

Abstract
TALEs targeting a promoter sequence and fused with a transcription activation domain (TAD) may be used to specifically induce the expression of a gene as a potential treatment for haploinsufficiency. This potential therapeutic approach was applied to increase the expression of frataxin in fibroblasts of Friedreich ataxia (FRDA) patients. FRDA fibroblast cells were nucleofected with a pCR3.1 expression vector coding for TALEFrat#8 fused with VP64. A twofold increase of the frataxin mRNA (detected by quantitative reverse transcription-PCR (qRT-PCR)) associated with a similar increase of the mature form of the frataxin protein was observed. The frataxin mRNA and protein were also increased by this TALE in the fibroblasts of the YG8R mouse model. The addition of 5-aza-2′-deoxycytidine (5-Aza-dC) or of valproic acid (VPA) to the TALE treatment did not produce significant improvement. Other TADs (i.e., p65, TFAP2α, SRF, SP1, and MyoD) fused with the TALEFrat#8 gene did not produce a significant increase in the frataxin protein. Thus the TALEFrat#8-VP64 recombinant protein targeting the frataxin promoter could eventually be used to increase the frataxin expression and alleviate the FRDA symptoms.
Keywords: Friedreich ataxia, frataxin, gene expression, TAL effector, transcription factor
Introduction
TALE proteins may be produced to target any nucleotide sequence starting with a thymidine.1,2,3,4 These TALE proteins can be fused with a transcription activation domain (TAD) to induce the expression of a gene located near the DNA attachment site.5,6,7 This could permit to specifically induce the expression of a gene to treat some hereditary and non-hereditary diseases, particularly haploinsufficiency diseases.
As an example of this new therapeutic approach, we have applied this strategy to Friedreich ataxia (FRDA), an autosomal recessive neurodegenerative and cardiac disease, caused by a trinucleotide (GAA) repeat expansion in the first intron of the frataxin gene located on chromosome 9.8 The mutation in the intron of the frataxin gene leads to a reduced expression without changing the protein. The mechanism of this pathology has been reviewed by Pandolfo et al.9,10 Neurons and cardiomyocytes are particularly sensitive to the reduction of frataxin.11,12,13 Thus neurological and cardiac symptoms appear during or before the second decade of life.14,15,16,17,18 There are also some systemic involvements, such as diabetes mellitus and scoliosis. Cardiomyopathy and associated arrhythmias lead to early death.16,19
Therefore, a potential treatment of FRDA is to increase the expression of frataxin in the patient cells using TALE proteins coupled with a TAD, e.g., four VP16 sequences (VP64), and specifically targeting the human frataxin promoter. Our research group has previously constructed 12 plasmids coding for various TALE proteins, which bind to various 12 nucleotide sequences in the promoter of the human frataxin gene.20 These TALE genes were under the control of a EF-1α promoter and fused with a nuclear localization signal and a TAD. These initial experiments used the VP64 TAD, which is made by four short VP16 sequences (Figure 1). These various TALEFrat-VP64 were tested for their capacity to induce the expression of a mCherry reporter gene placed downstream to the sequence of the proximal region of the frataxin promoter and under the control of a minimal nonfunctional cytomegalovirus promoter. TALEFrat#8-VP64 was among the three TALEs, which induced more strongly the expression of the reporter gene and was thus used for the present series of experiments.
Figure 1.

Schematic representation of pCR3.1-TALEFrat. (a) The TALE targeting the frataxin promoter is placed under the control of the cytomegalovirus (CMV) promoter in the pCR3.1-TALEFrat–VP64 plasmid. The TALE gene is fused with a nuclear localization signal (NLS) and the VP64 transcription activation domain (TAD). (b) A StuI restriction site was introduced in the pCR3.1-TALEFrat–p65 plasmid before the TAD and an XbaI site after the TAD. (c) The amino acid (aa) sequence of the TALEFrat#8-VP64 protein is presented. The repeat variable di-residues are over-lighted in gray. The VP64 sequence is included in a box.
In the present article, we have introduced TALEFrat#8-VP64 into an expression vector (pCR3.1) and the recombinant plasmid (Figure 1a,c) effectively increased the expression of the frataxin mRNA and protein in FRDA and in YG8R fibroblasts despite the presence of long GAA repeats in intron 1 of the frataxin gene. We are thus proposing that such a TALE coupled with a TAD could eventually be a therapeutic avenue to treat FRDA.
Results
The plasmid pCR3.1-TALEFrat#8–VP64 construct
Our previous article reported that several plasmids coding for various TALEFrat-VP64 targeting the frataxin promoter were able to increase the expression of the frataxin mRNA in the 293FT cell line, which contained a normal frataxin gene.20 To verify whether one of the best TALEs, i.e., TALEFrat#8-VP64, could also increase the expression of frataxin in FRDA cells, which have an expansion of the GAA trinucleotide repeat, we have modified the original lentiviral plasmid to produce a pCR3.1 expression vector (Figure 1a).
