

Abstract
Background
Apolipoprotein (APOE) e4 allele status has been linked to clinical presentation and progression in Alzheimer’s disease; however, evidence for a role of APOE e4 in Parkinson’s disease (PD) remains largely inconclusive. In this analysis we explored potential significant associations between APOE e4 allele status and characteristics of clinical presentation in patients with PD.
Methods
Data came from 424 subjects evaluated using the Uniform Data Set (UDS) assessment collected by the National Alzheimer’s Coordinating Center. Subjects had a known year of diagnosis of PD and experienced change in motor function prior to any change in cognition. Linear and logistic regression were used to model the association between APOE e4 carrier status and clinical characteristics including measures of cognitive decline and motor and neuropsychiatric symptoms. Amyloid burden was also evaluated for a subset of patients who died and consented to autopsy.
Results
Odds of dementia were higher in APOE e4 carriers (OR=5.15), and, on average, APOE e4 carriers scored two points worse on tests of episodic memory and the Clinical Dementia Rating Sum of Boxes assessment. There was little evidence to support an association between e4 carrier status and severity of motor features, and, of the four neuropsychiatric symptoms evaluated, only presence of hallucinations was significantly associated with APOE e4 carrier status (OR=5.29). Neuropathology data revealed higher frequencies of neuritic and diffuse amyloid plaques in APOE e4 carriers compared to non-carriers.
Conclusions
APOE e4 allele status is associated with dementia and severity of Alzheimer’s disease pathologic features in PD.
Keywords: Parkinson’s disease, APOE, neuropathology, clinical symptoms
INTRODUCTION
Apolipoprotein (APOE) e4 allele status has been linked to clinical presentation and progression in Alzheimer’s disease [1-3]; however, evidence for a role of APOE e4 in Parkinson’s disease (PD) remains largely inconclusive [4-6]. In 2012, researchers at the National Institute on Aging found that the frequency of the APOE e4 allele in subjects with PD was not statistically significantly different from dementia-free controls [5].
On the other hand, smaller studies, using different measures of cognition, have found evidence of an association between e4 carrier status and cognition in PD. Tsuang et al. found higher rates of APOE e4 allele frequency among subjects with PD dementia compared to non-PD cognitively normal subjects (OR=3.1) [7]. Similarly, Gomperts et al. found evidence of an association between APOE e4 and cognitive but not motor decline over time in PD [8]. Thus, additional research is needed to shed light on the seemingly complicated and conflicting associations observed between APOE and cognition in PD.
The National Alzheimer’s Coordinating Center (NACC) houses a large, multicenter database on subjects with a wide range of cognitive and motor features, several of whom have a clinical diagnosis of PD. Data collected include subject demographics, health history, performance on neuropsychological tests, as well as presence and severity of Parkinsonian and neuropsychiatric symptoms. Therefore, these data provide an excellent opportunity for studying clinical presentation of progressive neurodegenerative diseases such as PD. In this analysis, we tested the hypothesis that cognition, but not motor or neuropsychiatric symptoms in PD subjects, vary by APOE e4 allele status.
METHODS
Study sample
Data for this analysis were obtained from the NACC Uniform Data Set (UDS) funded by the National Institute on Aging [9,10]. All data came from the subject’s initial UDS visit, recorded between September 2005 and March 2013 at one of the 34 past and present Alzheimer’s Disease Centers (ADCs). Study inclusion criteria were (1) a diagnosis of PD, made by an ADC neurologist, at the initial UDS visit, (2) a known year of PD diagnosis, and (3) motor function was the first predominant change as opposed to cognition or behavior, as judged by a clinician. Research using the NACC database was approved by the University of Washington Institutional Review Board.
APOE e4 genotype, the main exposure of interest, was available for a subset of subjects and was categorized as either e4 carrier (hetero or homozygous) or non-carrier (no e4 alleles). Characteristics of clinical presentation used in this analysis included the presence of dementia, Clinical Dementia Rating Sum of Boxes score (CDR-SB), Logical Memory delayed score, motor symptoms measured by the Unified Parkinson’s Disease Rating Scale (UPDRS), and select neuropschiatric symptoms measured by the Neuropsychiatric Inventory (NPI-Q). Cognitive function was assessed as a continuous measure using the CDR-SB [11]. The CDR grades subjects’ cognitive and functional abilities in six domains: memory, orientation, judgment and problem solving, community affairs, home and hobbies, and personal care. The clinician, incorporating input from the subject’s co-participant, evaluates impairment in each domain as none (0), questionable or very mild (0.5), mild (1), moderate (2), or severe (3). The scores for each domain are summed to create a Sum of Box score ranging from 0 to 18, with higher scores indicating more severe impairment.
