

Abstract
Recent research has demonstrated that aberrant sphingolipid signaling is an important mechanism of chemo-resistance in solid tumors. Sphingosine kinase (Sphk), the primary enzyme metabolizing the sphingolipid ceramide into sphingosine-1-phosphate (S1P), is a primary mediator of breast cancer promotion, survival and chemoresistance. However, to date the mechanism of Sphk-mediated drug resistance is poorly understood. Using the dual sphingosine kinase isozyme inhibitor, SKI-II (4-[4-(4-chloro-phenyl)-thiazol-2-ylamino]-phenol), we explored the effects of sphingosine kinase inhibition on multi-drug-resistant breast cancer cells. We demonstrate that SKI-II alters endogenous sphingolipid signaling and decreases cancer proliferation, survival and viability. Furthermore, pharmacological inhibition of Sphk1/2 induced intrinsic apoptosis in these cells through modulation of the NF-κB pathway. SKI-II decreases NF-κB transcriptional activity through altered phosphorylation of the p65 subunit. Taken together, these results suggest that Sphk may be a promising therapeutic target in chemoresistant cancers.
Keywords: sphingolipids, chemoresistance, sphingosine kinase, NF-κB, breast cancer, ceramide, experimental therapeutics, sphingosine-1-phosphate
Introduction
NF-κB is a ubiquitously expressed transcription factor known to regulate a variety of cellular functions, including apoptosis, survival, and differentiation. It consists of five subunits: RelA (p65), c-Rel, RelB, NF-κB1 (p50/p105), NF-κB2 (p52/p100), that are sequestered in the cytoplasm under resting conditions (1). The NF-κB signaling cascade is activated through binding of death receptors on the cell surface, which are members of the tumor necrosis factor (TNF) superfamily that contain a death domain involved in protein-protein interactions (2). Upon ligand binding, the intracellular domain of the death receptor recruits a number of scaffolding proteins forming a ‘death inducing signaling complex’ or DISC. The DISC subsequently binds the inactive form of caspase-8 via the death domain (1). These pro-caspase-8 molecules are then cleaved to form active caspase-8, thus triggering a cascade that activates downstream caspases, including caspases 3, 6 and 7, which leads to apoptosis (3,4).
Upon TNF binding to the TNF receptor, inhibitory κ kinase (IκK) is activated and phosphorylates IκB, which in turn degrades and releases the transcription factor allowing the NF-κB to translocate to the nucleus and alter gene transcription (2). NF-κB is constitutively activated in cancer and mediates cancer progression and metastasis through regulation of a variety of specific genes, including the inhibitor of apoptosis (IAP) and matrix metalloproteinase (MMP) gene families (5). NF-κB gene transcription is tightly regulated through phosphorylation of the various subunits prior to nuclear binding (6). Each subunit has multiple phosphorylation binding sites, with p50 and p65 believed to the biologically relevant subunits for breast cancer. The p65 subunit is phosphorylated by several intracellular enzymes including protein kinase A (PKA), Akt, GSK1, and mitogen and stress activated protein kinase-1 (MSK1) (7,8).
Altered sphingolipid signaling is known to be a primary mechanism of solid tumor promotion, progression and survival (9,10). The enzyme sphingosine kinase (Sphk) converts the pro-apoptotic sphingolipid ceramide into its pro-survival metabolite sphingosine-1-phosphate (S1P) (11). Sphk induces cancer mitogenesis through both increased production of S1P as well as S1P-independent cellular mechanisms (9). The two isoforms of sphingosine kinase, Sphk1 and Sphk2, are differently regulated and were initially believed to have opposing roles in the cells (12). Recent evidence suggests that the isoforms have similar roles in certain cancers, including breast, and promote cancer survival and chemoresistance (13,14). However, to date the mechanism of Sphk induced drug resistance is not well understood, in part as a result of the scarcity of small molecular inhibitors targeting sphingosine kinase. Recently, crosstalk between the NF-κB and sphingolipid signaling pathways has been under investigation. It is now known that Sphk1 interacts with TRAF2 on the intracellular portion of the TNF receptor and that TNF can increase Sphk activity (15,16). Sphk also regulates TNF-induced expression of certain adhesion molecules (17). More recently, siRNA knockdown of Sphk1 in A549 lung cancer cells was shown to decrease phosphorylation and translocation of the p65/p50 complex to the nucleus (18). Our laboratory has recently demonstrated that isozyme selective inhibition of Sphk2 with ABC294640 can alter NF-κB transcriptional activity in metastatic cancer cells (19).
As mentioned above, only recently have small molecular inhibitors targeting Sphk been developed (20). In this study, we use the SKI-II, the first commercially available Sphk inhibitor, to pharmacologically target Sphk (21). While initially believed to selectively block Sphk1, it is now known to inhibit both Sphk1 and Sphk2 through competitive inhibition of the intermediate sphingosine (20). SKI-II was utilized to study the effects of sphingosine kinase inhibition on TNF-induced multidrug resistance in metastatic breast cancer cells.
