

Abstract
Hepatitis C virus afflicts approximately 180 million people worldwide, and the development of direct acting antivirals may offer substantial benefit compared to the current standard of care. Accordingly, prodrugs of 2′-deoxy-2′-fluoro-2′-C-methylguanosine monophosphate analogues were prepared and evaluated for their anti-HCV efficacy and tolerability. These prodrugs demonstrated >1000 fold greater potency than the parent nucleoside in a cell-based replicon assay as a result of higher intracellular triphosphate levels. Further optimization led to the discovery of the clinical candidate PSI-353661, which has demonstrated strong in vitro inhibition against HCV without cytotoxicity and equipotent activity against both the wild type and the known S282T nucleoside/tide resistant replicon. PSI-353661 is currently in preclinical development for the treatment of HCV.
Keywords: PSI-353661, hepatitis C virus, NS5B polymerase, antivirals, nucleoside, prodrug, phosphoramidate, triphosphate
Hepatitis C virus is a 9.6 kb positive-sense single-stranded RNA virus of the Flaviviridae family. Nearly 1.6% of the U.S. population and an estimated 180 million people worldwide are infected with HCV.1 Approximately 80% of infected individuals become chronically infected. Untreated HCV infections can progress to cirrhosis, hepatocellular carcinoma, and liver failure, a primary cause for liver transplantation.2 The current standard of care (SOC) for chronic HCV infection, PEG-IFNα and ribavirin, provides limited sustained virologic response (SVR) rates (e.g., ≤50% for genotype 1 patients) and can produce various undesirable side effects ranging from flu-like symptoms to severe adverse effects, including anemia, cardiovascular events, and psychiatric problems such as suicidal ideation.3 Consequently, there is a clear unmet medical need to discover new direct-acting antivirals (DAA) to effectively and safely treat chronic HCV infection.4
Three first generation nucleoside prodrugs (NM283, R1626, and RG7128) have been reported to show anti-HCV activity as direct-acting antiviral agents in clinical trials.5,6 Among them, only RG7128 is currently under development (phase IIb). RG7128 is a 3′,5′-diisobutyrate ester prodrug of β-d-2′-deoxy-2′-α-fluoro-2′-β-C-methylcytidine (PSI-6130) and has already established proof-of-concept in the clinic.7,8 In a 28 days combination study with SOC, RG7128 demonstrated efficacy in genotype 1, 2, and 3 patients and was the first direct-acting antiviral to show broad genotype coverage in the clinic.9
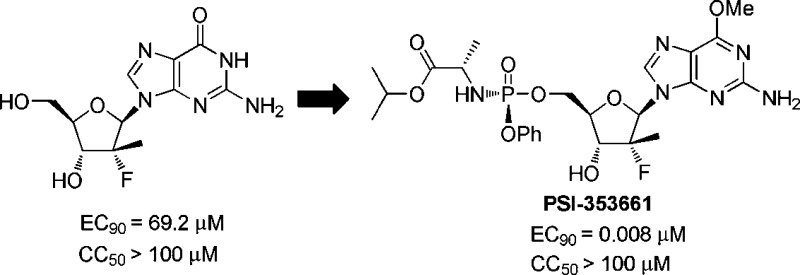
As part of our continuing efforts to discover novel anti-HCV agents, we were interested in finding second generation agents with improved potency, better resistance profile, enhanced pharmacokinetic properties to support QD dosing, and the potential for generating high concentrations of the active triphosphate in the liver. In addition to the cytidine analogue RG7128, we have investigated derivatives of 2-deoxy-2-α-fluoro-2-β-C-methylribofuranose with other nucleic acid bases.10−13 Among them, the guanosine analogue 1 was shown to be only weakly active (EC90 = 69.2 μM) in a HCV replicon assay, but its triphosphate 2 was found to be a potent inhibitor of the HCV NS5B polymerase (IC50 = 5.94 μM) (Figure 1).
Figure 1.

β-d-2′-Deoxy-2′-α-fluoro-2′-β-C-methylguanosine (1) and its anabolite triphosphate 2.
