

Abstract
Huntington's disease (HD) is a fatal autosomal dominant neurodegenerative disease caused by an increase in the number of polyglutamine residues in the huntingtin (Htt) protein. With the identification of the underlying basis of HD, therapies are being developed that reduce expression of the causative mutant Htt. RNA interference (RNAi) that seeks to selectively reduce the expression of such disease-causing agents is emerging as a potential therapeutic strategy for this and similar disorders. This study examines the merits of administering a recombinant adeno-associated viral (AAV) vector designed to deliver small interfering RNA (siRNA) that targets the degradation of the Htt transcript. The aim was to lower Htt levels and to correct the behavioral, biochemical, and neuropathological deficits shown to be associated with the YAC128 mouse model of HD. Our data demonstrate that AAV-mediated RNAi is effective at transducing greater than 80% of the cells in the striatum and partially reducing the levels (∼40%) of both wild-type and mutant Htt in this region. Concomitant with these reductions are significant improvements in behavioral deficits, reduction of striatal Htt aggregates, and partial correction of the aberrant striatal transcriptional profile observed in YAC128 mice. Importantly, a partial reduction of both the mutant and wild-type Htt levels is not associated with any notable overt neurotoxicity. Collectively, these results support the continued development of AAV-mediated RNAi as a therapeutic strategy for HD.
Introduction
Huntington's disease (HD) is an inherited neurodegenerative disease caused by an expansion of the CAG repeat in exon 1 of the huntingtin gene (HTT). The resulting extension of the polyglutamine tract in the N-terminal region confers a toxic gain-of-function to the mutant huntingtin protein (mHtt). mHtt toxicity may arise from the formation of insoluble mHtt-containing aggregates, transcriptional dysregulation, and perturbations in protein homeostasis, all of which can lead to neuronal death (Saudou et al., 1998; Zuccato et al., 2003; Schaffar et al., 2004; Benn et al., 2008). Pathological findings in patients with HD include cortical thinning and a striking progressive loss of striatal neurons (Rosas et al., 2002). Disease onset typically occurs during the third to fourth decade of life; symptoms include choreiform movements, impaired coordination, progressive dementia, and other psychiatric disturbances (Vonsattel et al., 1985). HD is ultimately fatal, with death occurring approximately 10–15 years after the onset of symptoms. Although the genetic basis of HD has been known for almost 20 years, current therapies are largely palliative and do not address the underlying cause of the disease. This is likely due in part to the fact that the etiology of this disease is complex, with detrimental effects observed in a wide variety of cellular processes. Hence, the focus of drug development has been directed at addressing the primary offending trigger, namely, the mutant HTT gene itself.
The potential for silencing mHtt expression as a therapeutic strategy for HD was first demonstrated in a conditional mouse model of the disease (Yamamoto et al., 2000). When the expression of mHtt was induced in these mice, pathological and behavioral aberrations became apparent. Subsequent tetracycline-mediated repression of the mHtt transgene reversed these abnormalities, indicating that a reduction of mHtt levels allowed protein clearance mechanisms within neurons to normalize mHtt-induced changes. Hence, therapeutic strategies that reduce mHtt levels could potentially halt disease progression and alleviate HD symptoms. Proposed approaches to blocking Htt expression include the use of antisense oligonucleotides (ASOs) as well as RNA interference (RNAi) that uses either duplex RNAs (dsRNAs) or chemically modified single-stranded RNAs (ssRNAs) (Harper et al., 2005; DiFiglia et al., 2007; Boudreau et al., 2009b; Drouet et al., 2009; Sah and Aronin, 2011; Matsui and Corey, 2012; Yu et al., 2012). We reported that the intracerebroventricular infusion of an ASO directed against mutant Htt mRNA yielded a therapeutic benefit in three distinct mouse models of HD (BACHD, YAC128, and R62) (Kordasiewicz et al., 2012). However, hurdles to translating this approach into the clinic may include the need to incorporate a device to facilitate repeated and chronic infusions of ASO into the CNS, and the need to adequately distribute the drug to target regions in a large brain.
To circumvent these potential issues with ASOs, we examined the merits of employing adeno-associated virus (AAV)-mediated expression of small interfering RNA (siRNA), which offers the potential for increased safety, increased efficiency, and longer-lasting efficacy. In nature, gene regulation by RNAi occurs through small RNAs known as microRNAs (miRNAs) (Ambros, 2004; Krol et al., 2010). MicroRNAs have emerged as powerful regulators of diverse cellular processes, and when delivered by viral vectors artificial miRNAs are continuously expressed, resulting in robust and sustained suppression of target genes. The elucidation of the mechanisms involved in miRNA processing has allowed scientists to co-opt the endogenous cellular RNAi machinery and direct the degradation of a target gene product with the use of artificial miRNAs (Davidson and Monteys, 2012).
As patients with HD express both mutant and wild-type Htt alleles, a majority of siRNA targeting sequences will likely degrade both alleles. However, non-allele-specific Htt silencing in HD mice has been shown to be well tolerated and can afford the same benefit as reducing mutant Htt alone (Boudreau et al., 2009b; Drouet et al., 2009; Kordasiewicz et al., 2012). Moreover, the partial and sustained suppression of wild-type Htt in the putamen of nonhuman primates after AAV-mediated RNAi reportedly did not have any untoward effects, which suggests that the adult brain can tolerate reduced levels of wild-type Htt (McBride et al., 2011; Grondin et al., 2012).
In the present study, we examined the therapeutic potential of AAV-mediated RNAi (conferred by artificial miRNAs) to provide sustained reduction of Htt levels and improvement of HD-associated abnormalities in the YAC128 transgenic mouse model of HD. Previous studies evaluating AAV-mediated RNAi for HD have employed the use of the R6/1- and N171-82Q fragment-based HD mouse models (Harper et al., 2005; Rodriguez-Lebron et al., 2005; Machida et al., 2006; Boudreau et al., 2009b). These models display an aggressive and rapidly progressing phenotype that makes long-term behavioral and functional testing challenging. By contrast, the yeast artificial chromosome (YAC) mouse model of HD harbors a mutant human Htt gene bearing 128 CAG repeats and exhibits characteristic HD pathology and progressive motor abnormalities. This model also exhibits age-dependent neuropathology, making it a useful mouse model with which to evaluate therapeutic interventions.
Materials and Methods
Animals
All procedures were performed according to a protocol approved by the Institutional Animal Care and Use Committee at Genzyme, a Sanofi Company (Department of Health and Human Services, NIH Publication 86-23). Mice used included YAC128 mice (a yeast artificial chromosome harboring the full-length human mutant HTT transgene with 128 CAG repeats on a pure FVB/NJ background) and FVB/NJ littermate mice (Slow et al., 2003; Van Raamsdonk et al., 2005). Both the YAC128 mice and FVB/NJ littermates were obtained from a Genzyme colony that was housed at Charles River Laboratories (Wilmington, MA). The mice were maintained on a 12-hr light/dark cycle with food and water available ad libitum. All behavioral testing was performed during the animals' light cycle (between the hours of 8 a.m. and 4 p.m.).
Plasmids and viral vectors
To generate recombinant AAV2/1 serotype vectors encoding an miRNA-based hairpin against the huntingtin gene (AAV2/1-miRNA-Htt), the cDNA for human HTT was cloned into a shuttle plasmid containing the AAV2 inverted terminal repeats (ITRs) and the 1.6-kb cytomegalovirus enhancer/chicken β-actin (CBA) promoter. Control vectors contained either an empty vector backbone (AAV2/1-Null) or expressed enhanced green fluorescent protein under the control of the same promoter (AAV2/1-eGFP). All viral vectors were generated by triple-plasmid cotransfection of human 293 cells, and the recombinant virions were column purified as previously described (Passini and Wolfe, 2001). The resulting titer of AAV2/1-miRNA-Htt was determined to be 4.5×1012 vector genomes (VG)/ml, and the titer of AAV2/1-Null was 2.3×1012 VG/ml, using quantitative PCR.