Materials and methods
Reagents
SKI-II (4-[4-(4-chloro-phenyl)-thiazol-2-ylamino]-phenol), etoposide and doxorubicin were purchased from Sigma-Aldrich. Dimethyl sulfoxide (DMSO) was purchased from Fisher Scientific.
Cell culture
ER-negative MCF-7TN-R and MDA-MB-231 cells were cultured as previously described (22,23). Briefly, the MDA-MB-231 cell line was obtained from the American Type Culture Collection (ATCC, Manassas, VA). The MCF-7TN-R cell line is a MCF-7 derived, ER/PR/HER2 negative, chemo-resistant breast cancer cell line generated in the laboratory (22,24,25). These cells were derived by growing MCF-7 cells in increasing concentrations of TNFα until resistance was established. The culture flasks were maintained in a tissue culture incubator in a humidified atmosphere of 5% CO2 and 95% air at 37°C.
Lipidomics analysis
Endogenous lipid levels were quantified by mass spectrometry (Lipidomics Core, Medical University of South Carolina) according to published methods (19,22,26). Briefly, cells were collected, fortified with internal standards and extracted with ethyl acetate/isopropyl alcohol. Electrospray ionization followed by tandem mass spectrometry (ESI/MS/MS) analyses of sphingoid bases, sphingoid base 1-phosphates, ceramides, and sphingomyelins were performed on a Thermo Finnigan TSQ 7000 triple quadrupole mass spectrometer.
Clonogenic survival assay
Colony assays were performed as described in previously published methods (22,24). Cells were plated in 6-well plates at a density of 1000 cells per well in full DMEM media. Twenty-four hours later, cells were treated with SKI-II (0.1–10 μM) and then monitored for colony growth. Ten days later the cells were fixed with 3% glutaraldehyde. Following fixation for 15 min, the plates were washed and stained with a 0.4% solution of crystal violet in 20% methanol for 30 min, washed with PBS, and dried. Colonies of ≥30 cells were counted as positive. Results were normalized to DMSO vehicle treated control cells.
Cell proliferation immunofluorescence assay
Proliferation assays were performed as previously described (19,26). Cells were plated at a density of 10,000 cells per well in a 96-well plate in 10% DMEM media and allowed to attach overnight. The following day, cells were treated with DMSO or SKI-II for 24 h. At endpoint, cells were fixed using 100 μl of 3.7% formaldehyde in PBS for 10 min. Formaldehyde was removed and cells were permeabilized using cold methanol for 5 min at room temperature and washed twice with PBS. One hundred μl of 3% FBS in PBS blocking buffer was then added. After 30 min, blocking buffer was removed and cells were incubated for 1 h with Ki-67 (BD Pharmingen, San Diego, CA) antibody. Cells were then washed with PBS and stained with DAPI nuclear stain for 5 min before imaging. For staining quantification, numbers of positively stained cells were expressed as a percentage of the total number of cells per field of view/image. The vehicle control was then set to 1 for comparison with SKI-II treatment.
Cell viability assay
Viability assays were performed as previously described (23,24). Briefly, cells were plated at a density of 7500 cells per well in a 96-well plate in phenol-free DMEM supplemented with 5% FBS and allowed to attach overnight. Cells were then treated with SKI-II (ranging from 10 nM to 100 μM) alone or in combination with etoposide or doxorubicin for 24 h. Following treatment, 20 μl of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, 5 mg/ml) reagent was incubated in each well for 4 h. Cells were lysed with 20% SDS in 50% dimethylformamide. The pH and absorbances were read on an ELx808 Microtek plate reader (Bio-Tek Instruments, Winooski, VT) at 550 nm, with a reference wavelength of 630 nm.
Cell death detection ELISA
The induction of apoptosis was determined as previously described using the nucleosome ELISA kit (Roche, Boulder, CO) (19). Cells were plated at 10,000 cells per well in 96-well plates and treated for 24 h with varying concentration of drug. The induction of apoptosis was determined by the amount of nucleosomes in the cytoplasm per the manufacturer’s protocol. Absorbances were read on an ELx808 Microtek plate reader (Bio-Tek Instruments) at 405 nm.
Caspase activation assay
Caspase activation was determined as previously described using the Caspase-Glo 9 Assay (Promega Corp., Madison, WI) per the manufacturer’s protocol (19). Cells were plated at a density of 10,000 cells per well in a 96-well plate in phenol-free DMEM supplemented with 5% FBS and allowed to attach overnight. Cells were then treated with indicated drug for 24 h. Following treatment, Caspase-Glo-9 Buffer, Caspase-Glo-9, substrate and MG-132 inhibitor reagent mix was added to cells in a 1:1 ratio with media for 35 min. Following incubation, luminescence was read in an Autoluminat Plus luminometer (Berthhold Technologies, Bad Wildbad, Germany).