Increased level of the frataxin pre-mRNA following the nucleofection of the plasmid pCR3.1-TALEFrat#8–VP64 in FRDA fibroblasts
The long GAA repeat in the FRDA frataxin gene has been reported to prevent the elongation of the pre-mRNA.21 Therefore, we verified whether the pCR3.1-TALEFrat#8–VP64 plasmid permitted the elongation of the frataxin pre-mRNA to pass the GAA repeats and was able to increase the expression of frataxin pre-mRNA in FRDA cells. The plasmid pCR3.1-TALEFrat#8–VP64 was thus nucleofected in FRDA fibroblasts. Control FRDA cells were either not nucleofected or nucleofected with a plasmid coding for green fluorescent protein (GFP). Five segments of the frataxin pre-mRNA (primary transcript) (Figure 2a) were quantified by quantitative reverse transcription-PCR (qRT-PCR) (Table 1 for primers). The results are presented as the number of frataxin pre-mRNA copies per μg of RNA (Figure 2b). The TALEFrat#8-VP64 had little influence on the expression of segment A (i.e., 5′-untranslated region and exon 1) of the frataxin pre-mRNA. In all FRDA cells (not nucleofected or nucleofected with GFP or with the TALE plasmid), there were marked decreases in the number of copies of the segment B (intron 1 upstream of the GAA repeats), segment C (intron 1 downstream of the GAA repeat), segment D (junction of intron 1 and exon 2), and segment E (junction of intron 2 and exon 3) relative to segment A. However, the TALEFrat#8-VP64 significantly increased (by about twofold) the segments B, C, D, and E as compared with the control FRDA cells not nucleofected or nucleofected with the GFP plasmid. Therefore, the TALEFrat#8-VP64 improved the elongation of the frataxin pre-mRNA.
Figure 2.

TALEFrat-VP64 permits the elongation of the frataxin pre-mRNA in Friedreich ataxia (FRDA) fibroblasts. Fibroblasts from a FRDA patient were nucleofected with pCR3.1-TALEFrat#8–VP64. Control cells were either not nucleofected (CONT) or nucleofected with the pCR3.1-GFP plasmid (GFP). (a) Five different segments of the frataxin pre-mRNA were amplified by quantitative reverse transcription-PCR: segment A = 5′ UTR/exon 1; segment B = intron 1 before the GAA repeat INT1(UP); segment C = intron 1 after the GAA repeat INT1(DOWN); segment D = a junction of intron 1 and exon 2 (INT1/EX2), segment E = a junction of intron 2 and exon 3 (INT2/EX3) were quantified by quantitative reverse transcription-PCR. (b,c) The results are expressed as the number of copies of frataxin primary transcript per μg of RNA. The TALEFrat#8-VP64 significantly increased the elongation of the immature mRNA as indicated by the increased detection of fragments B to D. In c, the results are from a different experiment than in b, the pCR3.1-TALEFrat#8–VP64 increased the expression of segments B (INT1(UP)) and C (INT1(DOWN)). The addition of 5-Aza (A) or valproic acid (V) to the TALE treatment did not further increase the expression of the frataxin pre-mRNA. All results are based on duplicates and representative of at least two different experiments. GFP, green fluorescent protein; UTR, untranslated region.
Table 1. Primers list used for qRT-PCR and genomic PCR in the present study.
A second experiment was done with the FRDA fibroblasts to verify whether the coadministration of a histone deacetylase inhibitor (i.e., valproic acid (VPA)) or of a DNA methyltransferase inhibitor (i.e., 5-aza-2′-deoxycytidine (5-Aza-dC)) could improve the elongation of the frataxin pre-mRNA induced by TALEFrat#8-VP64. In that experiment, the number of copies of the segment C (about 600 copies) was lower than that of the segment B (over 2,500 copies) (Figure 2c). However, the TALEFrat#8-VP64 increased by the number of copies of the segment B of the frataxin pre-mRNA located upstream of the GAA repeats by 2.4-fold and the segment C located downstream by 4.6-fold. This indicates that the TALEFrat#8-VP64 permits the elongation of the frataxin pre-mRNA to pass the GAA repeats.
The addition of VPA, 5-Aza-dC or a combination of both the drugs did not substantially increase the effect of the TALEFrat#8-VP64 on the elongation of the frataxin pre-mRNA (Figure 2c).
Increased levels of the mature frataxin mRNA by the nucleofection of the plasmid pCR3.1-TALEFrat#8–VP64 in FRDA fibroblasts
The number of mature frataxin mRNA copies was also measured by qRT-PCR. A segment (F) of mature mRNA corresponding to the junction of exons 3 and 4 was analyzed (Figure 3a). A significant increase (1.6-fold) in the number of copies of the mature mRNA was also observed in the presence of the TALEFrat#8-VP64 in the FRDA cells (Figure 3b). As previously observed for primary transcripts (Figure 2c), no significant influence on the number of frataxin mRNA copies was observed by adding epigenetic modifiers to the TALEFrat#8-VP64 (Figure 3b).
Figures 3.

TALEFrat-VP64 increases the mature frataxin mRNA in Friedreich ataxia (FRDA) fibroblasts. Fibroblasts from a FRDA patient were nucleofected with pCR3.1-TALEFrat#8–VP64. Control cells were either not nucleofected (CONT) or nucleofected with the pCR3.1-GFP plasmid. (a) A segment F of the frataxin mature mRNA corresponding to the junction of exons 3 and 4 (EX3 and EX4) was quantified by quantitative reverse transcription-PCR. (b) The results were expressed in number of mRNA copies per μg of RNA. The TALE consistently increased the mature mRNA segment by 1.6-fold. The addition of 5-Aza (A) or valproic acid (V) to the TALE treatment did not further improve the mRNA expression. All results are based on duplicates and representative of three different experiments. GFP, green fluorescent protein; UTR, untranslated region.
