

Summary
Autophagy is a key innate immune response to intracellular parasites that promotes their delivery to degradative lysosomes following detection in the cytosol or within damaged vacuoles. Like Listeria and Shigella, which use specific mechanisms to avoid autophagic detection and capture, the bacterial pathogen Francisella tularensis proliferates within the cytosol of macrophages without demonstrable control by autophagy. To examine how Francisella evades autophagy, we screened a library of F. tularensis subsp. tularensis Schu S4 HimarFT transposon mutants in GFP-LC3-expressing murine macrophages by microscopy for clones localised within autophagic vacuoles after phagosomal escape. Eleven clones showed autophagic capture at six hours post-infection, whose HimarFT insertions clustered to four genetic loci involved in lipopolysaccharidic and capsular O-antigen biosynthesis. Consistent with the HimarFT mutants, in-frame deletion mutants of two representative loci, FTT1236 and FTT1448c (manC), lacking both LPS and capsular O-antigen, underwent phagosomal escape but were cleared from the host cytosol. Unlike wild type Francisella, the O-antigen deletion mutants were ubiquitinated, and recruited the autophagy adaptor p62/SQSTM1 and LC3 prior to cytosolic clearance. Autophagy-deficient macrophages partially supported replication of both mutants, indicating that O-antigen-lacking Francisella are controlled by autophagy. These data demonstrate the intracellular protective role of this bacterial surface polysaccharide against autophagy.
Keywords: autophagy, O-antigen, Francisella, macrophage, capsule
Introduction
Detection of invading microbes by innate immune receptors and sensors is essential to the host’s ability to respond to and control infections by intracellular pathogens. Autophagy, a highly conserved process among eukaryotes, delivers cytoplasmic material to the lysosomal compartment for degradation. While initially described as a nonselective, bulk degradation pathway of cytosolic content up-regulated in response to nutrient deprivation, autophagy serves numerous other functions, including the removal of obsolete organelles, misfolded proteins, and the selective degradation of cytosolic pathogens (Yang et al., 2010). In its innate immune role, autophagy is used to degrade and clear invading organisms following their tagging as autophagic cargo via ubiquitination (Kraft et al., 2010). After initial targeting, the ubiquitinated cargo is recognized by autophagy adapter molecules such as p62/SQSTM1 or NDP52, which in turn enroll autophagy complexes via binding to Atg8/LC3-family proteins, thereby recruiting nascent autophagosomes (phagophores) (Pankiv et al., 2007, von Muhlinen et al., 2010, Zheng et al., 2009, Gal et al., 2009). Autophagosome elongation and closure around the targeted bacterium are orchestrated by various ubiquitin conjugation-like molecular complexes, including the ATG5-ATG12-ATG16L complex and LC3, and leads to sequestration of the pathogen from the cytosol and its subsequent degradation through lysosomal fusion.
Unlike intracellular pathogens that reside within a membrane-bound vacuole, bacteria that disrupt their original phagosome to reach, and replicate in, the host cell cytosol are targets of antibacterial autophagy. Therefore, their ability to survive and proliferate in this compartment must invoke specific mechanisms of autophagy avoidance. Indeed, the cytosolic pathogens Listeria monocytogenes and Shigella flexneri either modify their surface by recruiting host proteins to mask themselves from autophagic recognition (Dortet et al., 2011, Yoshikawa et al., 2009), or secrete a specific protein that interferes with the autophagic cascade on the bacterial surface (Ogawa et al., 2005). Similar to Listeria and Shigella, the tularemia-causing bacterium Francisella tularensis is a cytosolic pathogen that rapidly disrupts its phagosome upon uptake by macrophages (Clemens et al., 2004, Checroun et al., 2006). It then undergoes extensive replication between 4 to 16 hours post-infection without any apparent restriction from the host cell (Wehrly et al., 2009). While a fraction of cytosolic Francisella may translocate into autophagy-related vacuoles after several hours (> 16 h post-infection) in murine macrophages (Checroun et al., 2006), most bacteria successfully reside and proliferate within the cytosol for a protracted length of time without triggering an autophagy response. Additionally, replication-deficient Francisella mutants with eventual survival defects can still persist in the cytosol for up to 10 hours before recognition and clearance by antibacterial autophagy (Chong et al., 2012), arguing for an inherent ability to deflect targeting by selective autophagy. Based on these observations, we hypothesized that Francisella actively avoids autophagic recognition early during its infection cycle and set out to identify genes required for autophagy evasion after entry in the host cytosol. Here we report that F. tularensis strains that lack the surface polysaccharidic O-antigen are susceptible to autophagic capture upon entering the host cytosol, and consequently fail to survive and proliferate efficiently. This study ascribes an additional immune evasion function to Francisella O-antigen and describes a bacterial surface polysaccharide as a shield against autophagic recognition.
Results
Identification of F. tularensis mutants targeted by autophagy in murine macrophages
To identify Francisella genes required for evasion from antibacterial autophagy, we generated and screened a library of HimarFT transposon (Maier et al., 2006) insertion mutants of the highly virulent prototypical strain Schu S4 for clones localizing to autophagic vacuoles rapidly after phagosomal escape. Immortalized bone marrow-derived murine macrophages (iBMM) that stably express the autophagosome marker GFP-LC3 were infected in 96-well glass-bottom plates with individual transposon mutant clones at a multiplicity of infection (MOI) of 100:1. Approximately 6,400 individual insertion mutants were screened for colocalization with GFP-LC3 by fluorescence microscopy at 6 hours post-infection (h post-infection), a time point ensuring complete phagosomal escape but no substantial replication of bacteria. Eleven clones (0.17% of the mutants screened) triggered formation of GFP-LC3-positive vacuoles and were weakly stained by anti-Francisella LPS antibodies. All repeatedly showed detectable targeting to GFP-LC3-positive vacuoles (Fig. 1A) and to LC3-positive vacuoles in primary murine BMMs when counterstained with DAPI (Fig 4E). In addition to their localization to autophagic vacuoles, all 11 clones also exhibited increased uptake by iBMMs relative to wild type Schu S4 (Fig. 1A).
Figure 1.

HimarFT F. tularensis mutants in 4 genetic loci are targeted by autophagy. (A) Confocal micrographs of GFP-LC3 iBMMs infected with F. tularensis Schu S4 (Schu S4), and representative HimarFT clones (by clone number) at 6 h post-infection. DNA was stained with DAPI (gray), and GFP-LC3 is in green. Scale bar, 5 μm. (B) Diagram of the 11 HimarFT insertion loci identified in the autophagy screen. Each insertion is marked with a flag denoting the relative orientation of the kanamycin resistance cassette and the clone number. Interrupted genes are shown in gray. (C) Western blot analysis of HimarFT insertion mutant LPS O-antigen and O-antigen capsule. 1 × 107 bacterial equivalents were resolved and probed with antibodies against LPS O-antigen (LPS), capsule, or GroEL as a loading control.
Figure 4.

Capture of Francisella O-antigen mutants by autophagy in murine macrophages. (A), (C), and (E): Confocal micrographs of BMMs infected with Schu S4 (Schu S4), Schu S4ΔFTT1236 (ΔFTT1236) or Schu S4ΔmanC (ΔmanC) and labelled for the autophagic markers Ubiquitin (A), p62/SQSTM1 (C) and LC3 (E) at 2 h post-infection. DNA was stained with DAPI (red) and specific autophagic markers are in green. Insets are indicated with an arrowhead, and magnified to show bacterial colocalization with autophagic markers. Scale bars, 10 and 2 μm. (B), (D), and (F): Quantification of GFP-expressing F. tularensis Schu S4 (black), Schu S4ΔFTT1236 (red), Schu S4ΔmanC (blue), Schu S4ΔfevR (orange) and Schu S4ΔdipA (green) recruitment of (B) Ubiquitin, (D) p62/SQSTM1, or (F) LC3. Data are means ± SD (n=3).
Autophagy-targeted F. tularensis mutants are defective for O-antigen expression
Identification of the HimarFT insertion loci revealed that the genes disrupted in these mutants clustered to four genetic loci, all of which are involved in biosynthesis of the O-antigen, a polysaccharidic molecule that composes both the Francisella LPS and capsule (Fig 1B) (Prior et al., 2003, Raynaud et al., 2007, Gunn et al., 2007, Lai et al., 2010, Lindemann et al., 2011). Two genetic loci, the manCB operon and the FTT1235c-FTT1237 locus, harbored most HimarFt insertions, with respectively 5 and 4 clones (Fig. 1B), indicating that our screen reached saturation. The remaining mutants had transposon insertions in either the CMP-KDO synthase kdsB (FTT1478c) gene, or upstream of the FTT0804-capBCA locus (Fig. 1B). Using antibodies directed against either the Francisella LPS O-antigen or the Francisella O-antigen-derived capsule, we found that all mutants lacked both a complete LPS and capsular O-antigens (Fig. 1C), which share the same molecular identity (Apicella et al., 2010). Based on their LPS expression patterns (Fig. 1C), and by comparison to other characterized O-antigen mutants (Raynaud et al., 2007, Gunn et al., 2007, Lai et al., 2010), insertion mutants in the manBC, and FTT0804 loci likely possess a defective core polysaccharide, while the FTT1236, FTT1237 and kdsB mutants do not express any detectable O-antigen (Figs. 1B, C). Given the similar phenotypes of all eleven mutants identified in the screen, we generated in-frame deletion mutants of Schu S4 in either FTT1236 or manC, the most frequently identified genes in our screen, as mutants representative of each O-antigen phenotype for further characterization.