NF-κB-luciferase assay
As previously described, the cells were seeded in 24-well plates at a density of 5×105 cells/well in the same media and allowed to attach overnight (19,27,28). After 18 h, cells were transfected for 5 h in serum-free DMEM with 10 ng of pFC-NF-κB-luciferase plasmid, using 6 μl of Effectene (Qiagen) per μg of DNA. After 5 h the transfection medium was removed and replaced with phenol red-free DMEM supplemented with 5% CS-FBS containing vehicle, TNF, SKI-II, or TNF + SKI-II, and incubated at 37°C. After 18 h the medium was removed, and 100 μl of lysis buffer was added per well and then incubated for 15 min at room temperature. Cell debris was pelleted by centrifugation at 15,000 × g for 5 min. Cell extracts were normalized for protein concentration using reagent according to the manufacturers protocol (BioRad Laboratories). Luciferase activity for the cell extracts was determined using luciferase substrate (Promega Corp.) in an Autoluminat Plus luminometer (Berthhold Technologies).
Western blot analysis
Protein analysis was performed as described (19,26,29). Cells were plated at 50–60% confluency in 10-cm2 culture flasks in 10% DMEM for 48 h. They were then treated with DMSO or SKI-II for 1 h. Following treatment, cells were detached with PBS-EDTA and centrifuged. After removing supernatant, cells were lysed in 60–100 μl lysis buffer (Mammalian Protein Extraction Reagent, MPER, and Halt protease inhibitor (Pierce, Rockford, IL); and PhoSTOP phosphatase inhibitor (Roche). Lysed cells were centrifuged for 10 min at 12,000 G at 4°C to separate protein from cell debris. The supernatants were combined with loading buffer (5% 2-mercaptoethanol in 4X LDS Loading buffer, (Invitrogen), boiled for 5 min, and loaded onto a 4–12% Bis Tris polyacrylamide gel (Invitrogen) followed by polyacrylamide gel electrophoresis at 150 V for 1.25 h. Protein was transferred to nitrocellulose membranes using the iBlot (Invitrogen) transfer unit. Nitrocellulose membranes were blocked in 5% milk (BioRad Laboratories) Tris Buffered Saline with Tween-20 (TBS-T) for 1 h at room temperature. Cells were washed briefly with 1X TBS-T (USB, Cleveland, OH) and primary antibodies were diluted in 5% BSA (Bovine Serum Albumin, Sigma-Aldrich, St. Louis, MO) TBS-T according to manufacturer’s recommended dilutions. Antibodies for tubulin, p65 (C22B4), phospho-p65 (93H1), IκB (polyclonal), phospho-I IκB (14D4), IκK (L570) and phospho-IκK (16A6) (1:1000) were purchased from Cell Signaling Technology, Inc. (Beverly, MA). Membranes were incubated in primary antibody overnight at 4°C with gentle agitation. Secondary infrared conjugated antibodies (LI-COR Biosciences, Lincoln, NE) were diluted in 5% BSA-TBST solution at 1:10,000 and membranes were incubated for 1 h under gentle agitation at room temperature. Membranes were scanned using the LI-COR Odyssey imager and software (LI-COR Biosciences) to detect total and phosphorylated protein levels in cell lysates. Western blots and protein quantification were performed following three independent experiments, with representative blots shown. Protein levels were quantified using densitometry analyses.
Statistical analysis
Statistical analyses were performed as previously described (19,30). Briefly, the IC50 values were calculated from concentration-response curves using GraphPad Prism 5.0 (GraphPad software), using the equation:
assuming a standard slope, where the response goes from 10 to 90% of maximal as X increases over two log units. Differences in IC50 were compared using Student’s unpaired t-test with p<0.05 as the limit of statistical significance. Experiments comparing multiple concentrations to the control were tested with one-way ANOVA with Bonferroni post-test to compare individual concentrations. Combination effects were calculated using the formula: [X1a+X2b]/[(X1a+X2b)], wherein X1 is the effect of drug 1 at concentration a, X2 is the effect of drug 2 at concentration b and [X1a+X2b] is the effect of the combination of both drugs at concentrations a and b, respectively (31). All statistical analyses were done using GraphPad Prism 5.0 (GraphPad software).
Results
SKI-II blocks downstream S1P signaling in chemoresistant breast cancer
The metastatic breast cancer cell lines MDA-MB- 231 and MCF-7TN-R were chosen as models of drug resistance. The MDA-MB-231 cell line is a well-established, ER/PR/HER2 negative system that is endocrine therapy resistant, but chemotherapy sensitive, and is a model of de novo drug resistance (32). The MCF-7TN-R cell line was derived from ER-positive, chemosensitive MCF-7 cells through exposure to increasing concentrations of TNF until resistance was established (25,33). Similar to MDA-MB-231 cells, these are ER/PR/HER2 negative and hormone therapy resistant. MCF-7TN-R cells were previously shown to exhibit TNF-induced multidrug resistance to primary clinical chemotherapeutics paclitaxel, doxorubicin and etoposide (22,24). Therefore, the MCF-7TN-R cell is used as a model of acquired drug resistance.