Correspondence between the relative increase of the frataxin mRNA and protein expression induced by the plasmid pCR3.1-TALEFrat#8–VP64 in FRDA fibroblasts
The plasmid pCR3.1-TALEFrat#8–VP64 has no reporter gene to track the efficiency of the nucleofection within FRDA cells. The GFP plasmid was thus used as a reporter gene to evaluate the efficiency of the nucleofection process in FRDA cells (Figure 4a). A high level of GFP-positive FRDA cells (over 70%) was observed 24 hours after the nucleofection. In that experimental condition, the results of the mRNA presented in Figure 3b (exon 3/4) was normalized relative to three housekeeping RNAs (hypoxanthine phosphoribosyltransferase 1 (HPRT1), glyceraldehyde-3-phosphate dehydrogenase (GAPDH), and 18S ribosomal RNA (rRNA)) (Figure 4b). Increased expression of the mature frataxin mRNA produced by the plasmid pCR3.1-TALEFrat#8–VP64 alone was also observed with all the three normalization RNAs. These increases were variable depending on the housekeeping gene used for normalization (Figure 4b) showing respective increases of 1.8-, 2.9-, and 2.4-fold for HPRT1, GAPDH, and 18S rRNA. However, the addition of VPA or 5-Aza-dC alone or in combination did not improve the effect of the TALEFrat#8-VP64 plasmid, with the exception that the presence of 5-Aza-dC alone further increased the expression of frataxin mRNA (exon 3/4) but only when the results were normalized relative to the GAPDH and 18S rRNA. In summary, no important change of the frataxin mRNA expression was observed by the presence of the epigenetic modifiers to the FRDA cells in different experimental conditions.
Figure 4.
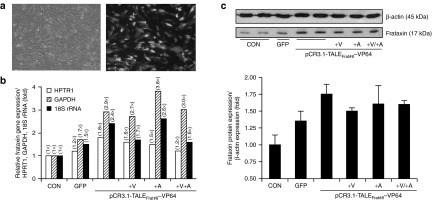
TALEFrat-VP64 increases the frataxin mRNA and the frataxin protein in Friedreich ataxia (FRDA) fibroblasts. The plasmid PCR3.1-TALEFrat#8–VP64 was nucleofected in FRDA fibroblasts. (a) Control cells were either not nucleofected (CONT) or nucleofected with a pCR3.1 plasmid coding for GFP. (b) The expression of the mature frataxin mRNA (i.e., the junction of exon 3 and exon 4) was quantified by reverse transcription-PCR. The results were normalized relative to three housekeeping genes (HPRT1, 18S rRNA, and GAPDH). The plasmid coding for the TALEFrat#8-VP64 increased the expression of frataxin more than the control plasmid coding for GFP. Valproic acid (V; 620 μmol/l), 5-aza-2′-deoxycytidine (A; 10 nmol/l) or a combination of both the drugs was added to the TALE treatment. Only 5-Aza further increased the frataxin expression but only when the results were normalized with the GAPDH mRNA. All results are based on duplicates. (c) The frataxin proteins from the same pool of cells as in b were quantified by western blot. The TALEFrat#8-VP64 alone increased the frataxin protein by 1.75-fold. However, the presence of valproic acid (+V) or 5-Aza (+A) alone or in combination did not result in an additional increase. All quantitative reverse transcription-PCR results are based on the three different experiments in duplicates. The western blot results are representative of two different experiments in duplicates. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; GFP, green fluorescent protein; HPRT1, hypoxanthine phosphoribosyltransferase 1; rRNA, ribosomal RNA.
Finally, we have analyzed the frataxin protein isolated from the same samples by western blot previously analyzed for mRNA. Of note, a significant increase (1.6- to 1.8-fold) of the mature frataxin protein (about 17 kDa) was observed when the results were internal standard protein (i.e., β-actin) (Figure 4c). The addition of VPA or 5-Aza-dC alone or in combination did not further increase the expression of the frataxin protein induced by the TALEFrat#8-VP64 plasmid (Figure 4c). Therefore, there was a clear correlation between the expression of the frataxin mRNA and frataxin protein confirming that the presence of the epigenetic modifiers did not have a great effect on the expression level of the frataxin gene.
Effects of nucleofection of various pCR3.1-TALEFrat#8–TADs in FRDA cells on the expression of the frataxin protein
To induce the expression of the frataxin gene, the TALEFrat#8 targeting the frataxin promoter was fused with a TAD. The VP64 TAD, i.e., four successive VP16 TADs, was used in all the previous experiments because it has been frequently used for various artificial transcription factors.22,23,24,25,26,27 We have made several constructs to replace the VP64 TAD (Figure 1b) by other well-known TADs such as p65, TFAP2α, SRF, SP1, and MyoD (Tables 2 and 3. Several of these TADs (TFAP2α, SRF, and SP1) were derived from transcription factors acting on the frataxin promoter. However, 50 hours after nucleofection with these different pCR3.1-TALEFrat#8 TADs in the FRDA cells, western blot analysis showed that only the pCR3.1-TALEFrat#8–VP64 was able to increase (1.6- to 1.9-fold) the frataxin protein expression (Figure 5a,b).
Table 2. Primers list used for cDNA and recombinant fusion TALE-TAD.

Figure 5.

Effects of various transcription activation domains (TADs) on the increase of frataxin protein by pCR3.1-TALEFrat#8–VP64. Different TADs were fused with the TALEFrat#8 gene. These plasmids were nucleofected in Friedreich ataxia fibroblasts. The frataxin protein was detected by western blot and the results were normalized with the β-actin protein. Only the VP64 TAD increased the expression of frataxin protein by 1.6- and 1.9-fold (two separate experiments). The other TADs (p65, TFAP2, SRF, SP1, and MyoD) did not increase the frataxin protein above the control level. All results are based on duplicates. MyoD, myogenic differentiation 1; SP1, Sp1 transcription factor; SRF, serum response factor.