Surgical procedures
Animals were anesthetized with 3% isoflurane and placed into a stereotaxic frame. Intracranial injections were performed as previously described (McBride et al., 2008). Briefly, 2 μl of the recombinant viral vector (AAV2/1-eGFP or AAV2/1-miRNA-Htt) was injected into the striatum (anteroposterior, +0.50; mediolateral, ±2.00; dorsoventral,−2.5 from bregma and dura; incisor bar, 0.0), using a 10-μl Hamilton syringe at the rate of 0.5 μl/min. The needle was left in place for 1 min after the completion of infusion. One hour before surgery and for 24 hr after surgery, the mice were administered ketoprofen (5 mg/kg) subcutaneously for analgesia.
Animal perfusion and tissue collection
The mice were perfused through the heart with phosphate-buffered saline (PBS) to remove all blood. The brains were cut sagittally along the midline, and the left hemisphere was postfixed in 4% paraformaldehyde followed by 30% sucrose and then sectioned into 20-μm sections, using a cryostat. The right hemisphere was cut along the coronal axis into 2-mm slabs, using a mouse brain matrix (Harvard Apparatus, Holliston, MA), and then flash-frozen in liquid nitrogen and stored at −80°C until use. For the analysis of Htt aggregates, the brains were postfixed in 4% paraformaldehyde for 48 hr, washed with PBS, and then sectioned into 40-μm coronal sections, using a vibrating microtome (Vibratome; Leica Biosystems, St. Louis, MO).
Cell culture and transfection
HEK293 cells were infected with 5×109 VG of either AAV2/1-eGFP or AAV2/1-miRNA-Htt and harvested 3 days later. RNA levels were measured by quantitative real-time RT-PCR. Total RNA was isolated with a TaqMan Cells-to-CT kit (Ambion, Austin, TX). Quantitative PCRs were conducted and analyzed on an ABI Prism 7500 sequence detector (Applied Biosystems/Life Technologies, Carlsbad, CA) as described previously (Winer et al., 1999). Expression levels were normalized to peptidylprolyl isomerase (PPIA) levels.
Quantitative real-time PCR (TaqMan)
RNA levels were measured by quantitative real-time RT-PCR. Brain tissue samples from brain slab 2 were used for all RT-PCR analyses. Total RNA was extracted with an RNeasy mini kit (Qiagen, Valencia, CA) and then reverse transcribed and amplified with a TaqMan One-Step RT-PCR master mix kit (Applied Biosystems/Life Technologies) according to the manufacturer's instructions. Quantitative RT-PCRs were conducted and analyzed on an ABI PRISM 7500 real-time PCR system (Applied Biosystems/Life Technologies). The expression levels of Htt mRNA were normalized to hypoxanthine-guanine phosphoribosyltransferase-1 (Hprt1) mRNA levels. Standard curves were generated with 5-fold serial dilutions of mouse brain cDNA. Each sample was run in duplicate. Relative gene expression was determined by using the standard curve or ΔΔCT method and normalizing to Hprt1 mRNA levels.
Western blotting
Tissues, at a final concentration of 50 mg/ml in T-PER lysis buffer (Pierce Biotechnology/Thermo Fisher Scientific, Rockford, IL) and containing complete protease inhibitor cocktail (Roche, Indianapolis, IN), were homogenized. The homogenates were cleared by centrifugation at 10,000×g for 6 min at 4°C. The protein concentration was measured by bicinchoninic acid (BCA) assay (Pierce Biotechnology/Thermo Fisher Scientific). Twenty to 30 μg of the homogenates was resolved on a 3–8% Novex Tris–acetate gel and then transferred to a nitrocellulose membrane. The membranes were probed with a mouse anti-huntingtin monoclonal antibody (Mab2166, 1:2000 dilution; Millipore, Bedford, MA) and rabbit polyclonal anti-β-tubulin antibody (1:750 dilution; Santa Cruz Biotechnology, Santa Cruz, CA). The membranes were then incubated with infrared secondary antibodies (1:20,000 dilution; Rockland, Gilbertsville, PA), and the proteins were visualized by quantitative fluorescence, using an Odyssey imager (LI-COR Biosciences, Lincoln, NE). To control for loading variances, Htt protein was normalized to β-tubulin and expressed as a percentage of untreated or saline-treated animals. Molecular weight markers were used to verify the identity of the proteins.
Flow cytometry and cell sorting
Single-cell suspensions were analyzed and isolated with a FACSAria II cell sorter (BD Biosciences San Jose, CA) with a 100-μm nozzle at the Genzyme Flow Cytometry Core Facility. Analysis of cells was performed by discriminating live single cells from debris by gating on the forward scatter (FWD-Sc) and side scatter (SSC). eGFP-positive cells were collected, using detector E with a 530/30 BP filter 505LP. The eGFP fluorescence data profile was displayed as a single-parameter histogram and sorting decisions were based on eGFP− and eGFP+. Sorted cells were collected in tissue culture medium containing 5% fetal bovine serum and plated onto 4-well chambered slides (Lab-Tek; Nalge Nunc International, Naperville, IL) at a concentration of 50,000 cells per well.
Immunohistochemistry
Vibratome sections were processed for immunostaining with EM48, an antibody that preferentially recognizes aggregated huntingtin (Gutekunst et al., 1999). The free-floating sections were first treated with dual endogenous enzyme block (Dako, Carpinteria, CA) for 30 min to block endogenous peroxidase activity. They were then washed with 0.01 M PBS (3 min) followed by three washes with 0.5% Triton X-100 in PBS (10 min each). Nonspecific sites were blocked by incubating the sections in Rodent Block M (Biocare Medical, Concord, CA) for 1 hr at room temperature. Sections were probed with the EM48 antibody (1:25 dilution in PBS; Millipore) by incubating at 4°C on a gentle rocker in a cold room overnight. The next day, sections were incubated with secondary antibody (MM HRP Polymer; Biocare Medical) for 1 hr at room temperature on a rocker. After three washes in PBS (15 min each) the signal was detected with a 3,3′-diaminobenzidine (DAB) peroxidase substrate kit (Vector Laboratories, Burlingame, CA). After washes, sections were mounted onto SuperFrost Plus slides, dried overnight, and coverslipped with Acrytol mounting medium.
Behavioral analysis
Accelerating Rotarod test
Motor coordination and motor learning were assessed on an accelerating Rotarod apparatus (AccuScan Instruments, Columbus, OH). Mice were trained on the Rotarod with three trials per day for three consecutive days. On the first training day, the Rotarod was set to accelerate from 0 to 5 rpm over 300 sec. Mice that fell off the rod before completion of the 300-sec time period were placed back on the rod until the full 300-sec period had expired. On the second and third days of training, the Rotarod was set to accelerate from 0 to 40 rpm over 300 sec, again requiring all mice to complete the full 300 sec on the rod. On the fourth day (test day), the mice were placed on the Rotarod set to accelerate from 0 to 40 rpm over 300 sec. Animals were not replaced after falling, and the latency to fall was recorded over three trials. Latency to fall was defined by the time elapsed until the animal fell from the Rotarod.
Porsolt swim test
Immobility in the Porsolt swim test was used as a measure of depression in rodents (Porsolt et al., 1977; Cryan et al., 2002a,b). The test was conducted by placing mice in individual glass cylinders (height, 20 cm; diameter, 10 cm) filled with water at 23°C up to a height of 15 cm. The mice were placed into the cylinders for a period of 7 min. The first 3 min of this period was considered an acclimation period, during which time no data were collected. During the last 4 min of the test session, the performance of the mice was scored by a blinded observer using a time-sampling technique to rate the predominant behavior over 10-sec intervals. Swimming and immobility behaviors were measured and recorded at the end of every 10 sec, which resulted in 24 data points per test. The percentage of time spent in an immobile state was calculated for each mouse.
Statistics
Mean values were used for statistical analyses. Data are expressed as means±SEM. For studies that used two groups, the Student t test was used for statistical comparison. For comparisons of more than two groups, one-way analysis of variance (ANOVA) was used followed by the Tukey multiple comparison post-hoc test (Prism; GraphPad Software, San Diego, CA). p<0.05 was considered a statistically significant difference.