Nucleoside analogues need to be phosphorylated to their corresponding triphosphate by host cellular kinases before they can bind to the HCV NS5B polymerase and inhibit RNA replication. However, many nucleoside analogues are poor substrates for nucleoside kinases that are responsible for their phosphorylation to their active triphosphate derivative.14 The tri-/di-/monophosphate species cannot be considered as possible drug candidates because of their instability and poor cellular permeability.15 Phosphoramidate prodrugs have been shown to enhance nucleoside potency in cell culture, presumably by increasing the intracellular concentrations of the nucleoside monophosphate, thus bypassing a potentially nonproductive first phosphorylation step.16−18 We therefore decided to investigate phosphoramidate derivatives of 2′-deoxy-2′-α-fluoro-2′-β-C-methylguanosine monophosphate.
The study of phosphoramidate prodrugs began with the investigation of the anti-HCV structure activity relationship at the 6-position of the purine base, keeping the substitution on the phosphoramidate moiety constant. A series of 6-(un)modified guanosine analogues 6a−k was synthesized starting from the previously reported19 ribonolactol 3 by preparing the 6-chloro nucleoside 5 as a key intermediate (Scheme 1).20 The phosphoramidate moiety was introduced onto each nucleoside by treatment with a phosphorochloridate reagent in the presence of N-methylimidazole.21 Each purine phosphoramidate derivative was prepared as a mixture of diastereomers at phosphorus.
Scheme 1. General Synthetic Pathway for the Phosphoramidates of Guanosine Analogues.

Reagents and conditions: (a) 4, DEAD, PPh3, THF, rt, 16 h. (b) for 6a, 2-mercaptoethanol, NaOMe, MeOH, Δ; for 6b to 6g, ROH, NaH or K2CO3, DME, Δ; for 6h−k, NHR′R′′, MeOH or EtOH, Δ. (c) 7, NMI, THF, 0 °C to rt.
Anti-HCV activity in the clone A replicon whole cell assay of the ethyl ester series 8−18 is summarized in Table 1.22−24 The 6-unsubstituted guanine phosphoramidate 8 showed low micromolar activity: approximately a 25-fold increase in potency over nucleoside 1. On the other hand, a number of the phosphoramidates having 6-alkoxy, -alkylsulfane, and -alkylamine substituents (double prodrug) demonstrated more dramatic improvements in potency against HCV. Especially, 6-methoxyguanosine analogue 9 and 6-amino substituted analogues 15 and 16 exhibited single digit nanomolar EC90 values, thus offering >5000-fold increased potency compared to the parent nucleoside 1.25
Table 1. HCV Clone A Replicon Data.
| compd | R1 | R2 | EC90 (μM) |
|---|---|---|---|
| 8 | −OH | Et | 2.38 |
| 9 | −OMe | Et | 0.0074 |
| 10 | −OEt | Et | 0.010 |
| 11 | −OPr | Et | 0.013 |
| 12 | −O(CH2)2OMe | Et | 0.020 |
| 13 | −O(CH2)2F | Et | 0.027 |
| 14 | −SEt | Et | 0.047 |
| 15 | −N(CH2)3 | Et | 0.0070 |
| 16 | −NH(cPr) | Et | 0.0098 |
| 17 | −NH(cBu) | Et | 0.024 |
| 18 | −NH(cPentyl) | Et | 0.026 |
Further SAR assessment focused on preferred C-6 substitutions on the base and small alkyl esters at the terminal carboxylate group of the phosphoramidate moiety (Table 2). A survey of substitutions at both the C-6 position of the base and the terminal ester showed that substituents that increased the overall lipophilicity of the molecule translated into increased cytotoxicity (data not shown). Consequently, we chose to minimize the overall lipophilicity and focus our efforts on the introduction of relatively small substituents both at