Francisella O-antigen mutants fail to survive in murine macrophages
Deletion of either FTT1236 or manC by allelic replacement abolished expression of the LPS and capsular O-antigen, which was fully restored by trans-complementation of each mutant with a plasmid-borne copy of the respective gene expressed under its own promoter (Fig. 2A). ΔFTT1236 and ΔmanC mutants previously generated in F. tularensis and F. novicida, respectively, display increased uptake by macrophages, as observed with our insertion mutants (Fig. 1A), and increased cytotoxicity (Lai et al., 2010, Lindemann et al., 2011). Previous studies have shown that O-antigen mutants of F. novicida do not enter host macrophages via looping phagocytosis (Lai et al., 2010), an uptake process of wild type bacteria that depends on the presence of active complement in the infection medium (Clemens, et al., 2012). To avoid any confounding effects possibly resulting from differential uptake mechanisms of the ΔFTT1236 and ΔmanC and wild type strains, our experiments were conducted in absence of active complement. Under these conditions, we examined the cytotoxic effects of our mutant strains on BMMs and confirmed increased cytotoxicity at 16, but not at 4 h post-infection (Fig. 2B), when using an MOI (50:1). However, decreasing the MOI to 5 or less for the mutants normalized uptake levels to those of Schu S4 at an MOI of 50 (Fig. 2C–D) and significantly decreased cytotoxicity at 16 h post-infection (Fig. 2B; p< 0.05). Using adjusted MOIs that avoid cytotoxic effects, we then examined the intracellular fate of the ΔFTT1236 and ΔmanC strains in murine BMMs. Unlike Schu S4 (MOI of 50), which grew by 2 Log over a 24 h period, the number of recoverable CFUs of both mutants (MOI of 1–3) progressively decreased, indicating their inability to survive intracellularly (Fig. 2C–D). Complementation of both mutations fully restored the bacteria’s ability to replicate (Fig. 2C–D). Hence, the failure of ΔFTT1236 and ΔmanC mutants to survive inside murine macrophages demonstrates that O-antigen expression is essential for survival and replication in these cells.
Figure 2.

Phenotypic characterization of in-frame O-antigen deletion mutants. (A) Western blots of whole bacterial lysates of wild type (Schu S4), ΔFTT1236 (ΔFTT1236), ΔmanC (ΔmanC), and their complemented strains (ΔFTT1236 + FTT1236, and ΔmanC + manC ). 1 × 107 bacteria equivalents were resolved and probed for either LPS O-antigen (LPS), or O-antigen capsule (capsule), and GroEL as a loading control. (B) Percent cytotoxicity of wild type Schu S4, Schu S4ΔFTT1236 (ΔFTT1236) and Schu S4ΔmanC (ΔmanC) at 4 and 16 h post-infection at an MOI of 5 (white bars) or 50 (black bars). Data are means ± SD (n=3; asterisks indicate a P value<0.05 by the unpaired student’s two-tailed t-test). ND, not determined. (C) and (D): Representative growth curves of wild type Schu S4 (black), Schu S4ΔFTT1236 and Schu S4ΔmanC (red), and their complemented strains (blue) in BMMs infected at the indicated MOIs.
Francisella O-antigen mutants are rapidly targeted by autophagy in the cytosol
Given that FTT1236 and manC mutants were selected based on their colocalization with LC3 (Fig. 1A), we next examined any interaction of these strains with the autophagic pathway in BMMs. First, the population of vacuolar GFP-expressing Schu S4 and mutants were monitored by colocalization with the late endosomal/lysosomal marker LAMP1 (Checroun et al., 2006) to assess their kinetics of phagosomal escape, as we could not use an established phagosomal integrity assay based on the detection of surface O-antigen (Checroun et al., 2006). Decrease in bacterial colocalization with LAMP1 in the first hour pi, a readout of phagosomal disruption, was comparable between Schu S4, the ΔFTT1236 and ΔmanC mutants and a ΔdipA mutant that is competent for phagosomal escape (Chong et al., 2012) (Fig. 3A and S1A), indicating that both O-antigen mutants are capable of normal phagosomal escape. In comparison, a phagosomal escape-deficient ΔfevR mutant (Wehrly et al., 2009) remained within LAMP1-positive vacuoles over a 10 h period (Fig. 3A and S1A). In agreement, ultrastructural analyses of these strains at 1 h post-infection showed an absence of phagosomal membranes around Schu S4 and both O-antigen mutants, while ΔfevR bacteria remained surrounded by intact membranes (Fig. 3B, and S2). Additionally, 79% and 92% of ΔFTT1236 (n=72) and ΔmanC (n=25) bacteria observed, respectively, lacked the electron-translucent layer normally seen around Schu S4 (78%, n=27) (Fig. 3B and S2), suggesting it is formed by the polysaccharidic O-antigen. Interestingly, while Schu S4 and the ΔdipA mutant remained cytosolic over a 10 h infection course, colocalization of both O-antigen mutants with LAMP1 increased after 1 h post-infection to reach 58 ± 7.9% of Δ FTT1236 and 48 ± 6.5% of ΔmanC bacteria by 10 h post-infection, indicating rapid re-entry of these mutants into the endosomal/lysosomal compartment. This early re-entry into LAMP1-positive vacuoles is unique to these O-antigen mutants and distinct from the autophagosomal capture described for the ΔdipA mutant, which is delayed until 16 h post-infection (Chong et al., 2012). Taken together, our results suggest that O-antigen mutants are quickly captured by autophagy following their initial escape into the cytosol.
Figure 3.

O-antigen deletion mutants escape their initial phagosome. (A) Colocalization studies of GFP-expressing F. tularensis Schu S4 (black circles), Schu S4ΔFTT1236 (red squares), Schu S4ΔmanC (blue triangles), Schu S4ΔfevR (orange diamonds), and Schu S4ΔdipA (green hexagones) with LAMP1. Data are means ± SD (n=3). (B) Representative transmission electron micrographs of wild type Schu S4 (Schu S4), Schu S4ΔfevR (ΔfevR), Schu S4ΔFTT1236 (ΔFTT1236) and Schu S4ΔmanC (ΔmanC) mutants at 1 h post-infection. Arrowheads indicate the presence of an intact phagosomal membrane. Scale bar, 500 nm.
To extend these findings, we examined recruitment of markers of the autophagic cascade in BMMs, such as ubiquitin, p62/SQSTM1 and LC3. Unlike Schu S4, the ΔfevR or ΔdipA strains, the O-antigen mutants displayed a rapidly increasing colocalization with ubiquitin (Fig. 4A–B), the autophagy receptor p62/SQSTM1 (Fig. 4C–D) and LC3 (Fig. 4E–F) between 1 and 2 h post-infection, which slowly decreased afterwards. Similar colocalization of mutants with these autophagy markers was observed in human blood monocyte-derived macrophages (Fig. S3), indicating that autophagic targeting of O-antigen mutants is comparable in both human and murine macrophages. Taken together, these results demonstrate that cytosolic Francisella lacking the O-antigen are rapidly recognized by the autophagy pathway and targeted to the lysosomal compartment. Since neither wild type bacteria, the ΔdipA nor ΔfevR mutants, which all express normal LPS O-antigen and capsule (Fig. S1C–D), were targeted by autophagy early after infection, this suggests that the surface O-antigen prevents Francisella from rapid autophagic recognition once it reaches the cytosol.
To confirm that autophagy targets cytosolic Francisella O-antigen mutants and not the remaining vacuolar fraction of bacteria (Fig. 3A), we first verified that the majority (> 85%) of ubiquitinated bacteria were not surrounded by LAMP-1-positive membranes (Fig. S4A). We then assessed autophagic targeting and capture of the O-antigen mutants using Bafilomycin-A1 (BAF), which delays escape of F. tularensis for up to 4 h post-infection (Chong et al., 2008). Pretreatment of BMMs with BAF increased the fraction of vacuolar, LAMP1-positive SchuS4 and O-antigen mutants at 2 h post-infection (Fig. S4B), confirming impaired phagosomal escape (Chong et al., 2008). Under these conditions, the association of both O-antigen mutants with ubiquitin, p62/SQSTM1 and LC3 was significantly reduced (Fig. S4C–E), indicating that phagosomal escape into the cytosol is required for autophagic recognition and targeting of these mutants. Hence, cytosolic Francisella lacking their surface O-antigen are targets of autophagy.
Autophagic targeting of the O-antigen mutants did not result from a general up-regulation of autophagy in response to infection with the particular strains, since BMM infections with either Schu S4 or the O-antigen mutants slightly increased autophagosome numbers to similar levels compared to mock-infected cells, as judged by the average number of LC3-positive puncta (Fig. S5A). Moreover, induction of autophagy via amino acid starvation (Fig. S5B) did not enhance autophagic capture of the O-antigen mutants (Fig. S5C). Taken together, these results indicate that autophagic capture of Francisella O-antigen mutants is a selective process that specifically recognizes Francisella lacking this surface polysaccharide.
