

Key Points
Systems biology approaches are required to advance our understanding of virus–host interactions, how these interactions cause disease and, ultimately, how to improve diagnostics, therapeutics and vaccines.
Over the past decade, the field of systems virology has evolved from using first-generation microarrays to the integration of multidimensional data sets. This has resulted in significant findings, including the identification of gene expression signatures that are predictive of viral pathogenesis and vaccine efficacy, insights into how viruses disrupt cellular metabolism, and the mapping of virus–host interactomes.
To fulfil its initial promise of revolutionizing our understanding of virus–host interactions, the field of systems virology must move beyond just the listing of molecules that are differentially expressed following viral infection; it must now look to define the relationships between key host molecules and their interactions with viral components.
Several key computational challenges must be addressed in order to move into this new phase of systems virology, including consideration of nonlinear relationships such as the dynamics of the system, the integration of multidimensional data sets and the identification of causal relationships.
Virologists, computer scientists and mathematicians must combine their skills and expertise in applying systems approaches to untangle the complex question of how viruses kill.
Supplementary information
The online version of this article (doi:10.1038/nrmicro3036) contains supplementary material, which is available to authorized users.
Subject terms: Virology, Genomics, Infectious diseases, Vaccines
Katze and colleagues provide an overview of the evolution of systems virology and the insights obtained from using such methodologies to study virus–host interactions. Combining systems, mathematical and computational approaches with traditional virology research will offer a better understanding of how viruses cause disease and will help in the development of therapeutics.
Supplementary information
The online version of this article (doi:10.1038/nrmicro3036) contains supplementary material, which is available to authorized users.
Abstract
High-throughput molecular profiling and computational biology are changing the face of virology, providing a new appreciation of the importance of the host in viral pathogenesis and offering unprecedented opportunities for better diagnostics, therapeutics and vaccines. Here, we provide a snapshot of the evolution of systems virology, from global gene expression profiling and signatures of disease outcome, to geometry-based computational methods that promise to yield novel therapeutic targets, personalized medicine and a deeper understanding of how viruses cause disease. To realize these goals, pipettes and Petri dishes need to join forces with the powers of mathematics and computational biology.
Supplementary information
The online version of this article (doi:10.1038/nrmicro3036) contains supplementary material, which is available to authorized users.
Main
Anyone who has taken an undergraduate virology course is familiar with subject matter focused on the structure of viral genomes and the molecular events associated with multistep viral life cycles. The field of virology has done a remarkable job of characterizing and categorizing viruses and defining the steps of viral attachment, entry, replication and release. Moreover, an understanding of viral protein function has paved the way for the development of antiviral drugs that target viral enzymatic activities. However, many of these drugs function poorly at best, and the virus-centric approach has not proved to be well suited for deciphering the complex and multifaceted virus–host interactions that underlie viral recognition, innate immune signalling and disease outcome. In the past decade, tools have become available to chart a new course, one directed at obtaining comprehensive systems-level views of the host response and the interplay between virus and host.
Systems virology is a phrase coined to describe the application of systems biology approaches to the field of virology1. Systems biology is highly interdisciplinary in character, requiring the combined talents of biologists, mathematicians and computer scientists, and the goal of the field is to gain comprehensive understanding of biological systems. In the case of systems virology, these biological systems can range from virus-infected cells to tissues to whole organisms. Systems-level analyses use high-throughput technologies to measure system-wide changes in biological components such as DNA, RNA, proteins and metabolites, and are dependent on the quality of both the resulting data sets (which are often noisy) and the subsequent data integration and modelling. Ideally, high-throughput data derived from these and other measurements are integrated and analysed using mathematical algorithms to generate predictive models of the system. When a model has been developed, subsequent experimental perturbations of the system (for example, the use of viral mutants or targeted inhibition of host genes or pathways) are used to yield refinements to the model and to increase its predictive capacity2,3,4 (Fig. 1).
Figure 1. The systems virology paradigm.
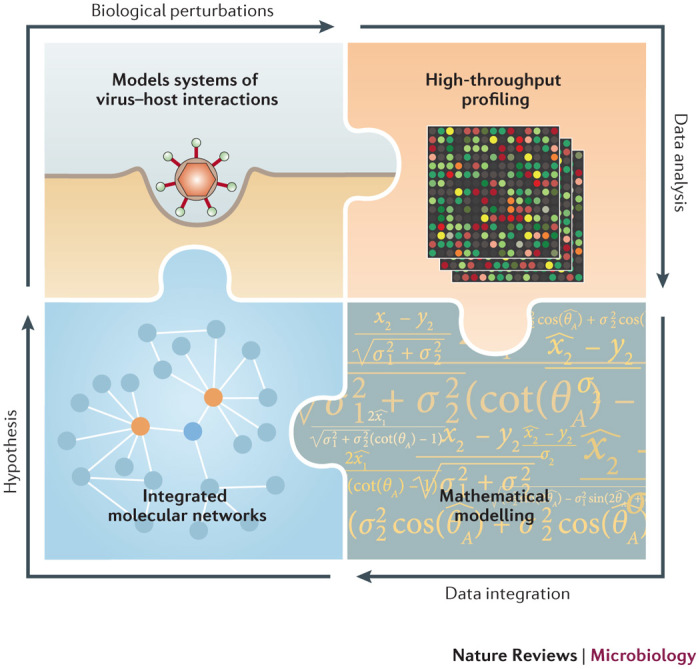
Appropriate experimental models and technologies are used to generate multidimensional data, including virology data. Analyses of such data, in combination with mathematical modelling, are used to generate comprehensive, integrated and predictive models of biological systems and virus–host interactions. Resulting predictions and hypotheses lead to subsequent experimental perturbations, model refinement and a deeper understanding of complex biological processes. These findings and outcomes can be used directly or further refined by the scientific community for various types of disease intervention.
This holistic, host-directed approach stands in contrast to the more traditional reductionist approaches that focus on a pre-determined small set of molecules (genes, proteins or metabolites). Although systems-level (or discovery-based) analyses are often criticized for not being hypothesis-driven, these analyses are increasingly being acknowledged as potent hypothesis generators. Moreover, for dynamic systems such as those involved in the host response to viral infection, systems-level analyses are considered the only way to understand emergent properties — that is, properties or biological outcomes that cannot be predicted by an understanding of the individual parts of a system alone, but rather become apparent only with knowledge of the specific organization of, and interactions between, components5. Because of this, systems virology is an essential and synergistic complement to traditional virology approaches.
This Review focuses on the host response to virus infection and discusses the evolution and significant findings of systems virology, including the identification of gene expression signatures that are predictive of viral pathogenesis and vaccine efficacy, insights into how viruses disrupt cellular metabolism, and the mapping of virus–host interactomes. These accomplishments did not come from a single experiment or study, but rather from a body of work undertaken over several years by different investigators. The field has seen a progression from genomics-based approaches to measurements of proteins and metabolites, and has also embraced the analysis of host genetic variation as a means to better understand disease processes rather than viewing such variation as a source of frustration. Moving forwards, systems virology must also embrace computational approaches that are capable of integrating this information to construct robust virus–host interaction models that incorporate multiple dimensions and scales6,7. We cite examples of studies that are moving in this direction and outline what the next phase of systems virology must encompass to reach its full potential.
Gene expression signatures
With the completion of the human genome project and the advent of microarrays capable of measuring RNA transcripts at a genome-wide scale, the first systems-level analyses became a reality. Over the past 12 years, DNA microarrays have evolved from hundreds of cDNAs spotted on nylon membranes to glass slides containing high-density oligonucleotides that encompass entire genomes (Box 1). Microarrays (then covering only 1,500 human genes) were first used in virology to evaluate the changes in cellular gene expression that occurred in a CD4+ T cell line infected with HIV8. Since then, the use of microarrays to evaluate changes in host gene expression in response to viral infection has become commonplace. In many cases, these studies remain small and narrowly focused, and although they provide glimpses into global responses, low sample numbers make it difficult to determine their reproducibility. In addition, these studies do not provide the robust data sets that are needed for computational modelling, which could result in deep insights into system architecture or behaviour. This shortcoming has perhaps fed scepticism regarding the ability of genome-wide expression profiling to yield transformative discoveries. Below, we provide examples of more comprehensive studies resulting in genomic signatures that have increased our understanding of both viral pathogenesis and the characteristics of the host response required for immune protection.
Signatures of highly pathogenic respiratory viruses. Influenza virus is well known for its ability to rapidly evolve new variants through genetic mutation and genome reassortment, yielding strains that can vary widely in virulence and transmissibility. Most strains cause mild respiratory disease, whereas others, such as the 1918 pandemic virus and highly pathogenic avian H5N1 influenza virus strains (such as A/VN/1203/04), can cause severe and often fatal infections9. The field has used DNA microarrays and functional analyses to define the virus–host interactions that regulate influenza virus pathogenesis10. These studies have identified gene expression signatures that correlate with virulence and have revealed that the timing and magnitude of the host response is a crucial determinant of the eventual outcome of infection.