Testing of pCR3.1-TALEFrat#8–VP64 in YG8R fibroblasts
Our ultimate aim is to test the TALEFrat#8 in an animal model of FRDA. The most appropriate model for this type of experiments is the YG8R mouse, which contains two KO mouse frataxin genes and a human transgene obtained from a FRDA patient. Therefore, this human gene contains a long GAA trinucleotide repeat and moreover, it is under the control of the human frataxin promoter.28,29,30 Therefore, the TALEFrat#8-VP64 should attach to the frataxin promoter of the human transgene. Initial experiments were done in a YG8R fibroblast cell line. The pCR3.1-TALEFrat#8–VP64 was nucleofected in these cells. Control cells were nucleofected with a GFP plasmid showing a lower efficiency (40–50%) of the nucleofection than in human FRDA cells (Figure 6a). The RNA and the proteins were extracted in parallel from each sample. The frataxin qRT-PCR for exons 3/4 results were normalized with the three housekeeping genes (HPRT1, GAPDH, and 18S rRNA). The frataxin mRNA was increased by 1.5-, 1.4-, and 1.9-fold, respectively (Figure 6b). The frataxin protein was quantified by western blot and normalized with the β-actin protein. The TALEFrat#8-VP64 plasmid increased the expression of the frataxin protein in the Y8GR cells by 1.4-fold (Figure 6c). The lower increase of the frataxin protein expression may be due to the lower nucleofection efficiency in these mouse cells than in the FRDA human fibroblasts.
Figure 6.

pCR3.1-TALEFrat#8–VP64 increases the frataxin mRNA and protein in YG8R fibroblasts. The plasmid pCR3.1-TALEFrat#8–VP64 was nucleofected in the YG8R fibroblasts. (a) Control cells were nucleofected with an eGFP plasmid to confirm the efficacy of the nucleofection. The RNA and the proteins were extracted in parallel from each sample. (b) The frataxin quantitative reverse transcription-PCR (i.e., the junction of exon 3 and exon 4) results were normalized with the three housekeeping genes (HPRT1, GAPDH, and 18S rRNA). The frataxin mRNA was increased respectively by 1.5-, 1.4-, and 1.9-fold. (c) The frataxin proteins were quantified by western blot and normalized with the β-actin protein. The TALEFrat#8-VP64 plasmid increased the expression of the frataxin protein in the YG8R cells by 1.4-fold. All results are based on duplicates. eGFP, enhanced green fluorescent protein; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; HPRT1, hypoxanthine phosphoribosyltransferase 1; rRNA, ribosomal RNA.
Discussion
The expansion of a trinucleotide (GAA) repeat in intron 1 of the frataxin gene is responsible for a reduction of the expression of that gene, which leads to the development of the symptoms in FRDA patients.8 Frataxin plays an important role in the mitochondrial metabolism of iron. The frataxin reduction thus in turn leads to a mitochondrial mis-function, characterized by accumulation of iron in the mitochondria and oxidative stress. This oxidative stress induces cell death, including that of neurons and cardiomyocytes. This progressive cell death results in several neurological and cardiac symptoms. Therefore, all FRDA symptoms are due to the reduction of frataxin protein.
Our present results open the door to a development of a new potential therapy to increase the expression of the frataxin gene, i.e., to engineer TALE genes, that are not only coding for proteins targeting specific sequences in the frataxin promoter but which are also coupled with a TAD.5,31,32,33 Most of our experiments have been done with a TAD, called VP64, which is made of four VP16 TAD from the HIV. Our previous results clearly demonstrated that several different TALEFrat-VP64 effectively increased the expression of a mCherry reporter gene placed under the control of the human frataxin promoter. Moreover, one of these TALEs, i.e., TALEFrat#8-VP64, increased the expression of the frataxin mRNA by two- to threefold in human 293FT cells. However, these experiments were done in a cell line, which contained a normal frataxin gene, i.e., a gene with no elongation of the trinucleotide repeat. These results left unanswered the question of whether this TALEFrat#8-VP64 protein would be able to increase the expression of the frataxin protein in FRDA patient cells, with an elongated GAA trinucleotide repeat.
Our present results clearly demonstrated that the TALEFrat#8-VP64 can increase the expression of frataxin even when there is an elongation of the GAA repeats. Indeed, the expression of the TALEFrat#8-VP64 protein in FRDA and YG8R fibroblasts led to increases of not only the segments of the frataxin pre-mRNA, which proceeds the GAA repeat in intron 1, but also of the segments located after the GAA repeat in intron 1. It has been previously hypothesized that the reduction of the frataxin expression in FRDA cells was due to a problem of elongation of the frataxin pre-mRNA.21 This elongation problem would be due to the epigenetic changes induced by the presence of the GAA repeats (for review, see Sandi et al.34). Our results indicate that the TALEFrat#8-VP64 protein permits to overcome this elongation problem. However, the epigenetic modifiers (5-Aza-dC and VPA) did not synergize with the TALE suggesting that the TALE protein itself modifies the chromatin around the GAA. Our results also demonstrated that it is not only the frataxin pre-mRNA and mature mRNA, which are increased by the TALE expression, but also and more importantly the frataxin protein. It is important to note that the mature form of the frataxin protein was increased, indicating that the immature frataxin protein produced by the mRNA can not only enter in the mitochondria but can also be processed, i.e., truncated to produce the shorter mature form.
