PCNA was observed in warts associated with low risk HPV infection (33). Recently, several proteins involved in DNA replication and repair had been localized to sites of HPV replication (34). These data suggest a model, where during HPV infection, expression of HPV proteins leads to the induction of cellular DNA replication components that are needed for an efficient viral replication, finally resulting in the activation of DNA repair pathways. The non-homologous end-joining protein, KU80, was also more highly expressed in HPV(+) versus HPV(−) tumors (Figure 1A and Table 2). Previous studies did not find differences in KU80 expression between HPV(+) and HPV(−) tumors using immunohistochemistry, but expression of KU80 associated with decreased survivalin HPV(−) HNSCC (35). Methodological (RPPA vs. IHC) and distinctions in patient characteristics (only OPSCC vs. all head and neck subsites) may explain the discrepant results. Given that nearly one-third of differentially expressed and the majority of proteins that have increased expression in HPV(+) tumors participate in various aspects of DNA repair (Table 2), the role of DNA damage and repair in HPV-associated head and carcinogenesis deserves further detailed investigation, which is currently a focus of our laboratory.
PIK3CA is one of the most frequently altered genes in HNSCC primarily by amplification or activating mutations. HPV(+) OPSCC have an increased incidence of mutations (15, 16), suggesting that PI3K signaling may be critical for HPV-associated tumor formation or progression. To begin understanding potential roles of PIK3CA mutations in HPV(+) OPSCC, RPPA data and PIK3CA mutation status were evaluated. We discovered activating PIK3CA mutations in 8 of 47(17%)oropharyngeal tumors, all from HPV(+) patients. The reason for the increased incidence of PIK3CA mutations in HPV(+)OPSCC remains unknown; however, PI3K signaling has been shown to be activated in response to exposure of cultured cells to HPV viral like particles, and active PI3K signaling is required for efficient HPV infection of HeLa and HaCaT cells (36, 37). Together, these data suggest that constitutive activation of PI3K pathway may be beneficial for HPV infection that could ultimately result in increased PIK3CA mutations observed in HPV(+) head and neck cancer.
RPPA demonstrated distinct expression profiles of HPV(+) tumors with and without PIK3CA mutations. One of the most interesting findings was that, despite harboring activating PIK3CA mutations, indications of the AKT signaling pathway activation were lacking in that group (Figure 3). As opposed to the lack of AKT activation, downstream targets of mTOR were up-regulated in PIK3CA mutant HPV(+) OPSCC (phospho-S6 phospho-4EBP1, phospho-STAT3, phospho-IRS1; Figure 3). Previous studies noted decreased AKT activation in 51 breast cancer cell lines with helical-domain PIK3CA mutations (i.e., E542K, E545K) and demonstrated an AKT-independent PIK3CA signaling mediated through PDK1 and SGK3 effectors (38–40). Our study expands this finding by demonstrating the lack of AKT activation in clinical specimens and HPV(+) cells expressing PIK3CA mutant. Furthermore, we show that mTOR activation and signaling remain robust in these tumors, indicating intact upstream PIK3CA signaling.
Initial reports implicated activating mutations in PIK3CA with more aggressive tumorigenicity (18, 26, 41). Likewise, PIK3CA mutations have been associated with cancer recurrence (42), metastasis (43), and worse prognosis (26). However, recent studies in breast and lung cancer have associated mutant PIK3CA with improved overall survival (44–46). In our cohort of OPSCC patients, mutations in PIK3CA associated with a better overall survival amongst the HPV(+) tumors with no deaths observed among the mutant PIK3CA group (Supplementary Fig. S1). Survival was not significantly different between the groups, which may be a reflection of the good survival of HPV(+) tumors in general and the modest number of patients on this study.