Our laboratory has previously demonstrated altered sphingolipid signaling profiles in both the MDA-MB-231 and MCF-7TN-R cell lines, including increased expression of S1P (19). Herein, we investigated the effect of SKI-II on endogenous sphingolipid signaling. As seen in Fig. 1, there is a clear decreasing trend in S1P levels following treatment with SKI-II in both MDA-MB-231 and MCF-7TN-R cell lines. For example, in MDA-MB-231 cells, SKI-II decreased S1P formation by 48.34±28.10%, and 34.86±19.12% in MCF-7TN-R cells. Alteration in ceramide protein levels were also observed, including a marked increase in sphinganine. These results are consistent with previously published studies of sphingosine kinase inhibitors in other cell lines (26,30).
Figure 1.
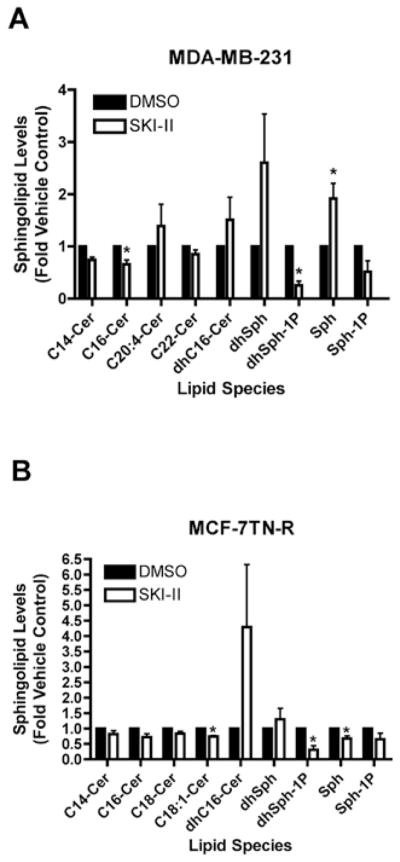
Pharmacologic inhibition of Sphk1/2 alters endogenous sphingolipid signaling. (A) MDA-MB-231 and (B) MCF-7TN-R cells were treated with either vehicle or SKI-II (10 μM) 24 h and measured for cellular levels of various sphingolipid species using ESI/MS/MS. Data points and error bars represent the mean ± SEM of three independent experiments.
We next investigated whether SKI-II could inhibit the downstream biological effects of Sphk1/2, including viability, survival, and proliferation. Using short-term viability assays, the IC50 value of SKI-II was determined in both endocrine and chemotherapy resistant cancer cell lines. SKI-II exhibited IC50 values of 11.77±2.17 μM (p<0.001) and 4.43±1.25 μM (p<0.001) in MDA-MB-231 and MCF-7TN-R cells, respectively (Fig. 2A). The IC50 values seen here are more efficacious than those previously published in the parental MCF-7 cell line, suggesting that Sphk is involved in the acquired resistance mechanisms of these cells. There is some debate as to the clinical relevance of short-term viability assays, with some studies demonstrating a poor predictive value between these and clinical models (34). Therefore, we determined the effects of sphingosine kinase inhibition on long-term metastatic cancer clonogenic survival to better determined the therapeutic potential of this target. Long-term treatment of SKI-II results in MDA-MB-231 and MCF-7TN-R IC50 values of 2.51±1.08 μM (p<0.001) and 2.70±1.05 μM (p<0.001), respectively (Fig. 2B). These results in the low micro-molar range are similar to those of current clinical therapeutics.
Figure 2.

Effect of SKI-II on metastatic cancer viability and survival. (A) MDA-MB-231 and MCF-7TN-R cells were plated at 7.5×103 cells per 96-well plate. The following day cells were treated with indicated concentrations of SKI-II for 24 h. Data are presented as percent of vehicle treated samples. Mean values of ± SEM of 5 different experiments in quadruplicate are reported. (B) MDA-MB-231 and MCF-7TN-R cells were plated at 500 cells per 60 mm2. The following day, cells were treated with SKI-II for 10–14 days. Data are presented as percent of vehicle treated samples. Mean values of ± SEM of 3 different experiments in duplicate are reported.
Inhibition of cancer proliferation is a necessary characteristic of any clinical chemotherapeutic. The effect of SKI-II on cancer proliferation was determined using Ki-67 immunofluorescence assays. Ki-67 is a nuclear protein expressed only during mitogenic phases of the cell cycle (35,36). As seen in Fig. 3, pharmacological inhibition of SKI-II has potent antiproliferative properties in MDA-MB-231 cells, decreasing Ki-67 staining by 80.23±4.87% (p<0.001). Of note, SKI-II was less effective in the MCF-7TN-R cell line, decreasing staining by 20.97±5.55% (p<0.001). This suggests that the primary viability effects of SKI-II may not be related to its anti-proliferative effects.
Figure 3.