The FRDA carriers express only 50% of the normal level of frataxin and yet are asymptomatic.10 However, the FRDA patients, who have both the alleles mutated, are producing frataxin levels, which are about 5–35% of the levels in healthy individuals.35 The lower the expression level, the more severe are the symptoms and the earlier are their developments. Therefore, the two- to threefold increases in the expression of the frataxin that we have observed could reduce or even completely prevent the symptoms in most of the FRDA patients. However, for a therapeutic application, the TALEFrat-VP64 will have to be expressed in most of the FRDA patient cells, especially the neurons and the cardiomyocytes to prevent the development of nervous system and cardiac symptoms. A potential solution is to couple the TALEFrat-VP64 protein with a cell penetrating peptide such as Tat and Pep-1.36,37,38,39 These fusion proteins could be delivered systemically; they would enter the cells and hopefully drive the expression of frataxin. The fusion proteins would however have to be readministered on a regular basis. Unfortunately, the TALE protein will also be immunogenic and thus sustained immunosuppression would have to be used or development of specific immunological tolerance to this protein would have to be developed.
The therapeutic approach that we are proposing with TALE proteins fused with a TAD for FRDA could also be used to treat other haploinsufficiency diseases in which the increased expression of a gene would produce beneficial effects.
Materials and methods
Culture of FRDA and YG8R fibroblasts. Fibroblasts (GM04078) were obtained from the Coriell Cell Repository (Camden, NJ) and originated from a symptomatic FRDA patient. This patient was homozygous for GAA expansion in the frataxin gene with alleles of 541 and 420 repeats. YG8R fibroblasts were derived from the YG8R mouse, which has both mouse frataxin genes KO but contain a human frataxin gene from a FRDA patient.28 The YG8R fibroblasts contain 190 + 90 GAA repeats. The human and mouse fibroblasts were both cultured at 37 °C in DMEM High glucose (Wisent, St-Bruno, Quebec, Canada), 10% FBS, 1X Pen/Strep, and 1X minimum essential amino acids medium.
Construction of PCR3.1-TALEFrat#8–VP64. We have previously engineered TALEFrat-VP64 genes20 in the original vector used by Zhang et al.6 VP64 is an artificial tetrameric repeat of four VP16 minimal TAD, amino acids 438-448 (DALDDFDLDML) uniprot/Q69113. To reduce the size of the original vector, a DNA fragment coding for one of these TALEs, i.e., TALEFrat#8-VP64, was cloned in the pCR3.1 vector (Invitrogen, Burlington, Ontario, Canada) digested KpnI/ApaI. Briefly, a 2.7 kb fragment was amplified by PCR with the Phusion enzyme (New England Biolabs, Ipswich, MA) with the following long primers: forward 5′-ggatccggtaccaccatgtcgcggacccggctcccttc-3′ containing BamHI/KpnI sites (underlined) and reverse 5′-gggcccttattatctagagtt aatcagcatgtccaggt-3′ containing an ApaI site (underlined). The amplification program was as follows: 98 °C for 90 seconds; 98 °C for 10 seconds, 55 °C for 20 seconds, 72 °C for 90 seconds for five cycles; 98 °C for 10 seconds, 60 °C for 20 seconds, 72 °C for 90 seconds for 30 cycles. At the end of the PCR reaction, a final step was done at 72 °C for 10 minutes with the addition of TAq DNA polymerase. The PCR reaction medium was electrophoresed on 1.2% agarose gel and a PCR DNA fragment of 2.7 kb was isolated from the gel and cloned in a TA cloning vector named pDrive (Qiagen, Valencia, CA). Finally, following the sequencing of the pDrive containing the TALEFrat#8-VP64, a 2.7 kb DNA fragment digested with BamHI/ApaIwas cloned in the pCR3.1 expression vector (Invitrogen) digested by the same restriction enzymes. The resulting construct referred as pCR3.1-TALEFrat#8–VP64 (Figure 1a) was used in the present study. Figure 1c gives the amino acid sequence of the resulting protein.
Fusion of TALEFrat#8 with various TADs. To construct different TALEFrat#8-TADs, we have initially made pCR3.1-TALEFrat#8–p65 (Figure 1b) by two consecutive PCR amplifications. First, using a template pCR3.1-TALEFrat#8–VP64, we amplified a fragment of 2.4 kb using the Phusion enzyme with p65 primers TA (SalI) and P1 (StuI) (primers are described in Table 2). A second PCR fragment was amplified from the purified p65 PCR fragment with primers TA (SalI) and P2 (XbaI) (Table 2). The resulting p65 PCR fragment (~2.5 kb) was cloned in the pCR3.1-TALEFrat#8–VP64 digested SalI/XbaI replacing VP64 by p65. The resulting plasmid contained two StuI restriction sites, the first site was already present in the pCR3.1vector and the second site was introduced by the primer P1 and was located before the TAD sequence. The first StuI restriction site was removed by site-directed mutagenesis using the Pfu Turbo DNA polymerase (Stratagene, Wilmington, DE). The remaining StuI site was present between the nuclear localization signal and the p65 nucleotide sequences described earlier (Figure 1b). Different nucleotide sequences coding for various TADs were cloned separately in the pCR3.1-TALEFrat#8–p65 digested with StuI and XbaI, a region located just before the stop codon (Figure 1b). Different TADs cDNA were generated by RT-PCR using RNA extracted from human endometrial cells.40 The PCR amplification products of the RT-PCR were purified, digested with StuI/XbaI, and cloned directly in the TALEFrat#8-p65 also digested by StuI/XbaI to substitute p65 by different TADs (amino acid sequences listed in Table 3).
Table 3. Recombinant fusion TALE-TAD.