This phenomenon was first demonstrated in studies that used a combination of mouse and macaque infection models and genome-wide transcriptional profiling to measure the host response to the reconstructed 1918 pandemic virus. In these animal models, the virus causes a rapidly fatal infection marked by severe lung pathology, intense neutrophil infiltration and the rapid and sustained induction of pro-inflammatory cytokine and chemokine genes11,12, an event often referred to as a cytokine storm13. By contrast, macaques infected with a highly pathogenic avian H5N1 influenza virus strain show a rapid and intense induction of interferon (IFN) and innate immune genes, but this eventually resolves as the animals recover14. Genomic analyses have also revealed that macaques infected with the 1918 pandemic strain15 and mice infected with H5N1 influenza virus A/VN/1203/04 show a strong induction of genes encoding inflammasome components (for example, NLRP3 (NOD-, LRR- and pyrin domain-containing 3) and interleukin-1β)16. This H5N1 virus is particularly virulent in mice, and although the inflammasome is part of the innate immune response to influenza A viruses17,18,19, the excessive activation of this response seems to be detrimental in this host.
As high-throughput data have accumulated in public databases, it has become possible to use this information to carry out meta-analyses. This strategy has been used to analyse data from a compendium of published studies that used mouse models to measure host transcriptional responses to lethal or non-lethal strains of influenza virus, respiratory syncytial virus or severe acute respiratory syndrome coronavirus (SARS CoV)20. Two alternative methods were used to generate gene expression signatures that are predictive of high or mild pathogenicity (defined here as 100% mortality and 100% survival, respectively). The first signature consists of 74 genes for which expression changes in the opposite direction in highly versus mildly pathogenic infections, with respect to mock infections (referred to as a 'digital' relationship). The second signature consists of 57 genes that are differentially expressed between highly and mildly pathogenic infections, without reference to mock infections (referred to as an 'analogue' relationship). Most genes in the analogue signature are differentially expressed during both lethal and non-lethal infections (compared with mock infections), but high pathogenicity corresponds with a higher degree of differential expression.
When the two signatures were tested for their ability to predict pathogenicity, the best predictor of a highly pathogenic infection was the analogue signature (Fig. 2). Significantly, this meta-analysis did not take into account the time after infection; that is, data from samples isolated at 1 day and at 5 days post-infection were treated equally. However, the majority of samples that were correctly identified as being from either a mildly or a highly pathogenic infection were from early or late time points, respectively. It is therefore likely that taking time post-infection into account would yield an even more accurate signature.
Figure 2. A 57-gene analogue signature predicts respiratory virus pathogenicity.

a | A meta-analysis was carried out on publically available transcriptional profiles of lung samples obtained from mice infected with influenza virus or severe acute respiratory syndrome coronavirus (SARS CoV). Two signatures predictive of the severity of infection (non-lethal versus lethal) were derived, a digital signal and an analogue signal. b–d | Each dot represents a gene expression profile; non-lethal infections are indicated by green dots, and lethal infections are indicated by red dots. Regions defined for positive identification of non-lethal (green square) and lethal (red square) infections are indicated. Classification results are shown for the entire gene expression profile (part b), the digital gene signature (part c) and the analogue gene signature (part d). The digital signature comprises a subset of genes (groups 1 and 2; 74 genes) for which expression changes in the opposite direction in lethal versus non-lethal infections, with respect to mock infection, and the analogue signature comprises a subset of genes (groups 3 and 4; 57 genes) that are differentially expressed between lethal and non-lethal infections without reference to mock infections. The analogue signature was more accurate at predicting lethal viral infection (true positives (TP) = 43%) than either the digital signature (TP = 1%) or the entire gene expression profile (TP = 0%).
From these studies, it is now apparent that highly pathogenic respiratory viruses induce or suppress the expression of many of the same genes as mildly pathogenic viruses, but to a greater degree (and with different kinetics). Therefore, knowing the identity of genes that are differentially expressed in response to infection provides only part of the information needed to predict pathogenicity. The magnitude and timing of the host response are crucial determinants of the eventual disease outcome and might have important implications for antiviral therapy. To date, efforts to target the host response with a range of anti-inflammatory drugs have been largely unsuccessful13, and it is likely that effective host-directed therapy will depend not only on the target, but also on exactly when elements of the host response are suppressed or enhanced. Moreover, these findings point to the need for computational approaches that can describe nonlinear relationships (see below) and account for various factors associated with large multivariate data sets21. This also suggests the need for a paradigm shift in biomarker discovery to one that looks at sets of quantitative molecular measurements.
Signatures of vaccine efficacy. The application of systems-level analyses to vaccine research — variously termed systems vaccinology22 or vaccinomics23 — has led to the identification of molecular signatures that are predictive of vaccine immunogenicity and to new insights into the mechanisms of action of vaccines. In one of the first large-scale uses of this strategy, gene expression profiling and computational methods were used to identify gene expression signatures predictive of the strength of human adaptive immune response to the yellow fever vaccine, YF-17D24.
Transcriptional profiling of peripheral blood mononuclear cells from vaccinated subjects revealed that YF-17D induces the expression of genes encoding proteins that are associated with viral recognition and with transcription factors that regulate type I IFNs. Although also characteristic of the transcriptional response to active viral infection, this response did not correlate with subsequent CD8+ T cell or neutralizing-antibody responses, which are thought to mediate protection. However, an alternative computational and classification method, discriminant analysis via mixed integer programming (DAMIP), identified a signature (consisting of complement system and stress response genes) that is highly accurate in predicting subsequent CD8+ T cell activation. This method also identified a separate signature (which includes tumour necrosis factor (TNF) receptor superfamily members) that accurately predicts neutralizing-antibody expression during infection. The robustness of these signatures was verified through the analysis of samples from a different group of subjects vaccinated with a different lot of vaccine, thereby identifying new correlates of vaccine immunogenicity24.
More recently, similar systems approaches have been used to evaluate innate and adaptive immune responses to vaccination against influenza viruses, with the goal of identifying early gene expression signatures that correlate with immunogenicity25. Over a 3-year period, a series of clinical studies was undertaken in which young adults were vaccinated with either inactivated influenza vaccine (TIV) or live-attenuated influenza vaccine (LAIV). Molecular signatures for predicting antibody responses were identified by combining gene expression profiling, antibody response data, real-time PCR analysis and DAMIP. The resulting predictive signature consisted of genes with known roles in antibody responses and other genes with previously unidentified roles in antibody or B cell responses. For example, one gene from the predictive signature, Camk4, encodes calcium–calmodulin-dependent protein kinase type IV (CaMKIV), a protein known to be involved in multiple immune system processes. However, it was not known whether this protein had a role in antibody responses25. To demonstrate the ability of the systems biology approaches used in this study to identify biologically significant targets, Camk4-knockout and wild-type mice were vaccinated with TIV, and the Camk4-knockout mice exhibited significantly higher antibody titres than wild-type mice 7, 14 and 28 days after vaccination, thus revealing that Camk4 is important in regulating B cell responses. Although further investigations are needed to confirm many of these signature predictions, these studies demonstrate that systems approaches can both identify biological targets and generate new testable hypotheses related to the mechanism of vaccine action.
An expanding view of the transcriptome. Until recently, transcriptional profiling depended on the use of microarrays to measure the expression of well-annotated protein-coding genes. The advent of next-generation sequencing (Box 2), however, has brought the ability to rapidly sequence the entire RNA complement of cells or tissues. This has led to a much expanded concept of the host transcriptome, as most recently revealed by the Encyclopedia of DNA Elements (ENCODE) project26. It is now apparent that as much as three-quarters of the human genome is capable of being transcribed and that cells contain vast numbers and varieties of non-protein-coding RNAs27. Some of these non-coding RNAs, such as microRNAs, have been well studied and are known to have roles in viral infection28. For most others, functionality is less clear, but there is growing evidence that long non-coding RNAs also play a part in transcriptional and epigenetic gene regulation and in disease29.
RNA sequencing (RNA-seq) analysis of the host response to SARS CoV infection has revealed the differential expression of a range of host long non-coding RNAs (>200 nucleotides long) in lung samples from virus-infected mice30. Many of these RNAs have similar expression patterns in vitro during influenza virus infection and during type I IFN treatment, suggesting that they are involved in regulating the innate immune response to a range of viruses30. Expanding these analyses to include the sequencing of small RNAs also revealed the differential expression of more than 200 small RNAs, such as small nucleolar RNAs (snoRNAs) and PIWI-interacting RNAs (piRNAs), in response to SARS CoV and influenza virus infections31. Similarly, RNA-seq has revealed that an HIV-infected CD4+ T cell line exhibits differential expression of host microRNAs, snoRNAs and pseudogenes compared with uninfected cells32. Viral mRNA constitutes a surprisingly large portion of the total RNA in HIV-infected CD4+ T cells (in this study, nearly 40% by 24 hours after infection), and reads mapping to the viral genome have revealed novel viral RNA splice variants. A correlative analysis that combined mRNA-seq and small RNA-seq data suggested additional roles for host microRNAs in T cell activation and in transcriptional and cell cycle regulation during HIV infection33.