Deficits in episodic memory were assessed using the Logical Memory IIA-Delayed Story Units Recalled test, which tests subjects’ story retention ability [12]. The only tests of executive function in the UDS neuropsychological battery are the Trail Making A and B tests, which require preserved motor function [13]. As many PD subjects have substantial motor impairment, we were not able to assess executive function independent of motor impairment.
Motor features were evaluated using Levy summary scores derived from the UPDRS [14]. The Levy A score is the sum of the Levodopa-responsive symptoms, including facial expression, rigidity, and bradykinesia domains. The A score ranges from 0 to 80. The Levy B score, ranging from 0 to 20, is the sum of the Levodopa-nonresponsive symptoms of speech and axial impairment [15]. Subjects who did not receive the UPDRS or had any ‘untestable’ items were considered to be missing the Levy A and/or B score.
Neuropsychiatric symptoms were evaluated with the NPI-Q, in which physicians interview the co-participant about the subject’s symptoms [16,17]. For this analysis, we included the items on depression or dysphoria, hallucinations, anxiety, and nighttime behavior (awake during the night, rise too early, or take excessive naps), since these symptoms are commonly associated with Alzheimer’s disease and dementia with Lewy bodies [18-21].
Statistical analysis
Linear and logistic regression were used to estimate the association between APOE e4 carrier status and each individual characteristic of clinical presentation and cognitive decline. Generalized estimating equations (GEE) were used to account for clustering of subjects within an ADC [22]. An independent correlation structure with robust standard errors was employed. Since symptoms generally worsen as PD progresses, the analytic sample was split according to years since PD diagnosis: subjects whose initial UDS visit was less than five years after their first PD diagnosis were grouped into the <5 years group, while the 5+ group comprised subjects whose initial UDS visit took place five or more years after PD diagnosis. This dichotomy allowed for a stratified analysis, where associations between e4 carrier status and clinical presentation could be evaluated separately for disease periods with potentially different patterns and severity of symptoms.
Each clinical characteristic was an outcome measurement in a model with APOE e4 status as the main predictor, resulting in nine separate regressions for each strata. Models were run both with and without adjustment for age at PD diagnosis, years since PD diagnosis, sex, and education. Frequencies of reported use of an antipsychotic medication or dopaminergic agonist (ropinirole, pramipexole, rotigotine) were compared between APOE e4 carrier groups using a Chi-squared test. A significant difference in the reported use of either class of medication would prompt inclusion of these indicators in the models for neuropsychiatric symptoms.
The GEE approach assumes that all missing data are missing completely at random, meaning that there are no patterns or trends making some subjects more likely than others to be missing data. While this is likely a reasonable assumption for the missing data on APOE genotype, we performed both a complete case analysis and an analysis using multiple imputation in order to avoid overlooking potential biases introduced by these missing data.
In the complete case analysis, only subjects with non-missing data on APOE e4 allele status and the outcome measure were included in each regression model. For the imputation model, missing values were estimated using multiple imputation with chained equations [23,24]. The cognitive scores, neuropsychiatric symptoms, Levy scores, years since PD diagnosis, age at PD diagnosis, sex, and education were used to estimate missing APOE e4 status and measures of clinical presentation. The imputation step was run 20 times, yielding 20 complete data sets. Coefficients and standard errors were combined using Rubin’s rules [25], producing a single regression coefficient with corresponding confidence intervals. The complete case analysis was considered the main analysis and the imputation model a sensitivity analysis.
Neuropathology
APOE genotype has been linked to levels of amyloid beta in cerebrospinal fluid (CSF) and brain deposition captured via PET imaging [26,27]. Although CSF biomarker and imaging data were not available to evaluate amyloid burden, neuropathological data were available for a small number of subjects included in the analytic sample who died and had an autopsy report (n=29). Using Fisher’s exact test, we tested for statistically significant differences in CERAD neuritic plaque frequency (none or sparse vs. moderate or frequent) [28], as well as diffuse plaque frequency (none or sparse vs. moderate or frequent) observed at autopsy by e4 carrier status. Braak & Braak neurofibrillary stage [29] was also examined in order to evaluate the overall severity of AD neuropathologic features. Finally, Lewy body pathology was evaluated according to criteria from the Consortium on Dementia with Lewy Bodies [30,31].