Varying anti-proliferative effects of Sphk inhibition in de novo acquired drug resistance. (A) MDA-MB-231 cells and (B) MCF-7TN-R cells were treated with vehicle or SKI-II (10 μM) for 48 h. Following treatment, cells were fixed and stained with anti-Ki-67 (red) and nuclei counter stained with DAPI (blue). (A) Representative images of cells at ×250. (B) Quantification of cells positive for Ki-67 staining from 10 fields of view per treatment. Data is represented as percent positive cells as compared to total cells normalized to vehicle control. Bars represent mean values ± SEM of three independent experiments.
Pharmacological inhibition of sphingosine kinase-1/2 enhances chemotherapeutic-induced intrinsic apoptosis in chemoresistant cancer
Given the decreased anti-proliferative effect of SKI-II in MCF-7TN-R cells compared to MDA-MB-231 cells, we examined whether SKI-II induced apoptosis to exert its anti-viability properties. MCF-7TN-R cells were treated with SKI-II and analyzed for cytoplasmic histone-associated oligonucleosome DNA fragments. As seen in Fig. 4A, treatment with SKI-II resulted in treatment with SKI-II increased apoptosis by 11.90±2.96-fold (p<0.05) compared to vehicle control (Fig. 4A). This finding is interesting given that the MCF-7TN-R cell line is resistant to TNF-induced apoptosis. Given that sphingolipids are known to regulate both intrinsic and extrinsic apoptotic pathways, we investigated the effect of SKI-II on the downstream apoptotic protein caspase-9, a commonly used marker for intrinsic cell death. As seen in Fig. 4B, pharmacological inhibition of Sphk1/2 resulted in a corresponding increase in caspase-9 activity, suggesting that SKI-II-induced apoptosis is at least partially through the intrinsic cell death pathway.
Figure 4.

Pharmacological inhibition of Sphk1/2 induced intrinsic apoptosis in chemoresistance cancer. MCF-7TN-R cells were plated at 7.5×103 cells per 96-well plate. The following day, cells were treated with SKI-II (50 μM) for 24 h and measured for (A) fragmented DNA oligonucleotides using ELISA assays and (B) analyzed for caspase-9 activation. Mean values of ± SEM of four different experiments in duplicate are reported.
The ability of therapeutic agents to act synergistically is beneficial in the treatment of any cancer. Therefore, we determined whether pharmacological inhibition of Sphk1/2 enhanced the effects of the clinical chemotherapeutics doxorubicin and etoposide. MCF-7TN-R breast cancer cells were pre-treated for 1 h with 10 μM of SKI-II followed by 24 h of treatment with a chemotherapeutic agent and compared to isolated treatment with each drug. Following treatment, cells were measured for apoptosis, caspase-9 activity and cell viability.
As expected, etoposide and doxorubicin also induced apoptosis through the intrinsic pathway. Pre-treatment with SKI-II enhanced the apoptotic effect of both etoposide and doxorubicin in MCF-7TN-R cells (Fig. 5). Treatment with SKI-II alone showed modest induction of apoptosis and caspase-9 compared to etoposide and doxorubicin alone. However, treatment with SKI-II and etoposide increased apoptosis by 20.99±4.83 fold (p<0.05) and caspase-9 activity by 4.97±0.74 (p<0.05) fold. Similarly, following SKI-II and doxorubicin treatment, apoptosis increased by 33.33±9.94-fold (p<0.05) and caspase-9 activity by 5.26±1.22-fold (p<0.05). These results suggest that combination treatment with sphingosine kinase inhibition can synergistically enhance the apoptotic effects of clinical chemotherapeutics.
Figure 5.
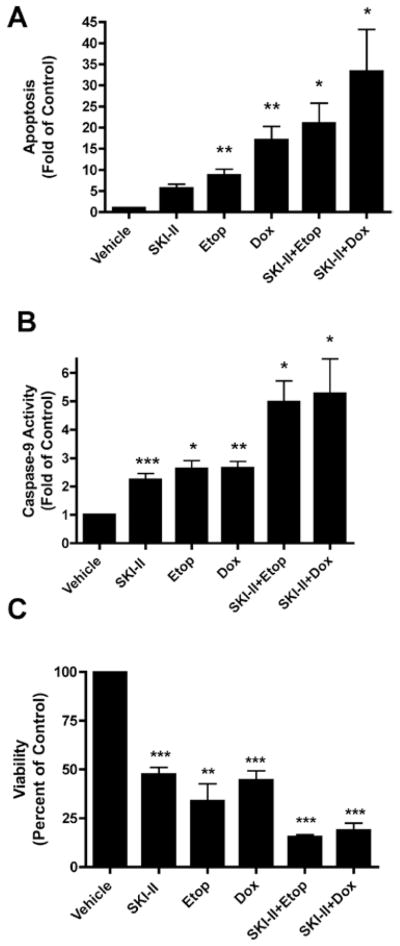
Enhanced efficacy of clinical chemotherapeutics in combination with Sphk inhibition in drug resistant breast cancer cells. MCF-7TN-R cells were plated at 7.5×103 cells per 96-well plate. The following day, cells were treated with SKI-II (10 μM), chemotherapeutic (etoposide 15 μM, doxorubicin, 0.1 μM), or SKI-II + chemotherapeutic for 24 h. Cells were then measured for (A) fragmented DNA oligonucleotides using ELISA assays, (B) analyzed for caspase-9 activation and (C) measured for viability using MTT analyses. Mean values of ± SEM of 4 different experiments in duplicate are reported.