Together, these studies attest to the power of RNA-seq to provide entirely new views of the transcriptional landscape and to highlight the previously unanticipated changes in transcription that occur in response to viral infection. Such insights are not limited to host transcription, as a combination of RNA-seq and mass spectrometry recently revealed that human cytomegalovirus (HCMV; a 240 kb DNA virus) produces hundreds of previously unidentified transcripts and short proteins that might have functional, regulatory or antigenic properties34. Although the functional significance of changes in non-coding-RNA expression are only beginning to be examined29, a better grasp of non-coding-RNA expression and function will certainly be necessary for a complete understanding of viral pathogenesis and the innate and adaptive immune responses, and for a more general view of gene regulation in the context of viral infection. Unfortunately, despite the advances that can be made using RNA-seq, the extensive computing infrastructure needed to handle large data files and the computational prowess required to align, assemble and analyse short sequence reads continues to put the approach out of reach for most laboratories.
Box 1: Microarrays.
DNA microarrays are widely used for global transcriptome profiling and remain the workhorse technology of systems biology. Microarrays can trace at least part of their ancestry to the Southern blot, in which DNA fragments are immobilized on a filter membrane for subsequent detection by hybridization of labelled DNA, and Southern himself has provided a detailed description of the many early technological developments that together led to the first commercial microarrays93. Over the past 15 years, the technology has progressed from cDNAs deposited on membranes by spotting robots, to commercial microarrays consisting of tens of thousands of oligonucleotides on glass slides or other solid supports. Today, major commercial providers of microarrays include Affymetrix, Agilent Technologies, Illumina and NimbleGen, with platforms varying in the length of oligonucleotide used, the number of oligonucleotides representing each gene, and the methods used for oligonucleotide synthesis and attachment to solid supports. Although microarrays are best known for their use in profiling the expression of protein-coding genes, the technology has also been adapted for profiling of microRNA expression, DNA methylation and SNPs, as well as for promoter analysis and the detection of genome-wide DNA copy number variation. Methods have also been developed to extract RNA from formalin-fixed paraffin-embedded (FFPE) samples, a technique that has opened the door for microarray-based gene expression profiling of the large numbers of clinical samples that have been archived using this preservation method. Microarrays can now provide considerably more information about virus–host interactions than simply the differential expression of protein-coding genes in response to viral infection94,95,96,97.
Box 2: Next-generation sequencing.
Next-generation sequencing is a massively parallel sequencing-by-synthesis approach that has replaced the first-generation automated Sanger sequencing method for genome analysis98. The approach generates hundreds of millions of bases of high-quality DNA sequence per run and has enabled the rapid sequencing of entire genomes. It is also used for transcriptome profiling, an approach referred to as RNA sequencing (RNA-seq)99. This has a number of advantages over microarray-based profiling, including the ability to identify and quantify rare or as-yet-undiscovered transcripts and the ability to provide information on alternative splicing, genetic variation, and gene and exon boundaries. The method used for the construction of the cDNA library determines the type of transcripts to be sequenced; these include small RNAs (<200 bases; for example, microRNAs, small nucleolar RNAs and PIWI-interacting RNAs), mRNAs (various types of polyadenylated transcripts) or whole transcriptomes (polyadenylated and non-polyadenylated transcripts). After sequence reads are generated, they are aligned to a known reference genome, or if no reference genome is available, sequences can be assembled de novo. Because of the short length of sequence reads, the alignment and assembly of sequence information is a significant computational challenge100. Additional potential applications of next-generation sequencing that are relevant to systems virology include the detection of binding events involving protein–DNA (ChIP–seq (chromatin immunoprecipitation followed by sequencing)) or protein–RNA (RIP–seq and CLIP–seq (crosslinking immunoprecipitation followed by high-throughput sequencing)) and DNA methylation profiling (Methyl–seq)59,101,102.
Given the advantages of next-generation sequencing, some have suggested that it will soon replace the use of microarrays for transcriptomic profiling. However, the enormous amount of data generated by next-generation sequencing requires an extensive information technology and computational infrastructure for data processing, storage and analysis. These costs and complexities, coupled with the cost of the sequencing itself, make it likely that microarrays will continue to be widely used for some time.
Beyond the transcriptome
Of course, gene expression profiling provides only one measure of the host response to infection. In recent years, advances in systems-wide technologies have facilitated a 'multi-omics' approach that includes proteomics, metabolomics, lipidomics and elucidating virus–host protein interactomes. All these measurements are adding to our understanding of the host response to viral infection, and new abilities to evaluate the role of host genetics and epigenetics are adding additional layers of complexity.
Alterations in cellular metabolism. Viruses have long been known to cause changes in host metabolism; however, the full extent of such changes was not clear until systems approaches were used to evaluate the metabolomic reprogramming that results from HCMV infection35. Using liquid chromatography–mass spectrometry to directly measure the levels of more than 160 different metabolites, it was discovered that HCMV infection induces large increases in numerous metabolites, including glycolytic and tricarboxylic acid (TCA) cycle intermediates, amino acids, NADH and pyrimidine. The metabolic signature induced by HCMV infection is readily distinguishable from the signature associated with the transition of quiescent cells into the G1 phase of the cell cycle, revealing the replacement of cellular metabolic homeostasis with an HCMV-specific metabolic programme. Systems-level metabolic flux profiling produced a first-of-a-kind metabolic map showing linkages between compounds and quantitative information about metabolic activity. The map is biochemically revealing, indicating that there is a global upregulation of metabolism by HCMV, with the greatest increase in the TCA cycle and its efflux to feed fatty acid biosynthesis36.
To determine whether this reprograming of the host metabolome is cell type or virus specific, fibroblast and epithelial cells were used to compare the host response to two strains of HCMV and two strains of herpes simplex virus type 1 (HSV-1)37. All four viruses produced significant changes in approximately 50% of the metabolome. Interestingly, the changes are consistent across different strains of the same virus and across cell types, but differ markedly between HCMV and HSV-1, demonstrating that these viruses induce distinct metabolic programmes. The findings derived from these systems-level approaches generated the hypotheses needed to drive additional focused studies in which more traditional methods were used to investigate the molecular mechanisms underlying the virus-specific hijacking of the host metabolome and the relevance of these mechanisms to potential therapeutic interventions38,39,40,41,42.
Genomic and lipidomic analyses have also revealed that infection of primary bone marrow-derived macrophages with mouse cytomegalovirus results in a downregulation of metabolites involved in the cholesterol metabolism pathway43. The lowering of cholesterol levels is mediated through the IFN-dependent downregulation of sterol regulatory element-binding protein 2 (SREBP2), a transcription factor that regulates sterol biosynthesis. Pharmacological or RNAi-mediated inhibition of the sterol pathway was shown to result in increased protection against viral infection in cell culture and in mice, demonstrating the potential benefit of targeting a host metabolic pathway as an antiviral strategy.
Virus–host interactomes. Based on the premise that viral proteins interact with cellular factors to promote efficient viral replication and pathogenesis, system-wide small interfering RNA (siRNA) or short hairpin RNA (shRNA) screens, yeast two-hybrid libraries and bioinformatic methods are being used to construct and describe virus–host interactomes and in turn identify cellular targets for therapeutic intervention. Such interactomes have been generated for numerous viruses, including influenza virus, HIV, dengue virus, hepatitis C virus (HCV), herpesviruses and SARS CoV, and have yielded 'hit lists' of cellular factors that might be important in viral pathogenesis44,45,46,47,48,49,50,51. Although the number of interactions identified by these studies is impressive, few of the genes identified have been subjected to functional analyses to confirm their role in viral replication. Moreover, there is little overlap in the host factors identified by the different screens. This could be due to a variety of factors, including variation in the screening systems, cell types and viruses used, as well as differences in the methods applied to identify interacting partners. The benefits and shortcomings of these studies, along with factors affecting their outcome, have been the subject of several detailed reviews52,53,54,55.
To fully realize the collective information residing in this vast collection of data, it will be necessary to develop computational and mathematical methods which are capable of fully integrating interactomes (as well as their associated metadata) that have been constructed using different methods or obtained from different biological systems. In a first attempt at such integration, meta-analysis of virus–host interactome data for five distinct viruses identified both common and virus-specific human protein targets56. Common targets include proteins involved in the cell cycle, apoptosis, the unfolded-protein response and nuclear transport. Many of the common host targets identified are multifunctional hubs — that is, they have multiple functions or roles within the cell. When coupled with the fact that many viral proteins interact with multiple host proteins, this combination of factors might explain how viruses, with their relatively small genomes, are capable of dysregulating so many aspects of host biology. Unfortunately, because these common host targets often have numerous cellular functions, focusing on them as drug targets might be problematic52.
In an interesting twist on these studies, virus–host interactome data, together with data on host transcriptional changes resulting from the expression of 123 viral ORFs derived from DNA tumour viruses, were used to predict cellular genomic variations (for example, mutations, deletions or translocations) that can give rise to cancer57. By defining the rewiring of cellular networks and pathways caused by the viral proteins and identifying a list of host proteins that are central to the rewiring, the systematic identification of host targets of DNA tumour viruses was found to be as successful as traditional large-scale cataloguing of tumour mutations for cancer gene identification. This suggests that disease phenotypes, whether resulting from viral infection or cancer, might be the result of network perturbations rather than individual genetic or genomic variations.