RESULTS
Of the nearly 600 subjects with a diagnosis of PD at the initial UDS visit and a known year of diagnosis, 424 (71%) experienced a decline in motor function prior to any decline in cognition or behavior. Approximately 55% of these subjects had APOE e4 genotype data available; there were 61 APOE e4 carriers and 171 non-carriers. There were also 192 subjects for whom APOE e4 genotype data were not available. A Chi-squared test comparing the proportion of demented subjects with APOE e4 genotype known vs. unknown was performed. The test yielded a p-value of .74, suggesting that subjects with dementia were not more or less likely to have a known APOE e4 genotype compared to non-demented subjects.
More than half of the subjects were diagnosed with PD five or more years prior to their first UDS visit; however, the proportion falling into each group was fairly even between e4 carriers and non-carriers (Supplemental Figure 1).
Subject demographics and clinical characteristics are displayed in Table 1. Overall, the groups were quite similar. APOE e4 carriers were slightly older than non-carriers and more often men. Across, all groups, the main reason for participation in the ADC study was to participate in research; a much smaller portion (7-13%) enrolled in order to obtain a clinical evaluation, thus suggesting that the sample was not comprised mainly of subjects who enrolled in order to evaluate a cognitive complaint.
Table1.
Subject characteristics by APOE e4 carrier status
| Characteristic | APOE e4 carriersa (n=61) | APOE e4 non-carriers (n=171) | APOE e4 carrier status unknown (n=192) |
|---|---|---|---|
| Age at PD diagnosis | |||
| <50 | 5 (8%) | 18 (11%) | 15 (8%) |
| 50-59 | 11 (18%) | 49 (29%) | 43 (22%) |
| 60-69 | 21 (34%) | 48 (28%) | 79 (41%) |
| 70-79 | 17 (28%) | 45 (26%) | 44 (23%) |
| 80+ | 7 (12%) | 11 (6%) | 11 (6%) |
| Age at initial visit | |||
| <50 | 2 (3%) | 5 (3%) | 1 (1%) |
| 50-59 | 4 (7%) | 28 (16%) | 13 (7%) |
| 60-69 | 16 (26%) | 51 (30%) | 76 (39%) |
| 70-79 | 29 (48%) | 60 (35%) | 75 (39%) |
| 80+ | 10 (16%) | 27 (16%) | 27 (14%) |
| Years between visit and diagnosis | |||
| <5 | 28 (46%) | 74 (43%) | 74 (39%) |
| 5+ | 33 (54%) | 97 (57%) | 118 (61%) |
| Sex | |||
| Male | 48 (79%) | 124 (73%) | 145 (76%) |
| Female | 13 (21%) | 47 (26%) | 47 (24%) |
| Years of education | |||
| High school only | 10 (16%) | 35 (21%) | 41 (21%) |
| 1-4 years of college | 33 (54%) | 84 (49%) | 82 (43%) |
| Some graduate school | 18 (30%) | 52 (30%) | 69 (36%) |
| Race | |||
| White | 59 (96%) | 163 (95%) | 180 (94%) |
| Black | 1 (2%) | 4 (2%) | 9 (4%) |
| Asian | 1 (2%) | 2 (1%) | 2 (1%) |
| Multiracial | 0 (0%) | 2 (1%) | 1 (1%) |
| Reason for participation | |||
| Participate in research study | 57 (93%) | 148 (87%) | 174 (91%) |
| Clinical evaluation | 4 (7%) | 23 (13%) | 18 (9%) |
| First predominant motor symptomb | |||
| Gait disorder | 10 (20%) | 24 (21%) | 37 (23%) |
| Falls | 0 (0%) | 0 (0%) | 6 (4%) |
| Tremor | 20 (39%) | 51 (45%) | 66 (40%) |
| Slowness | 21 (41%) | 38 (34%) | 54 (33%) |
| Family history of dementiac | |||
| No | 34 (56%) | 100 (59%) | 119 (63%) |
| Yes | 27 (44%) | 70 (41%) | 70 (37%) |
| Antipsychotic medication used | |||
| No | 153 (89%) | 51 (84%) | 166 (86%) |
| Yes | 18 (11%) | 10 (16%) | 25 (13%) |
| Dopaminergic agonist use | |||
| No | 36 (59%) | 98 (57%) | 111 (58%) |
| Yes | 25 (41%) | 73 (43%) | 81 (42%) |
Includes 7 APOE e4 homozygotes
10 APOE e4 carriers, 58 non-carriers, and 29 with e4 carrier status unknown were missing information on the first predominant motor symptom
1 APOE e4 non-carrier and 3 with unknown e4 carrier status were missing information on family history of dementia
1 subject with unknown APOE e4 carrier status was missing data on antipsychotic medication use
There was also no difference in frequency of antipsychotic medication or dopaminergic agonist use among APOE e4 carriers and non-carriers (χ2, p= 0.2272 and p=0.8168, respectively). Therefore, adjustment for these factors in modeling neuropsychiatric symptoms among APOE e4 carriers and non-carriers was determined to be unnecessary.