Results
AAV2/1-miRNA-Htt reduces Htt expression in vitro
A targeting sequence that was previously shown to effectively target mouse and human Htt mRNAs (McBride et al., 2008) was embedded into an artificial miRNA backbone and cloned into a previral vector. This plasmid was engineered to express the miRNA targeting Htt and an enhanced GFP (eGFP) reporter gene under the transcriptional control of a chicken β-actin (CBA) promoter (pSP70-CBA-eGFP) as illustrated in Fig. 1A. The candidate targeting sequence, which was selected from the Htt coding region, was 5′-TAGACAATGATTCACACGGT-3′. High-titer recombinant AAV2/1 serotype vectors encoding the targeting sequence (AAV2/1-miRNA-Htt) and control vectors (AAV2/1-eGFP and AAV2/1-Null) were generated and their gene-silencing activities tested by infecting human embryonic kidney (HEK) 293 cells. Cells were infected with 5×109 VG of AAV vector. Using fluorescence-activated cell-sorting (FACS) analysis, we confirmed that this dose resulted in greater than 90% infection efficiencies in HEK293 cells after infection with AAV-eGFP-miRNA-Htt (Supplementary Fig. S1; supplementary data are available online at www.liebertpub.com/hum). Cells infected with AAV2/1-eGFP did not show any reduction of endogenous Htt levels when analyzed by real-time PCR 3 days postinfection; however, cells infected with AAV2/1-miRNA-Htt exhibited an approximately 40% reduction in Htt mRNA levels (Fig. 1B).
FIG. 1.
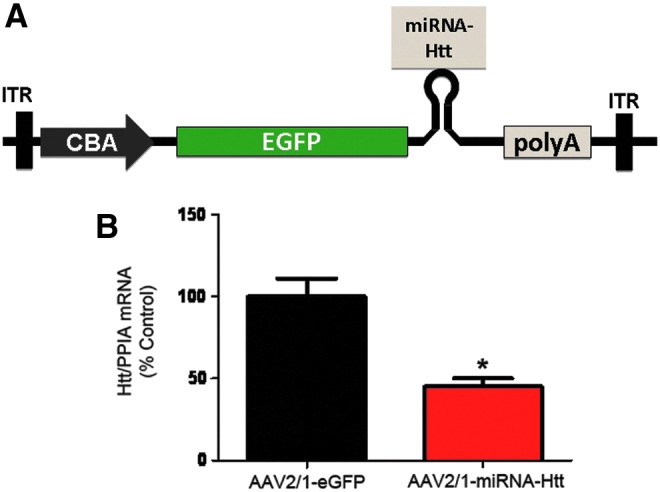
AAV2/1-miRNA-Htt mediates the reduction of Htt levels in vitro. (A) Schematic of the previral construct used to generate AAV2/1-miRNA-Htt. The plasmid was designed to express GFP and an miRNA sequence against Htt under the transcriptional control of the chicken β-actin (CBA) promoter. ITR, inverted terminal repeat; eGFP, enhanced green fluorescent protein. (B) Quantitative PCR analysis evaluating Htt mRNA levels in HEK293 cells 48 hr after AAV-2/1-miRNA-Htt treatment. PPIA served as a normalization control gene. Values are given as means±SEM. *p<0.05. Color images available online at www.liebertpub.com/hum
AAV2/1-miRNA-Htt injection into YAC128 mice results in widespread striatal transduction and reduction of Htt mRNA
After verification of the ability of AAV2/1-miRNA-Htt to suppress Htt mRNA levels in vitro, we next evaluated the ability of this vector to silence Htt expression in the striatum of YAC128 mice. To determine the percent transduction of cells within the striatum after intrastriatal injections of AAV2/1-miRNA-Htt, FACS was employed. Adult YAC128 mice received bilateral intrastriatal injections of AAV2/1-eGFP-miRNA-Htt (4.5×1012 VG/ml) or the control vector, AAV2/1-eGFP (5.6×1012 VG/ml). One month after injection the striatal region of each animal was microdissected and eGFP-containing versus non-eGFP-containing cells were sorted and quantified by FACS analysis (Fig. 2A and B). The data showed that greater than 80% of the striatum was transduced by our vector as demonstrated by the presence of eGFP within a majority of the sorted striatal cells (Fig. 2C).
FIG. 2.

Widespread striatal transduction and Htt reduction after intrastriatal injection of AAV2/1-miRNA-Htt into YAC128 mice. (A) Flow cytometric scatter profile of striatal cells with eGFP. Forward light scatter A (FSC-A) represents relative cell size, area and SSC-A represents relative cell complexity, area with each dot representing one cell. (B) Dot plot based on FSC-A versus FITC-A analysis. Dead cells were sorted out and eGFP+ and eGFP− cells were selected. GFP expression from these cells was evaluated, quantified, and shown in (C), a fluorescence plot of eGFP fluorescence intensity collected with a 530/30BP filter 505LP. (D) Fluorescence microscopy showing widespread eGFP expression throughout the striatum after intracranial administration of AAV2/1-eGFP-miRNA-Htt. (E) Quantitative PCR analysis evaluating Htt mRNA levels in the striatum 1 and 5 moths after injection of AAV2/1-miRNA-Htt or AAV2/1-null control vector. PPIA served as a normalization control gene. Values are given as means±SEM. *p<0.05. AAV2/1-miRNA-Htt-treated YAC128 mice (n=8) showed an approximately 50% reduction in Htt mRNA levels in the striatum when compared with AAV2/1-Null-injected mice (n=8 per time point) at 1 and 5 months posttreatment.
To evaluate the ability of our vector to reduce Htt in vivo and to monitor the longevity of the response, adult mice received bilateral intrastriatal injections of AAV2/1-miRNA-Htt (4.5×1012 VG/ml) (n=16, n=8+8 per time point) or the AAV2/1-Null control vector (2.3×1012 VG/ml) (n=8), and the brains were analyzed 1 or 5 months posttreatment. Fluorescence microscopy analysis of brain sections from mice treated with AAV2/1-miRNA-Htt at both time points showed widespread eGFP fluorescence throughout the entire striatum and surrounding brain regions, consistent with our FACS analysis suggesting almost complete striatal transduction (Fig. 2D). The levels of eGFP expression in the brains of mice attained at 1 month posttreatment appeared undiminished at the 5-month time point (Fig. 2D). The striatal level of mutant human Htt mRNA was significantly reduced in the AAV2/1-miRNA-Htt-injected mice when compared with AAV2/1-Null-treated controls, and an equivalent extent of reduction (approximately 45%; p<0.01) was noted at both time points (Fig. 2E). We also evaluated striatal levels of endogenous mouse Htt mRNA and found that levels were significantly reduced after AAV2/1-miRNA-Htt treatment when compared with AAV2/1-Null-treated controls. An equivalent extent of reduction (approximately 45%; p<0.01) was noted at both time points (Supplementary Fig. S2).
AAV2/1-miRNA-Htt injection into YAC128 mice does not cause overt toxicity in the brain
To determine whether injections of AAV2/1-miRNA-Htt and the consequent reduction of Htt conferred neurotoxicity and inflammation, cellular morphology and integrity of striatal sections were examined by hematoxylin and eosin (H&E) staining. Levels of the neuroinflammatory markers glial fibrillary acidic protein (GFAP, a marker of astrocytes) and Iba-1 (a marker of microglia) were also examined 1 and 5 months posttreatment. Analysis by H&E staining showed no remarkable histopathological changes in the injected brain regions (Fig. 3A, panels a–c). No notable increases in either the number of GFAP-positive astrocytes (visualized by immunohistochemistry) or the levels of GFAP mRNA (quantitated by qPCR) were observed in the injected regions 1 or 5 months postinjection when compared with AAV2/1-Null-treated animals (Fig. 3A, panels d–f and Fig. 3B). However, an increase in the number of activated microglia, as evidenced by an increase in Iba-1 immunostaining (Fig. 3A, panel h) and Iba-1 mRNA levels in the striatum were noted at 1 month postinjection (Fig. 3C). Interestingly, at 5 months postinjection, microglial activation returned to control levels (Fig. 3A, panel i and Fig. 3C), suggesting that the response was transient. As the AAV2/1-Null control vector used in this study did not harbor an eGFP gene, we speculate that the expression of eGFP from AAV2/1-miRNA-Htt was likely responsible for the transient microglial activation. Taken together, these results corroborate and extend the findings that AAV2/1-miRNA-Htt is capable of mediating sustained Htt silencing not only in vitro but also in the striatum of YAC128 mice. Moreover, partial suppression of Htt levels for up to 5 months does not lead to overt toxicity or neuroinflammation in the mouse brain.