Modulation of the NF-κB signaling through inhibition of Sphk1/2
The ability of Sphk1/2 inhibition to induce apoptosis in these TNF resistant cells led us investigate the mechanism of cell death. Our laboratory previously discovered increased basal p65 transcriptional activity in MCF-7TN-R cells compared to the parental MCF-7 cell line. Therefore, we hypothesized that Sphk1/2 inhibition may be targeting this pathway to restore chemosensitivity to these cells. Both the parental and acquired resistant cell lines were utilized to determine affect of Sphk1/2 signaling on death receptor cascades. Pharmacological inhibition of Sphk1/2 dose-dependently decreased p65 promoter transcriptional activity in both MCF-7 and MCF-7TN-R cells, suggesting Sphk1/2 is involved in endogenous NF-κB gene regulation (Fig. 6A).
Figure 6.

SKI-II inhibits NF-κB transcriptional activity. (A) MCF-7 and (B) MCF-7TN-R cells were transiently transfected with pFC-NF-κB-luciferase plasmid. Following transfection, cells were treated with vehicle, TNFα, SKI-II or SKI-II + TNFα. Cells treated with TNFα were set to 1. Data points and error bars represent the mean ± SEM of three independent experiments. (C) MCF-7 cells were treated with vehicle or SKI-II (10 μM) for 6 h and analyzed by western blotting for phosphorylated and total IκB and p65. Data shown are representative of three independent experiments.
The NF-κB subunit p65 has several phosphorylation sites, which are differentially activated by specific kinase cascades. The PI3K/Akt signaling pathway can activate p65 at the Ser365 phosphorylation site. SKI-II has previously been shown to decrease Akt activation in breast cancer cells. Therefore, we hypothesized that SKI-II decreased phosphorylation of p65 to exert its transcriptional effect. As seen in Fig. 6C, treatment with SKI-II markedly blocked TNF-induced phosphorylation of the p65 subunit at the Ser365 site, without significant altering upstream total or phosphorylation protein levels of IKB. Taken together, these results show that pharmacological inhibition of Sphk1/2 blocks p65 transcription activity through decreased phosphorylation at the Ser365 activation site.
Discussion
Acquired chemoresistance is an important mechanism of clinical treatment failure in cancer. Recent studies have demonstrated the importance of sphingolipid signaling in cancer drug resistance (14). In this study, we show that inhibition of both Sphk1 and Sphk2 with the small molecule inhibitor SKI-II decreases viability, proliferation, and survival in both de novo and acquired drug resistance models of metastatic breast cancer. Furthermore, we demonstrate that inhibition of Sphk1/2 can induce apoptosis through the intrinsic cell death pathway. Of note, combination treatments of SKI-II with clinical chemotherapy enhances the apoptotic potential of doxorubicin and etoposide in a synergistic fashion.
While the NF-κB signaling pathway has long been known to promote cancer progression, drug resistance and metastasis, targeting this pathway in an effective and non-toxic fashion has proven difficult. Herein, we demonstrate that pharmacological inhibition of Sphk1/2 blocks NF-κB transcriptional activation through decreased phosphorylation of the p65 subunit. This mechanism likely accounts for the increased efficacy SKI-II and in combination with chemotherapeutics in acquired-drug-resistance breast cancer cells. SKI-II is well-tolerated in vivo, and these results suggest that combination therapy may allow for a decrease in systemically toxic chemotherapeutics to reach the same anti-tumor effects.
While we have demonstrated one mechanism of SKI-II-induced NF-κB signaling blockade, further investigation is necessary to further understand the crosstalk between the sphingolipid and NF-κB pathways. Sphk/S1P signaling is known to activate a number of intracellular kinase signaling cascades, including Ras, Akt, c-Jun N-terminal Kinase (JNK), Rho and ERK. Many of these kinase cascades are also known to phosphorylate the various Rel family subunits. It is possible that further primary effects of Sphk inhibition on these kinases have downstream secondary effects on the NF-κB phosphorylation. Furthermore, pharmacokinetic studies are needed to elucidate the mechanism of synergy between Sphk inhibition and traditional chemotherapies. The studies presented suggest that pharmacological targeting of Sphk1/2 may be a promising therapeutic avenue in the treatment of drug resistant cancer.