Our data suggest that mutant PIK3CA in HPV(+) tumors preferentially activates the mTOR pathway, while not activating AKT. Activation of mTOR through the PI3K pathway typically involves AKT effects on TSC1/2. Since PIK3CA activating mutations were associated with increased markers of mTOR activity without increased phospho-AKT, these data suggest that an additional PI3K target may be mediating mTOR activation. For example, it has been shown that TSC2 is a target for PKC and MAP kinases (47). In addition, Prostaglandin F2α did not increase AKT phosphorylation, but activated mTOR/S6K and phosphorylated 4EBP1 (48). We also noticed differential activation of mTORC1 and mTORC2 by PIK3CA mutants in the presence of HPV. mTORC1 activates downstream pathways important for protein synthesis (S6K, eIF4E), but suppresses AKT signaling, whereas mTORC2 activates AKT (49). These data are consistent with PIK3CA mutants preferentially activating mTORC1 in the presence of HPV and are further supported by our finding that PKCa, which is phosphorylated and activated by mTORC2, has decreased phosphorylation in HPV(+) tumors with PIK3CA mutations (Table 3). In addition, we found that expression of the HPV E6 and E7 oncogenes in the presence of an endogenous PIK3CA mutant decreased phosphorylation of AKT (Figure 3E). Further mechanisms through which HPV oncoproteins inhibit AKT phosphorylation are needed to be explored.
Cell survival assays performed with BEZ-235, a dual PIK3/mTOR inhibitor, and MK-2206, a pan-AKT inhibitor, further demonstrated the AKT-independent effect of PIK3CA overexpression in HPV(+) OPSCC (Supplemental Fig. S3). Currently, both drugs are in Phase II clinical trials involving solid tumors with PIK3CA activating mutations.
In summary, our study highlights signaling differences between HPV(+) and HPV(−) OPSCC and identified dysregulated pathways: 1) cell cycle, 2) DNA repair, 3) apoptosis, 4) receptor, kinase, signaling and 5) PI3K/AKT/mTOR. In addition, these data suggest the need for more detailed and accurate preclinical testing in specific tumor subtypes, including HPV(+) OPSCC, since there is increasing acknowledgment that oncogenic mutations, such as PIK3CA, may behave differently in various cell types.
Supplementary Material
Translational Relevance.
Although response and cure of advanced stage HPV-associated HNSCC is high under current therapeutic regimens, patients treated with multi-modality therapy can be saddled with lifelong morbidity. Swallowing and speech dysfunction, accelerated carotid stenosis, xerostomia, neck muscle fibrosis, mandibular osteoradionecrosis, accelerated dental decay, and lymphedema are a few of described side effects that in culmination have been reported to affect up to 50% of survivors. Therefore, therapeutic de-escalation, potentially by targeting dysregulated pathways, for HPV(+) HNSCC is one of the major goals of the head and neck oncology community. Here, we identified molecular pathways differentially regulated in HPV(+) and HPV(−) head and neck cancer. We also found that PIK3CA activating mutations occurred more frequently in HPV(+) than in HPV(−) OPSCC, and that these mutations corresponded with mTOR, but not AKT activation. Given the challenges associated with clinical use of PI3K/AKT/mTOR inhibitors, improved understanding of mutant PIK3CA signaling in HPV-associated OPSCC may contribute to new therapeutic options.
Acknowledgments
This work was supported by funds from the Department of Surgery and the Division of Otolaryngology at Yale University, the Barry Baker Laboratory for Head and Neck Oncology and the Department of Otolaryngology at Vanderbilt University and from the Percy Memorial Award (AAO-HNSF). RPPA analysis was supported by MD Anderson Head and Neck SPORE P50CA097007 and CCSG grant P30 CA016672.
The authors thank Dr. John H. Lee for LXSN and 16 E6/E7 LXSN constructs and Tomas Carey for UMSCC47 cells.
Footnotes
The authors report no conflicts of interest
Conception and design: Yarbrough W.G., Issaeva N., Mills G.B.
Acquisition of data: Sewell A., Brown B., Biktasova A., Lu Y., Tyson D.R., Issaeva N.
Analysis and interpretation of data: Yarbrough W.G., Issaeva N., Tyson D.R., Sewell A., Mills G.B.
Writing, review, and/or revision of the manuscript: Yarbrough W.G., Issaeva N., Mills G.B., Sewell A.
Study supervision: Yarbrough W.G., Issaeva N.