C-6 of the base and at the terminal ester group. For the 6-unmodified guanosine series (19, 20, and 21), improved anti-HCV activity was observed when the terminal carboxylate ester substituent was either iPr, cyclopentyl, or cyclohexyl, affording EC90 values of ≤0.75 μM. None of the 6-OH (natural guanosine) analogues showed cytotoxicity up to 100 μM against an expanded cell panel. Most of the 6-alkoxy analogues demonstrated a >1000-fold increase in potency compared to the guanosine analogue 1. Among the 6-alkoxy derivatives, isopropyl ester 23 and methyl ester 24 showed no significant cytotoxicity. Alkylamine substitution at the 6-position of the base also produced derivatives with potent anti-HCV activity. Of the alkylamine derivatives, the 6-N-azetidine 28 gave the best cytotoxicity profile relative to the other phosphoramidates in the alkylamine series. Several analogues (9, 19, 20, 23, 24, and 28) were tested further against the known nucleoside resistant mutant, S282T, which has demonstrated reduced sensitivity to the 2′-methyl containing nucleoside analogues RG7128 and NM283.26 It is noteworthy that all of these purine nucleosides were shown to be equipotent against both the wild type and the S282T mutant replicon (Table 2). Based on anti-HCV replicon potency, cytotoxicity profile, and structural diversity, a set of four guanosine phosphoramidate analogues, 20, 23, 24, and 28, were selected for further study.
Table 2. Anti-HCV Activity and Cytotoxicity.
| HCV inhibition EC90 (μM) |
cytotoxicity CC50 (μM) |
|||||||
|---|---|---|---|---|---|---|---|---|
| compd | R1 | R2 | WT | S282T | Huh7 | BxBC3 | HepG2 | CEM |
| 8 | −OH | Et | 2.38 | >100 | >100 | >100 | >100 | |
| 19 | −OH | iPr | 0.75 | 0.61 | >100 | >100 | >100 | >100 |
| 20 | −OH | cPentyl | 0.18 | 0.12 | >100 | >100 | >100 | >100 |
| 21 | −OH | cHexyl | 0.446 | >100 | >100 | >100 | >100 | |
| 22 | −OMe | Me | 0.0081 | >100 | 65 | 71 | 20 | |
| 9 | −OMe | Et | 0.0074 | 0.011 | >100 | 67 | 62 | 55 |
| 23 | −OMe | iPr | 0.020 | 0.024 | >100 | >100 | >100 | >100 |
| 24 | −OEt | Me | 0.016 | 0.016 | >100 | >100 | >100 | 74 |
| 10 | −OEt | Et | 0.010 | 47 | 68 | 61 | 37 | |
| 25 | −OEt | iPr | 0.020 | 59 | >100 | >100 | 93 | |
| 26 | −OPr | Me | 0.0146 | 39 | 39 | 45 | 17 | |
| 27 | −OPr | iPr | 0.033 | 49 | >100 | >100 | 44 | |
| 28 | −N(CH2)3 | Me | 0.090 | 0.04 | >100 | >100 | >100 | 75 |
| 15 | −N(CH2)3 | Et | 0.0070 | 84 | 92 | 75 | 39 | |
| 29 | −N(CH2)3 | iPr | 0.019 | 53 | 74 | 100 | 74 | |
| 30 | −NH(cPr) | Me | 0.0082 | >100 | >100 | >100 | 34 | |
| 16 | −NH(cPr) | Et | 0.0098 | >100 | >100 | >100 | 45 | |
| 31 | −NH(cPr) | iPr | 0.0088 | 96 | 98 | >100 | >100 | |
Since some compounds previously reported to inhibit the HCV replicon also inhibited cell growth27 and exhibited mitochondrial toxicity,28 compounds 20, 23, 24, and 28 were evaluated for effects on cell growth and for mitochondrial toxicity. When evaluated for their cytostatic potential, compounds 20, 23, and 28 showed no significant effect on cell doubling time up to 100 μM; however, the 6-ethoxy ether analogue 24 was shown to retard cell growth by 5 h at 50 μM. When mitochondrial toxicity (CC90) was evaluated by incubating CEM and HepG2 cells with compounds 20, 23, and 28 for 14 days and then measuring the levels of mitochondrial COXII DNA (mtDNA) and rDNA (rDNA) using real time PCR, all three compounds showed no measurable mitochondrial toxicity up to 100 μM in both cell lines.