As the presence of multiple diagnoses could also influence the results, the frequencies of probable AD and dementia with Lewy bodies (DLB), in addition to PD, were calculated. Upon investigation, only two subjects had probable AD (one APOE e4 non-carrier and one with e4 status unknown). Several more subjects did have a diagnosis of DLB in addition to PD (13 APOE e4 non-carriers, nine e4 carriers, and six who did not have APOE e4 allele information). A Chi-square test was performed to determine whether or not the frequency of a DLB diagnosis varied across the e4 allele status groups. The test yielded a p-value of 0.46, thus, inclusion of these subjects is unlikely to influence our comparisons of APOE e4 carriers and non-carriers.
Regression model results
In the complete case analysis, adjusted and unadjusted models yielded similar results. The complete case adjusted models are presented in Table 2. In subjects seen within five years of their PD diagnosis, the odds of dementia were approximately 5 times higher for e4 carriers compared to non-carriers, and for those seen 5+ years after their diagnosis, the odds were 3 times higher. Statistically significant differences in cognitive function were also seen in the CDR-SB, where the e4 carriers averaged nearly 2 points worse than non-carriers and recalled fewer story units on the Logical Memory Delayed test. APOE e4 carriers evaluated within five years of their diagnosis also had higher odds of hallucinations compared to non-carriers (OR=5.29, 95% CI=2.47-19.00); however, the odds of hallucinations were similar for carriers and non-carriers evaluated 5+ years after their diagnosis. There were no statistically significant differences for other selected neuropsychiatric symptoms or Levy A and B scores.
Table 2.
Adjusted analysis of association between APOE e4 allele status (at least one e4 vs. no e4 alleles) and each clinical characteristic at first ADC visit using complete cases
| Clinical characteristic | <5 years since PD diagnosis | 5+ years since PD diagnosis | ||||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| n | OR | 95% CI | p-value | n | OR | 95% CI | p-value | |
| Dementia | 102 | 5.15 | (1.52,17.40) | .008 | 130 | 3.54 | (1.27,9.92) | .02 |
| Depression | 100 | 1.30 | (0.62,2.74) | .48 | 130 | 1.02 | (0.51,2.03) | .96 |
| Hallucinations | 100 | 5.29 | (1.47,19.00) | .01 | 130 | 1.69 | (0.54,5.32) | .37 |
| Anxiety | 100 | 1.37 | (0.72,2.61) | .34 | 130 | 2.16 | (0.82,5.66) | .12 |
| Nighttime behaviors | 102 | 1.67 | (0.67,4.17) | .28 | 130 | 1.53 | (0.77,3.04) | .22 |
|
| ||||||||
| n | β | 95% CI | p-value | n | β | 95% CI | p-value | |
|
| ||||||||
| CDR Sum of Boxes score | 102 | 1.81 | (0.94,2.67) | <.0001 | 130 | 1.99 | (0.12,3.86) | .04 |
| Logical memory delayed | 93 | -1.89 | (-3.62,-0.16) | .03 | 123 | -1.69 | (-2.87,-0.51) | .005 |
| Levy A score | 99 | 2.74 | (-0.91, 6.39) | .14 | 128 | 2.24 | (-1.88,6.36) | .29 |
| Levy B score | 99 | 0.72 | (-0.57, 2.01) | .27 | 123 | 0.86 | (-0.46,2.18) | .20 |
Adjusted for years since PD diagnosis, age at PD diagnosis, sex, and education
In the model run with multiple imputed data sets, the results for those evaluated within five years of their PD diagnosis were similar to those obtained from the complete case analysis: odds of dementia and hallucinations were increased for e4 carriers, and e4 carriers performed worse on the CDR-SB and Logical Memory test compared to non-carriers (Supplemental Table 1). However, for those evaluated five or more years after their diagnosis, the results from the imputation model differed from the results obtained from the complete case analysis: using multiple imputation, only the odds of dementia differed by APOE e4 carrier status.