Abbreviations
- BCL-2
B-cell lymphoma 2
- BH3
Bcl-2 homology domain 3
- CER
ceramide
- DAB
diaminobenzidine
- DAP
I 4′,6-diamidino-2-phenylindole
- DCC
dicyclohexylcarbodiimide
- IAP
inhibitor of apoptosis
- IκB
inhibitory κB
- IκK
inhibitory κB kinase
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NF-κB
nuclear factor κB
- Sphk1
sphingosine kinase-1
- Sphk2
sphingosine kinase-2
- SKI
sphingosine kinase inhibitor
- S1P
sphingosine-1-phosphate
- PKC
protein kinase C
References
- 1.Bours V, Bentires-Alj M, Hellin AC, et al. Nuclear factor-kappa B, cancer, and apoptosis. Biochem Pharmacol. 2000;60:1085–1089. doi: 10.1016/s0006-2952(00)00391-9. [DOI] [PubMed] [Google Scholar]
- 2.Hayden MS, Ghosh S. Signaling to NF-kappaB. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 3.Baldwin AS., Jr Series introduction: the transcription factor NF-kappaB and human disease. J Clin Invest. 2001;107:3–6. doi: 10.1172/JCI11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS., Jr NF-kappaB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol. 1999;19:5785–5799. doi: 10.1128/mcb.19.8.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakshatri H, Bhat-Nakshatri P, Martin DA, Goulet RJ, Jr, Sledge GW., Jr Constitutive activation of NF-kappaB during progression of breast cancer to hormone-independent growth. Mol Cell Biol. 1997;17:3629–3639. doi: 10.1128/mcb.17.7.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Viatour P, Merville MP, Bours V, Chariot A. Phosphorylation of NF-kappaB and IkappaB proteins: implications in cancer and inflammation. Trends Biochem Sci. 2005;30:43–52. doi: 10.1016/j.tibs.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 7.Mattioli I, Sebald A, Bucher C, et al. Transient and selective NF-kappa B p65 serine 536 phosphorylation induced by T cell costimulation is mediated by I kappa B kinase beta and controls the kinetics of p65 nuclear import. J Immunol. 2004;172:6336–6344. doi: 10.4049/jimmunol.172.10.6336. [DOI] [PubMed] [Google Scholar]
- 8.Sovak MA, Bellas RE, Kim DW, et al. Aberrant nuclear factor-kappaB/Rel expression and the pathogenesis of breast cancer. J Clin Invest. 1997;100:2952–2960. doi: 10.1172/JCI119848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Antoon JW, Beckman BS. Sphingosine kinase: a promising cancer therapeutic target. Cancer Biol Ther. 2011;11:647–650. doi: 10.4161/cbt.11.7.14921. [DOI] [PubMed] [Google Scholar]
- 10.Efroni S, Schaefer CF, Buetow KH. Identification of key processes underlying cancer phenotypes using biologic pathway analysis. PLoS One. 2007;2:e425. doi: 10.1371/journal.pone.0000425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Spiegel S, Merrill AH., Jr Sphingolipid metabolism and cell growth regulation. FASEB J. 1996;10:1388–1397. doi: 10.1096/fasebj.10.12.8903509. [DOI] [PubMed] [Google Scholar]
- 12.Maceyka M, Sankala H, Hait NC, et al. SphK1 and SphK2, sphingosine kinase isoenzymes with opposing functions in sphingolipid metabolism. J Biol Chem. 2005;280:37118–37129. doi: 10.1074/jbc.M502207200. [DOI] [PubMed] [Google Scholar]
- 13.Hait NC, Oskeritzian CA, Paugh SW, Milstien S, Spiegel S. Sphingosine kinases, sphingosine 1-phosphate, apoptosis and diseases. Biochim Biophys Acta. 2006;1758:2016–2026. doi: 10.1016/j.bbamem.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 14.Meacham WD, Antoon JW, Burow ME, Struckhoff AP, Beckman BS. Sphingolipids as determinants of apoptosis and chemoresistance in the MCF-7 cell model system. Exp Biol Med (Maywood) 2009;234:1253–1263. doi: 10.3181/0902-MR-77. [DOI] [PubMed] [Google Scholar]
- 15.Xia P, Wang L, Gamble JR, Vadas MA. Activation of sphingosine kinase by tumor necrosis factor-alpha inhibits apoptosis in human endothelial cells. J Biol Chem. 1999;274:34499–34505. doi: 10.1074/jbc.274.48.34499. [DOI] [PubMed] [Google Scholar]
- 16.Xia P, Wang L, Moretti PA, et al. Sphingosine kinase interacts with TRAF2 and dissects tumor necrosis factor-alpha signaling. J Biol Chem. 2002;277:7996–8003. doi: 10.1074/jbc.M111423200. [DOI] [PubMed] [Google Scholar]