FIG. 3.

Sustained lowering of Htt levels in YAC128 mice by AAV2/1-miRNA-Htt does not cause overt neuroinflammation. (A) Panels a–c: Hematoxylin and eosin (H&E) staining of striatal tissue sections 1 or 5 months after AAV-miRNA-Htt injection. Panels d–i: GFAP and Iba-1 immunohistochemical staining of sections from YAC128 mice treated with AAV2/1-miRNA-Htt or AAV2/1-Null vector. All photographs were exposure-matched for accurate comparisons. Scale bar: 0.25 mm. (B) Striatal levels of GFAP mRNA levels by qPCR 1 or 5 months after the injection of AAV2/1-miRNA-Htt. (C) Iba-1 mRNA levels by qPCR 1 or 5 months after the injection of AAV2/1-miRNA-Htt. Values are given as means±SEM. *Significant difference, p<0.05; ANOVA. Color images available online at www.liebertpub.com/hum
Striatal delivery of AAV2/1-miRNA-Htt corrects the aberrant behavioral and neurochemical profiles in YAC128 mice
The impact of the AAV-mediated reduction of mutant Htt levels on the well-characterized phenotypic deficits that are present in the YAC128 mouse model of HD was also examined. Age-matched (2 months old) YAC128 and wild-type littermate mice received bilateral intrastriatal injections of either AAV2/1-miRNA-Htt or AAV2/1-Null vector and were then killed 3 months after treatment (Fig. 4A). As expected, an analysis of brain sections demonstrated eGFP expression throughout the entire striatum and surrounding regions, as previously observed (Fig. 4B). Western blot analysis of brain homogenates showed that the levels of both mutant human and endogenous mouse Htt proteins were significantly reduced in the striata of AAV2/1-miRNA-Htt-injected YAC128 and wild-type mice (approximately 55% reduction; p<0.01) when compared with AAV2/1-Null-treated controls (Fig. 4C and D). Real-time quantitative PCR analysis indicated that a commensurate reduction in mRNA levels was also attained (data not shown).
FIG. 4.

Striatal administration of AAV2/1-miRNA-Htt reduces behavioral deficits in YAC128 mice. (A) Illustration of experimental timeline. Two-month-old YAC128 (YAC) and wild-type (WT) mice received bilateral striatal injections of either AAV2/1-miRNA-Htt (YAC, n=8; WT, n=8) or AAV2/1-Null control (YAC, n=8; WT, n=8) and were subjected to a Rotarod test and the Porsolt swim test at 4 and 5 months of age, respectively. All mice were killed at 5 months of age, and tissues were then collected for biochemical and histological analyses. (B) Fluorescence microscopy showing eGFP expression in the striatum 3 months posttreatment. (C and D) Mouse and human Htt protein levels by Western blot 3 months after AAV2/1-miRNA-Htt treatment. (E) Accelerating Rotarod test 2 months after the injection of AAV2/1-miRNA-Htt. (F) Time spent immobile in the Porsolt swim test 3 months after the injection of AAV2/1-miRNA-Htt. Values are given as means±SEM. *Significant difference, p<0.05; ANOVA followed by Tukey post-hoc test.
YAC128 mice have been reported to exhibit motor coordination deficits (which can be revealed by the Rotarod test) and a depressive phenotype (using the Porsolt swim test) beginning at 3 months of age (Slow et al., 2003; Van Raamsdonk et al., 2007). Rotarod testing of AAV2/1-Null-treated YAC128 mice 2 months postinjection showed significant motor coordination deficits when compared with AAV2/1-Null- or AAV2/1-miRNA-Htt-treated wild-type littermates (ANOVA, p<0.01) (Fig. 4E). However, YAC128 mice that had been treated with AAV2/1-miRNA-Htt showed performance levels that were indistinguishable from those of wild-type mice (ANOVA, Tukey post-hoc test; wild-type Htt vs. YAC128 Htt, p=not significant [NS]; wild-type Null vs. YAC128 Null, p<0.05). Hence, a partial lowering of mutant Htt levels was sufficient to correct the motor deficits of YAC128 mice. There were no significant differences in Rotarod performance between wild-type mice that received AAV2/1-miRNA-Htt and wild-type mice that received AAV2/1-Null.
Previous reports indicated that YAC128 mice (beginning at 3 months of age) exhibit a depressive phenotype that can be detected by the Porsolt swim test (Pouladi et al., 2009). Animals are deemed to exhibit a depressive state if they are immobile for an extended period when placed into a container of water. Using a basic swim speed test (in which swim latency to reach a platform was measured), researchers have demonstrated that this depressive phenotype in the Porsolt swim test is unrelated to the swimming ability of YAC128 mice and is independent of the well-documented motor coordination deficits observed in this model (Pouladi et al., 2009, 2012). Two-month-old YAC128 and wild-type littermate mice were injected with AAV2/1-miRNA-Htt or AAV2/1-Null vector and tested 3 months later in the Porsolt swim test. Untreated YAC128 mice displayed an increased period of time in an immobile state when compared with either AAV2/1-miRNA-Htt-treated YAC mice or AAV2/1-Null-treated wild-type animals (Fig. 4F; ANOVA, p<0.05). Again, there were no significant differences in the performance of wild-type mice that received either AAV2/1-miRNA-Htt or AAV2/1-Null. YAC128 mice that had been injected with AAV2/1-miRNA-Htt spent significantly less time in an immobile state than did AAV2/1-Null-treated controls. Indeed, the performance of AAV2/1-miRNA-Htt-treated YAC128 mice was similar to that of their wild-type littermates, suggesting a near-complete correction of this aberrant phenotype (ANOVA, Tukey post-hoc test; YAC Htt vs. YAC Null, p<0.05) (Fig. 4F).
Transcriptional dysregulation of a number of genes enriched in the striatum has been observed in HD brains (Richfield et al., 1995; Augood et al., 1997; Sugars et al., 2004; Desplats et al., 2006). This aberration is also evident in YAC128 mice, as illustrated in particular by their significantly lower striatal levels of DARPP-32 (dopamine- and cAMP-regulated phosphoprotein Mr 32 kDa) and D1 dopamine receptors compared with those of wild-type animals (Pouladi et al., 2012). To examine whether the suppression of Htt levels in YAC128 mice corrected this altered transcriptional profile, real-time quantitative PCR analysis was performed on striatal tissues of YAC128 mice that had been treated at 2 months of age with AAV2/1-miRNA-Htt and analyzed 3 months later. An analysis of brain extracts of AAV2/1-Null-treated YAC128 mice indicated that the mRNA levels of DARPP-32 and D1 dopamine receptor (D1R) were significantly lower when compared with age-matched wild-type controls (ANOVA, Tukey post-hoc test; wild-type Null vs. YAC128 Null, p<0.05) (Fig. 5A and B). YAC128 mice that were administered AAV2/1-miRNA-Htt exhibited higher levels of DARPP-32 and D1R mRNA than those treated with AAV2/1-Null vector; however, these levels were still lower than those observed in the wild-type controls (ANOVA, Tukey post-hoc test; wild-type Null vs. YAC Htt, p=NS). Thus, the AAV2/1-miRNA-Htt-mediated reduction of Htt levels in YAC128 mice conferred a partial correction of the aberrant striatal transcriptional profile. It is possible that examination at later time points (greater than 5 months posttreatment) may reveal a more complete correction of this aberrant profile.