O-antigen mutants display reduced viability following phagosomal escape
Previous studies have reported that Francisella mutants in FTT1236, manC and other genes required for O-antigen LPS and capsule biosynthesis are hypercytotoxic in macrophages (Lai et al., 2010, Lindemann et al., 2011), likely due to an increased propensity to lyse in the cytosol and activate AIM2-mediated pyroptosis (Peng et al., 2011). A correlate to this increased fragility is that such mutants may die in the cytosol and be subsequently cleared by autophagy, as we previously demonstrated in the case of a Schu S4 ΔdipA mutant (Chong et al., 2012), in addition to triggering activation of the AIM2 inflammasome. To examine whether the survival defect displayed by Schu S4 ΔFTT1236 and ΔmanC mutants (Fig. 2C–D) is due to either a lack of intrinsic viability and/or to autophagic killing, we first assessed the physical integrity of the ΔFTT1236 and ΔmanC mutants compared to the wild type strain in both the infection inocula and in BMMs following phagosomal escape (90 min post-infection), using the membrane-impermeant dye propidium iodide (PI), either applied to the bacterial suspension or delivered to the host cell cytosol using digitonin permeabilization of infected cells (Chong et al., 2012). Since these measurements were performed using two different methodologies, ratios between wild type and mutant percentages of PI-positive bacteria (integrity index) were established to allow for meaningful comparison. In both inocula and cytosolic populations of bacteria, the ΔFTT1236 (integrity index values of 0.33 ± 0.12 and 0.27 ± 0.05, respectively) and ΔmanC (integrity index values of 0.31 ± 0.13 and 0.28 ± 0.10, respectively) mutants displayed decreased physical integrity (Fig. 5), indicating that a significant number of the O-antigen-deficient bacteria were non-viable. Notably, these ratios did not change between the inocula (Fig. 5A) and the cytosolic population of bacteria (Fig 5B), suggesting that the O-antigen mutants are not significantly more sensitive to the phagosomal environment than the wild type strain. Taken together, these results indicate that a fraction of the O-antigen mutants are non-viable, which may account for the survival defects observed in BMMs (Fig. 2C and D).
Figure 5.
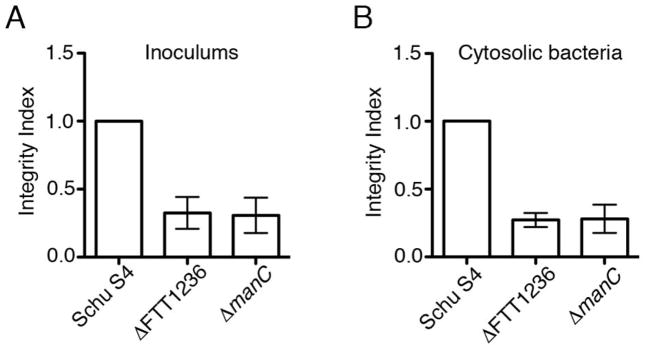
Viability of O-antigen mutants. Integrity index representing propidium iodide (PI) staining of wildtype Schu S4 (Schu S4), Schu S4ΔFTT1236 (ΔFTT1236), and Schu S4ΔmanC (ΔmanC) bacteria in (A) infection inocula, or (B) cytosolic bacteria at 90 min post-infection. Data are means ± SD (n=3).
Viable cytosolic O-antigen mutants are killed by Atg5-dependent autophagy
To determine the contribution of autophagic capture to the intracellular killing of the ΔFTT1236 and ΔmanC mutants, we examined their fate in control Atg5flox/flox and autophagy-deficient Atg5flox/flox-Lyz-Cre BMMs (Zhao et al., 2008) by assessing their intracellular trafficking and growth. Comparable numbers of the ΔFTT1236 mutant were ubiquitinated in control Atg5flox/flox (43.1 ± 9.2%) and Atg5-deficient BMMs (58.3 ± 11.4%) at 2 h post-infection (Fig. 6A), and the percentage of ubiquitinated ΔmanC bacteria was slightly yet non-significantly increased in the Atg5flox/flox-Lyz-Cre BMMs (51.9 ± 11.2%) relative to control BMMs (33.9 ± 2.7%) (Fig. 6A). Yet, capture of both mutants within LC3-positive structures was abolished in Atg5flox/flox-Lyz-Cre BMMs (Fig. 6B), consistent with Atg5 function in the autophagic cascade. This demonstrates that autophagic capture of the O-antigen mutants is an Atg5-dependent process subsequent to their ubiquitination.
Figure 6.

F. tularensis O-antigen mutants replicate within Atg5-deficient macrophages. (A) Colocalization of wildtype Schu S4 (Schu S4), Schu S4ΔFTT1236 (ΔFTT1236), and Schu S4ΔmanC (ΔmanC) with ubiquitin (Ub) in either Atg5flox/flox (white bars) or Atg5flox/flox-Lyz-Cre (black bars) BMMs at 2 hours post-infection. Data are means ± SD (n=3). ND, not detected. (B) Colocalization of wild type Schu S4 (Schu S4), Schu S4ΔFTT1236 (ΔFTT1236) and Schu S4ΔmanC (ΔmanC) with LC3 at 2 h post-infection in either Atg5flox/flox (white bars) or Atg5flox/flox-Lyz-Cre (black bars) BMMs. Data are means ± SD (n=3). ND, not detected. (C): Quantification of nonviable ubiquitin (Ub)-positive bacteria by PI staining at 2 hours post-infection in either Atg5flox/flox (white bars) or Atg5flox/flox-Lyz-Cre (black bars) BMMs. Data are means ± SD (n=3); ND, not detected. (D) and (E): Representative growth curves of wild type Schu S4 (circles) and O-antigen mutant bacteria (squares) within control Atg5flox/flox (filled symbols), or knockout Atg5flox/flox-Lyz-Cre (open symbols) BMMs. Arrows mark times of increased O-antigen mutant survival in Atg5flox/flox-Lyz-Cre BMMs compared to control Atg5flox/flox BMMs (P value<0.05 by the unpaired student’s two-tailed t-test). (F) Confocal micrographs of wild type Schu S4 (Schu S4) Schu S4ΔFTT1236 (ΔFTT1236) and Schu S4ΔmanC (ΔmanC) in Atg5flox/flox or Atg5flox/flox-Lyz-Cre BMMs at 16 h post-infection. DNA is stained with DAPI (gray), scale bar, 10 μm. (G) Single-cell replication analysis of wild type Schu S4 (Schu S4) Schu S4ΔFTT1236 (ΔFTT1236) and Schu S4ΔmanC (ΔmanC) in Atg5flox/flox or Atg5flox/flox-Lyz-Cre BMMs at 16 h post-infection. The percent of infected macrophages with either 1–5 (white bars), 6–20 (gray bars), or >20 (black bars) bacteria were counted. Data are means ± SD (n=3). (H) Cytotoxicity induced by wild type Schu S4, Schu S4ΔFTT1236, and Schu S4ΔmanC infections at 16 h post-infection in either Atg5flox/flox (white bars) or Atg5flox/flox-Lyz-Cre (black bars) BMMs. Data are means ± SD (n=3; asterisks indicate a P value<0.05 by the unpaired student’s two-tailed t-test).
To examine whether the failure of the O-antigen mutants to replicate intracellularly (Fig. 2C and D) was also Atg5-dependent, we compared their ability to grow in the Atg5flox/flox-Lyz-Cre and control Atg5flox/flox BMMs. While intracellular growth of Schu S4 was unchanged in control and autophagy-deficient BMMs, deletion of Atg5 significantly increased the recovery of viable mutants, by 3 Logs at 16 h post-infection for the ΔFTT1236 strain and by 0.6 Log by 10 h post-infection for the ΔmanC strain compared to control BMMs (Fig. 6D–E, arrows). This recovery indicates that autophagy further restricts viability of the O-antigen mutants. Interestingly, the ΔFTT1236 strain, which does not express residual LPS O-antigen unlike the ΔmanC strain (Fig. 2A), was killed more efficiently in wild type BMMs and exhibited greater CFU recovery in the autophagy-deficient cells than the ΔmanC mutant (Fig. 6D–E). This suggests that the residual LPS O-antigen expressed by the ΔmanC strain renders it less vulnerable to targeting by autophagy. For both strains, however, CFU enumeration did not show restoration of mutant intracellular growth to wild-type levels, and even showed decreased viability of the ΔFTT1236 and ΔmanC mutants after 16 and 10 h post-infection, respectively (Fig. 6D–E). A microsocopy-based analysis of single cells infected with the O-antigen-deficient strains revealed, however, that both mutants were capable of significant replication in Atg5flox/flox-Lyz-Cre but not in Atg5flox/flox BMMs (Fig. 6F–G). Moreover, quantification of replication in these cells showed that deletion of Atg5 allowed a shift of the intracellular mutant populations towards replication, with a majority of intact infected Atg5flox/flox-Lyz-Cre BMMs containing more than 20 bacteria at 16 h post-infection compared to control BMMs, which had 5 or fewer (Fig. 6G). Hence, autophagy deficiency confirms that after phagosomal escape, some O-antigen mutants are intracellularly viable and reveals that they are capable of replication in the cytosol, demonstrating that autophagy acts as a bactericidal mechanism against the O-antigen mutants.