To assess the ability of prodrugs 20, 23, and 28 to survive exposure in the gastrointestinal tract and preferentially deliver the nucleotide monophosphate to the liver, stability in simulated gastrointestinal fluid (SGF), simulated intestinal fluid (SIF), human plasma, and human liver S9 fraction, a surrogate in vitro model for liver stability, was studied. As shown in Table 3, the derivatives with bulky ester groups, 20 and 23, provided better stability in human plasma as well as in SGF and SIF when compared to the methyl ester analogue 28. Each compound exhibited short half-lives in liver S9 fraction, indicating that each phosphoramidate had the potential for rapid conversion to the desired nucleotides in the liver.
Table 3. Stability of Key Compounds 20, 23, and 28.
| stability, t1/2 (h) |
||||
|---|---|---|---|---|
| compd | SGF | SIF | human plasma | S9 |
| 20 | 15 | >20 | >24 | 0.2 |
| 23 | 14 | >20 | >24 | 0.2 |
| 28 | >20 | >20 | 11.6 | 0.4 |
Since triphosphate levels are correlated to the in vivo potency of nucleos(t)ide analogues,29in vitro triphosphate production of the remaining compounds 20, 23, and 28 in primary human hepatocytes was assessed and compared to that produced in the replicon clone A cells (Table 4). In vitro analysis of triphosphate 2 production was accomplished by incubating each compound and then extracting triphosphate 2 from the cells and quantifying by HPLC analysis. Of the three compounds studied, compound 23 produced the highest level of triphosphate in primary human hepatocytes. Triphosphate levels for each of the compounds in clone A cells correlated well with anti-HCV replicon potency. Therefore, one might anticipate that triphosphate levels observed in primary human hepatocytes in vitro provide an indication of anti-HCV potency in human hepatocytes in vivo.
Table 4. Cellular Triphosphate Levels of Key Compounds 20, 23, and 28.
| cellular TP levelsa (mM) |
||
|---|---|---|
| compd | clone A (at 48 h) | primary human hepatocytes (at 24 h) |
| 20 | 0.06 | 0.04 |
| 23 | 0.12 | 0.12 |
| 28 | 0.16 | 0.05 |
Incubated at 100 μM compound concentration.
Since 23 was developed as a liver targeted nucleotide prodrug, the relative liver to plasma exposure and the production of active triphosphate 2 in the liver was evaluated when 23 was administered in vivo. To accomplish this, tritiated phosphoramidate nucleotide 33 was prepared by first bromination of 23 at C-8 of the purine base followed by reductive tritiation (Scheme 2). The tritiated derivative 33 was administered orally to rats (60 μCi total dose in peanut oil), and plasma samples were collected at 1 and 6 h after dosing. In addition, after administration of tritiated derivative 33, livers were removed at 1 and 6 h postdosing and levels of triphosphate and key metabolites were determined from the liver extracts. Parent prodrug and metabolites were measured in plasma and liver extracts by HPLC using radiodetection. As has been described for other nucleotide phosphoramidates,16 metabolism of phosphoramidate 23 proceeds first to the diacid intermediate 34 and then to the monophosphate 35, to the diphosphate 36, and finally to the active triphosphate 2 (Scheme 3). With radiolabeled 33, we were able to measure the levels of each of these productive metabolites in both the liver and plasma (Table 5). In plasma after 1 h, each of the diastereomers of 23 is evident, as is the diacid 34 and the guanosine nucleoside 1. Nucleoside 1 presumably arises from decomposition of the monophosphate. At the 6 h time point, the major metabolite in plasma is the nucleoside 1, and very little of the parent prodrugs or intermediate metabolite 34 is present. In the liver at 1 h, the mono-, di-, and triphosphates are observed in addition to the intermediate metabolite 34 and the nucleoside 1. At 6 h postdose, liver extracts still show the presence of the mono-, di-, and triphosphates and intermediate metabolite 34 in addition to large amounts of 1. To determine the liver to plasma ratio, the total metabolites observed in the liver were compared to the total metabolites observed in plasma (Table 5). The liver to plasma ratio was determined to be 3.5/1 and 4.8/1 at 1 and 6 h postdose, respectively, thus supporting the liver targeting properties of 23. In addition, the presence of diacid 34, mono-, di-, and triphosphate in the liver indicates that 23 is able to reach the liver and achieve conversion to the active triphosphate as designed.