FIG. 5.
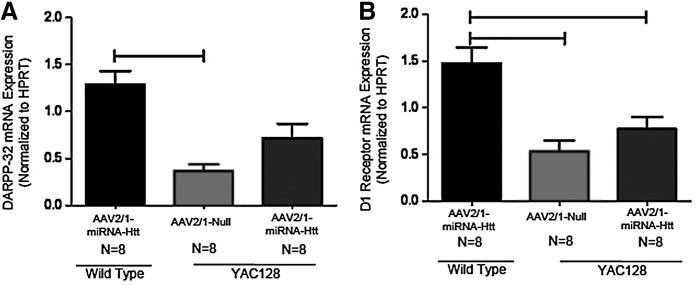
Treatment with AAV2/1-miRNA-Htt partially corrects the transcriptional dysregulation of DARPP-32 and D1 receptor in YAC128 mice. DARPP-32 and D1 receptor mRNA levels in the striatum of YAC128 and wild-type (WT) mice were assessed by qPCR 3 months after the injection of either AAV2/1-miRNA-Htt (YAC, n=8; WT, n=8) or AAV2/1-Null control (YAC, n=8; WT, n=8). (A) Striatal DARPP-32 mRNA levels in YAC128 and FVB wild-type littermate mice after AAV2/1-Null or AAV2/1-miRNA-Htt treatment. (B) Striatal D1 receptor mRNA levels in YAC128 and FVB wild-type littermate mice after AAV2/1-Null or AAV2/1-miRNA-Htt treatment. Values are given as means±SEM. *Significant difference, p<0.05; ANOVA followed by Tukey post-hoc test.
Together, these results corroborate earlier in vitro and in vivo findings that AAV2/1-miRNA-Htt is capable of mediating a sustained lowering of Htt levels. Importantly, we show that this reduction in striatal Htt levels in YAC128 mice results in measurable improvements in motor function and behavior as well as a partial correction of the well-characterized transcriptional dysregulation in the striatum.
Striatal delivery of AAV2/1-miRNA-Htt reduces Htt aggregates in the brains of YAC128 mice
The appearance of Htt aggregates and inclusion bodies in the CNS is a neuropathological hallmark of HD. Lowering the levels of these aggregates in HD mice has been correlated with notable improvements in pathology (Harper et al., 2005; Rodriguez-Lebron et al., 2005). The YAC128 mouse model reportedly exhibits significant and widespread accumulation of Htt aggregates in the striatum by 12 months of age (Slow et al., 2003; Pouladi et al., 2012). Immunohistochemical staining of brain sections of 6-, 9-, and 24-month-old YAC128 mice in our colony, using the anti-Htt antibody EM48, also showed evidence of aggregates in both the striatum and cortex as early as 6 months of age that progressed over time (Fig. 6A). Twelve-month-old tissues (16-μm frozen sections) were also analyzed and shown to have a similar extent of aggregates as the 24-month-old cohort; however, nonspecific background staining in our frozen sections was significantly higher than in the Vibratome sections (data not shown).
FIG. 6.

Intracranial administration of AAV2/1-miRNA-Htt ameliorates motor deficits and reduces mutant Htt aggregates in the striatum of aged YAC128 mice. (A) Immunohistochemical staining of YAC128 mouse brain sections showing mutant Htt aggregates in the striatum. Aggregates were observed in 6, 9, 12 (data not shown), and 24-month-old YAC128 mice. Wild-type mice exhibited no aggregates at all ages tested. (B) An illustration of the experimental timeline for testing AAV2/1-miRNA-HTT in aged YAC128 and wild-type mice. Seven-month-old mice received bilateral intrastriatal injections of AAV2/1-miRNA-Htt (YAC, n=6; WT, n=4) or AAV2/1-GFP control (YAC, n=6; WT, n=4) and were then subjected to behavioral testing at 10 months of age. Brains were harvested 5 months postinjection (when the mice were 12 months old). (C) Performance of aged YAC128 mice on the Rotarod test 3 months after injection of AAV-miRNA-Htt. (D) EM48 immunohistochemical analysis of brain sections of AAV2/1-miRNA-Htt- or AAV2/1-eGFP-treated YAC128 mice 5 months posttreatment.
To examine whether the AAV-mediated reduction of mutant Htt levels lowered the extent of accumulation of Htt aggregates in the brains of postsymptomatic YAC128 mice (7 months old) and, in turn, correct the motor and behavioral deficits, mice were subjected to bilateral intrastriatal injections of either AAV2/1-miRNA-Htt or AAV2/1-GFP. The animals underwent Rotarod testing 3 months postinjection (the animals were 10 months old) and killed 5 months postinjection (when the mice were 12 months old) (Fig. 6B). AAV2/1-miRNA-Htt-treated YAC128 mice on the Rotarod exhibited a level of competency that was comparable to that of their wild-type littermates (Fig. 6C; ANOVA, Tukey post-hoc test; p=NS). However, these results did not reach statistical significance because of the low numbers of mice used in the study (wild-type, n=4; YAC128, n=6). Immunohistochemical staining of striatal sections with the EM48 antibody showed the presence of significantly fewer Htt aggregates in AAV2/1-miRNA-Htt-treated than in AAV2/1-GFP-treated YAC128 mice (Fig. 6D). The brains of AAV-miRNA-Htt-treated YAC128 mice were essentially indistinguishable from those of wild-type mice.
To confirm that the observed reduction in aggregates with AAV2/1-miRNA-Htt was not due to neuronal loss or potential neurotoxicity, H&E staining as well as immunohistochemical staining for NeuN (neuronal marker), GFAP (astrocytic marker), and Iba-1 (microglial marker) were performed on adjacent sagittal brain sections. Compared with AAV-2/1-Null-injected controls, AAV2/1-miRNA-Htt-injected animals showed the same abundance of NeuN-positive neurons in the striatum by fluorescence microscopy. Using light microscopy, H&E staining of adjacent coronal brain sections also appeared normal, providing supporting evidence of this lack of neuronal loss; however, because stereology was not performed we could not quantify these results. In addition, as observed in our earlier studies, we did not detect an increase in either GFAP or Iba-1 staining in AAV2/1-miRNA-Htt-treated mice 5 months postinjection.
Discussion
The present study supports the merits of lowering mutant Htt levels as a therapeutic strategy for HD and demonstrates that the partial reduction of mutant Htt in the striatum produces behavioral, biochemical, and neuropathological improvements in a full-length transgenic mouse model of HD, the YAC128 mouse model. Previous efforts at evaluating this therapeutic strategy were performed on mouse models harboring fragments of the mutant HTT gene, such as R6/1 and N171-82Q HD mice (Harper et al., 2005; Rodriguez-Lebron et al., 2005; Machida et al., 2006; Boudreau et al., 2009b). A partial reduction in the levels of the mutant Htt conferred a modest survival benefit in some of the more severe models, such as the N171-82Q HD mouse model, but not in others (e.g., the R6/1 mouse model) (Harper et al., 2005; Rodriguez-Lebron et al., 2005; Machida et al., 2006; Boudreau et al., 2009b). Motor improvements were also noted in these studies using the Rotarod and stride-length tests; however, the severity of these models precluded the long-term evaluation of treatment on behavioral, neuropathological, and biochemical aberrations.
In the present study, we used the YAC128 mouse model (this model harbors a mutant human HTT gene containing 128 CAG repeats), which develops progressive motor abnormalities and age-dependent neuropathology. Compared with other HD mouse models, YAC128 mice are well suited for testing therapeutic efficacy because they recapitulate the salient genetic and clinical features of the human disease. The natural history of disease-related changes in YAC128 mice is well defined, and the animals exhibit phenotypically uniform disease characteristics that have low interanimal variability. YAC128 mice develop an altered striatal transcriptional profile, a trait that is not observed in other, similar mouse models, such as BACHD mice (Pouladi et al., 2012), and show age-dependent striatal neurodegeneration. As such, the testing of therapeutic interventions and the measurement of outcomes in this model may have more predictive value for clinical translation (Slow et al., 2003).