These results predict that autophagy recognizes viable cytosolic O-antigen mutants in addition or not to a non-viable population. To test this hypothesis, we examined the proportion of cytosolic ubiquitinated bacteria (Fig. S4A) for physical integrity via digitonin-mediated delivery of PI in either Atg5flox/flox or Atg5flox/flox-Lyz-Cre BMMs. In both the Atg5flox/flox and Atg5flox/flox-Lyz-Cre BMMs, approximately one third of ΔFTT1236 (30.5 ± 6.9% and 34.0 ± 10.7%, respectively) and ΔmanC (36.4 ± 7.2% and 33.0 ± 15.6%, respectively) bacteria were PI-positive (Fig. 6C), indicating that some autophagy-targeted bacteria were viable and that autophagic capture of the O-antigen mutants is both a killing mechanism and a clearing process of non-viable bacteria.
Since the cytotoxic effects O-antigen mutants have on macrophages depend upon the MOI and intracellular bacterial load (Fig. 2B), we tested whether the decrease in recoverable CFUs observed in Atg5flox/flox-Lyz-Cre BMMs after 10 or 16 h post-infection (Fig. 6D and E) was due to increased cytotoxicity. When compared between control Atg5flox/flox and autophagy-deficient Atg5flox/flox-Lyz-Cre BMMs, cytotoxicity levels in infected BMMs after 16 hours were 2.8 and a 12.6-fold higher in autophagy-deficient than in control BMMs for the ΔmanC and ΔFTT1236 mutants, respectively (Fig. 6H) and likely caused a loss of infected cells and CFUs (Fig. 6D–E). This indicates that restoration of intracellular growth of these mutants (Fig. 6F–G) promotes their cytotoxic effect via increased intracellular numbers (Fig. 6G–H), a condition that can be otherwise mimicked by high MOIs (Fig. 2B; (Lai et al., 2010, Lindemann et al., 2011)). This finding is also consistent with the previous demonstration that the cytotoxic effects of O-antigen mutants limit the capacity of these mutants to replicate in human macrophages (Lindemann et al., 2011). Additionally, the increased cytoxicity of the O-antigen mutants in autophagy-deficient BMMs is not simply due to prolonged residence in the cytosol, as a purine auxotroph, replication-deficient ΔpurMCD Francisella strain does not induce cytotoxicity despite its extended presence in the macrophage cytoplasm (Maier et al., 2007; Chong et al., 2012).
Discussion
Based on Francisella’s ability to thrive for protracted periods of time in the cytosol of host cells, we have tested the hypothesis that this pathogen employs specific mechanisms of autophagy avoidance. An unbiased genetic screen revealed that a surface polysaccharide, the O-antigen, contributes to this phenotype early in infection, by protecting F. tularensis from autophagic detection once it reaches the cytosol. The F. tularensis O-antigen is a component of both its LPS and capsule (Apicella et al., 2010), both of which have other known immune-evasion properties. Francisella LPS is anti-inflammatory (Hajjar et al., 2006), largely due to its unique lipid A structure (Phillips et al., 2004, Gunn et al., 2007) and both the LPS and capsule contribute to resistance against complement-mediated lysis (Sorokin et al., 1996, Lindemann et al., 2011). Consistent with its multiple roles in interactions with the host, the demonstration of the O-antigen’s protective function against autophagy was complicated by the deleterious consequences of its absence on bacterial viability and resulting hypercytotoxicity. Nonetheless, our data demonstrate that the viable population of cytosolic O-antigen-deficient bacteria is subjected to efficient autophagic detection and killing in macrophages. Our results further extend the innate immune evasion functions of the O-antigen by assigning it an intracellular protective role against antibacterial autophagy.
Whether the LPS or the capsular fraction of O-antigen, or both, are protective against autophagy remains to be determined. Consistent with LPS and capsular O-antigens having the same molecular identity and sharing common biosynthetic pathways (Apicella et al., 2010), we were unable to genetically uncouple their expression, as any mutant tested was deficient in expression of both forms. Unless it is associated to a unique molecular pattern, it would be surprising that the LPS O-antigen is solely protective against autophagy, given the ubiquitous existence of polysaccharidic O-antigens among Gram-negative bacteria. Interestingly, greater than 95% of Francisella lipid A lacks O-antigen (Wang et al., 2006), suggesting that most of it on the bacterial surface is expressed as a component of capsular material. We therefore speculate that capsule is responsible for shielding Francisella from autophagic recognition, as supported by the absence of the capsule-like electron-translucent layer around O-antigen mutants that normally surrounds cytosolic Francisella (Fig. 3B, S2; (Clemens et al., 2012)). In addition to the O-antigen component of the capsule, the Francisella genome encodes a conserved capBCA operon showing weak homology to the Bacillus anthracis poly-D-glutamic acid capsule locus (Larsson, et al., 2005). In-frame deletion of various genes of the capBCA operon did not however result in autophagic targeting of F. tularensis (data not shown). These observations are consistent with our hypothesis that the O-antigen constituent of the capsule, but not putative poly-D-glutamic acid components, is necessary for autophagy avoidance.
Whereas other mutant strains of Francisella, such as ΔdipA, are targeted by antibacterial autophagy, their detection and capture is delayed for greater than 16 hours, long after they have accessed the host cytosol (Chong et al., 2012). Unlike the O-antigen mutants in this study, ΔdipA mutant replication is not restored in Atg5-deficient macrophages (Chong et al., 2012), indicating that autophagy does not limit their growth, but that they die prior to targeting. It is possible that as the ΔdipA mutant dies in the cytosol, it eventually sheds its previously intact O-antigen or capsule (Fig. S1C) and exposes or releases molecules that can trigger the autophagic cascade. Similarly, the fraction of wild type Francisella that translocates into autophagic vacuoles (FCVs) late in their cycle within murine macrophages (Checroun et al., 2006) may also alter the composition of its surface, thereby permitting autophagic capture. In this context, the intrinsic lack of surface O-antigen on the ΔFTT1236 and ΔmanC mutants explains their immediate detection and autophagic capture, compared to the protracted detection of a ΔdipA mutant (Chong et al., 2012). Yet, we cannot exclude that Francisella expresses additional anti-autophagic molecules to permit long-term cytosolic survival, as our screen focused on genes required for early autophagy avoidance.
A possible explanation to early autophagic targeting of Francisella O-antigen mutants could invoke a response to uncharacterized bacterial molecules released into the host cytosol, as a result of the bacterium’s compromised integrity (Fig. 5). Indeed, various F. novicida hypercytotoxic mutants, including a ΔfopA strain, display increased intracellular bacteriolysis, resulting in activation of the AIM2 inflammasome in response to the release of bacterial DNA (Peng et al., 2011). Yet, a ΔfopA mutant in F. tularensis exhibits the same intracellular fate as a ΔdipA mutant (Chong et al., 2012), indicating that it is not subject to early autophagic capture despite its compromised integrity. If early autophagic capture was a response to cytosolic release of bacterial molecules, our screen would also have identified mutants with defects unrelated to O-antigen. Yet, only O-antigen mutants were identified, which strongly argues that rapid autophagic capture of Francisella results from the lack of this surface molecule and not generic compromised bacterial integrity.
We therefore propose that the Francisella surface O-antigen shields bacterial molecules that would otherwise be sensed by innate cytosolic surveillance components and would trigger ubiquitin tagging and autophagic capture. Uptake of O-antigen mutants could also induce alternative intracellular signaling that contribute to autophagic capture. In support of differential signaling following phagocytosis of these mutants, infection with a F. novicida ΔlpcC O-antigen mutant, whose LPS defects are similar to that of a ΔmanC mutant (Lai, et al., 2010), up-regulates phosphorylation of the kinase Erk2 and the phospholipase A2 (PLA2) compared to wild-type bacteria (Nakayau, et al., 2013). Regardless of the yet-to-be-defined molecular cascade leading to autophagic recognition of Francisella O-antigen mutants, our data supports the concept that expression of the O-antigen is required to prevent autophagic recognition and capture of cytosolic Francisella.
A recent study has shown that F. tularensis induces non-selective macroautophagy to acquire nutrients during infection, via an Atg5-independent process that is not accompanied by Francisella autophagic capture (Steele et al., 2013). While these findings may appear contradictory to our results, they are nonetheless consistent with the protective role of the surface O-antigen against selective recognition and capture of Francisella. Taken together, Steele et al and our studies emphasize that Francisella both positively and negatively modulates non-selective and selective arms of the autophagy pathway to ensure its survival and promote proliferation in the macrophage cytosol.
While surface modification is a common strategy for autophagy evasion, as exemplified by Shigella and Listeria (Ogawa et al., 2005, Yoshikawa et al., 2009), our study is first to attribute anti-autophagic properties to a bacterial surface polysaccharide. In the case of Shigella, the Type III secretion effector IscB competes with host ATG5 for binding to surface-exposed VirG, thereby interfering with the autophagic cascade (Ogawa et al., 2005). Listeria coats its surface with host proteins via the actin-based motility protein ActA (Yoshikawa et al., 2009) and InlK (Dortet et al., 2011), thereby disguising itself as a host organelle. We propose that once Francisella escapes the phagosome, the thick capsular O-antigen prevents exposure of surface molecules to the host cytosol that would otherwise be recognized by innate immune sensors, targeting the bacteria for autophagic elimination. Recently, LRSAM1, an E3 ubiquitin ligase with bacteria-binding properties was shown to initiate recruitment of early components of the autophagic cascade on cytosolic Salmonella (Huett et al., 2012). Future studies will take advantage of the efficient targeting of Francisella O-antigen mutants to identify the bacterial surface molecules recognized and the autophagic sensors involved in antibacterial autophagy.