Nucleofection of the various pCR3.1-TALEFrat#8–TADs in FRDA and in YG8R fibroblasts. Ten microgram of each of the various plasmids pCR3.1-TALEFrat#8–TADs were nucleofected in FRDA (passages 3–10) and YG8R fibroblasts in six well plates at around 80% confluence. The nucleofection solution (normal human dermal fibroblasts-adult) was used with the Amaxa nucleofector (Amaxa Nucleofector System; Lonza Walkersville,Walkersville, MD) using the program P-022. The cells were treated with trypsin-EDTA and pelleted by centrifugation in the conical tube. After centrifugation, the supernatant was discarded, the cell pellet was washed once with 1X Hank's balanced salt solution, centrifuged again, resuspended in 100 μl of the nucleofection solution and then nucleofected. The cells were then resuspended in 3 ml of complete growth culture medium and grown for 50 hours with several medium changes. The frataxin mRNA level was determined in these cells by qRT-PCR and the protein level by western blot. To evaluate the efficiency of the nucleofection in the human and mouse fibroblasts, a control GFP plasmid (provided in the Lonza nucleofection kit) was used at 5 μg/nucleofection for 500,000–750,000 cells. The fluorescence was visualized using a Zeiss Axiovert 100-Inverted microscope (Zeiss, Oberkochen, Germany). Images were captured and integrated using the Northern Exposure program (Empix Imaging, Mississauga, Ontario, Canada).
Treatment with epigenetic inhibitors of FRDA fibroblasts nucleofected with pCR3.1-TALEFrat#8–VP64. Eighteen hours after the nucleofection with pCR3.1-TALEFrat#8–VP64, the FRDA fibroblasts were treated during 48 hours either with the histone deacetylase inhibitor (VPA) (Sigma-Aldrich, Oakville, Ontario, Canada) at 620 μmol/l, with the DNA methyltransferase inhibitor (5-Aza-dC) (Sigma-Aldrich) at 10 nmol/l or with a combination of VPA 310 μmol/l and 5-Aza-dC at 5 nmol/l. The stock solutions of VPA at 250 mmol/l and 5-Aza-dC at 30 mmol/l in phosphate-buffered saline (PBS) 1X were prepared as previously described.41 The final drug concentrations used were those proven effective on HEK 293T cells by that group. The culture medium containing these drugs was replaced at every 12 hours. Cells not exposed to these drugs were also used as control. All cells were harvested for analysis of the frataxin mRNA by qRT-PCR or of the frataxin protein by western blot.
qRT-PCR for the frataxin mRNA. RT-PCR were done using a technique previously described by Luu-The et al.42 Briefly, the total RNA was extracted from one well of six well plates using RNeasy Mini kit (Qiagen, Valencia, CA). RNA quality was assessed with an Agilent 2100 Bioanalyzer (Agilent Technologies, Mississauga, Ontario, Canada). First-strand cDNA synthesis was obtained using 5 μg of isolated RNA. The frataxin mRNA was amplified with primers designed from GenBank sequence NM_000144 (Table 1). Quantification of the frataxin primary transcripts (Figure 2) and mRNA (Figure 3) of the frataxin gene was done by the determination of the Cp using the second derivative method described by Luu-The (2005)42 and the results were expressed as number of copies per μg of total RNA. The PCR results for exons 3–4 (Figures 4b and 6b) were also normalized with three housekeeping genes which are HPRT1, GAPDH, and 18S rRNA. This was done by calculating the ratio of the number of frataxin copies per μg of total relative to the number of copies of these three housekeeping genes per μg total RNA. The results were finally expressed as fold increase relative to the non-nucleofected cells.
Protein extraction and frataxin analysis by western blot. FRDA and YG8R fibroblasts nucleofected in six well plates were directly lysed to extract proteins as previously described.43 Briefly, the cells attached at the bottom of a well were rinsed twice with PBS and lysed with 200 μl of extraction buffer (50 mmol/l Tris-HCl pH 7.5, 1 mmol/l EDTA, 1 mmol/l dithiothreitol, 1 mmol/l PMSF, 1% sodium dodecyl sulfate). The plate was placed on a rocking surface to permit complete cell lysis. About 200 μl of this viscous lysis solution was transferred in a 1.5 ml microtube. The proteins were precipitated by adding a mixture of methanol (600 μl), chloroform (200 μl), and water (500 μl), followed by vigorous vortexing (15 seconds). This suspension was finally centrifuged for 3 minutes at full speed (14,000 RPM). The pellet at the interface was recuperated by discarding the aqueous phase. Methanol (800 μl) was added and mixed by inversion. The suspension was again centrifuged at full speed for 3 minutes. The supernatant was discarded and the pellet was dried by lyophilization during a few minutes. The pellet was then dissolved in protein sodium dodecyl sulfate-polyacrylamide gel loading buffer 1X and heated for 5 minutes in boiling water. The proteins were quantified as previously described.43 For western blot analysis, about 5 μg of protein were loaded in each well and electrophoresed on a 12% sodium dodecyl sulfate-polyacrylamide gel. The proteins were then electrotransferred onto a 0.45 mm nitrocellulose membrane (Bio-Rad, Hercules, CA). This membrane was cut into two sections: the upper part (proteins range over 30 kDa) for the detection of the β-actin (45 kDa) and the lower part for the detection of mature frataxin (~15–17 kDa).
Membrane sections were blocked with 5% (wt/vol) non-fat dry milk (bovine lacto transfer technique optimizer (BLOTTO)) dissolved in PBS containing 0.05% Tween-20 and then incubated for 1 hour. A mouse monoclonal antibody against β-actin (Sigma-Aldrich) diluted at 1/5,000 was added to the upper section. A mouse monoclonal antibody against frataxin (MitoSciences, Eugene, OR) diluted at 3 μg/ml was added to the lower section. After 1 hour incubation with the primary antibody, the membranes were washed three times for 10 minutes in PBS-Tween and incubated for 1 hour with a goat anti-mouse antibody coupled with peroxidase (Jackson Laboratory, Bar Harbor, ME) diluted in BLOTTO at 1/10,000. This was followed by three washes (10 minutes each) with PBS-Tween. The membranes were then treated for 1 minute with Renaissance reagent (NEN Life Science Products, PerkinElmer, Boston, MA). The signal intensity of the immunoreaction was quantified by densitometry using a Multimage Light Cabinet equipped with AlphaImager 2000 software (Cell Biosciences, Santa Clara, CA). The ratios of frataxin to β-actin were used to estimate the increase of the frataxin in different experimental conditions.