Scheme 2. Synthesis of Radiolabeled Prodrug.

Reagents and conditions: (a) NBS, dioxane, rt, 5 h. (b) 3H2, 10% Pd/C, Et3N, MeOH, rt, 1 h.
Scheme 3. Activation Pathway for Prodrug 23.

Table 5. Radio Counts of Tritiated 23 (33) and Metabolites in Rat Plasma and Liver Samples.
| peak areab (cpm) |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| rat samplesa | sample (volume or weight) | 1 | 34 | 35 | 36 | 2 | 23a | 23b | total (cpm/mL or cpm/g) | |
| plasma | 1 h | 1.33 mL | 1376 | 920 | ND | ND | ND | 1160 | 1112 | 3426 |
| 6 h | 1.00 mL | 3848 | 288 | ND | ND | ND | 272 | 368 | 4776 | |
| liver | 1 h | 0.75 g | 4224 | 1648 | 1828 | 988 | 384 | ND | ND | 12096 |
| 6 h | 0.70 g | 11512 | 572 | 1920 | 1620 | 460 | ND | ND | 22977 | |
Dose: 60 μCi total dose in 2.9 mL of peanut oil.
ND = not detected.
As a result of its strong in vitro potency against HCV, good metabolic stability, very clean in vitro toxicity profile, and high liver to plasma ratio in a rat in vivo study, the 6-methoxyguanosine analogue 23 was chosen for further study. Phosphoramidate 23 is a mixture of diastereomers (23a:23b, ∼1:1.8 ratio) at the phosphorus center.30 The diastereomers of 23 were separated by simulated moving bed chromatography to provide the two pure isomers 23a and 23b. The anti-HCV activity of each diastereomer against the wild type clone A replicon and the S282T mutant replicon was determined, and diastereomer 23b was shown to be 3.4-fold more potent than diastereomer 23a and again retained activity in the mutant replicon (Table 6). The more potent diastereomer 23b also produced about 3-fold higher concentrations of triphosphate in both clone A and primary human hepatocytes than did 23a, consistent with its in vitro potency in clone A cells.
Table 6. Comparison of the Diastereomers 23a and 23b.
| HCV inhibition EC90 (μM) |
cellular TP levelsa (mM) |
|||
|---|---|---|---|---|
| compd | WT | S282T | clone A (at 48 h) | primary human hepatocytes (at 24 h) |
| 23a | 0.027 | 0.038 | 0.07 | 0.14 |
| 23b | 0.008 | 0.011 | 0.17 | 0.44 |
Incubated at 25 μM compound concentration for clone A cells and 100 μM compound concentration for primary human hepatocytes, respectively.
Pure 23b was crystallized in dichloromethane and hexane solution, and a single crystal X-ray structure of 23b was obtained unambiguously, confirming the configuration of the phosphorus center as Sp (see Supporting Information). Subsequently, it was demonstrated that diastereomer 23b could be selectively crystallized directly from the diastereomeric mixture without prior chromatographic separation. Based on its superior overall potency and safety profile, compound 23b (PSI-353661) was selected as a clinical development candidate.