Experimental Procedures
Bacterial Strains and Culture Conditions
All F. tularensis strains were grown on modified Mueller-Hinton (MMH) agar plates [Mueller-Hinton medium supplemented with 0.1% glucose (Sigma), 0.025% ferric pyrophosphate (Sigma) and 2% IsoVitaleX (Becton Dickinson)] for 3 days at 37°C under 7% CO2. MMH medium was supplemented with either 10 μg/ml kanamycin (Sigma) or 8% sucrose (Sigma) when required. All manipulations of F. tularensis strain SchuS4 and derivative strains were performed in a Biosafety Level 3 facility according to standard operating procedures approved by the Rocky Mountain Laboratories Institutional Biosafety Committee.
The F. tularensis subsp. tularensis Schu S4 strain, the SchuS4ΔdipA (ΔFTT0369c) and SchuS4ΔfevR (ΔFTT0383) mutants have been described previously (Wehrly et al., 2009). In-frame deletions of FTT1236 and manC (FTT1448c) were generated in the F. tularensis subsp. tularensis SchuS4 strain using the SacB-assisted allelic replacement suicide vector pJC84, as described previously (Wehrly et al., 2009), and were designed to preserve the integrity of surrounding genes and avoid any polar effects. To engineer an in-frame deletion of FTT1236, a 5′ 1074 bp fragment containing the start codon of FTT1236 was generated by PCR amplification using the primers RC53 and RC54 (Supporting Information, Table S1) and a 3′ 960 bp fragment containing the last four codons and the stop codon of FTT1236 was amplified using primers RC55 and RC56 (Table S1). Primers RC59 and RC60 (Table S1) were used to check for the presence of the deleted allele and primers RC57 and RC58 (Table S1) verified allelic replacement within the correct chromosomal locus by PCR. To construct an in-frame deletion of manC (FTT1448c), a 5′ 1262 bp fragment containing the start codon of manC was generated by PCR amplification using the primers TW979 and TW980 (Table S1) and a 3′ 1237 bp fragment containing the last four codons and the stop codon of manC was amplified using primers TW981 and TW982 (Table S1). In both cases, the amplified hemi-fragments were cloned into the BamHI and SalI sites of pJC84 using the In-Fusion® PCR Cloning System (Clontech, Palo Alto, CA) and fully sequenced. For the FTT1236 deletion, Primers TW985 and TW986 were used to confirm replacement at the correct locus, and primers TW983 and TW984 confirmed deletion of the gene (Table S1). For both constructs, PCR was used to confirm the loss of the sacB gene as described previously (Chong et al., 2012). Independent clones with the correct in-frame deletion were isolated and used for further studies.
Genetic complementation of the deletion mutants was performed in trans by reintroducing a full-length copy of the chromosomally deleted genes carried on a pFNLTP6 derivative under the control of its own promoter. To construct the complementation plasmid for the ΔFTT1236 mutant, primers RC134 and RC135 (Table S1) were used to amplify a 1329 bp fragment encompassing a 223 bp promoter region and the FTT1236 open reading frame. To construct the complementation plasmid for the ΔmanC mutant, a 1627 bp fragment comprising the FTT1448c open reading frame and 214 bp upstream of the start codon was amplified using primers RC141 and RC142 (Table S1). The resulting fragments were cloned into pFNLTP6 using the In-Fusion® PCR Cloning System between EcoRI and BamHI restriction sites to generate pFNLTP6-FTT1236 and pFNLTP6-manC, respectively, which were confirmed by sequencing using primers JC884 and JC946 (Table S1) prior to electroporation into the deletion strains. To generate GFP-expressing bacteria, the plasmid pFNLTP6groE-gfp (Maier et al., 2004) was introduced in Francisella strains by electroporation as described (Kampen et al., 2004, Maier et al., 2004).
Primary macrophage culture and infection
Murine bone marrow-derived macrophages (BMMs) were harvested from C57BL/6J (Jackson Laboratories, Bar Harbor, ME), C57BL/6J Atg5flox/flox and Atg5flox/flox-Lyz-Cre (Zhao et al., 2007) mice femurs and differentiated as described (Chong et al., 2008). All animal rearing, handling and experimental methods were conducted under protocols approved by the RML Institutional Animal Care and Use Committee (IACUC). Human monocyte-derived macrophages (MDMs) were generated from peripheral blood monocytes and differentiated as previously described (Chong et al., 2012). Anonymized volunteers provided the human blood cells, after written informed consent, under protocols approved by the NIH Clinical Center Institutional Review Board.
Macrophage infections were performed as described (Chong et al., 2008) at the indicated MOIs. When required, BMMs were treated for 1 h prior to infection with 100nM Bafilomycin-A1 (BAF; AG Scientific, Inc.), or DMSO (Sigma) control vehicle, which was maintained throughout the duration of the experiment. To induce autophagy by starvation, macrophages were washed extensively (>3 times) with HBSS (Life Technologies) and the media was replaced with HBSS containing 1 g/L D-glucose (Sigma) for 4 hours prior to the processing time point.
Immortalization and transduction of murine bone marrow-derived macrophages
To generate an autophagy reporter cell system, murine BMMs were immortalized and transduced to express GFP-LC3 as follows. Moloney Murine Leukemia Virus (MMULV)-derived retroviral particles containing the v-mycMC29 oncogene were produced using the Ecotropic Phoenix packaging system and pBMNZ-v-mycMC29, a pBMN-Z derivative carrying a v-mycMC29 encoding fragment derived from pΔRM (Overell et al., 1988) kindly provided by Drs Jacques Peschon and Ed Miao, and Fugene 6 (Promega) according to the manufacturer’s recommendations. Bone marrow cells were harvested and cultured for 2 days as described above and replated at a density of 1.2 × 105 cells/well in a 6-well plate. After 6 h, cells were transduced by spinfection (1,800 × g for 2 h at room temperature) with 0.45 μm-filtered retroviral supernatants supplemented with 4 μg/ml polybrene (Sigma), and allowed to recover at 37°C with 7% CO2 for 1 h before the retroviral supernatants were replaced with fresh BMM medium. Immortalized BMMs (iBMMs) were expanded and CD11b+/CD14+ cells were positively selected using rat anti-mouse CD11b (AbD Serotec) and rat anti-mouse CD14 (clone 159010; R&D Systems) antibodies and a Dynabeads™ Sheep anti-rat IgG kit (Life Technologies) according to the manufacturer’s instructions. iBMMs were then transduced with GFP-LC3-encoding retroviral particles generated using pJC110 and 293T cells as previously described (Checroun et al., 2006). GFP-LC3-expressing iBMMs were selected and expanded under G418 (Life Technologies; 200 μg/ml) selection for 7–10 days.
Construction and screening of a F. tularensis Schu S4 HimarFT library
A library of random transposon insertion mutants was generated by electroporation of F. tularensis subsp. tularensis SchuS4 with pHimarH3, a suicide vector containing the HimarFT transposon (Maier et al., 2007). Approximately 2×1010 Schu S4 harvested from a mid-log phase MMH broth culture were electroporated with 1 μg pHimar H3 as previously described (Wehrly et al., 2009), incubated at 37°C for 3.5 hours before plating for selection on MMH agar supplemented with 10 μg/mL kanamycin. After a 3-day incubation at 37°C, 7% CO2, 9,500 individual kanamycin-resistant clones were picked and suspended in 200 μl MMH broth in 96-well plates, with the first well (A1) of each plate inoculated with wild-type Schu S4. The plates were incubated at 37°C with 7% CO2 without agitation for 3 days to allow growth of the bacteria to late-log phase, after which cultures were supplemented with a final concentration of 10% (w/v) glycerol for storage at −80°C.
To screen for HimarFT insertion clones targeted by autophagy, 96-well plates were replicated on rectangular MMH agar plates using the 96-pin library copier tool (V&P Scientific) and grown at 37°C, 7% CO2 for 3 days prior to infection. One day before infection, GFP-LC3 iBMMs were seeded in 96-well glass bottom plates (Matrical Bioscience) (2×104 cells/well in 200 μl) in BMM medium without G418. On the day of infection, individual HimarFT clones were suspended in 200 μl of MMH broth of a 96-well round-bottom plate using the 96-pin library copier tool. A reference OD600nm was determined for well A1, which served to calculate the MOI (100). GFP-LC3-iBMMs were infected with 100 μl of bacterial suspensions in BMM medium as previously described (Chong et al., 2008). At 1 h post-infection, 100 μl of BMM medium supplemented with 100 μg/ml gentamicin was added to each well (50 μg/ml final concentration) of the infected plates to kill extracellular bacteria, and maintained for the duration of the experiment. At 6 h post-infection, the wells were washed twice with PBS and processed for immunostaining as described (Chong et al., 2008). After blocking for 15 min in 60 μl of 10% horse serum, 0.02% saponin, PBS pH 7.4, wells were incubated for 30 min with a 1:1,000 dilution in the same buffer of a mouse monoclonal anti-F. tularensis LPS antibody (clone 1.B.288; US Biological) directly conjugated to Alexa Fluor™ 568 using an Alexa Fluor™ 568 Antibody Labeling Kit (Life Technologies), and washed twice in PBS. The stained plates were then screened by epifluorescence microscopy for clones that co-localized with GFP-LC3 signals using a Carl Zeiss (Thornwood, NY) MicroImaging LSM 5 LIVE confocal laser scanning microscope.