Acknowledgments
The YG8R fibroblasts were kindly provided by Mark Pook, Brunel University, Uxbridge, UK. This project was supported by grants from the Canadian Association of Familial Ataxia, from the Association Française contre les Ataxies Familiales, and from the Canadian Institute of Health Research. The authors declared no conflict of interest.
References
- Boch J, Scholze H, Schornack S, Landgraf A, Hahn S, Kay S, et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326:1509–1512. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326:1501. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- Cermak T, Doyle EL, Christian M, Wang L, Zhang Y, Schmidt C, et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39:e82. doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, Joung JK. FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol. 2012;30:460–465. doi: 10.1038/nbt.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Cong L, Zhou Y, Cunniff MM, Feng G, Zhang F. A transcription activator-like effector toolbox for genome engineering. Nat Protoc. 2012;7:171–192. doi: 10.1038/nprot.2011.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Cong L, Lodato S, Kosuri S, Church GM, Arlotta P. Efficient construction of sequence-specific TAL effectors for modulating mammalian transcription. Nat Biotechnol. 2011;29:149–153. doi: 10.1038/nbt.1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg A, Lohmueller JJ, Silver PA, Armel TZ. Engineering synthetic TAL effectors with orthogonal target sites. Nucleic Acids Res. 2012;40:7584–7595. doi: 10.1093/nar/gks404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, et al. Friedreich's ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- Pandolfo M. Iron and Friedreich ataxia. J Neural Transm Suppl. 2006. pp. 143–146. [DOI] [PubMed]
- Pandolfo M. Friedreich ataxia. Handb Clin Neurol. 2012;103:275–294. doi: 10.1016/B978-0-444-51892-7.00017-6. [DOI] [PubMed] [Google Scholar]
- Becker E, Richardson DR. Frataxin: its role in iron metabolism and the pathogenesis of Friedreich's ataxia. Int J Biochem Cell Biol. 2001;33:1–10. doi: 10.1016/s1357-2725(00)00067-4. [DOI] [PubMed] [Google Scholar]
- Wallis J, Shaw J, Wilkes D, Farrall M, Williamson R, Chamberlain S, et al. Prenatal diagnosis of Friedreich ataxia. Am J Med Genet. 1989;34:458–461. doi: 10.1002/ajmg.1320340327. [DOI] [PubMed] [Google Scholar]
- Rötig A, de Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, et al. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat Genet. 1997;17:215–217. doi: 10.1038/ng1097-215. [DOI] [PubMed] [Google Scholar]
- Babady NE, Carelle N, Wells RD, Rouault TA, Hirano M, Lynch DR, et al. Advancements in the pathophysiology of Friedreich's Ataxia and new prospects for treatments. Mol Genet Metab. 2007;92:23–35. doi: 10.1016/j.ymgme.2007.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JM, Schapira AH. Friedreich's Ataxia: disease mechanisms, antioxidant and Coenzyme Q10 therapy. Biofactors. 2003;18:163–171. doi: 10.1002/biof.5520180219. [DOI] [PubMed] [Google Scholar]
- Harding AE. Friedreich's ataxia: a clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain. 1981;104:589–620. doi: 10.1093/brain/104.3.589. [DOI] [PubMed] [Google Scholar]
- Lynch DR, Farmer JM, Balcer LJ, Wilson RB. Friedreich ataxia: effects of genetic understanding on clinical evaluation and therapy. Arch Neurol. 2002;59:743–747. doi: 10.1001/archneur.59.5.743. [DOI] [PubMed] [Google Scholar]
- Pandolfo M. Molecular pathogenesis of Friedreich ataxia. Arch Neurol. 1999;56:1201–1208. doi: 10.1001/archneur.56.10.1201. [DOI] [PubMed] [Google Scholar]
- Singh G, Binstadt BA, Black DF, Corr AP, Rummans TA. Electroconvulsive therapy and Friedreich's ataxia. J ECT. 2001;17:53–54. doi: 10.1097/00124509-200103000-00011. [DOI] [PubMed] [Google Scholar]
- Tremblay JP, Chapdelaine P, Coulombe Z, Rousseau J. Transcription activator-like effector proteins induce the expression of the frataxin gene. Hum Gene Ther. 2012;23:883–890. doi: 10.1089/hum.2012.034. [DOI] [PubMed] [Google Scholar]
- Kim E, Napierala M, Dent SY. Hyperexpansion of GAA repeats affects post-initiation steps of FXN transcription in Friedreich's ataxia. Nucleic Acids Res. 2011;39:8366–8377. doi: 10.1093/nar/gkr542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa FC, Fedosyuk H, Neades R, de Los Rios JB, Barbas CF, 3rd, Peterson KR. Induction of fetal hemoglobin in vivo mediated by a synthetic γ-globin zinc finger activator. Anemia. 2012;2012:507894. doi: 10.1155/2012/507894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnenat L, Schwimmer LJ, Barbas CF., 3rd Drug-inducible and simultaneous regulation of endogenous genes by single-chain nuclear receptor-based zinc-finger transcription factor gene switches. Gene Ther. 2008;15:1223–1232. doi: 10.1038/gt.2008.