Neuropathology
Autopsy data were available for 10 e4 carriers and 19 non-carriers (Table 3). Among e4 carriers, 90% had moderate to frequent neuritic plaques, while only 42% of non-carriers had moderate to frequent neuritic plaques (p=0.02). Similarly, frequency of diffuse plaques was higher in e4 carriers compared to non-carriers: 80% of carriers and 37% of non-carriers had moderate or frequent diffuse plaques (p=0.05). Differences were also observed for Braak stage where e4 carriers had higher frequencies of advanced tau pathology (70% vs. 42% with Braak stage III-IV and 20% vs. 11% with Braak stage V-VI). Figure 1 shows the frequencies of both CERAD neuritic plaque scores and Braak stage among APOE e4 carriers and non-carriers. While APOE e4 non-carries has a wide range of CERAD scores and Braak stages, the e4 carriers tended to have high scores for both features, suggesting a higher burden of AD pathology in carriers than non-carriers overall. Yet, it is important to note that some APOE e4 non-carriers had very high levels of both amyloid and tau pathology.
Table 3.
Frequency (%) of neuropathologic characteristics in clinical PD
| Characteristic | APOE e4 carriers (n=10) | APOE e4 non-carriers (n=19) |
|---|---|---|
| Neuritic plaque frequency | ||
| None | 0 (0%) | 7 (36%) |
| Low | 1 (10%) | 4 (21%) |
| Moderate | 4 (40%) | 3 (16%) |
| Frequent | 5 (50%) | 5 (26%) |
| Diffuse plaque frequency | ||
| None | 1 (10%) | 6 (32%) |
| Sparse | 1 (10%) | 6 (32%) |
| Moderate | 1 (10%) | 3 (16%) |
| Frequent | 7 (70%) | 4 (21%) |
| Braak stage | ||
| 0 | 0 (0%) | 1 (5%) |
| I-II | 1 (10%) | 8 (42%) |
| III-IV | 7 (70%) | 8 (42%) |
| V-VI | 2 (20%) | 2 (11%) |
| Lewy body pathology | ||
| Not present | 0 (0%) | 0 (0%) |
| Brainstem predominant | 2 (20%) | 2 (11%) |
| Limbic | 2 (20%) | 6 (32%) |
| Diffuse | 6 (60%) | 9 (48%) |
| Unspecified | 0 (0%) | 2 (11%) |
Figure 1.