References
- 1.Jemal A, Simard EP, Dorell C, Noone A-M, Markowitz LE, Kohler B, et al. Annual Report to the Nation on the Status of Cancer, 1975–2009, Featuring the Burden and Trends in Human Papillomavirus (HPV)–Associated Cancers and HPV Vaccination Coverage Levels. Journal of the National Cancer Institute. 2013 doi: 10.1093/jnci/djs491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chaturvedi AK, Goedert JJ. Human papillomavirus genotypes among women with HIV: implications for research and prevention. Aids. 2006;20:2381–3. doi: 10.1097/01.aids.0000253366.94072.b4. [DOI] [PubMed] [Google Scholar]
- 3.Mellin H, Friesland S, Lewensohn R, Dalianis T, Munck-Wikland E. Human papillomavirus (HPV) DNA in tonsillar cancer: clinical correlates, risk of relapse, and survival. International journal of cancer Journal international du cancer. 2000;89:300–4. [PubMed] [Google Scholar]
- 4.Chaturvedi AK, Engels EA, Pfeiffer RM, Hernandez BY, Xiao W, Kim E, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2011;29:4294–301. doi: 10.1200/JCO.2011.36.4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ang KK, Harris J, Wheeler R, Weber R, Rosenthal DI, Nguyen-Tan PF, et al. Human papillomavirus and survival of patients with oropharyngeal cancer. The New England journal of medicine. 2010;363:24–35. doi: 10.1056/NEJMoa0912217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Argiris A, Li Y, Forastiere A. Prognostic factors and long-term survivorship in patients with recurrent or metastatic carcinoma of the head and neck. Cancer. 2004;101:2222–9. doi: 10.1002/cncr.20640. [DOI] [PubMed] [Google Scholar]
- 7.Schlecht NF, Burk RD, Adrien L, Dunne A, Kawachi N, Sarta C, et al. Gene expression profiles in HPV-infected head and neck cancer. The Journal of pathology. 2007;213:283–93. doi: 10.1002/path.2227. [DOI] [PubMed] [Google Scholar]
- 8.Kaczkowski B, Morevati M, Rossing M, Cilius F, Norrild B. A Decade of Global mRNA and miRNA Profiling of HPV-Positive Cell Lines and Clinical Specimens. The open virology journal. 2012;6:216–31. doi: 10.2174/1874357901206010216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smeets SJ, Braakhuis BJ, Abbas S, Snijders PJ, Ylstra B, van de Wiel MA, et al. Genome-wide DNA copy number alterations in head and neck squamous cell carcinomas with or without oncogene-expressing human papillomavirus. Oncogene. 2006;25:2558–64. doi: 10.1038/sj.onc.1209275. [DOI] [PubMed] [Google Scholar]
- 10.Wilting SM, Smeets SJ, Snijders PJ, van Wieringen WN, van de Wiel MA, Meijer GA, et al. Genomic profiling identifies common HPV-associated chromosomal alterations in squamous cell carcinomas of cervix and head and neck. BMC medical genomics. 2009;2:32. doi: 10.1186/1755-8794-2-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Slebos RJ, Jehmlich N, Brown B, Yin Z, Chung CH, Yarbrough WG, et al. Proteomic analysis of oropharyngeal carcinomas reveals novel HPV-associated biological pathways. International journal of cancer Journal international du cancer. 2013;132:568–79. doi: 10.1002/ijc.27699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lohavanichbutr P, Houck J, Fan W, Yueh B, Mendez E, Futran N, et al. Genomewide gene expression profiles of HPV-positive and HPV-negative oropharyngeal cancer: potential implications for treatment choices. Archives of otolaryngology--head & neck surgery. 2009;135:180–8. doi: 10.1001/archoto.2008.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–60. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agrawal N, Frederick MJ, Pickering CR, Bettegowda C, Chang K, Li RJ, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science. 2011;333:1154–7. doi: 10.1126/science.1206923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lui VW, Hedberg ML, Li H, Vangara BS, Pendleton K, Zeng Y, et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer discovery. 2013;3:761–9. doi: 10.1158/2159-8290.CD-13-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yarbrough WG, Whigham A, Brown B, Roach M, Slebos R. Phosphoinositide kinase-3 status associated with presence or absence of human papillomavirus in head and neck squamous cell carcinomas. International journal of radiation oncology, biology, physics. 2007;69:S98–101. doi: 10.1016/j.ijrobp.2007.04.082. [DOI] [PubMed] [Google Scholar]