Host genetics and epigenetics. Host genetic variation has typically been thought of as a confounding factor that limits the ability to draw conclusions from data obtained using outbred (for example, human and nonhuman primate) populations. More recently, attempts are being made to better understand how genetic diversity influences infection outcome and how knowledge of genetic diversity can be incorporated into the construction of robust and predictive network models. One particularly exciting example is the Collaborative Cross mouse resource. The Collaborative Cross is a unique panel of multiparental recombinant inbred mouse strains designed to capture the level of genetic diversity found in outbred populations; this provides a resource for systematically identifying individual and multiple host genetic traits that contribute to complex immune phenotypes and disease outcome (Box 3).
As an example, a genetically diverse panel of pre-Collaborative Cross mice (not fully inbred) determined to have severe or mild responses to influenza virus infection was used to identify expression quantitative trait loci (eQTLs) associated with the host response to infection. Twenty-one high-confidence eQTLs were identified, 17 of which were confirmed using mice from the eight Collaborative Cross founder strains58. Many of these genes have known functions related to immunity or the host response to infection, such as Ifi27l2a (IFNα-inducible 27-like 2A), Clec16a, Pde7a (high-affinity cyclic AMP-specific 3′,5′-cyclic phosphodiesterase 7A) and Tcf7l1 (transcription factor 7-like 1), whereas for others, such as the serine/threonine protein kinase gene Sik1 and sentrin-specific protease 5 (Senp5), their role in influenza virus infection is not yet clear. Structural equation modelling was used to identify potential regulatory relationships between additional genes and the validated eQTLs, suggesting that these genes and corresponding subnetworks have important roles in the host response by either promoting a protective (mild) or a pathologic (severe) response to influenza virus infection, depending on the specific genetics of the host. Thus, using this genetically diverse population, high-confidence gene candidates involved in regulating the host response to influenza virus infection were identified, allowing for future investigations into their utility as therapeutic targets.
It is also becoming apparent that epigenetic mechanisms have a role in regulating the outcome of viral infection, and methods for genome-scale mapping of DNA methylation59 and histone modification60 are now available. Such epigenetic modifications contribute to chromatin structure and organization, which in turn influence transcriptional activity, the immune response61 and viral latency62. Viruses can also use epigenetic control mechanisms to their advantage; the NS1 protein of H3N2 influenza virus, for example, acts as a histone mimic to suppress the expression of antiviral genes63. Characterizing and understanding epigenetic mechanisms might therefore be an essential requirement for the construction of gene regulatory networks64,65.
Box 3: The Collaborative Cross.
The Collaborative Cross is a genetic reference population of recombinant inbred (RI) mice derived from eight laboratory mouse strains50. Such populations are used to study complex traits and are most beneficial when they contain large numbers of lines that exhibit substantial and uniform genetic variation. In the case of the Collaborative Cross, five inbred strains (A/J, C57BL/6J, 129S1/SvImJ, NOD/ShiLtJ and NZO/HlLtJ) and three wild-derived strains (CAST/EiJ, PWK/PhH and WSB/EiJ) are being used to generate a large panel of RI lines through a randomized funnel breeding scheme103, resulting in hundreds of independent RI lines in which genetic variation is randomly and uniformly distributed throughout the genome. When they are established, RI lines can be intercrossed among themselves to generate recombinant intercross (RIX) lines or backcrossed to other non-Collaborative Cross lines to generate RI backcross (RIB) lines. RIX lines maximize genetic diversity, and because they are outbred, they are better models of human populations and can provide information on gene dominance and epistasis104. RIB lines can be used, for example, to evaluate whether specific allele combinations alter the phenotype associated with a given target gene (such as a dominant targeted gene knockout). For example, a comparison between viral titre in the lungs and the extent of airway inflammation for a panel of pre-Collaborative Cross (not fully inbred) mice demonstrates the impact of genetic diversity on influenza virus disease phenotypes (see the figure; each diamond represents a different mouse). Phenotypes range from high viral titre and high inflammation to low viral titre and little inflammation; in addition, some animals show unique phenotypes, such as a high viral titre with little accompanying inflammation. The Collaborative Cross is being developed as a resource for the biomedical research community, and genotype information and mouse line availability is publically available at University of North Carolina (UNC) Systems Genetics, Chapel Hill, USA.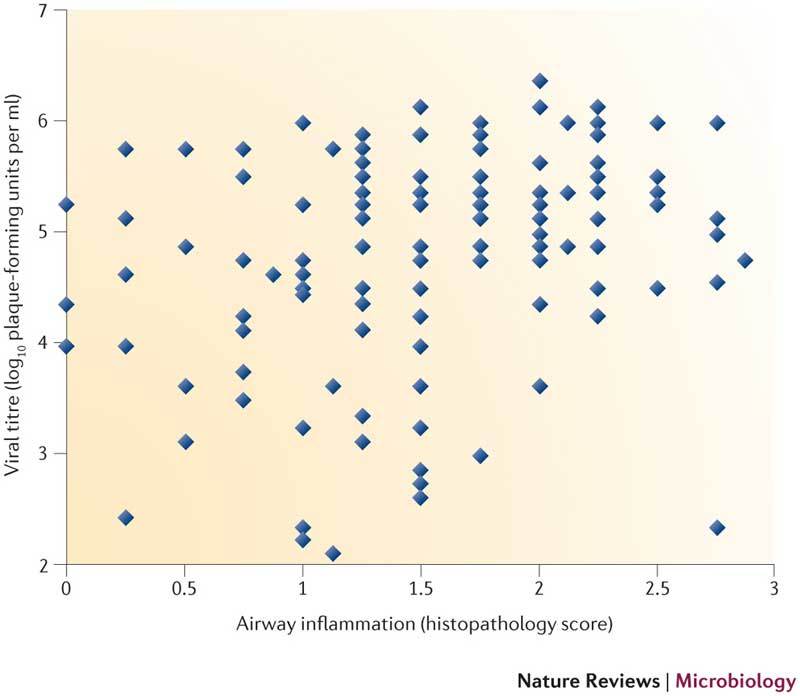
Putting the pieces together
As more and more high-throughput data become available, systems virology is poised to enter a new phase to fulfil its initial promise of revolutionizing our understanding of virus–host interactions. To do this, the field must move beyond just the listing of molecules that are differentially expressed on viral infection. Instead, the relationships between key molecules must be defined. Such relationships may be cause-and-effect relationships (for example, transcription factors and their target genes), the result of co-expression, or due to genetic or direct physical interactions. Here, we give examples of several methods that are being used to further our understanding of virus–host interaction networks, and we then discuss key computational challenges that must be addressed.
Network modelling and analysis explores the relationships among molecules and the structure and organization of these relationships to predict the behaviour of the network or system. For example, the context likelihood of relatedness (CLR) method is used to predict genes that are highly interconnected (referred to as hubs) or that exhibit a high degree of betweenness centrality (referred to as bottlenecks). Genes with high betweenness centrality exhibit fewer connections than hub genes, but because they are located between (and connect or bridge) multiple subnetworks, they can have a powerful role in controlling network signalling (Fig. 3). Bottleneck genes often function as key genes in the regulation of disease progression and are therefore attractive targets for further experimentation66,67. An alternative method, co-regulation network analysis (PCluster)68, has been combined with genome-wide expression profiling and yeast two-hybrid analysis to identify relationships between gene expression and direct physical interactions, revealing previously unrecognized roles for several cellular and viral proteins in the host response to H1N1 influenza virus69. These proteins include a network of RNA-binding proteins, components of the WNT signalling pathway and viral polymerase subunits. However, as these types of analyses often only infer correlations between network components, additional studies are required to verify model predictions.
Figure 3. Co-regulation networks.

a | A schematic showing one bottleneck gene and two hub genes in a hypothetical network. Hubs are highly connected to other nodes in the network. Bottlenecks have a high degree of betweenness centrality and connect or act as bridges between subnetworks. b | The context likelihood of relatedness method was used to infer functional associations between differentially expressed genes responding to H5N1 influenza virus infection in mice. Genes (nodes) are coloured according to the ratio of gene expression values for high-dose infection to low-dose infection. Red indicates higher expression and blue indicates lower expression in the high dose than in the low dose. The complexity of these networks does not allow a visual determination of bottlenecks. Rather, the betweenness centrality is calculated for each node, and the size of the node is relative to this value. The larger nodes are major bridges between different parts of the network. c | A major hub from a separate subnetwork within the H5N1 influenza virus co-regulatory network.
These network analysis methods have also been used to analyse the network topology of networks derived from proteomic and lipidomic profiling data. For example, such an analysis identified two mitochondrial fatty acid oxidation enzymes, DCI (also known as ECI1) and HADHB, as bottleneck proteins and possible targets through which HCV disrupts cellular metabolic homeostasis70. The importance of DCI (and of cellular metabolic homeostasis in general) during HCV infection was then confirmed by additional means, which included pharmacological inhibition of fatty acid oxidation and targeted siRNA knockdown techniques, both of which demonstrated that DCI is required for productive HCV infection in hepatoma cell lines71,72. Similarly, an analysis of the interaction networks between host proteins and HCV core and NS4B proteins has been used to identify host proteins that constitute potential anti-HCV therapeutic targets, including α-enolase, paxillin and a solute carrier protein (SLC25A5)73. A better understanding of network topology will not only provide the opportunity to identify potential targets for therapeutic intervention, but also offer insights into possible off-target effects on network signalling that might be induced by drug treatment.