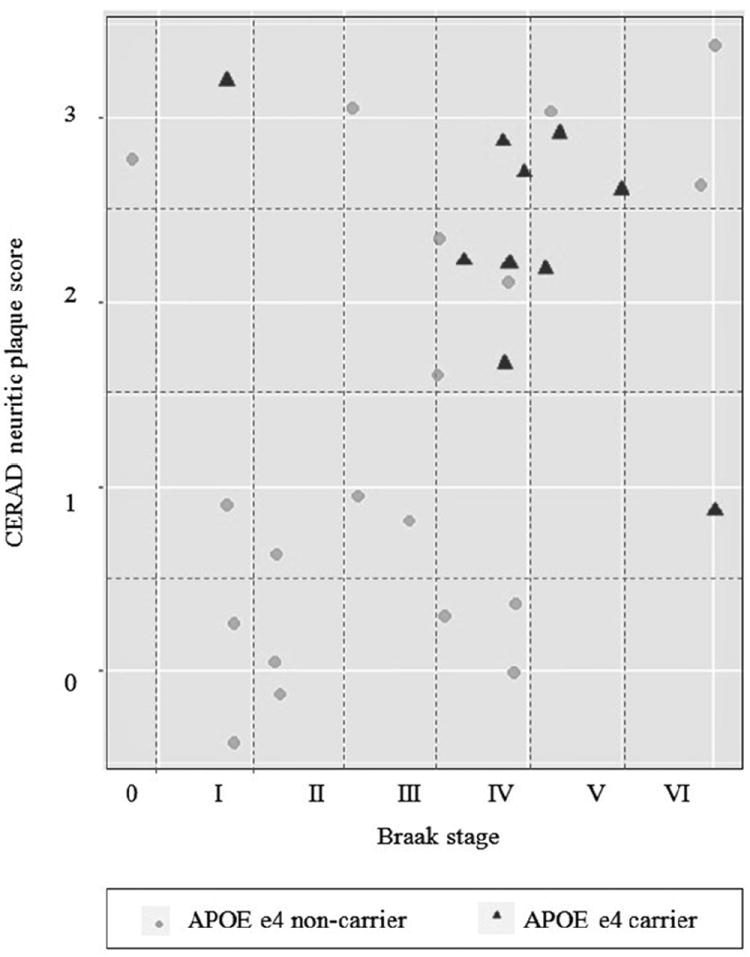
Distribution of neurofibrillary tangles and neuritic plaques by APOE e4 carrier status
While all autopsied subjects had Lewy body pathology, there were also trends towards differences in the location of the Lew body pathology. Overall, e4 carriers had higher proportions of diffuse and brainstem predominat Lewy bodies compared to non-carriers, who had higher proportions of subjects with limbic and unspecified Lewy bodies.
DISCUSSION
Our study found that the odds of dementia in PD was 5 times higher in APOE e4 carriers compared to non-carriers. In addition, we found increased odds of hallucinations for e4 carriers within the first five years of diagnosis; however, APOE e4 status was not associated with other neuropsychiatric or motor features. Despite having a small sample of PD subjects with neuropathology data, these limited results do suggest an increased burden of amyloid and tau pathology in APOE e4 carriers with PD compared to e4 non-carriers with PD. As all autopsied subjects had LB pathology, there is strong evidence for co-pathologic burden of AD and LBs, especially among e4 carriers.
The findings of our study are similar to those of recently published analyses of APOE in clinical PD. Irwin et al. examined prevalence of dementia in a cohort of PD patients and found APOE e4 allele carrier status to be a significantly associated with dementia (OR=4.19) [32]. Tsuang et al. compared rates of e4 allele status among subjects with PD dementia (PDD) and those with normal cognition and found a strong association between PDD and the presence of an e4 allele when compared to controls [7]. While the control group in this study comprised normally cognitive subjects and our study compared PD subjects with and without impairment, both studies provide evidence of an association between e4 allele status and dementia in PD. Similar to Gomperts et al., we did not find evidence to suggest a difference in motor symptom among e4 carriers and non-carriers [8].
This study has both strengths and limitations. First, the analytic sample comprised subjects from a relatively large prospective national cohort. All UDS visit data were obtained using standardized forms, diagnostic criteria, and test administration. In addition, by limiting our sample to only those subjects who first presented with motor symptoms, we had a more homogenous sample of PD patients. In addition, we applied multiple techniques to handle missing data and potential confounding by years since diagnosis and other clinical correlates. On the other hand, these PD patients were seen at Alzheimer’s Disease Centers and may not be similar to other PD patients. Still, most subjects reported enrolling in order to participate in research, not to obtain a clinical evaluation. In addition, the percent of subjects reporting a family history of dementia was similar across the APOE allele status groups (37-44%).
It is also possible that use of non-uniform methods of diagnosis influenced our findings for although the PD diagnosis was confirmed by the ADC neurologist, the initial PD diagnosis was most made before the subject enrolled at the ADC, meaning that different diagnostic criteria could have been applied. Multiple diagnostic criteria could also have been used to diagnosis dementia. The UDS Coding Guidebook specifies that dementia should be diagnosed “in accordance with standard criteria for dementia of the Alzheimer’s type or non-Alzheimer’s dementing disorders.” Thus, we cannot be certain that all subjects were diagnosed using the same diagnostic criteria, only that they met at least one set of standardized criteria. In addition, due to lack of data on onset of neuropsychiatric symptoms, we are unable to tell whether the presence of hallucinations were related to APOE e4 through biological mechanisms independent of cognition, or whether they were a consequence of cognitive decline. Finally, we did not have access to ante-mortem biomarkers, images, or information pertaining to subject characteristics and symptoms at the time of PD onset.