Using YAC128 mice, we demonstrated that AAV2/1-mediated expression of an miRNA targeting mutant human Htt led to a significant reduction in striatal levels of Htt mRNA and protein. Associated with the lowering of this offending entity were significant improvements in function as assessed by Rotarod and Porsolt swim tests 5 months posttreatment as well as a significant reduction in Htt aggregates within the striatum. It is notable that the level of Htt reduction observed in our studies was only approximately 40% of control, suggesting that a partial reduction of mutant Htt was sufficient to produce a significant therapeutic benefit in this mouse model. Moreover, 80% transduction of the striatum with our AAV vector led to only a partial Htt reduction. A similar phenomenon was seen in HEK293 cells in culture, in which approximately 90–95% transduction efficiencies yielded only a consequent 40–50% reduction in endogenous Htt levels (see Supplementary Fig. S1). These findings are consistent with previous studies in rodents and primates showing only a partial reduction of Htt levels using comparable strategies of miRNA-based silencing (McBride et al., 2008, 2011; Boudreau et al., 2009b; Grondin et al., 2012). There are a number of potential hypotheses as to why miRNA produces only partial target knockdown in the transduced region. The miRNA stem–loop format used to mediate Htt silencing requires processing by the cell before generating functional small interfering RNAs. This requirement for cellular processing may thus set natural limits on the extent of RNA silencing imparted by miRNA-based hairpins. A report by Boudreau and colleagues (2009a) described the improved safety of miRNA-based platforms for therapeutic silencing in the mammalian brain and highlighted the improved toxicity profiles of miRNAs compared with traditional short hairpin structures. This improvement in safety could be due to the reliance of miRNA on endogenous cellular processing mechanisms (Boudreau et al., 2009a). Despite these proposed hypotheses, the exact mechanisms behind miRNA-based Htt silencing in the brain are still unknown; however, the data presented here suggest that partial Htt reduction can achieve therapeutic benefits, at least in a mouse model of HD.
As the functional role of Htt remains unclear, there is an obvious concern associated with deploying therapeutic strategies that confer non-allele-specific silencing. Our studies indicated that partial lowering of endogenous mouse Htt in the CNS of wild-type mice, as well as diseased YAC128 mice, for up to 5 months was well tolerated. Administration of AAV-miRNA-Htt reduced wild-type mouse and mutant human Htt by approximately 40%, thus allowing for the preservation of at least 60% of wild-type Htt levels while still maintaining the therapeutic benefits of silencing mutant toxic Htt. No overt toxicity or aberrant behaviors were observed. The current data are consistent with previous studies showing a similar lack of toxicity after non-allele-specific Htt silencing in HD mice and wild-type mice for up to 9 months after treatment (Boudreau et al., 2009b). The safety of the partial suppression of wild-type Htt has also been reported in nonhuman primates (McBride et al., 2011; Grondin et al., 2012), providing further confidence that the partial lowering of levels of normal Htt may not lead to significant detrimental consequences. This study further demonstrates that partial suppression (∼40%) of both mutant and wild-type Htt in the YAC128 mice was therapeutic, as evidenced by their performance in a variety of behavioral tests, and was not associated with any obvious adverse issues. Previous reports had suggested a role for Htt in embryogenesis and postnatal neurogenesis (Bhide et al., 1996; Reiner et al., 2003; Cattaneo et al., 2005). However, to date, several preclinical studies demonstrate that partially reducing wild-type Htt levels in adult brain appears to be well tolerated in both mouse and nonhuman primates (Boudreau et al., 2009b; McBride et al., 2011; Grondin et al., 2012). Clearly, more stringent investigations of the safety of the long-term RNAi-mediated silencing of wild-type Htt levels in the adult primate brain will be needed before clinical testing.
Although it may be possible to design siRNAs that specifically target disease-linked single-nucleotide polymorphisms (SNPs) on the mutant Htt transcript, it remains uncertain whether this specificity will be conserved in a clinical setting (Liu et al., 2008; van Bilsen et al., 2008; Lombardi et al., 2009; Pfister et al., 2009; Seyhan, 2011). Unfortunately, no one SNP has yet been identified that would allow for the specific silencing of the mutant Htt allele in all patients with HD. Therefore, the clinical application of allele-specific targeting may require the development of unique inhibitory RNA sequences on a patient-by-patient basis. On the basis of our current data that partial suppression of both mutant and wild-type Htt is both safe and efficacious (at least at the time points examined), a non-allele-specific therapeutic approach may be feasible as a first-generation therapeutic strategy for the treatment of HD.
A notable hallmark of HD pathology in both mouse models and human patients is the presence of mutant Htt-immunoreactive aggregates (DiFiglia et al., 1997; Scherzinger et al., 1997). The precise role of aggregates within the cascade of pathophysiological events in HD continues to be a matter of debate (Lansbury and Lashuel, 2006) and the suggestion of a causal relationship between mutant Htt aggregates and disease remains controversial (Bates, 2003; Arrasate et al., 2004). However, there is consensus that the formation of insoluble protein aggregates confers an increased burden on cellular degradative processes (Yamamoto et al., 2000). We observed that YAC128 mice displayed widespread striatal aggregates as early as 6 months of age (earlier than previously reported) and injection of AAV2/1-miRNA-Htt at 7 months of age (after aggregates had already formed) significantly reduced the number of EM48-positive Htt aggregates within the striatum to nearly wild-type levels. These data suggest that AAV2/1-miRNA-Htt treatment may diminish the available pool of Htt, significantly alleviating the burden of mutant Htt aggregates and thus potentially contributing to the functional improvements noted in this mouse model.
In addition to the substantial removal of Htt aggregates, AAV-miRNA-Htt treatment also conferred a behavioral benefit in YAC128 mice. YAC128 mice begin to exhibit deficits on the Rotarod test starting at 3 months of age, and by 7 months they show severe impairment (Slow et al., 2003). YAC128 mice treated with AAV2/1-miRNA-Htt at 7 months of age (when motor coordination would be significantly impaired) showed improvements on the Rotarod test, suggesting a reversal of established motor deficits was obtained. Although reductions in Htt aggregates have been reported previously (N171 and R6/2 fragment models) (Rodriguez-Lebron et al., 2005; Machida et al., 2006), this is the first report to demonstrate amelioration of aggregates and concomitant behavioral improvements in a full-length mouse model of HD. We also observed a significant improvement in the Porsolt swim test after AAV-miRNA-Htt injection into the striatum. This is the first report to show an improvement in this depressive phenotype after AAV-RNAi and, importantly, these results suggest that reduction of Htt levels in the striatum was sufficient to improve the depressive phenotype exhibited in the YAC128 model. Finally, when 7-month-old YAC128 mice were treated with AAV-miRNA-Htt (postsymptomatic treatment) we found a significant reduction in Htt aggregates in the striatum and a potential reversal in the motor coordination deficit exhibited by this model. These data suggest that postsymptomatic treatment with AAV2/1-miRNA-Htt may alleviate the mHtt burden within cells and provide a therapeutic benefit even after mHtt aggregates have formed.
Suppression of striatal Htt also resulted in a modest correction of DARPP-32 and D1 receptor mRNA levels, two transcripts that decline progressively with age in YAC128 mice and human patients with HD. However, a more complete correction of this aberration might have been realized if the animals were analyzed at a later time point (greater than 3 months postinjection). Mutant Htt is known to confound a number of cellular processes, leading to neuronal dysfunction and transcriptional dysregulation (Cha, 2000). If the affected neurons were accorded sufficient time to recover from the insult, a more robust improvement in the striatal transcription profile may have been realized. Transcriptional dysregulation may also be due to local as well as distal neuropathology and thus more global reduction of Htt may be required to confer a complete correction of this phenotype.