Identification of HimarFT insertion sites
The insertion loci of the HimarFT clones identified as being targeted by autophagy were amplified by PCR or cloned out for identification of the disrupted gene(s). For each clone, genomic DNA was isolated from fresh colonies scraped from MMH agar using the Puregene Yeast/Bact. II kit (Qiagen Inc.) according to the manufacturer’s instructions. For most clones, the HimarFT insertion locus was amplified by PCR first using the randomized primer TW642, and TW913 (Table S1), a primer specific for the HimarFT transposon, and purified with the QIAquick PCR purification kit (Qiagen). A second round of nested PCR was used to specifically amplify the insertion loci using TW644 (Table S1), which annealed to specific adaptor sequence on the 5′ end of TW642, and the nested transposon-specific primer TW914 (Table S1). After the second round of PCR, the products were gel purified using the QIAquick Gel extraction kit (Qiagen) and sequenced with primer TW915 (Table S1). Clone 52B6 was initially amplified with a different randomized primer, TW646 (Table S1), and the transposon-specific TW681 primer (Table S1). The second round of amplification for 52B6 used primers TW644 and the nested primer TW647 (Table S1). The product of the second round of PCR was sequenced with primer TW943 (Table S1). Cloning and subsequent sequencing of recovered plasmid identified the insertion loci for clones 25D9 and 36D8. 500 ng each of 25D9 and 36D8 genomic DNA were digested overnight at 37°C with NdeI, and SpeI, respectively, and purified with the QIAQuick PCR purification kit (Qiagen Inc.). The eluted DNA was then ligated with T4 DNA Ligase (New England Biolabs) at 4°C for 72 hours. The ligated DNA was transformed into chemically competent OneShot PIR1 E. coli (Life Technologies) and plated on LB agar supplemented with 50 μg/ml kanamycin. Plasmid DNA was then recovered from the transformants using the Nucleospin Plasmid QuickPure Kit (Macherey-Nagel, Bethlehem, PA) and sequenced using TW915.
Bacterial viability assay
To assess the viability of Francisella in our infection inoculums, several colonies of GFP-expressing bacteria were scraped from MMH agar plates and suspended in MMH broth for calculation of MOI based on optical density as described previously (Chong et al., 2008). From each bacterial suspension, an aliquot of 1 × 109 cells was pelleted and washed once with PBS, followed by re-suspension in 150 μl PBS containing 2.6μM propidium iodide (PI; Life Technologies), and incubated at room temperature for 15 minutes. The PI-stained bacteria were diluted 1:10 in PBS, 2 μl of which were placed on a 35-mm, glass bottom live cell imaging dish (WillCo Wells BV), and topped with a glass coverslip for immediate counting under fluorescence. For viability analysis of cytosolic F. tularensis, BMMs infected with GFP-expressing bacteria were permeabilized with 50 μg/ ml digitonin (Sigma) in KHM buffer (Checroun et al., 2006) for 1 min at room temperature, immediately washed three times with KHM buffer and incubated for 12 min at 37°C in KHM buffer containing 2.6 μM PI. The infected coverslips were then fixed with 3% paraformaldehyde in PBS, and when required, incubated with antibodies directed against mouse mono- and poly-ubiquitinylated conjugates (FK2, Enzo Life Sciences), and Alexa Fluor Cy5-conjugated secondary antibodies (Life Technologies) to detect ubiquitinated cytosolic bacteria. For the dual ubiquitin-PI counts, at least 100 total bacteria were counted over the course of three independent experiments, and the data are presented as the mean ± SD.
To calculate the integrity index of cytosolic F. tularensis and the infection inoculums, total PI-positive bacteria were counted in triplicate from 100 bacteria and averaged to determine the percentage of nonviable bacteria in the sample. The integrity index was derived from the ratio of the average percentage of non-viable wild type Francisella to the average percentage of non-viable mutant bacteria. The data presented is the mean integrity index calculated from three independent experiments ± their standard deviation.
Microscopy
For fluorescence microscopy, infected BMMs on 12 mm glass coverslips were processed for immunostaining as described previously (Chong et al., 2008). Primary antibodies used were: mouse monoclonal anti-F. tularensis LPS (US Biological), rat monoclonal anti-mouse LAMP1 (clone 1D4B, developed by J.T. August and obtained from the Developmental Studies Hybridoma Band; developed under the auspices of the NICHD and maintained by The University of Iowa, Iowa City, IA), rabbit polyclonal anti-human LAMP1 (Novus Biologicals), mouse monoclonal anti-mono- and poly-ubiquitinylated conjugates (FK2, Enzo Life Sciences), guinea pig polyclonal anti-SQSTM1 (Progen Biotechnik), mouse monoclonal anti-LC3 (MBL International) and rabbit polyclonal anti-human LC3 (MBL International). Secondary antibodies used were: Alexa Fluor™ 568-conjugated donkey anti-mouse, Alexa Fluor™ 568-conjugated goat anti-rabbit, Alexa Fluor™ 568-conjugated goat anti-rat, Alexa Fluor™ 568-conjugated goat anti-rabbit, and Alexa Fluor™ 568-conjugated goat anti-guinea pig antibodies (Life Technologies). When required, macrophage nuclei and bacteria were stained with DAPI (100 ng/ml; Life Technologies) in distilled water for 10 min at room temperature after incubation with secondary antibodies. Samples were observed on a Carl Zeiss Axio Imager epifluorescence microscope equipped with a Plan-APOCHROMAT 63×/1.4 objective for quantitative analysis. For co-localization studies, at least 100 bacteria were counted per replicate for each experimental condition, with the exception of the dual ubiquitin-LAMP1 counts, for which at least 100 total bacteria were counted. For single-cell replication assays and LC3 puncta counts, at least 100 intact infected BMMs were counted per replicate for each condition. Confocal images of 1024 × 1024 pixels were acquired using a Carl Zeiss LSM 710 confocal laser scanning microscope and assembled using Adobe Photoshop CS3.
For transmission electron microscopy (TEM), BMMs on 12 mm Aclar coverslips were processed as described previously (Chong et al., 2008). Sections were viewed in a Hitachi H7500 transmission electron microscope (Hitachi) at 80 kV. Images were acquired with a Hamamatsu XR-100 digital camera system (AMT) and assembled in Adobe Photoshop CS3.
O-antigen and protein analysis
To analyze O-antigen and capsule expression by Western blotting, pellets of freshly grown bacteria resuspended in MMH broth were lysed in sample buffer (0.035 M Tris-HCl–sodium dodecyl sulfate (SDS), pH 6.8, 30% glycerol, 10% SDS, 10% dithiothreitol, 0.012% bromophenol blue) and boiled for 10 min. Based on OD600nm equivalents, 1 × 107 bacteria were resolved on 12% or 8% SDS-PAGE for LPS or capsule analysis, respectively. After transfer onto Hybond-ECL nitrocellulose membranes (GE Healthcare Biosciences) using a semidry apparatus, membranes were processed for Western Blotting analysis as previously described (Chong et al., 2008) using either a mouse monoclonal anti-Francisella LPS O-antigen antibody (1:1 × 106 dilution; US Biological), or a mouse monoclonal anti-Francisella capsule antibody (clone 11B7, 1:10,000; gift of Michael Apicella, University of Iowa, Iowa City, IA). To verify equal loading between samples, blots were stripped using Restore Western Blot Stripping Buffer (Thermo Scientific) according to manufacturer’s instructions, and probed overnight at 4°C with rabbit polyclonal anti-E. coli GroEL antibodies (1:10,000; Enzo Life Sciences).
Determination of bacterial intracellular replication
Intracellular growth of Schu S4 and its derivatives was monitored by enumerating the number of colony-forming units (CFU) recovered from macrophages lysed in PBS containing 1% saponin (Sigma) as described previously (Chong et al., 2008). The number of viable intracellular bacteria per well was determined in triplicate for each time point.
Determination of host cell cytotoxicity
To quantitate cytotoxic effects of F. tularensis on BMMs via lactate dehydrogenase (LDH) release, BMMs were plated and infected as described (Chong et al., 2008) in triplicate wells. At the indicated times, 50 μl of cell culture supernatant was collected and sampled in triplicate for quantification of LDH using the Cytotox96 Non-radioactive Cytotoxicity Assay kit (Promega) according to the manufacturer’s instructions. Cytotoxicity levels were compared to lysed, uninfected positive control wells corresponding to 100% of LDH release.
Statistical Analyses
All data are presented as mean ± SD from three independent experiments, unless otherwise stated. Statistical analyses were performed using an unpaired, two-tailed Student t-test. *p < 0.05, **p<0.01.
Supplementary Material
Figure S1. Intracellular trafficking and O-antigen expression of Schu S4 mutants. (A) and (B): Confocal micrographs of BMMs infected with Francisella strains and stained for either LAMP1(A) or cellular markers of autophagy (B) at 2 h post-infection. Wild type Schu S4 (Schu S4), Schu S4ΔFTT1236 (ΔFTT1236) Schu S4ΔmanC (ΔmanC), Schu S4ΔfevR (ΔfevR) and Schu S4ΔdipA (ΔdipA) DNA was stained with DAPI (gray) and specific cellular markers are shown in red. Insets are indicated with a white box (A) or arrows (B) and magnified to show bacteria (Ft), and LAMP1, Ubiquitin (Ub), p62/SQSTM1 (p62) or LC3 in red. Scale bars, 10 and 2 μm. (C) and (D): Western blots of whole cell lysates of wild type Schu S4 (Schu S4) Schu S4ΔfevR (ΔfevR), Schu S4ΔdipA (ΔdipA). 1 × 107 bacterial equivalents were resolved and probed for either LPS O-antigen (C), or O-antigen capsule (D), and GroEL was used as a loading control.