In conclusion, a series of β-d-2′-deoxy-2′-α-fluoro-2′-β-C-methylguanosine phosphoramidate analogues were prepared and their SAR as inhibitors of HCV replication was studied. Most of the prodrugs showed superior anti-HCV potency relative to the parent guanosine analogue 1, often by more than 1000-fold. After a comprehensive evaluation that included cytotoxicity, resistance profile, cytostatic activity, metabolic stability, and cellular triphosphate production, the single diastereomer 23b (PSI-353661) was chosen as a preclinical development candidate for the treatment of HCV infection. PSI-353661 exhibited strong inhibition of HCV in the cell-based replicon assay, equipotent activity against both the wild type and S282T resistant replicons, and ability to produce high concentrations of the active triphosphate in primary human hepatocytes.
Acknowledgments
We thank Dr. Patrick Carroll, University of Pennsylvania for X-ray analysis, and WuXi AppTec and Moravek Biochemicals for their contribution to the work described in this manuscript.
Supporting Information Available
Experimental procedures and analytical data for all new compounds. X-ray structure and cif data for 23b. HPLC radiochromatograms and triphosphate measurement of active prodrugs. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Lavanchy D. The global burden of hepatitis C. Liver Int. 2009, 29S174–81. [DOI] [PubMed] [Google Scholar]
- Zeuzem S.; Berg T.; Moeller B.; Hinrichsen H.; Mauss S.; Wedemeyer H.; Sarrazin C.; Hueppe D.; Zehnter E.; Manns M. P. Expert opinion on the treatment of patients with chronic hepatitis C. J. Viral Hepatitis 2009, 16, 75–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neukam K.; Macias J.; Mira J. A.; Pineda J. A. A review of current anti-HCV treatment regimens and possible future strategies. Expert Opin. Pharmacother. 2009, 10, 417–433. [DOI] [PubMed] [Google Scholar]
- Schinazi R. F.; Bassit L.; Gavegnano C. HCV drug discovery aimed at viral eradication. J. Viral Hepatitis 2010, 17, 77–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman P. A.; Lam A. M.; Murakami E. Nucleoside analogue inhibitors of hepatitis C viral replication: recent advances, challenges and trends. Future Med. Chem. 2009, 1, 1429–1452. [DOI] [PubMed] [Google Scholar]
- Carroll S. S.; La Femina R. L.. Nucleoside analogue inhibitors of hepatitis C viral replication. In Antiviral Research; La Femina R. L., Ed.; American Society for Microbiology: Washington, DC, 2009; pp153−166. [Google Scholar]
- Stuyver L. J.; McBrayer T. R.; Tharnish P. M.; Clark J. L.; Hollecher L.; Lostia S.; Nachman T.; Grier J.; Bennett M. A.; Xie M.-Y.; Schinazi R. F.; Morrey J. D.; Julander J. L.; Furman P. A.; Otto M. J. Inhibition of hepatitis C replicon RNA synthesis by β-d-2′-deoxy-2′-fluoro-2′-C-methylcytidine: a specific inhibitor of hepatitis C virus replication. Antiviral Chem. Chemother. 2006, 17, 79–87. [DOI] [PubMed] [Google Scholar]
- Murakami E.; Niu C.; Bao H.; Steuer H. M.; Whitaker T.; Nachman T.; Sofia M. A.; Wang P.; Otto M. J.; Furman P. A. The mechanism of action of β-d-2′-deoxy-2′-fluoro-2′-C-methylcytidine involves a second metabolic pathway leading to β-d-2′-deoxy-2′-fluoro-2′-C-methyluridine 5′-triphosphate, a potent inhibitor of the hepatitis C virus RNA-dependent RNA polymerase. Antimicrob. Agents Chemother. 2008, 52, 458–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Pogam S.; Kang H.; Seshaadri A.; Kosaka A.; Hu S.; Ewing A.; Yan J.-M.; Beard A.; Symons J.; Cammack N.; Nájera I.. No evidence of R7128 drug resistance after up to 4 weeks treatment of GT 1, 2 and 3 hepatitis C virus infected individuals. 44th Annual Meeting of the European Association for the Study of the Liver; Copenhagen, Denmark, April, 2009; Abstr. No. 839. [Google Scholar]