Despite these limitations, this study adds evidence to support statistically and clinically significant associations between APOE e4 allele status and disease symptoms in PD patients. The knowledge that cognitive symptoms are more likely to develop in PD patients with at least one e4 allele can inform clinicians and patients on how to treat and plan for disease progression, as well as inform biomarker research and clinical trial design.
Supplementary Material
{kind=link}
Acknowledgments
The authors thank the study participants, clinicians, and other ADC personnel who made this research possible.
The National Alzheimer’s Coordinating Center is supported by NIA cooperative UO1 AG016976. I Litvan reports funding from R01AG024040.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Reiman EM. In this issue: genetics, cholesterol, and the risk of Alzheimer’s disease. J Clin Psychiatry. 2005;66:938–9. doi: 10.4088/jcp.v66n0719. [DOI] [PubMed] [Google Scholar]
- 2.Cosentino S, Scarmeas N, Helzner E, Glymour MM, Brandt J, Albert M, et al. APOE epsilon 4 allele predicts faster cognitive decline in mild Alzheimer disease. Neurology. 2008;70:1842–9. doi: 10.1212/01.wnl.0000304038.37421.cc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim J, Basak JM, Holtzman DM. The role of apolipoprotein E in Alzheimer’s disease. Neuron. 2009;63:287–303. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campbell MC, Markham J, Flores H, Hartlein JM, Goate AM, Cairns NJ, et al. Principal component analysis of PiB distribution in Parkinson and Alzheimer diseases. Neurology. 2013;81:520–7. doi: 10.1212/WNL.0b013e31829e6f94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Federoff M, Jimenez-Rolando B, Nalls MA, Singleton AB. A large study reveals no association between APOE and Parkinson’s disease. Neurobiol Dis. 2012;46:389–92. doi: 10.1016/j.nbd.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kurz MW, Dekomien G, Nilsen OB, Larsen JP, Aarsland D, Alves G. APOE alleles in Parkinson disease and their relationship to cognitive decline: a population-based, longitudinal study. J Geriatr Psychiatry Neurol. 2009;22:166–70. doi: 10.1177/0891988709332945. [DOI] [PubMed] [Google Scholar]
- 7.Tsuang D, Leverenz JB, Lopez OL, Hamilton RL, Bennett DA, Schneider JA, et al. APOE ε4 increases risk for dementia in pure synucleinopathies. JAMA Neurol. 2013;70:223–8. doi: 10.1001/jamaneurol.2013.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gomperts SN, Locascio JJ, Rentz D, Santarlasci A, Marquie M, Johnson KA, et al. Amyloid is linked to cognitive decline in patients with Parkinson disease without dementia. Neurology. 2013;80:85–91. doi: 10.1212/WNL.0b013e31827b1a07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beekly DL, Ramos EM, Lee WW, Deitrich WD, Jacka ME, Wu J, et al. The National Alzheimer’s Coordinating Center (NACC) database: the Uniform Data Set. Alzheimer Dis Assoc Disord. 2007;21:249–58. doi: 10.1097/WAD.0b013e318142774e. [DOI] [PubMed] [Google Scholar]
- 10.Morris JC, Weintraub S, Chui HC, Cummings J, Decarli C, Ferris S, et al. The Uniform Data Set (UDS): clinical and cognitive variables and descriptive data from Alzheimer Disease Centers. Alzheimer Dis Assoc Disord. 2006;20:210–6. doi: 10.1097/01.wad.0000213865.09806.92. [DOI] [PubMed] [Google Scholar]
- 11.Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology. 1993;43:2412–4. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- 12.Wechsler D. WMS-R: Wechsler Memory Scale--Revised manual. Psychological Corp.: Harcourt Brace Jovanovich; 1987. [Google Scholar]
- 13.Reitan RM, Wolfson D. The Halstead-Reitan neuropsychological test battery: Theory and clinical interpretation. 2. Neuropsychology Press; 1993. [Google Scholar]
- 14.Fahn S, Marsden C, Calne D, Goldstein M. Recent Developments in Parkinson’s Disease. Florham Park, NJ: Macmillan Health Care Information; 1987. [Google Scholar]