Figure S2. Representative transmission electron micrographs of wild type Schu S4 (Schu S4), Schu S4ΔFTT1236 (ΔFTT1236) Schu S4ΔmanC (ΔmanC), and Schu S4ΔfevR (ΔfevR), mutants at 1 h post-infection. Scale bar, 500 nm.
Figure S3. Capture of Francisella O-antigen mutants by autophagy in human macrophages. (A), (C), and (E): Confocal micrographs of MDMs infected with wild type Schu S4 (Schu S4), Schu S4 ΔFTT1236 (ΔFTT1236) or Schu S4ΔmanC (ΔmanC), showing recruitment of (A) Ubiquitin (Ub), (C) p62/SQSTM1 (p62), or (E) LC3 at 2 h post-infection around the O-antigen mutants stained with DAPI (gray), and specific cellular markers (red). Insets are indicated with arrows and magnified to show bacteria. Scale bars,10 and 2 μm. (B), (D), and (F): Quantification of autophagy markers recruitment to Schu S4 (black), Schu S4ΔFTT1236 (red), and Schu S4ΔmanC (blue) with (B) Ubiquitin, (D) p62/SQSTM1, or (F) LC3. Data are means ± SD (n=3).
Figure S4. Phagosomal escape is required for autophagic targeting. (A) Colocalization of ubiquitinated wild type Schu S4 (Schu S4), Schu S4ΔFTT1236 (ΔFTT1236) and Schu S4ΔmanC (ΔmanC) with the phagosomal marker LAMP1 at 2 h post-infection. Data are means ± SD (n=3; asterisks indicate a P value<0.05 by the unpaired student’s two-tailed t-test). ND, not detected. (B), (C), (D) and (E): Effect of bafilomycin-A1 (BAF) treatment on intracellular trafficking of O-antigen mutants. Recruitment of LAMP1 (B), Ubiquitin (C), p62/SQSTM1 (D) or LC3 (E) at 2 h post-infection to wild type Schu S4, Schu S4ΔFTT1236, and Schu S4ΔmanC in BMMs treated with either no (white bars) or 100 nM (black bars) BAF. Data are means ± SD (n=3; asterisks indicate a P value<0.05 by the unpaired student’s two-tailed t-test).
Figure S5. Autophagy induction of infected macrophages. (A) Quantification of LC3 puncta in uninfected BMMs (Mock) or BMMs infected with wild type Schu S4 (Schu S4), Schu S4ΔFTT1236 (ΔFTT1236) and Schu S4ΔmanC (ΔmanC) at 2 h post-infection. Data are means ± SD (n=3). (B) Quantification of LC3 puncta in uninfected BMMs subjected (black bar) or not (white bar) to amino-acid starvation. Data are means ± SD (n=3; asterisks indicate a P value<0.01 by the unpaired student’s two-tailed t-test). Quantification of endogenous LC3 with wild type Schu S4 (Schu S4), Schu S4ΔFTT1236 (ΔFTT1236) and Schu S4ΔmanC (ΔmanC) at 2 h post-infection in untreated (white bars) or starved (black bars) macrophages. Data are means ± SD (n=3).
Acknowledgments
We thank members of the lab for their assistance with generating the Francisella-HimarFT insertion library, Michael Apicella for the monoclonal 11B7 antibody against Francisella capsule, helpful suggestions and critical reading of the manuscript, Thomas Zahrt for the pHimarH3, Jacques Peschon and Ed Miao for pBMN-Z-v-mycMC29, and Leigh Knodler for critical reading of the manuscript. We also thank Stephanie Lathrop and Olivia Steele-Mortimer for providing human peripheral blood monocytes. This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Allergy and Infectious Diseases and by NIAID grant U54 AI057160 to H. W. Virgin.
Footnotes
The authors declare that they have no conflict of interest.
References
- Apicella MA, Post DM, Fowler AC, Jones BD, Rasmussen JA, Hunt JR, et al. Identification, characterization and immunogenicity of an O-antigen capsular polysaccharide of Francisella tularensis. PLoS One. 2010;5:e11060. doi: 10.1371/journal.pone.0011060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Checroun C, Wehrly TD, Fischer ER, Hayes SF, Celli J. Autophagy-mediated reentry of Francisella tularensis into the endocytic compartment after cytoplasmic replication. Proc Natl Acad Sci U S A. 2006;103:14578–14583. doi: 10.1073/pnas.0601838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong A, Wehrly TD, Child R, Hansen B, Hwang S, Virgin HW, Celli J. Cytosolic clearance of replication-deficient mutants reveals Francisella tularensis interactions with the autophagic pathway. Autophagy. 2012;8:1342–1356. doi: 10.4161/auto.20808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong A, Wehrly TD, Nair V, Fischer ER, Barker JR, Klose KE, Celli J. The early phagosomal stage of Francisella tularensis determines optimal phagosomal escape and Francisella pathogenicity island protein expression. Infect Immun. 2008;76:5488–5499. doi: 10.1128/IAI.00682-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens DL, Lee BY, Horwitz MA. Virulent and avirulent strains of Francisella tularensis prevent acidification and maturation of their phagosomes and escape into the cytoplasm in human macrophages. Infect Immun. 2004;72:3204–3217. doi: 10.1128/IAI.72.6.3204-3217.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens DL, Lee BY, Horwitz MA. O-antigen-deficient Francisella tularensis Live Vaccine Strain mutants are ingested via an aberrant form of looping phagocytosis and show altered kinetics of intracellular trafficking in human macrophages. Infect Immun. 2012;80:952–967. doi: 10.1128/IAI.05221-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dortet L, Mostowy S, Samba-Louaka A, Gouin E, Nahori MA, Wiemer EA, et al. Recruitment of the major vault protein by InlK: a Listeria monocytogenes strategy to avoid autophagy. PLoS pathogens. 2011;7:e1002168. doi: 10.1371/journal.ppat.1002168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gal J, Strom AL, Kwinter DM, Kilty R, Zhang J, Shi P, et al. Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism. J Neurochem. 2009;111:1062–1073. doi: 10.1111/j.1471-4159.2009.06388.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunn JS, Ernst RK. The structure and function of Francisella lipopolysaccharide. Ann N Y Acad Sci. 2007;1105:202–218. doi: 10.1196/annals.1409.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajjar AM, Harvey MD, Shaffer SA, Goodlett DR, Sjostedt A, Edebro H, et al. Lack of in vitro and in vivo recognition of Francisella tularensis subspecies lipopolysaccharide by Toll-like receptors. Infect Immun. 2006;74:6730–6738. doi: 10.1128/IAI.00934-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huett A, Heath RJ, Begun J, Sassi SO, Baxt LA, Vyas JM, et al. The LRR and RING domain protein LRSAM1 is an E3 ligase crucial for ubiquitin-dependent autophagy of intracellular Salmonella Typhimurium. Cell host & microbe. 2012;12:778–790. doi: 10.1016/j.chom.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Q, Lee BY, Bowen R, Dillon BJ, Som SM, Horwitz MA. A Francisella tularensis live vaccine strain (LVS) mutant with a deletion in capB, encoding a putative capsular biosynthesis protein, is significantly more attenuated than LVS yet induces potent protective immunity in mice against F. tularensis challenge. Infect Immun. 2010;78:4341–4355. doi: 10.1128/IAI.00192-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kampen H, Rotzel DC, Kurtenbach K, Maier WA, Seitz HM. Substantial rise in the prevalence of Lyme borreliosis spirochetes in a region of western Germany over a 10-year period. Appl Environ Microbiol. 2004;70:1576–1582. doi: 10.1128/AEM.70.3.1576-1582.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol. 2010;12:836–841. doi: 10.1038/ncb0910-836. [DOI] [PubMed] [Google Scholar]
- Lai XH, Shirley RL, Crosa L, Kanistanon D, Tempel R, Ernst RK, et al. Mutations of Francisella novicida that alter the mechanism of its phagocytosis by murine macrophages. PLoS One. 2010;5:e11857. doi: 10.1371/journal.pone.0011857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson P, Oyston PCF, Chain P, Chu MC, Duffield M, Fuxelius HH, et al. The complete genome sequence of Francisella tularensis, the causative agent of tularemia. Nat Genet. 2005;37:153–159. doi: 10.1038/ng1499. [DOI] [PubMed] [Google Scholar]