Although much can be learned from the construction and topological analysis of host–pathogen interaction networks using samples from whole tissues, the heterogeneity of cell types present in most tissues means that such networks provide a generalized picture of the changes that occur in the host during the course of infection. For example, it is difficult to delineate from these types of networks the signalling events that might occur between infected lung epithelial cells and cells of the immune system, both inside and outside the infected tissue. These intercellular interactions are also controlled by signal transduction pathways, which communicate signals from the extracellular environment to intracellular effector processes. To fully interpret the data, we need a much better understanding of intercellular signalling processes, the cells that are involved and the directionality of their effects on infection outcome.
A few studies have begun to explore cell type-specific and intercellular signalling on a system-wide scale. For example, flow cytometry and gene expression data from bronchoalveolar lavage (BAL) fluid from young adult and aged macaques infected with 2009 pandemic H1N1 influenza virus were analysed in conjunction with data from the Immune Response In Silico (IRIS) database. This database contains cell type-specific gene expression patterns associated with various types of immune cells. By computationally comparing differentially expressed genes in BAL with cell type-specific gene expression patterns in the IRIS database, it was possible to identify genes associated with specific immune cell types, including activated dendritic cells, CD4+ and CD8+ T cells and naive B cells. In particular, genes associated with B cell and T cell markers were more highly upregulated in young adult animals74.
Recent studies of mouse models of intestinal inflammation induced by TNF treatment provide another good example of how systems approaches can be used to evaluate signalling between cell types in a complex tissue environment75,76. In these studies, the authors combined flow cytometry measurements and phosphoprotein, cytokine and chemokine expression data from various immune cell types over time and under diverse conditions and were thus able to construct statistically robust multivariate regression models that related the phosphoprotein signals, cytokines, chemokines and cell types to specific phenotypes. These models helped to elucidate key molecular and cellular processes governing epithelial cell apoptosis and proliferation in response to TNF treatment. For example, monocyte chemotactic protein 1 (MCP1; also known as CCL2) was predicted by the model to be especially protective against TNF-induced apoptosis, and this hypothesis was confirmed by treating with an MCP1-specific antibody before TNF administration. The model also indicated that plasmacytoid dendritic cells might be particularly important for inducing apoptosis, and depleting these cells from mice under conditions that normally produced the most severe epithelial cell apoptosis did indeed revert the TNF-induced phenotype to the mildest outcome.
In addition to constructing network models that span intra- and intercellular signalling processes, it will be necessary to consider nonlinear relationships such as how the network functions over time (that is, the dynamics of the system). This is particularly true in light of evidence (discussed above) that the magnitude and timing of the host response to respiratory viruses are crucial determinants of the eventual disease outcome. Similar evidence is accumulating that the outcome of HIV infection is also related to the activation dynamics of host gene regulation77,78. However, high-throughput data are typically static and often not adequate for modelling dynamic systems. To help overcome this limitation in analysing network dynamics, inference methodologies are being devised to reinterpret activity differences caused by system perturbations as differences in observation time. For example, changes in pathogen-induced gene expression that are associated with genetic variability, whether in the pathogen (for example, mutant viruses) or the host (as occurs in the Collaborative Cross mouse model), can potentially be used to indirectly infer the crucial dynamics of a system without having to measure the system over time. This innovative approach has only recently become feasible with major breakthroughs in the theory of dynamic systems and in geometrical high-dimensional analysis methodology79,80.
As virology continues to transition into a more quantitative science, increasing attention must be paid not only to network dynamics but also to other nonlinear interactions, such as cooperative or synergistic relationships, which characterize so much of biology. Current biomarker discovery and the identification of molecular predictors of adjuvant, vaccine or, more generally, drug efficacy are largely unsuccessful because nonlinear interactions between molecules, as well as genetic diversity in populations, are not taken into sufficient consideration. Geometrical methods (that is, methods used to identify the structure in data by identifying spatial and temporal relationships) are increasingly being used in the analysis of high-throughput molecular data. In particular, novel combinations of geometrical methods, such as those based on singular value decomposition (SVD) and multidimensional scaling (MDS)81,82, are beginning to be used in systems virology to better understand the nonlinear interactions between variables and isolate those interactions from biological noise.
Liver biopsies were carried out on patients who were infected with HCV and had received liver transplants, and SVD–MDS analysis of transcriptomic data from these biopsy samples, in combination with categorical analysis (to take into account variables such as age, time post-transplantation and fibrosis score), has been used to identify a molecular signature for patients at risk of developing severe fibrosis83. SVD–MDS and co-abundance networks (which relate molecules to each other on the basis of their abundance profiles) were also used to integrate proteomic and metabolomic data sets obtained from the same cohort of patients. This strategy identified a potential role for oxidative stress in rapid fibrosis progression after the transplant surgery and identified serum metabolites that might prove useful as biomarkers for predicting progression to fibrosis84. This understanding of network structure can now be used to simulate human liver metabolism using novel flux balance modelling approaches to better comprehend and eventually treat disease85. Additional geometrical approaches have been described and should prove useful for effectively bridging different technologies86,87 and for integrating diverse types of data, thereby enabling better analysis of the data that are already available in public databases and repositories. Finally, geometrical methods, as well as links between geometry, information theory and probability theory7, will also help to identify causal relationships88, which is clearly a currently unmet challenge. Unlike purely statistical approaches, geometrical methods can be used to integrate different individual measures for the purposes of comparison and combination into coherent objects that identify relationships between genes, transcripts or proteins.
Conclusions
After having been extensively hyped as a paradigm shift, systems-level approaches have since been criticized for failing to rapidly fulfil their initial grand promises. Standing in the way have been numerous technical, experimental and mathematical hurdles. However, as discussed in this Review, significant progress is being made, and new computational approaches are leading the way. Also of importance is the ever-growing availability of high-throughput data in public databases; indeed, data sharing is crucial to the future success of systems virology (Box 4). To date, bench work has been the necessary precedent to computational approaches, but we are now at a point when there are sufficient data available that computational methods can be the starting point for making discoveries, generating hypotheses and, in turn, guiding targeted bench work. A prime example of this is the flourishing of virtual screening methods in drug discovery; these methods depend on systematic drug characterization efforts and on public databases holding functional genomics information obtained under standard experimental conditions89,90. Moreover, the information gained from systems approaches forms the basis for what has been termed P4 (personalized, predictive, preventive and participatory) medicine91. In the case of infectious disease, genetic information on the individual and the pathogen, disease-predictive molecular signatures, targeted risk reduction and prophylactic measures, and active patient participation will merge into a new approach to medical care92.
In conclusion, contemporary virology cannot afford to simply catalogue myriad circumstantial observations. Systems-level approaches provide the opportunity to assemble the incomplete puzzle of biology in a meaningful way that will advance our understanding of how viruses cause disease and lead to improved patient care. Although there will always be a need for traditional microbiology, the success of undergraduates in this day and age will depend on their ability to combine traditional skills with systems approaches and mathematics. The time is ripe, the data are here, and the mathematics to put them together is coming along. Take heed of the words on Plato's doorstep: “αγεωμετρητος μηδεις εισιτω” (“Let no one ignorant of geometry enter”).
Box 4: The importance of data sharing.