- 17.Xia P, Gamble JR, Rye KA, et al. Tumor necrosis factor-alpha induces adhesion molecule expression through the sphingosine kinase pathway. Proc Natl Acad Sci USA. 1998;95:14196–14201. doi: 10.1073/pnas.95.24.14196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Billich A, Bornancin F, Mechtcheriakova D, Natt F, Huesken D, Baumruker T. Basal and induced sphingosine kinase 1 activity in A549 carcinoma cells: function in cell survival and IL-1beta and TNF-alpha induced production of inflammatory mediators. Cell Signal. 2005;17:1203–1217. doi: 10.1016/j.cellsig.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 19.Antoon JW, White MD, Slaughter EM, et al. Targeting NFκB mediated breast cancer chemoresistance through selective inhibition of sphingosine kinase-2. Cancer Biol Ther. 2011;11:678–689. doi: 10.4161/cbt.11.7.14903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.French KJ, Upson JJ, Keller SN, Zhuang Y, Yun JK, Smith CD. Antitumor activity of sphingosine kinase inhibitors. J Pharmacol Exp Ther. 2006;318:596–603. doi: 10.1124/jpet.106.101345. [DOI] [PubMed] [Google Scholar]
- 21.French KJ, Schrecengost RS, Lee BD, et al. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003;63:5962–5969. [PubMed] [Google Scholar]
- 22.Antoon JW, Liu J, Gestaut MM, Burow ME, Beckman BS, Foroozesh M. Design, synthesis, and biological activity of a family of novel ceramide analogues in chemoresistant breast cancer cells. J Med Chem. 2009;52:5748–5752. doi: 10.1021/jm9009668. [DOI] [PubMed] [Google Scholar]
- 23.Antoon JW, Liu J, Ponnapakkam AP, Gestaut MM, Foroozesh M, Beckman BS. Novel D:-erythro N-octanoyl sphingosine analogs as chemo- and endocrine-resistant breast cancer therapeutics. Cancer Chemother Pharmacol. 2010;65:1191–1195. doi: 10.1007/s00280-009-1233-0. [DOI] [PubMed] [Google Scholar]
- 24.Struckhoff AP, Bittman R, Burow ME, et al. Novel ceramide analogs as potential chemotherapeutic agents in breast cancer. J Pharmacol Exp Ther. 2004;309:523–532. doi: 10.1124/jpet.103.062760. [DOI] [PubMed] [Google Scholar]
- 25.Weldon CB, Parker AP, Patten D, et al. Sensitization of apoptotically-resistant breast carcinoma cells to TNF and TRAIL by inhibition of p38 mitogen-activated protein kinase signaling. Int J Oncol. 2004;24:1473–1480. [PubMed] [Google Scholar]
- 26.Antoon JW, Meacham WD, Bratton MR, et al. Pharmacological inhibition of sphingosine kinase isoforms alters estrogen receptor signaling in human breast cancer. J Mol Endocrinol. 2011;46:205–216. doi: 10.1530/JME-10-0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bratton MR, Frigo DE, Vigh-Conrad KA, et al. Organochlorine-mediated potentiation of the general coactivator p300 through p38 mitogen-activated protein kinase. Carcinogenesis. 2009;30:106–113. doi: 10.1093/carcin/bgn213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boue SM, Tilghman SL, Elliott S, et al. Identification of the potent phytoestrogen glycinol in elicited soybean (Glycine max) Endocrinology. 2009;150:2446–2453. doi: 10.1210/en.2008-1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burow ME, Weldon CB, Tang Y, et al. Differences in susceptibility to tumor necrosis factor alpha-induced apoptosis among MCF-7 breast cancer cell variants. Cancer Res. 1998;58:4940–4946. [PubMed] [Google Scholar]
- 30.Antoon JW, White MD, Meacham WD, et al. Antiestrogenic effects of the novel sphingosine kinase-2 inhibitor ABC294640. Endocrinology. 2010;151:5124–5135. doi: 10.1210/en.2010-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bindslev N. Drug-Acceptor Interactions : Modeling Theoretical Tools to Test and Evaluate Experimental Equilibrium Effects. Co-Action Pub; Haggeby, Sweden: 2008. [Google Scholar]
- 32.Neve RM, Chin K, Fridlyand J, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weldon CB, Scandurro AB, Rolfe KW, et al. Identification of mitogen-activated protein kinase kinase as a chemoresistant pathway in MCF-7 cells by using gene expression microarray. Surgery. 2002;132:293–301. doi: 10.1067/msy.2002.125389. [DOI] [PubMed] [Google Scholar]
- 34.Brown JM, Wouters BG. Apoptosis, p53, and tumor cell sensitivity to anticancer agents. Cancer Res. 1999;59:1391–1399. [PubMed] [Google Scholar]
- 35.Kill IR. Localisation of the Ki-67 antigen within the nucleolus. Evidence for a fibrillarin-deficient region of the dense fibrillar component. J Cell Sci. 1996;109:1253–1263. doi: 10.1242/jcs.109.6.1253. [DOI] [PubMed] [Google Scholar]
- 36.Starborg M, Gell K, Brundell E, Hoog C. The murine Ki-67 cell proliferation antigen accumulates in the nucleolar and heterochromatic regions of interphase cells and at the periphery of the mitotic chromosomes in a process essential for cell cycle progression. J Cell Sci. 1996;109:143–153. doi: 10.1242/jcs.109.1.143. [DOI] [PubMed] [Google Scholar]