- 17.Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, et al. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Molecular cancer therapeutics. 2006;5:2512–21. doi: 10.1158/1535-7163.MCT-06-0334. [DOI] [PubMed] [Google Scholar]
- 18.Samuels Y, Velculescu VE. Oncogenic mutations of PIK3CA in human cancers. Cell cycle. 2004;3:1221–4. doi: 10.4161/cc.3.10.1164. [DOI] [PubMed] [Google Scholar]
- 19.Boyer SN, Wazer DE, Band V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer research. 1996;56:4620–4. [PubMed] [Google Scholar]
- 20.Troy JD, Weissfeld JL, Youk AO, Thomas S, Wang L, Grandis JR. Expression of EGFR, VEGF, and NOTCH1 Suggest Differences in Tumor Angiogenesis in HPV-Positive and HPV-Negative Head and Neck Squamous Cell Carcinoma. Head and neck pathology. 2013 doi: 10.1007/s12105-013-0447-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Slebos RJ, Yi Y, Ely K, Carter J, Evjen A, Zhang X, et al. Gene expression differences associated with human papillomavirus status in head and neck squamous cell carcinoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12:701–9. doi: 10.1158/1078-0432.CCR-05-2017. [DOI] [PubMed] [Google Scholar]
- 22.Tao R, Gong J, Luo X, Zang M, Guo W, Wen R, et al. AMPK exerts dual regulatory effects on the PI3K pathway. Journal of molecular signaling. 2010;5:1. doi: 10.1186/1750-2187-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Panetta D, Biedi C, Repetto S, Cordera R, Maggi D. IGF-I regulates caveolin 1 and IRS1 interaction in caveolae. Biochemical and biophysical research communications. 2004;316:240–3. doi: 10.1016/j.bbrc.2004.02.037. [DOI] [PubMed] [Google Scholar]
- 24.Chen J, Capozza F, Wu A, Deangelis T, Sun H, Lisanti M, et al. Regulation of insulin receptor substrate-1 expression levels by caveolin-1. Journal of cellular physiology. 2008;217:281–9. doi: 10.1002/jcp.21498. [DOI] [PubMed] [Google Scholar]
- 25.Hutchinson L. Targeted therapies: Activated PI3K/AKT confers resistance to trastuzumab but not lapatinib. Nature reviews Clinical oncology. 2010;7:424. doi: 10.1038/nrclinonc.2010.113. [DOI] [PubMed] [Google Scholar]
- 26.Ogino S, Nosho K, Kirkner GJ, Shima K, Irahara N, Kure S, et al. PIK3CA mutation is associated with poor prognosis among patients with curatively resected colon cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009;27:1477–84. doi: 10.1200/JCO.2008.18.6544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang L, Huang J, Yang N, Greshock J, Liang S, Hasegawa K, et al. Integrative genomic analysis of phosphatidylinositol 3′-kinase family identifies PIK3R3 as a potential therapeutic target in epithelial ovarian cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13:5314–21. doi: 10.1158/1078-0432.CCR-06-2660. [DOI] [PubMed] [Google Scholar]
- 28.Freudlsperger C, Burnett JR, Friedman JA, Kannabiran VR, Chen Z, Van Waes C. EGFR-PI3K AKT-mTOR signaling in head and neck squamous cell carcinomas: attractive targets for molecular-oriented therapy. Expert opinion on therapeutic targets. 2011;15:63–74. doi: 10.1517/14728222.2011.541440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jameson MJ, Beckler AD, Taniguchi LE, Allak A, Vanwagner LB, Lee NG, et al. Activation of the insulin-like growth factor-1 receptor induces resistance to epidermal growth factor receptor antagonism in head and neck squamous carcinoma cells. Molecular cancer therapeutics. 2011;10:2124–34. doi: 10.1158/1535-7163.MCT-11-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nature reviews Cancer. 2011;11:9–22. doi: 10.1038/nrc2982. [DOI] [PubMed] [Google Scholar]
- 31.Kim JH, Yoon MS, Chen J. Signal transducer and activator of transcription 3 (STAT3) mediates amino acid inhibition of insulin signaling through serine 727 phosphorylation. The Journal of biological chemistry. 2009;284:35425–32. doi: 10.1074/jbc.M109.051516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–7. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 33.Lu S, Syrjanen K, Havu VK, Syrjanen S. Expression of PCNA is associated with the presence of HPV DNA in skin warts. Archives of dermatological research. 1996;289:35–9. doi: 10.1007/s004030050149. [DOI] [PubMed] [Google Scholar]