- 15.Levy G, Tang MX, Cote LJ, Louis ED, Alfaro B, Mejia H, et al. Motor impairment in PD: relationship to incident dementia and age. Neurology. 2000;55:539–44. doi: 10.1212/wnl.55.4.539. [DOI] [PubMed] [Google Scholar]
- 16.Cummings JL, Mega M, Gray K, Rosenberg-Thompson S, Carusi DA, Gornbein J. The Neuropsychiatric Inventory: comprehensive assessment of psychopathology in dementia. Neurology. 1994;44:2308–14. doi: 10.1212/wnl.44.12.2308. [DOI] [PubMed] [Google Scholar]
- 17.Kaufer DI, Cummings JL, Ketchel P, Smith V, MacMillan A, Shelley T, et al. Validation of the NPI-Q, a brief clinical form of the Neuropsychiatric Inventory. J Neuropsychiatry Clin Neurosci. 2000;12:233–9. doi: 10.1176/jnp.12.2.233. [DOI] [PubMed] [Google Scholar]
- 18.Ganguli M, Du Y, Dodge HH, Ratcliff GG, Chang C-CH. Depressive symptoms and cognitive decline in late life: a prospective epidemiological study. Arch Gen Psychiatry. 2006;63:153–60. doi: 10.1001/archpsyc.63.2.153. [DOI] [PubMed] [Google Scholar]
- 19.Holroyd S, Currie L, Wooten GF. Prospective study of hallucinations and delusions in Parkinson’s disease. J Neurol Neurosurg Psychiatry. 2001;70:734–8. doi: 10.1136/jnnp.70.6.734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee AH, Weintraub D. Psychosis in Parkinson’s disease without dementia: common and comorbid with other non-motor symptoms. Mov Disord Off J Mov Disord Soc. 2012;27:858–63. doi: 10.1002/mds.25003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kurtis MM, Rodriguez-Blazquez C, Martinez-Martin P ELEP Group. Relationship between sleep disorders and other non-motor symptoms in Parkinson’s disease. Parkinsonism Relat Disord. 2013;12:1152–5. doi: 10.1016/j.parkreldis.2013.07.026. [DOI] [PubMed] [Google Scholar]
- 22.Liang K-Y, Zeger SL. Longitudinal data analysis using generalized linear models. Biometrika. 1986;73:13–22. [Google Scholar]
- 23.Little RJA, Rubin DB. Statistical Analysis with Missing Data. 2. Wiley-Interscience; 2002. [Google Scholar]
- 24.Van Buuren S. Multiple imputation of discrete and continuous data by fully conditional specification. Stat Methods Med Res. 2007;16:219–42. doi: 10.1177/0962280206074463. [DOI] [PubMed] [Google Scholar]
- 25.Rubin DB, Schenker N. Multiple imputation in health-care databases: an overview and some applications. Stat Med. 1991;10:585–98. doi: 10.1002/sim.4780100410. [DOI] [PubMed] [Google Scholar]
- 26.Storandt M, Head D, Fagan AM, Holtzman DM, Morris JC. Toward a multifactorial model of Alzheimer disease. Neurobiol Aging. 2012;33:2262–71. doi: 10.1016/j.neurobiolaging.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Morris JC, Roe CM, Xiong C, Fagan AM, Goate AM, Holtzman DM, et al. APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010;67:122–31. doi: 10.1002/ana.21843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, et al. The Consortium to Establish a Registry for Alzheimer’s Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer’s disease. Neurology. 1991;41:479–86. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- 29.Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol (Berl) 2006;112:389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): Report of the consortium on DLB international workshop. Neurology. 1996;47:1113–1124. doi: 10.1212/wnl.47.5.1113. [DOI] [PubMed] [Google Scholar]
- 31.McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lew bodies: Third report of the DLB Consortium. Neurology. 2005;65:1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 32.Irwin DJ, White MT, Toledo JB, Xie SX, Robinson JL, Van Deerlin V, et al. Neuropathologic substrates of Parkinson disease dementia. Ann Neurol. 2012;72:587–98. doi: 10.1002/ana.23659. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.