- Lindemann SR, Peng K, Long ME, Hunt JR, Apicella MA, Monack DM, et al. Francisella tularensis Schu S4 O-antigen and capsule biosynthesis gene mutants induce early cell death in human macrophages. Infect Immun. 2011;79:581–594. doi: 10.1128/IAI.00863-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier TM, Casey MS, Becker RH, Dorsey CW, Glass EM, Maltsev N, et al. Identification of Francisella tularensis Himar1-based transposon mutants defective for replication in macrophages. Infect Immun. 2007;75:5376–5389. doi: 10.1128/IAI.00238-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier TM, Havig A, Casey M, Nano FE, Frank DW, Zahrt TC. Construction and characterization of a highly efficient Francisella shuttle plasmid. Appl Environ Microbiol. 2004;70:7511–7519. doi: 10.1128/AEM.70.12.7511-7519.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maier TM, Pechous R, Casey M, Zahrt TC, Frank DW. In vivo Himar1-based transposon mutagenesis of Francisella tularensis. Appl Environ Microbiol. 2006;72:1878–1885. doi: 10.1128/AEM.72.3.1878-1885.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayasu ES, Tempel R, Cambronne XA, Petyuk VA, Jones MB, Gritsenko MA, et al. Comparative phosphoproteomics reveals components of host cell invasion and post-transcriptional regulation during Francisella infection. Mol Cell Proteomics. 2013 doi: 10.1074/mcp.M113.029850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C. Escape of intracellular Shigella from autophagy. Science. 2005;307:727–731. doi: 10.1126/science.1106036. [DOI] [PubMed] [Google Scholar]
- Overell RW, Weisser KE, Cosman D. Stably transmitted triple-promoter retroviral vectors and their use in transformation of primary mammalian cells. Mol Cell Biol. 1988;8:1803–1808. doi: 10.1128/mcb.8.4.1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007;282:24131–24145. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- Peng K, Broz P, Jones J, Joubert LM, Monack D. Elevated AIM2-mediated pyroptosis triggered by hypercytotoxic Francisella mutant strains is attributed to increased intracellular bacteriolysis. Cellular microbiology. 2011;13:1586–1600. doi: 10.1111/j.1462-5822.2011.01643.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips NJ, Schilling B, McLendon MK, Apicella MA, Gibson BW. Novel modification of lipid A of Francisella tularensis. Infect Immun. 2004;72:5340–5348. doi: 10.1128/IAI.72.9.5340-5348.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior JL, Prior RG, Hitchen PG, Diaper H, Griffin KF, Morris HR, et al. Characterization of the O antigen gene cluster and structural analysis of the O antigen of Francisella tularensis subsp. tularensis. J Med Microbiol. 2003;52:845–851. doi: 10.1099/jmm.0.05184-0. [DOI] [PubMed] [Google Scholar]
- Raynaud C, Meibom KL, Lety MA, Dubail I, Candela T, Frapy E, Charbit A. Role of the wbt locus of Francisella tularensis in lipopolysaccharide O-antigen biogenesis and pathogenicity. Infect Immun. 2007;75:536–541. doi: 10.1128/IAI.01429-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorokin VM, Pavlovich NV, Prozorova LA. Francisella tularensis resistance to bactericidal action of normal human serum. FEMS immunology and medical microbiology. 1996;13:249–252. doi: 10.1111/j.1574-695X.1996.tb00246.x. [DOI] [PubMed] [Google Scholar]
- Steele S, Brunton J, Ziehr B, Taft-Benz S, Moorman N, Kawula T. Francisella tularensis harvests nutrients derived via ATG5-independent autophagy to support intracellular growth. PLoS Pathog. 2013;8:e1003562. doi: 10.1371/journal.ppat.1003562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Muhlinen N, Thurston T, Ryzhakov G, Bloor S, Randow F. NDP52, a novel autophagy receptor for ubiquitin-decorated cytosolic bacteria. Autophagy. 2010;6:288–289. doi: 10.4161/auto.6.2.11118. [DOI] [PubMed] [Google Scholar]
- Wang X, Ribeiro AA, Guan Z, McGrath SC, Cotter RJ, Raetz CR. Structure and biosynthesis of free lipid A molecules that replace lipopolysaccharide in Francisella tularensis subsp. novicida. Biochemistry. 2006;45:14427–14440. doi: 10.1021/bi061767s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wehrly TD, Chong A, Virtaneva K, Sturdevant DE, Child R, Edwards JA, et al. Intracellular biology and virulence determinants of Francisella tularensis revealed by transcriptional profiling inside macrophages. Cell Microbiol. 2009;11:1128–1150. doi: 10.1111/j.1462-5822.2009.01316.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010;12:814–822. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa Y, Ogawa M, Hain T, Yoshida M, Fukumatsu M, Kim M, et al. Listeria monocytogenes ActA-mediated escape from autophagic recognition. Nat Cell Biol. 2009;11:1233–1240. doi: 10.1038/ncb1967. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Fux B, Goodwin M, Dunay IR, Strong D, Miller BC, et al. Autophagosome-independent essential function for the autophagy protein Atg5 in cellular immunity to intracellular pathogens. Cell Host Microbe. 2008;4:458–469. doi: 10.1016/j.chom.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, Thackray LB, Miller BC, Lynn TM, Becker MM, Ward E, et al. Coronavirus replication does not require the autophagy gene ATG5. Autophagy. 2007;3:581–585. doi: 10.4161/auto.4782. [DOI] [PubMed] [Google Scholar]
- Zheng YT, Shahnazari S, Brech A, Lamark T, Johansen T, Brumell JH. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol. 2009;183:5909–5916. doi: 10.4049/jimmunol.0900441. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Intracellular trafficking and O-antigen expression of Schu S4 mutants. (A) and (B): Confocal micrographs of BMMs infected with Francisella strains and stained for either LAMP1(A) or cellular markers of autophagy (B) at 2 h post-infection. Wild type Schu S4 (Schu S4), Schu S4ΔFTT1236 (ΔFTT1236) Schu S4ΔmanC (ΔmanC), Schu S4ΔfevR (ΔfevR) and Schu S4ΔdipA (ΔdipA) DNA was stained with DAPI (gray) and specific cellular markers are shown in red. Insets are indicated with a white box (A) or arrows (B) and magnified to show bacteria (Ft), and LAMP1, Ubiquitin (Ub), p62/SQSTM1 (p62) or LC3 in red. Scale bars, 10 and 2 μm. (C) and (D): Western blots of whole cell lysates of wild type Schu S4 (Schu S4) Schu S4ΔfevR (ΔfevR), Schu S4ΔdipA (ΔdipA). 1 × 107 bacterial equivalents were resolved and probed for either LPS O-antigen (C), or O-antigen capsule (D), and GroEL was used as a loading control.
Figure S2. Representative transmission electron micrographs of wild type Schu S4 (Schu S4), Schu S4ΔFTT1236 (ΔFTT1236) Schu S4ΔmanC (ΔmanC), and Schu S4ΔfevR (ΔfevR), mutants at 1 h post-infection. Scale bar, 500 nm.
Figure S3. Capture of Francisella O-antigen mutants by autophagy in human macrophages. (A), (C), and (E): Confocal micrographs of MDMs infected with wild type Schu S4 (Schu S4), Schu S4 ΔFTT1236 (ΔFTT1236) or Schu S4ΔmanC (ΔmanC), showing recruitment of (A) Ubiquitin (Ub), (C) p62/SQSTM1 (p62), or (E) LC3 at 2 h post-infection around the O-antigen mutants stained with DAPI (gray), and specific cellular markers (red). Insets are indicated with arrows and magnified to show bacteria. Scale bars,10 and 2 μm. (B), (D), and (F): Quantification of autophagy markers recruitment to Schu S4 (black), Schu S4ΔFTT1236 (red), and Schu S4ΔmanC (blue) with (B) Ubiquitin, (D) p62/SQSTM1, or (F) LC3. Data are means ± SD (n=3).
Figure S4. Phagosomal escape is required for autophagic targeting. (A) Colocalization of ubiquitinated wild type Schu S4 (Schu S4), Schu S4ΔFTT1236 (ΔFTT1236) and Schu S4ΔmanC (ΔmanC) with the phagosomal marker LAMP1 at 2 h post-infection. Data are means ± SD (n=3; asterisks indicate a P value<0.05 by the unpaired student’s two-tailed t-test). ND, not detected. (B), (C), (D) and (E): Effect of bafilomycin-A1 (BAF) treatment on intracellular trafficking of O-antigen mutants. Recruitment of LAMP1 (B), Ubiquitin (C), p62/SQSTM1 (D) or LC3 (E) at 2 h post-infection to wild type Schu S4, Schu S4ΔFTT1236, and Schu S4ΔmanC in BMMs treated with either no (white bars) or 100 nM (black bars) BAF. Data are means ± SD (n=3; asterisks indicate a P value<0.05 by the unpaired student’s two-tailed t-test).
Figure S5. Autophagy induction of infected macrophages. (A) Quantification of LC3 puncta in uninfected BMMs (Mock) or BMMs infected with wild type Schu S4 (Schu S4), Schu S4ΔFTT1236 (ΔFTT1236) and Schu S4ΔmanC (ΔmanC) at 2 h post-infection. Data are means ± SD (n=3). (B) Quantification of LC3 puncta in uninfected BMMs subjected (black bar) or not (white bar) to amino-acid starvation. Data are means ± SD (n=3; asterisks indicate a P value<0.01 by the unpaired student’s two-tailed t-test). Quantification of endogenous LC3 with wild type Schu S4 (Schu S4), Schu S4ΔFTT1236 (ΔFTT1236) and Schu S4ΔmanC (ΔmanC) at 2 h post-infection in untreated (white bars) or starved (black bars) macrophages. Data are means ± SD (n=3).