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran AS, Sun X, Lizardi PM, Blancafort P. Reprogramming epigenetic silencing: artificial transcription factors synergize with chromatin remodeling drugs to reactivate the tumor suppressor mammary serine protease inhibitor. Mol Cancer Ther. 2008;7:1080–1090. doi: 10.1158/1535-7163.MCT-07-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh JC, Cheng YC, Su MC, Moore M, Choo Y, Klug A. Tailor-made zinc-finger transcription factors activate FLO11 gene expression with phenotypic consequences in the yeast Saccharomyces cerevisiae. PLoS ONE. 2007;2:e746. doi: 10.1371/journal.pone.0000746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran A, Parikh S, Liu Y, Cuevas BD, Johnson GL, Futscher BW, et al. Re-activation of a dormant tumor suppressor gene maspin by designed transcription factors. Oncogene. 2007;26:2791–2798. doi: 10.1038/sj.onc.1210072. [DOI] [PubMed] [Google Scholar]
- Lund CV, Blancafort P, Popkov M, Barbas CF., 3rd Promoter-targeted phage display selections with preassembled synthetic zinc finger libraries for endogenous gene regulation. J Mol Biol. 2004;340:599–613. doi: 10.1016/j.jmb.2004.04.057. [DOI] [PubMed] [Google Scholar]
- Al-Mahdawi S, Pinto RM, Varshney D, Lawrence L, Lowrie MB, Hughes S, et al. GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics. 2006;88:580–590. doi: 10.1016/j.ygeno.2006.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Mahdawi S, Pinto RM, Ismail O, Varshney D, Lymperi S, Sandi C, et al. The Friedreich ataxia GAA repeat expansion mutation induces comparable epigenetic changes in human and transgenic mouse brain and heart tissues. Hum Mol Genet. 2008;17:735–746. doi: 10.1093/hmg/ddm346. [DOI] [PubMed] [Google Scholar]
- Pook MA, Al-Mahdawi S, Carroll CJ, Cossée M, Puccio H, Lawrence L, et al. Rescue of the Friedreich's ataxia knockout mouse by human YAC transgenesis. Neurogenetics. 2001;3:185–193. doi: 10.1007/s100480100118. [DOI] [PubMed] [Google Scholar]
- Morbitzer R, Römer P, Boch J, Lahaye T. Regulation of selected genome loci using de novo-engineered transcription activator-like effector (TALE)-type transcription factors. Proc Natl Acad Sci USA. 2010;107:21617–21622. doi: 10.1073/pnas.1013133107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Römer P, Recht S, Strauss T, Elsaesser J, Schornack S, Boch J, et al. Promoter elements of rice susceptibility genes are bound and activated by specific TAL effectors from the bacterial blight pathogen, Xanthomonas oryzae pv. oryzae. New Phytol. 2010;187:1048–1057. doi: 10.1111/j.1469-8137.2010.03217.x. [DOI] [PubMed] [Google Scholar]
- Li L, Piatek MJ, Atef A, Piatek A, Wibowo A, Fang X, et al. Rapid and highly efficient construction of TALE-based transcriptional regulators and nucleases for genome modification. Plant Mol Biol. 2012;78:407–416. doi: 10.1007/s11103-012-9875-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandi C, Al-Mahdawi S, Pook MA. Epigenetics in Friedreich's Ataxia: Challenges and Opportunities for Therapy. Genet Res Int. 2013;2013:852080. doi: 10.1155/2013/852080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandolfo M. Friedreich ataxia: new pathways. J Child Neurol. 2012;27:1204–1211. doi: 10.1177/0883073812448534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- Morris MC, Depollier J, Mery J, Heitz F, Divita G. A peptide carrier for the delivery of biologically active proteins into mammalian cells. Nat Biotechnol. 2001;19:1173–1176. doi: 10.1038/nbt1201-1173. [DOI] [PubMed] [Google Scholar]
- Noguchi H, Matsushita M, Kobayashi N, Levy MF, Matsumoto S. Recent advances in protein transduction technology. Cell Transplant. 2010;19:649–654. doi: 10.3727/096368910X508744. [DOI] [PubMed] [Google Scholar]
- Münst B, Patsch C, Edenhofer F. Engineering cell-permeable protein. J Vis Exp. 2009. p. pii: 1627. [DOI] [PMC free article] [PubMed]
- Chapdelaine P, Kang J, Boucher-Kovalik S, Caron N, Tremblay JP, Fortier MA. Decidualization and maintenance of a functional prostaglandin system in human endometrial cell lines following transformation with SV40 large T antigen. Mol Hum Reprod. 2006;12:309–319. doi: 10.1093/molehr/gal034. [DOI] [PubMed] [Google Scholar]
- Bultmann S, Morbitzer R, Schmidt CS, Thanisch K, Spada F, Elsaesser J, et al. Targeted transcriptional activation of silent oct4 pluripotency gene by combining designer TALEs and inhibition of epigenetic modifiers. Nucleic Acids Res. 2012;40:5368–5377. doi: 10.1093/nar/gks199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luu-The V, Paquet N, Calvo E, Cumps J. Improved real-time RT-PCR method for high-throughput measurements using second derivative calculation and double correction. BioTechniques. 2005;38:287–293. doi: 10.2144/05382RR05. [DOI] [PubMed] [Google Scholar]
- Chapdelaine P, Vignola K, Fortier MA. Protein estimation directly from SDS-PAGE loading buffer for standardization of samples from cell lysates or tissue homogenates before Western blot analysis. BioTechniques. 2001;31:478, 480, 482. doi: 10.2144/01313bm04. [DOI] [PubMed] [Google Scholar]