- 34.Gillespie KA, Mehta KP, Laimins LA, Moody CA. Human papillomaviruses recruit cellular DNA repair and homologous recombination factors to viral replication centers. Journal of virology. 2012;86:9520–6. doi: 10.1128/JVI.00247-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moeller BJ, Yordy JS, Williams MD, Giri U, Raju U, Molkentine DP, et al. DNA repair biomarker profiling of head and neck cancer: Ku80 expression predicts locoregional failure and death following radiotherapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17:2035–43. doi: 10.1158/1078-0432.CCR-10-2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raff AB, Woodham AW, Raff LM, Skeate JG, Yan L, Da Silva DM, et al. The evolving field of human papillomavirus receptor research: a review of binding and entry. Journal of virology. 2013;87:6062–72. doi: 10.1128/JVI.00330-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Surviladze Z, Sterk RT, DeHaro SA, Ozbun MA. Cellular entry of human papillomavirus type 16 involves activation of the phosphatidylinositol 3-kinase/Akt/mTOR pathway and inhibition of autophagy. Journal of virology. 2013;87:2508–17. doi: 10.1128/JVI.02319-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vasudevan KM, Barbie DA, Davies MA, Rabinovsky R, McNear CJ, Kim JJ, et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer cell. 2009;16:21–32. doi: 10.1016/j.ccr.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer research. 2008;68:6084–91. doi: 10.1158/0008-5472.CAN-07-6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Long X, Muller F, Avruch J. TOR action in mammalian cells and in Caenorhabditis elegans. Current topics in microbiology and immunology. 2004;279:115–38. doi: 10.1007/978-3-642-18930-2_8. [DOI] [PubMed] [Google Scholar]
- 41.Samuels Y, Diaz LA, Jr, Schmidt-Kittler O, Cummins JM, Delong L, Cheong I, et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer cell. 2005;7:561–73. doi: 10.1016/j.ccr.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 42.He Y, Van’t Veer LJ, Mikolajewska-Hanclich I, van Velthuysen ML, Zeestraten EC, Nagtegaal ID, et al. PIK3CA mutations predict local recurrences in rectal cancer patients. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:6956–62. doi: 10.1158/1078-0432.CCR-09-1165. [DOI] [PubMed] [Google Scholar]
- 43.Akagi I, Miyashita M, Makino H, Nomura T, Hagiwara N, Takahashi K, et al. Overexpression of PIK3CA is associated with lymph node metastasis in esophageal squamous cell carcinoma. International journal of oncology. 2009;34:767–75. doi: 10.3892/ijo_00000202. [DOI] [PubMed] [Google Scholar]
- 44.Kalinsky K, Jacks LM, Heguy A, Patil S, Drobnjak M, Bhanot UK, et al. PIK3CA mutation associates with improved outcome in breast cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:5049–59. doi: 10.1158/1078-0432.CCR-09-0632. [DOI] [PubMed] [Google Scholar]
- 45.Di Cosimo S, Baselga J. Phosphoinositide 3-kinase mutations in breast cancer: a “good” activating mutation? Clinical cancer research : an official journal of the American Association for Cancer Research. 2009;15:5017–9. doi: 10.1158/1078-0432.CCR-09-1173. [DOI] [PubMed] [Google Scholar]
- 46.Loi S, Haibe-Kains B, Majjaj S, Lallemand F, Durbecq V, Larsimont D, et al. PIK3CA mutations associated with gene signature of low mTORC1 signaling and better outcomes in estrogen receptor-positive breast cancer. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:10208–13. doi: 10.1073/pnas.0907011107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tee AR, Manning BD, Roux PP, Cantley LC, Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Current biology : CB. 2003;13:1259–68. doi: 10.1016/s0960-9822(03)00506-2. [DOI] [PubMed] [Google Scholar]
- 48.Arvisais EW, Romanelli A, Hou X, Davis JS. AKT-independent phosphorylation of TSC2 and activation of mTOR and ribosomal protein S6 kinase signaling by prostaglandin F2alpha. The Journal of biological chemistry. 2006;281:26904–13. doi: 10.1074/jbc.M605371200. [DOI] [PubMed] [Google Scholar]
- 49.Sabatini DM. mTOR and cancer: insights into a complex relationship. Nature reviews Cancer. 2006;6:729–34. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 50.Wright A, Wait R, Begum S, Crossett B, Nagy J, Brown K, et al. Proteomic analysis of cell surface proteins from Clostridium difficile. Proteomics. 2005;5:2443–52. doi: 10.1002/pmic.200401179. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.