The rapid dissemination of high-throughput omics data sets to the scientific community is an important element for the future success of systems virology. The complexity of virus–host interactions can be unravelled only through meta-analyses that rely on advanced computational and mathematical approaches and the integration of large, coherent and systematic multi-dimensional data sets. Owing to the immense amounts of time, expertise, manpower and expense that are required to produce these data sets, it is imperative for the systems virology community to share data in a timely manner. In most cases, owing to the richness of information contained in these data sets, this can be done with little chance of compromising one's own research. Two of the US National Institute of Allergy and Infectious Diseases (NIAID)-sponsored Bioinformatics Resource Centers, ViPR (Virus Pathogen Resource) and IRD (Influenza Research Database), are charged with capturing, publically sharing, storing, integrating and visualizing systems-wide high-throughput omics data sets that detail the host response to viral infection. Importantly, in addition to capturing data types such as transcriptomic, proteomic, metabolomic, lipidomic and ChIP–seq (chromatin immunoprecipitation followed by sequencing) data, the goal of these databases is to capture extensive metadata that provide users with the experimental details needed to facilitate data interpretation. The practice of linking metadata to several forms of the omics data (be it raw, analysed or modelled) enables ViPR and IRD to meet the demands of a range of end users (see the figure). For instance, at their portals, users can search for information on individual genes, download lists of differentially expressed genes or link metadata to raw data for doing meta-analysis. All laboratories, as a service to the community, should be committed to sharing these types of data sets, whether through organization-specific public websites, the appropriate NIAID Bioinformatics Resource Center or a combination of both.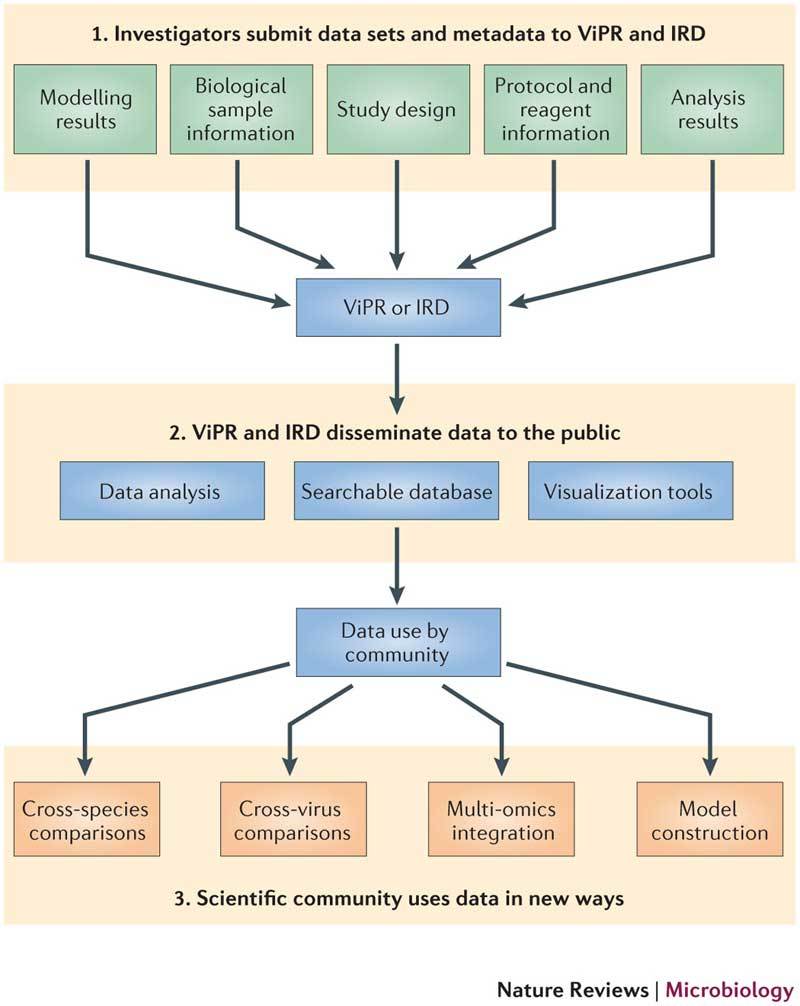
Acknowledgements
The authors thank L. Josset for generating the networks in figure 3, and M. Heise and M. Ferris for providing the data used in box 3. Research in the author's laboratory is supported by Public Health Service grants R2400011172, R2400011157, P30DA015625, P51RR00166 and U54AI081680, and by federal funds from the US National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contract HHSN272200800060C.
Glossary
- Complement system
Blood proteins that react with one another in a cascade to aid the ability of phagocytic cells to eliminate microorganisms. Complement proteins also have a role in the development of inflammation.
- Small nucleolar RNAs
(snoRNAs). RNAs that guide the modification (for example, methylation or pseudouridylation) of other RNAs, particularly ribosomal RNAs.
- PIWI-interacting RNAs
(piRNAs). Small RNAs that are thought to be involved in gene silencing through the formation of ribonucleoprotein complexes with PIWI proteins.
- RIP–seq
Immunoprecipitation of RNA-binding proteins followed by high-throughput sequencing of the bound RNA.
- CLIP–seq
(Crosslinking immunoprecipitation followed by high-throughput sequencing). A screening method used to identify RNA sequences that interact with either RNA-binding proteins or other RNAs.
- Metabolic flux profiling
A measurement approach that uses liquid chromatography–tandem mass spectrometry to quantify the rate of conversion of biochemical molecules in a metabolic network after perturbing the system. Systems-level metabolic flux profiling is a high-throughput approach to quantifying changes in metabolic activity.
- Short hairpin RNA
(shRNA). A type of RNA that forms a tight hairpin which has the ability to silence gene expression through RNAi.
- Unfolded-protein response
A cellular stress response to the accumulation of unfolded proteins in the ER. The response is characterized by a signal transduction pathway that aims to restore homeostasis by limiting protein biosynthesis and increasing the abundance of molecular chaperones involved in protein folding.
- Expression quantitative trait loci
(eQTLs). Genomic loci, as identified by gene expression profiling, that regulate mRNA expression. eQTLs are mapped by computationally connecting DNA sequence variation with variation in gene expression, providing information on how host genetics affects the function of molecular networks.
- Structural equation modelling
A multivariate analysis technique for testing and estimating causal relationships among variables.
- Betweenness centrality
A measure of the location of a gene in a network. Genes with high betweenness centrality, referred to as bottleneck genes, are located between and therefore connect different portions of the network (that is, different subnetworks).
- Epistasis
The phenomenon in which the effects of one gene are modified by one or more other genes.
- Network topology
The arrangement and connections of the various components of a network.
Biographies
G. Lynn Law received her Ph.D. in chemistry at Washington State University, Pullman, USA, and has expertise in the fields of transcriptional and translational control in eukaryotic systems. During the past 5 years, she has been a senior research scientist in Michael Katze's laboratory at the University of Washington, Seattle, where she is the project manager of the Systems Virology Center, sponsored by the US National Institute of Allergy and Infectious Diseases (NIAID) Systems Biology for Infectious Diseases research programme.
Marcus J. Korth received his Ph.D. in microbiology from the University of Washington, Seattle, USA, for work on the role of fimbrial adhesins in Escherichia coli pathogenesis. He is now a senior research scientist in Michael Katze's laboratory at the University of Washington and is interested in applying high-throughput technologies to the study of viral pathogenesis.
Arndt G. Benecke is a tenured researcher with the Centre National de la Recherche Scientifique (CNRS) at the Université Pierre et Marie Curie, Paris, France. He has strong expertise in the biochemistry, mathematics and bioinformatics of gene expression regulation in higher eukaryotes in the context of host–pathogen interactions and immune responses. His research focuses on transcription regulation and functional genomics using both novel bioinformatics and geometrical analysis and new representation strategies.
Michael G. Katze received his Ph.D. in microbiology from Hahnemann University Hospital, Philadelphia, USA, and is now Professor of Microbiology at the University of Washington, Seattle, USA, where he serves as Associate Director at the Washington National Primate Research Center and Head of the centre's Division of Nonhuman Primate Systems Biology. His research interests are focused on applying systems biology approaches to the study of virus–host interactions and the innate immune response.
Related links
FURTHER INFORMATION
PowerPoint slides
Competing interests
The authors declare no competing financial interests.