- Clark J. L.; Hollecker L.; Mason J. C.; Stuyver L. J.; Tharnish P. M.; Lostia S.; McBrayer T. R.; Schinazi R. F.; Watanabe K. A.; Otto M. J.; Furman P. A.; Stec W. J.; Patterson S. E.; Pankiewicz K. W. Design, synthesis, and antiviral activity of 2′-deoxy-2′-fluoro-2′-C-methyl-cytidine, a potent inhibitor of hepatitis C virus replication. J. Med. Chem. 2005, 48, 5504–5508. [DOI] [PubMed] [Google Scholar]
- Clark J. L.; Mason J. C.; Hollecker L.; Stuyver L. J.; Tharnish P. M.; McBrayer T. R.; Otto M. J.; Furman P. A.; Schinazi R. F.; Watanabe K. A. Synthesis and antiviral activity of 2′-deoxy-2′-fluoro-2′-C-methyl purine nucleosides as inhibitors of hepatitis C virus RNA replication. Bioorg. Med. Chem. Lett. 2006, 16, 1712–1715. [DOI] [PubMed] [Google Scholar]
- Wang P.; Sofia M. J.; Chun B.-K.; Du J.; Rachakonda S.; Steuer H. M.; Murakami E.; Bao H.; Nagarathnam D.; Otto M. J.; Furman P. A.. d-2′-Deoxy-2′-fluoro-2′-C-methyl-ribofuranosyl nucleosides: Effect of base modifications on antihepatitis C virus activity. 238th ACS National Meeting, Washington, DC, August, 2009; MEDI 100. [Google Scholar]
- Sofia M. J.; Bao D.; Chang W.; Du J.; Nagarathnam D.; Rachakonda S.; Reddy P. G.; Ross B. S.; Wang P.; Zhang H.-R.; Bansal S.; Espiritu C.; Keilman M.; Lam A. M.; Micolochick Steuer H. M.; Niu C.; Otto M. J.; Furman P. A. Discovery of a β-d-2′-deoxy-2′-α-fluoro-2′-β-C-methyluridine nucleotide prodrug (PSI-7977) for the treatment of hepatitis C virus. J. Med. Chem. 2010, 53, 7202–7218. [DOI] [PubMed] [Google Scholar]
- Oeberg Bo. Rational design of polymerase inhibitors as antiviral drugs. Antiviral Res. 2006, 71, 90–95. [DOI] [PubMed] [Google Scholar]
- Hecker S. J.; Erion M. D. Prodrugs of phosphates and phosphonates. J. Med. Chem. 2008, 51, 2328–2345. [DOI] [PubMed] [Google Scholar]
- Mehellou Y.; Balzarini J.; McGuigan C. Aryloxy phosphoramidate triesters: a technology for delivering monophosphorylated nucleosides and sugars into cells. ChemMedChem 2009, 4, 1779–1791. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Madela K.; Aljarah M.; Gilles A.; Brancale A.; Zonta N.; Chamberlain S.; Vernachio J.; Hutchins J.; Hall A.; Ames B.; Gorovits E.; Ganguly B.; Kolykhalov A.; Wang J.; Muhammad J.; Patti J. M.; Henson G. Design, synthesis and evaluation of a novel double pro-drug: INX-08189. A new clinical candidate for hepatitis C virus. Bioorg. Med. Chem. Lett. 2010, 20, 4850–4854. [DOI] [PubMed] [Google Scholar]
- McGuigan C.; Gilles A.; Madela K.; Aljarah M.; Holl S.; Jones S.; Vernachio J.; Hutchins J.; Ames B.; Bryant K. D.; Gorovits E.; Ganguly B.; Hunley D.; Hall A.; Kolykhalov A.; Liu Y.; Muhammad J.; Raja N.; Walters R.; Wang J.; Chamberlain S.; Henson G. Phosphoramidate ProTides of 2′-C-methylguanosine as highly potent inhibitors of hepatitis C virus. study of their in vitro and in vivo properties. J. Med. Chem. 2010, 53, 4949–4957. [DOI] [PubMed] [Google Scholar]
- Wang P.; Chun B.-K.; Rachakonda S.; Du J.; Khan N.; Shi J.; Stec W.; Cleary D.; Ross B. S.; Sofia M. J. An efficient and diastereoselective synthesis of PSI-6130: a clinically efficacious inhibitor of HCV NS5B polymerase. J. Org. Chem. 2009, 74, 6819–6824. [DOI] [PubMed] [Google Scholar]
- The improved synthesis of the intermediate 5 will be published elsewhere.