We, as well as others, have shown that lowering of mutant Htt may also be realized by using antisense oligonucleotides (ASOs) or synthetic siRNAs (Wang et al., 2005; Kordasiewicz et al., 2012). Although these approaches were also able to provide some measure of correction of the disease characteristics in HD mice, because the moieties are unable to efficiently traverse the blood–brain barrier and the effects are transient (e.g., 4-week half-life for ASOs), delivery may require the deployment of a device for chronic and repeated infusions into the brain. The use of AAV-mediated gene transfer vectors would circumvent this issue as they can confer sustained gene silencing over a significant period. For example, AAV vectors have been shown to facilitate gene expression in nonhuman primate brains for up to 10 years and in human brains, up to 6 years (the longest time point evaluated) posttreatment (Bankiewicz et al., 2006; Christine et al., 2009). Moreover, the growing clinical experience with delivering recombinant AAV vectors into the brain coupled with advances in both vectorology and CNS delivery portend the therapeutic potential of this strategy for neurodegenerative diseases (LeWitt et al., 2011; Mingozzi and High, 2011; Mittermeyer et al., 2012).
In summary, this study confirms similar findings in other mouse models of HD, by demonstrating that AAV-mediated RNAi could significantly improve HD-related behavioral abnormalities in the YAC128 mouse model of HD. Furthermore, it also extends these findings as this is the first report to show that the AAV-mediated delivery of an miRNA targeting Htt can lead to sustained suppression of Htt levels, correction of cellular and neuropathological aberrations, and improvements in motor and behavioral deficits in a transgenic mouse model of HD without overt toxicity.
Off-target effects associated with the targeting sequence as well as perturbations of the endogenous microRNA (miRNA) processing pathway will need to be extensively investigated before clinical testing. Work has begun to address these concerns, and a number of potential solutions are now being implemented to mitigate these potential risks (Boudreau et al., 2011). The careful design of RNAi sequences that minimizes their interaction with off-targets, along with extensive long-term safety and tolerability studies, will be critical before transitioning this practice into the clinic. Advancement in AAV vectorology continues to encourage the clinical testing of this drug delivery system, and there are several ongoing clinical studies employing centrally delivered AAV for neurologic indications (Janson et al., 2002; Leone et al., 2012; Mittermeyer et al., 2012). The data generated here are suggestive of a transformative therapy for HD. With a growing understanding of the mechanism of RNAi, AAV-mediated RNAi has the potential to treat not only HD but also a number of other toxic gain-of-function neurodegenerative disorders.
Supplementary Material
Acknowledgments
The authors thank John Lydon (Pathology Department); Juanita Campos-Rivera (Molecular Immunology); Leah Curtin, JoAnne Fagan, Sara Savage, David Lee-Parritz, and Matthew Flegal (Comparative Medicine Department); and Brenda Burnham, Shelley Nass, and Denise Woodcock (Molecular Biology Vector Core) from Genzyme for technical assistance with some of the studies.
Author Disclosure Statement
Lisa M. Stanek, Sergio P. Sardi, Bryan Mastis, Amy R. Richards, Christopher M. Treleaven, Tatyana Taksir, Kuma Misra, Seng H. Cheng, and Lamya S. Shihabuddin are paid employees of Genzyme, a Sanofi company. No other competing interests exist.
References
- Ambros V. (2004). The functions of animal microRNAs. Nature 431, 350–355 [DOI] [PubMed] [Google Scholar]
- Arrasate M., Mitra S., Schweitzer E.S., et al. (2004). Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431, 805–810 [DOI] [PubMed] [Google Scholar]
- Augood S.J., Faull R.L., and Emson P.C. (1997). Dopamine D1 and D2 receptor gene expression in the striatum in Huntington's disease. Ann. Neurol. 42, 215–221 [DOI] [PubMed] [Google Scholar]
- Bankiewicz K.S., Forsayeth J., Eberling J.L., et al. (2006). Long-term clinical improvement in MPTP-lesioned primates after gene therapy with AAV-hAADC. Mol. Ther. 14, 564–570 [DOI] [PubMed] [Google Scholar]
- Bates G. (2003). Huntingtin aggregation and toxicity in Huntington's disease. Lancet 361, 1642–1644 [DOI] [PubMed] [Google Scholar]
- Benn C.L., Sun T., Sadri-Vakili G., et al. (2008). Huntingtin modulates transcription, occupies gene promoters in vivo, and binds directly to DNA in a polyglutamine-dependent manner. J. Neurosci. 28, 10720–10733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhide P.G., Day M., Sapp E., et al. (1996). Expression of normal and mutant huntingtin in the developing brain. J. Neurosci. 16, 5523–5535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau R.L., Martins I., and Davidson B.L. (2009a). Artificial microRNAs as siRNA shuttles: Improved safety as compared with shRNAs in vitro and in vivo. Mol. Ther. 17, 169–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau R.L., McBride J.L., Martins I., et al. (2009b). Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington's disease mice. Mol. Ther. 17, 1053–1063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudreau R.L., Spengler R.M., and Davidson B.L. (2011). Rational design of therapeutic siRNAs: Minimizing off-targeting potential to improve the safety of RNAi therapy for Huntington's disease. Mol. Ther. 19, 2169–2177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattaneo E., Zuccato C., and Tartari M. (2005). Normal huntingtin function: An alternative approach to Huntington's disease. Nat. Rev. Neurosci. 6, 919–930 [DOI] [PubMed] [Google Scholar]
- Cha J.H. (2000). Transcriptional dysregulation in Huntington's disease. Trends Neurosci. 23, 387–392 [DOI] [PubMed] [Google Scholar]
- Christine C.W., Starr P.A., Larson P.S., et al. (2009). Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology 73, 1662–1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryan J.F., Markou A., and Lucki I. (2002a). Assessing antidepressant activity in rodents: Recent developments and future needs. Trends Pharmacol. Sci. 23, 238–245 [DOI] [PubMed] [Google Scholar]
- Cryan J.F., Page M.E., and Lucki I. (2002b). Noradrenergic lesions differentially alter the antidepressant-like effects of reboxetine in a modified forced swim test. Eur. J. Pharmacol. 436, 197–205 [DOI] [PubMed] [Google Scholar]
- Davidson B.L., and Monteys A.M. (2012). Singles engage the RNA interference pathway. Cell 150, 873–875 [DOI] [PubMed] [Google Scholar]
- Desplats P.A., Kass K.E., Gilmartin T., et al. (2006). Selective deficits in the expression of striatal-enriched mRNAs in Huntington's disease. J. Neurochem. 96, 743–757 [DOI] [PubMed] [Google Scholar]
- DiFiglia M., Sapp E., Chase K.O., et al. (1997). Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science 277, 1990–1993 [DOI] [PubMed] [Google Scholar]
- DiFiglia M., Sena-Esteves M., Chase K., et al. (2007). Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc. Natl. Acad. Sci. U.S.A. 104, 17204–17209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouet V., Perrin V., Hassig R., et al. (2009). Sustained effects of nonallele-specific Huntingtin silencing. Ann. Neurol. 65, 276–285 [DOI] [PubMed] [Google Scholar]
- Grondin R., Kaytor M.D., Ai Y., et al. (2012). Six-month partial suppression of Huntingtin is well tolerated in the adult rhesus striatum. Brain 135, 1197–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutekunst C.A., Li S.H., Yi H., et al. (1999). Nuclear and neuropil aggregates in Huntington's disease: Relationship to neuropathology. J. Neurosci. 19, 2522–2534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper S.Q., Staber P.D., He X., et al. (2005). RNA interference improves motor and neuropathological abnormalities in a Huntington's disease mouse model. Proc. Natl. Acad. Sci. U.S.A. 102, 5820–5825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janson C., McPhee S., Bilaniuk L., et al. (2002). Clinical protocol: Gene therapy of Canavan disease: AAV-2 vector for neurosurgical delivery of aspartoacylase gene (ASPA) to the human brain. Hum. Gene Ther. 13, 1391–1412 [DOI] [PubMed] [Google Scholar]