References
- 1.Aderem A, et al. A systems biology approach to infectious disease research: innovating the pathogen-host research paradigm. mBio. 2011;2:e00325-10. doi: 10.1128/mBio.00325-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tisoncik JR, Katze MG. What is systems biology? Future Microbiol. 2010;5:139–141. doi: 10.2217/fmb.09.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Katze MG. Systems Biology. 2013. [Google Scholar]
- 4.Macilwain C. Systems biology: evolving into the mainstream. Cell. 2011;144:839–841. doi: 10.1016/j.cell.2011.02.044. [DOI] [PubMed] [Google Scholar]
- 5.Diercks A, Aderem A. Systems approaches to dissecting immunity. Curr. Top. Microbiol. Immunol. 2013;363:1–19. doi: 10.1007/82_2012_246. [DOI] [PubMed] [Google Scholar]
- 6.Lauffenburger DA. The multiple dimensions of integrative biology. Integr. Biol. (Camb.) 2012;4:9. doi: 10.1039/C1IB90041B. [DOI] [PubMed] [Google Scholar]
- 7.Lesne A. Multiscale analysis of biological systems. Acta Biotheor. 2013;61:3–19. doi: 10.1007/s10441-013-9170-z. [DOI] [PubMed] [Google Scholar]
- 8.Geiss GK, et al. Large-scale monitoring of host cell gene expression during HIV-1 infection using cDNA microarrays. Virology. 2000;266:8–16. doi: 10.1006/viro.1999.0044. [DOI] [PubMed] [Google Scholar]
- 9.Fukuyama S, Kawaoka Y. The pathogenesis of influenza virus infections: the contributions of virus and host factors. Curr. Opin. Immunol. 2011;23:481–486. doi: 10.1016/j.coi.2011.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Korth, M. J., Tchitchek, N., Benecke, A. G. & Katze, M. G. Systems approaches to influenza-virus host interactions and the pathogenesis of highly virulent and pandemic viruses. Semin. Immunol. 4 Dec 2012 (doi:10.1016/j.smim.2012.11.001). [DOI] [PMC free article] [PubMed]
- 11.Kash JC, et al. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature. 2006;443:578–581. doi: 10.1038/nature05181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kobasa D, et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature. 2007;445:319–323. doi: 10.1038/nature05495. [DOI] [PubMed] [Google Scholar]
- 13.Tisoncik JR, et al. Into the eye of the cytokine storm. Microbiol. Molec Biol. Rev. 2012;76:16–32. doi: 10.1128/MMBR.05015-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baskin CR, et al. Early and sustained innate immune response defines pathology and death in nonhuman primates infected by highly pathogenic influenza virus. Proc. Natl Acad. Sci. USA. 2009;106:3455–3460. doi: 10.1073/pnas.0813234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cilloniz C, et al. Lethal influenza virus infection in macaques is associated with early dysregulation of inflammatory related genes. PLoS Pathog. 2009;5:e1000604. doi: 10.1371/journal.ppat.1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cilloniz C, et al. Lethal dissemination of H5N1 influenza virus is associated with dysregulation of inflammation and lipoxin signaling in a mouse model of infection. J. Virol. 2010;84:7613–7624. doi: 10.1128/JVI.00553-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thomas PG, et al. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity. 2009;30:566–575. doi: 10.1016/j.immuni.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ichinohe T, Lee HK, Ogura Y, Flavell R, Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. J. Exp. Med. 2009;206:79–87. doi: 10.1084/jem.20081667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Allen IC, et al. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. 2009;30:556–565. doi: 10.1016/j.immuni.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang ST, Tchitchek N, Ghosh D, Benecke A, Katze MG. A chemokine gene expression signature derived from meta-analysis predicts the pathogenicity of viral respiratory infections. BMC Syst. Biol. 2011;5:202. doi: 10.1186/1752-0509-5-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khondoker MR, et al. Multi-factorial analysis of class predication error: estimating optimal number of biomarkers for various classification rules. J. Bioinform. Comput. Biol. 2010;08:945–965. doi: 10.1142/S0219720010005063. [DOI] [PubMed] [Google Scholar]
- 22.Pulendran B, Li S, Nakaya HI. Systems vaccinology. Immunity. 2010;33:516–529. doi: 10.1016/j.immuni.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Poland GA, Ovsyannikova IG, Kennedy RB, Haralambieva IH, Jacobson RM. Vaccinomics and a new paradigm for the development of preventive vaccines against viral infections. OMICS. 2011;15:625–636. doi: 10.1089/omi.2011.0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Querec TD, et al. Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nature Immunol. 2009;10:116–125. doi: 10.1038/ni.1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakaya HI, et al. Systems biology of vaccination for seasonal influenza in humans. Nature Immunol. 2011;12:786–795. doi: 10.1038/ni.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dunham I, et al. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Djebali S, et al. Landscape of transcription in human cells. Nature. 2012;489:101–108. doi: 10.1038/nature11233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Skalsky RL, et al. The viral and cellular microRNA targetome in lymphoblastoid cell lines. PLoS Pathog. 2012;8:e1002484. doi: 10.1371/journal.ppat.1002484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nature Rev. Genet. 2009;10:155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 30.Peng X, et al. Unique signatures of long noncoding RNA expression in response to virus infection and altered innate immune signaling. mBio. 2010;1:e00206-10. doi: 10.1128/mBio.00206-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peng X, et al. Integrative deep sequencing of the mouse lung transcriptome reveals differential expression of diverse classes of small RNAs in response to respiratory virus infection. mBio. 2011;2:00198-11. doi: 10.1128/mBio.00198-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chang ST, et al. Next-generation sequencing reveals HIV-1-mediated suppression of T cell activation and RNA processing and regulation of noncoding RNA expression in a CD4+ T cell line. mBio. 2011;2:e00134-11. doi: 10.1128/mBio.00134-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chang ST, et al. Next-generation sequencing of small RNAs from HIV-infected cells identifies phased microRNA expression patterns and candidate novel microRNAs differentially expressed upon infection. mBio. 2013;4:e00549-12. doi: 10.1128/mBio.00549-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stern-Ginossar N, et al. Decoding human cytomegalovirus. Science. 2012;338:1088–1093. doi: 10.1126/science.1227919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Munger J, Bajad SU, Coller HA, Shenk T, Rabinowitz JD. Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathog. 2006;2:e132. doi: 10.1371/journal.ppat.0020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Munger J, et al. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nature Biotech. 2008;26:1179–1186. doi: 10.1038/nbt.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vastag L, Koyuncu E, Grady SL, Shenk TE, Rabinowitz JD. Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog. 2011;7:e1002124. doi: 10.1371/journal.ppat.1002124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McArdle J, Schafer XL, Munger J. Inhibition of calmodulin-dependent kinase kinase blocks human cytomegalovirus-induced glycolytic activation and severely attenuates production of viral progeny. J. Virol. 2011;85:705–714. doi: 10.1128/JVI.01557-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Spencer CM, Schafer XL, Moorman NJ, Munger J. Human cytomegalovirus induces the activity and expression of acetyl-coenzyme A carboxylase, a fatty acid biosynthetic enzyme whose inhibition attenuates viral replication. J. Virol. 2011;85:5814–5824. doi: 10.1128/JVI.02630-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu ST, et al. Synaptic vesicle-like lipidome of human cytomegalovirus virions reveals a role for SNARE machinery in virion egress. Proc. Natl Acad. Sci. USA. 2011;108:12869–12874. doi: 10.1073/pnas.1109796108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McArdle J, Moorman NJ, Munger J. HCMV targets the metabolic stress response through activation of AMPK whose activity is important for viral replication. PLoS Pathog. 2012;8:e1002502. doi: 10.1371/journal.ppat.1002502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grady SL, Hwang J, Vastag L, Rabinowitz JD, Shenk T. Herpes simplex virus 1 infection activates poly(ADP-ribose) polymerase and triggers the degradation of poly(ADP-ribose) glycohydrolase. J. Virol. 2012;86:8259–8268. doi: 10.1128/JVI.00495-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Blanc M, et al. Host defense against viral infection involves interferon mediated down-regulation of sterol biosynthesis. PLoS Biol. 2011;9:e1000598. doi: 10.1371/journal.pbio.1000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pfefferle S, et al. The SARS-coronavirus-host interactome: identification of cyclophilins as target for pan-coronavirus inhibitors. PLoS Pathog. 2011;7:e1002331. doi: 10.1371/journal.ppat.1002331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brass AL, et al. Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008;319:921–926. doi: 10.1126/science.1152725. [DOI] [PubMed] [Google Scholar]
- 46.Krishnan MN, et al. RNA interference screen for human genes associated with West Nile virus infection. Nature. 2008;455:242–245. doi: 10.1038/nature07207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sessions OM, et al. Discovery of insect and human dengue virus host factors. Nature. 2009;458:1047–1050. doi: 10.1038/nature07967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brass AL, et al. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell. 2009;139:1243–1254. doi: 10.1016/j.cell.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Q, et al. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc. Natl Acad. Sci. USA. 2009;106:16410–16415. doi: 10.1073/pnas.0907439106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.The Complex Trait Consortium. The Collaborative Cross, a community resource for the genetic analysis of complex traits. Nature Genet.36, 1133–1137 (2004). [DOI] [PubMed]
- 51.Uetz P, et al. Herpesviral protein networks and their interaction with the human proteome. Science. 2006;311:239–242. doi: 10.1126/science.1116804. [DOI] [PubMed] [Google Scholar]
- 52.Friedel CC, Haas J. Virus–host interactomes and global models of virus-infected cells. Trends Microbiol. 2011;19:501–508. doi: 10.1016/j.tim.2011.07.003. [DOI] [PubMed] [Google Scholar]