- McGuigan C.; Hassan-Abdallah A.; Srinivasan S.; Wang Y.; Siddiqui A.; Daluge S. M.; Gudmundsson K. S.; Zhou H.; McLean E. W.; Peckham J. P.; Burnette T. C.; Marr H.; Hazen R.; Condreay L. D.; Johnson L.; Balzarini J. Application of phosphoramidate ProTide technology significantly improves the antiviral potency of carbocyclic adenosine derivatives. J. Med. Chem. 2006, 49, 7215–7226. [DOI] [PubMed] [Google Scholar]
- In addition to l-alanine, other amino acid derivatives were also prepared and studied (no data shown). However, the phosphoramidates of l-alanine were strongly preferred to the others in terms of anti-HCV potency in vitro.
- For previously reported 6-modified guanine nucleotide analogues with anti-HCV activity, see:Gunic E.; Girardet J.-L.; Ramasamy K.; Stoisavljevic-Petkov V.; Chow S.; Yeh L.-T.; Hamatake R. K.; Raney A.; Hong Z. Cyclic monophosphate prodrugs of base-modified 2′-C-methyl ribonucleosides as potent inhibitors of hepatitis C virus RNA replication. Bioorg. Med. Chem. Lett. 2007, 17, 2452–2455. [DOI] [PubMed] [Google Scholar]
- Lambe C. U.; Averett D. R.; Paff M. T.; Reardon J. E.; Wilson J. G.; Krenitsky T. A. 2-Amino-6-methoxypurine arabinoside: an agent for T-cell malignancies. Cancer Res. 1995, 55, 3352–3356. [PubMed] [Google Scholar]
- The 5′-triphosphate of 6b (6-methoxyguanosine analogue) was synthesized according to the reported procedure:Ludwig J. A new route to nucleoside 5′-triphosphates. Acta Biochim. Biophys. Acad. Sci. Hung. 1981, 16, 131–133. We found that the 6b-TP did not significantly inhibit the HCV NS5B polymerase (IC50 > 100 μM), indicating the represented compounds are double prodrugs.. [PubMed] [Google Scholar]
- Sarrazin C.; Zeuzem S. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 2010, 138, 447–462. [DOI] [PubMed] [Google Scholar]
- Hocek M.; Naus P.; Pohl R.; Votruba I.; Furman P. A.; Tharnish P. M.; Otto M. J. Cytostatic 6-arylpurine nucleosides. 6. SAR in anti-HCV and cytostatic activity of extended series of 6-hetarylpurine ribonucleosides. J. Med. Chem. 2005, 48, 5869–5873. [DOI] [PubMed] [Google Scholar]
- Fleischer R.; Boxwell D.; Sherman K. E. Nucleoside analogues and mitochondrial toxicity. Clin. Infect. Dis. 2004, 38, e79–80. [DOI] [PubMed] [Google Scholar]
- Ma H.; Jiang W.-R.; Robledo N.; Leveque V.; Ali S.; Lara-Jaime T.; Masjedizadeh M.; Smith D. B.; Cammack N.; Klumpp K.; Symons J. Characterization of the metabolic activation of hepatitis C virus nucleoside inhibitor β-d-2′-Deoxy-2′-fluoro-2′-C-methylcytidine (PSI-6130) and identification of a novel active 5′-triphosphate species. J. Biol. Chem. 2007, 282, 29812–29820. [DOI] [PubMed] [Google Scholar]
- When compound 23 was prepared by the method described in the Supporting Information. The ratio varies depending on the reaction conditions. Selective synthetic preparation of 23b will be reported elsewhere in the near future.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.