- Kordasiewicz H.B., Stanek L.M., Wancewicz E.V., et al. (2012). Sustained therapeutic reversal of Huntington's disease by transient repression of huntingtin synthesis. Neuron 74, 1031–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krol J., Loedige I., and Filipowicz W. (2010). The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 11, 597–610 [DOI] [PubMed] [Google Scholar]
- Lansbury P.T., and Lashuel H.A. (2006). A century-old debate on protein aggregation and neurodegeneration enters the clinic. Nature 443, 774–779 [DOI] [PubMed] [Google Scholar]
- Leone P., Shera D., McPhee S.W., et al. (2012). Long-term follow-up after gene therapy for Canavan disease. Sci. Transl. Med. 4, 165ra163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeWitt P.A., Rezai A.R., Leehey M.A., et al. (2011). AAV2-GAD gene therapy for advanced Parkinson's disease: A double-blind, sham-surgery controlled, randomised trial. Lancet Neurol. 10, 309–319 [DOI] [PubMed] [Google Scholar]
- Liu W., Kennington L.A., Rosas H.D., et al. (2008). Linking SNPs to CAG repeat length in Huntington's disease patients. Nat. Methods 5, 951–953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombardi M.S., Jaspers L., Spronkmans C., et al. (2009). A majority of Huntington's disease patients may be treatable by individualized allele-specific RNA interference. Exp. Neurol. 217, 312–319 [DOI] [PubMed] [Google Scholar]
- Machida Y., Okada T., Kurosawa M., et al. (2006). rAAV-mediated shRNA ameliorated neuropathology in Huntington disease model mouse. Biochem. Biophys. Res. Commun. 343, 190–197 [DOI] [PubMed] [Google Scholar]
- Matsui M., and Corey D.R. (2012). Allele-selective inhibition of trinucleotide repeat genes. Drug Discov. Today 17, 443–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride J.L., Boudreau R.L., Harper S.Q., et al. (2008). Artificial miRNAs mitigate shRNA-mediated toxicity in the brain: Implications for the therapeutic development of RNAi. Proc. Natl. Acad. Sci. U.S.A. 105, 5868–5873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride J.L., Pitzer M.R., Boudreau R.L., et al. (2011). Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington's disease. Mol. Ther. 19, 2152–2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mingozzi F., and High K.A. (2011). Therapeutic in vivo gene transfer for genetic disease using AAV: Progress and challenges. Nat. Rev. Genet. 12, 341–355 [DOI] [PubMed] [Google Scholar]
- Mittermeyer G., Christine C.W., Rosenbluth K.H., et al. (2012). Long-term evaluation of a phase 1 study of AADC gene therapy for Parkinson's disease. Hum. Gene Ther. 23, 377–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passini M.A., and Wolfe J.H. (2001). Widespread gene delivery and structure-specific patterns of expression in the brain after intraventricular injections of neonatal mice with an adeno-associated virus vector. J. Virol. 75, 12382–12392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister E.L., Kennington L., Straubhaar J., et al. (2009). Five siRNAs targeting three SNPs may provide therapy for three-quarters of Huntington's disease patients. Curr. Biol. 19, 774–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porsolt R.D., Bertin A., and Jalfre M. (1977). Behavioral despair in mice: A primary screening test for antidepressants. Arch. Int. Pharmacodyn. Ther. 229, 327–336 [PubMed] [Google Scholar]
- Pouladi M.A., Graham R.K., Karasinska J.M., et al. (2009). Prevention of depressive behaviour in the YAC128 mouse model of Huntington disease by mutation at residue 586 of huntingtin. Brain 132, 919–932 [DOI] [PubMed] [Google Scholar]
- Pouladi M.A., Stanek L.M., Xie Y., et al. (2012). Marked differences in neurochemistry and aggregates despite similar behavioural and neuropathological features of Huntington disease in the full-length BACHD and YAC128 mice. Hum. Mol. Genet. 21, 2219–2232 [DOI] [PubMed] [Google Scholar]
- Reiner A., Dragatsis I., Zeitlin S., and Goldowitz D. (2003). Wild-type huntingtin plays a role in brain development and neuronal survival. Mol. Neurobiol. 28, 259–276 [DOI] [PubMed] [Google Scholar]
- Richfield E.K., Maguire-Zeiss K.A., Cox C., et al. (1995). Reduced expression of preproenkephalin in striatal neurons from Huntington's disease patients. Ann. Neurol. 37, 335–343 [DOI] [PubMed] [Google Scholar]
- Rodriguez-Lebron E., Denovan-Wright E.M., Nash K., et al. (2005). Intrastriatal rAAV-mediated delivery of anti-huntingtin shRNAs induces partial reversal of disease progression in R6/1 Huntington's disease transgenic mice. Mol. Ther. 12, 618–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas H.D., Liu A.K., Hersch S., et al. (2002). Regional and progressive thinning of the cortical ribbon in Huntington's disease. Neurology 58, 695–701 [DOI] [PubMed] [Google Scholar]
- Sah D.W., and Aronin N. (2011). Oligonucleotide therapeutic approaches for Huntington disease. J. Clin. Invest. 121, 500–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saudou F., Finkbeiner S., Devys D., and Greenberg M.E. (1998). Huntingtin acts in the nucleus to induce apoptosis but death does not correlate with the formation of intranuclear inclusions. Cell 95, 55–66 [DOI] [PubMed] [Google Scholar]
- Schaffar G., Breuer P., Boteva R., et al. (2004). Cellular toxicity of polyglutamine expansion proteins: Mechanism of transcription factor deactivation. Mol. Cell 15, 95–105 [DOI] [PubMed] [Google Scholar]
- Scherzinger E., Lurz R., Turmaine M., et al. (1997). Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 90, 549–558 [DOI] [PubMed] [Google Scholar]
- Seyhan A.A. (2011). RNAi: A potential new class of therapeutic for human genetic disease. Hum. Genet. 130, 583–605 [DOI] [PubMed] [Google Scholar]
- Slow E.J., van Raamsdonk J., Rogers D., et al. (2003). Selective striatal neuronal loss in a YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 12, 1555–1567 [DOI] [PubMed] [Google Scholar]
- Sugars K.L., Brown R., Cook L.J., et al. (2004). Decreased cAMP response element-mediated transcription: An early event in exon 1 and full-length cell models of Huntington's disease that contributes to polyglutamine pathogenesis. J. Biol. Chem. 279, 4988–4999 [DOI] [PubMed] [Google Scholar]
- van Bilsen P.H., Jaspers L., Lombardi M.S., et al. (2008). Identification and allele-specific silencing of the mutant huntingtin allele in Huntington's disease patient-derived fibroblasts. Hum. Gene Ther. 19, 710–719 [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk J., Murphy J.M., Slow E., et al. (2005). Selective degeneration and nuclear localization of mutant huntingtin in the YAC128 mouse model of Huntington disease. Hum. Mol. Genet. 14, 3823–3835 [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk J.M., Metzler M., Slow E., et al. (2007). Phenotypic abnormalities in the YAC128 mouse model of Huntington disease are penetrant on multiple genetic backgrounds and modulated by strain. Neurobiol. Dis. 26, 189–200 [DOI] [PubMed] [Google Scholar]
- Vonsattel J.P., Myers R.H., Stevens T.J., et al. (1985). Neuropathological classification of Huntington's disease. J. Neuropathol. Exp. Neurol. 44, 559–577 [DOI] [PubMed] [Google Scholar]
- Wang Y.L., Liu W., Wada E., et al. (2005). Clinico-pathological rescue of a model mouse of Huntington's disease by siRNA. Neurosci. Res. 53, 241–249 [DOI] [PubMed] [Google Scholar]
- Winer J., Jung C.K.S., Shackel I., Williams P.M. (1999). Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Analytical Biochemistry 270, 41–49 [DOI] [PubMed] [Google Scholar]
- Yamamoto A., Lucas J.J., and Hen R. (2000). Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell 101, 57–66 [DOI] [PubMed] [Google Scholar]
- Yu D., Pendergraff H., Liu J., et al. (2012). Single-stranded RNAs use RNAi to potently and allele-selectively inhibit mutant huntingtin expression. Cell 150, 895–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C., Tartari M., Crotti A., et al. (2003). Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat. Genet. 35, 76–83 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.