- 53.Ma-Lauer Y, Lei J, Hilgenfeld R, von Brunn A. Virus–host interactomes — antiviral drug discovery. Curr. Opin. Virol. 2012;2:614–621. doi: 10.1016/j.coviro.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Panda D, Cherry S. Cell-based genomic screening: elucidating virus–host interactions. Curr. Opin. Virol. 2012;2:778–786. doi: 10.1016/j.coviro.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stertz S, Shaw ML. Uncovering the global host cell requirements for influenza virus replication via RNAi screening. Microbes Infect. 2011;13:516–525. doi: 10.1016/j.micinf.2011.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Meyniel-Schicklin L, de Chassey B, Andre P, Lotteau V. Viruses and interactomes in translation. Mol. Cell. Proteomics. 2012;11:M111.014738. doi: 10.1074/mcp.M111.014738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rozenblatt-Rosen O, et al. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature. 2012;487:491–495. doi: 10.1038/nature11288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bottomly D, et al. Expression quantitative trait loci for extreme host response to influenza A in pre-collaborative cross mice. G3 (Bethesda) 2012;2:213–221. doi: 10.1534/g3.111.001800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Laird PW. Principles and challenges of genomewide DNA methylation analysis. Nature Rev. Genet. 2010;11:191–203. doi: 10.1038/nrg2732. [DOI] [PubMed] [Google Scholar]
- 60.Garcia BA. Mass spectrometric analysis of histone variants and post-translational modifications. Front. Biosci. (Schol. Ed.) 2009;1:142–153. doi: 10.2741/s14. [DOI] [PubMed] [Google Scholar]
- 61.Knight JC. Genomic modulators of the immune response. Trends Genet. 2012;29:74–83. doi: 10.1016/j.tig.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ernberg I, Karimi M, Ekstrom TJ. Epigenetic mechanisms as targets and companions of viral assaults. Ann. NY Acad. Sci. 2011;1230:E29–E36. doi: 10.1111/j.1749-6632.2011.06357.x. [DOI] [PubMed] [Google Scholar]
- 63.Marazzi I, et al. Suppression of the antiviral response by an influenza histone mimic. Nature. 2012;483:428–433. doi: 10.1038/nature10892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Benecke A. Gene regulatory network inference using out of equilibrium statistical mechanics. HFSP J. 2008;2:183–188. doi: 10.2976/1.2957743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Berg J. Out-of-equilibrium dynamics of gene expression and the Jarzynski equality. Phys. Rev. Lett. 2008;100:188101. doi: 10.1103/PhysRevLett.100.188101. [DOI] [PubMed] [Google Scholar]
- 66.McDermott JE, Taylor RC, Yoon H, Heffron F. Bottlenecks and hubs in inferred networks are important for virulence in Salmonella typhimurium. J. Comput. Biol. 2009;16:169–180. doi: 10.1089/cmb.2008.04TT. [DOI] [PubMed] [Google Scholar]
- 67.Girvan M, Newman MEJ. Community structure in social and biological networks. Proc. Natl Acad. Sci. USA. 2002;99:7821–7826. doi: 10.1073/pnas.122653799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Segal E, et al. Module networks: identifying regulatory modules and their condition-specific regulators from gene expression data. Nature Genet. 2003;34:166–176. doi: 10.1038/ng1165. [DOI] [PubMed] [Google Scholar]
- 69.Shapira SD, et al. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell. 2009;139:1255–1267. doi: 10.1016/j.cell.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Diamond DL, et al. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. 2010;6:e1000719. doi: 10.1371/journal.ppat.1000719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McDermott JE, et al. Topological analysis of protein co-abundance networks identifies novel host targets important for HCV infection and pathogenesis. BMC Syst. Biol. 2012;6:28. doi: 10.1186/1752-0509-6-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rasmussen AL, et al. Systems virology identifies a mitochondrial fatty acid oxidation enzyme, dodecenoyl coenzyme A delta isomerase, required for hepatitis C virus replication and likely pathogenesis. J. Virol. 2011;85:11646–11654. doi: 10.1128/JVI.05605-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tripathi LP, et al. Network based analysis of hepatitis C virus core and NS4B protein interactions. Mol. Biosyst. 2010;6:2539–2553. doi: 10.1039/c0mb00103a. [DOI] [PubMed] [Google Scholar]
- 74.Josset L, et al. Increased viral loads and exacerbated innate host response in aged macaques infected with 2009 pandemic H1N1 influenza A virus. J. Virol. 2012;86:11115–11127. doi: 10.1128/JVI.01571-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lau KS, et al. In vivo systems analysis identifies spatial and temporal aspects of the modulation of TNF-α-induced apoptosis and proliferation by MAPKs. Sci. Signal. 2011;4:ra16. doi: 10.1126/scisignal.2001338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lau KS, et al. Multi-scale in vivo systems analysis reveals the influence of immune cells on TNF-α-induced apoptosis in the intestinal epithelium. PLoS Biol. 2012;10:e1001393. doi: 10.1371/journal.pbio.1001393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Benecke A, Gale M, Jr, Katze MG. Dynamics of innate immunity are key to chronic immune activation in AIDS. Curr. Opin. HIV AIDS. 2012;7:79–85. doi: 10.1097/COH.0b013e32834dde31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Benecke AG. Critical dynamics in host–pathogen systems. Curr. Top. Microbiol. Immunol. 2013;363:235–259. doi: 10.1007/82_2012_260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Soulé C. Graphic requirements for multistationarity. ComPlexUs. 2003;1:123–133. doi: 10.1159/000076100. [DOI] [Google Scholar]
- 80.Cinquin O, Demongeot J. High-dimensional switches and the modelling of cellular differentiation. J. Theor. Biol. 2005;233:391–411. doi: 10.1016/j.jtbi.2004.10.027. [DOI] [PubMed] [Google Scholar]
- 81.Becavin C, Tchitchek N, Mintsa-Eya C, Lesne A, Benecke A. Improving the efficiency of multidimensional scaling in the analysis of high-dimensional data using singular value decomposition. Bioinformatics. 2011;27:1413–1421. doi: 10.1093/bioinformatics/btr143. [DOI] [PubMed] [Google Scholar]
- 82.Becavin C, Benecke A. New dimensionality reduction methods for the representation of high dimensional 'omics' data. Expert Rev. Mol. Diagnost. 2011;11:27–34. doi: 10.1586/erm.10.95. [DOI] [PubMed] [Google Scholar]
- 83.Rasmussen AL, et al. Early transcriptional programming links progression to hepatitis C virus-induced severe liver disease in transplant patients. Hepatology. 2012;56:17–27. doi: 10.1002/hep.25612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Diamond DL, et al. Proteome and computational analyses reveal new insights into the mechanisms of hepatitis C virus-mediated liver disease posttransplantation. Hepatology. 2012;56:28–38. doi: 10.1002/hep.25649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sonnenschein N, et al. A network perspective on metabolic inconsistency. BMC Syst. Biol. 2012;6:41. doi: 10.1186/1752-0509-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ponnapalli SP, Saunders MA, Van Loan CF, Alter O. A higher-order generalized singular value decomposition for comparison of global mRNA expression from multiple organisms. PLoS ONE. 2011;6:e28072. doi: 10.1371/journal.pone.0028072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huang SS, Fraenkel E. Integrating proteomic, transcriptional, and interactome data reveals hidden components of signaling and regulatory networks. Sci. Signal. 2009;2:ra40. doi: 10.1126/scisignal.2000350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Luksza M, Lassig M, Berg J. Significance analysis and statistical mechanics: an application to clustering. Phys. Rev. Lett. 2010;105:220601. doi: 10.1103/PhysRevLett.105.220601. [DOI] [PubMed] [Google Scholar]
- 89.Josset L, et al. Gene expression signature-based screening identifies new broadly effective influenza A antivirals. PLoS ONE. 2010;5:e13169. doi: 10.1371/journal.pone.0013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yang W-LR, Lee Y-E, Chen M-H, Chao K-M, Huang C-YF. In-silico drug screening and potential target identification for hepatocellular carcinoma using Support Vector Machines based on drug screening result. Gene. 2013;518:201–208. doi: 10.1016/j.gene.2012.11.030. [DOI] [PubMed] [Google Scholar]
- 91.Hood L, Friend SH. Predictive, personalized, preventive, participatory (P4) cancer medicine. Nature Rev. Clin. Oncol. 2011;8:184–187. doi: 10.1038/nrclinonc.2010.227. [DOI] [PubMed] [Google Scholar]
- 92.Bengoechea JA. Infection systems biology: from reactive to proactive (P4) medicine. Int. Microbiol. 2012;15:55–60. doi: 10.2436/20.1501.01.158. [DOI] [PubMed] [Google Scholar]
- 93.Southern EM. DNA microarrays. History and overview. Methods Mol. Biol. 2001;170:1–15. doi: 10.1385/1-59259-234-1:1. [DOI] [PubMed] [Google Scholar]
- 94.Gorreta F, Carbone W, Barzaghi D. Molecular Profiling. 2012. pp. 89–105. [DOI] [PubMed] [Google Scholar]
- 95.Li W, Ruan K. MicroRNA detection by microarray. Anal. Bioanal. Chem. 2009;394:1117–1124. doi: 10.1007/s00216-008-2570-2. [DOI] [PubMed] [Google Scholar]
- 96.Fouse SD, Nagarajan RP, Costello JF. Genome-scale DNA methylation analysis. Epigenomics. 2010;2:105–117. doi: 10.2217/epi.09.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lam C-W, Lau K-C, Tong S-F. Microarrays for personalized genomic medicine. Adv. Clin. Chem. 2010;52:1–18. doi: 10.1016/S0065-2423(10)52001-8. [DOI] [PubMed] [Google Scholar]
- 98.Metzker ML. Sequencing technologies — the next generation. Nature Rev. Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 99.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nature Rev. Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Martin JA, Wang Z. Next-generation transcriptome assembly. Nature Rev. Genet. 2011;12:671–682. doi: 10.1038/nrg3068. [DOI] [PubMed] [Google Scholar]
- 101.Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nature Rev. Genet. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.König J, Zarnack K, Luscombe NM, Ule J. Protein–RNA interactions: new genomic technologies and perspectives. Nature Rev. Genet. 2012;13:77–83. doi: 10.1038/nrg3141. [DOI] [PubMed] [Google Scholar]
- 103.Collaborative-Cross-Consortium. The genome architecture of the Collaborative Cross mouse genetic reference population. Genetics190, 389–401 (2012). [DOI] [PMC free article] [PubMed]
- 104.Zou F, et al. Quantitative trait locus analysis using recombinant inbred intercrosses: theoretical and empirical considerations. Genetics. 2005;170:1299–1311. doi: 10.1534/genetics.104.035709. [DOI] [PMC free article] [PubMed] [Google Scholar]