

Abstract
Specific missense mutations in the CACNA1A gene, which encodes a subunit of voltage-gated CaV2.1 channels, are associated with familial hemiplegic migraine type 1 (FHM1), a rare monogenic subtype of common migraine with aura. We used transgenic knock-in (KI) mice harboring the human pathogenic FHM1 mutation S218L to study presynaptic Ca2+ currents, EPSCs, and in vivo activity at the calyx of Held synapse. Whole-cell patch-clamp recordings of presynaptic terminals from S218L KI mice showed a strong shift of the calcium current I–V curve to more negative potentials, leading to an increase in basal [Ca2+]i, increased levels of spontaneous transmitter release, faster recovery from synaptic depression, and enhanced synaptic strength despite smaller action-potential-elicited Ca2+ currents. The gain-of-function of transmitter release of the S218L mutant was reproduced in vivo, including evidence for an increased release probability, demonstrating its relevance for glutamatergic transmission. This synaptic phenotype may explain the misbalance between excitation and inhibition in neuronal circuits resulting in a persistent hyperexcitability state and other migraine-relevant mechanisms such as an increased susceptibility to cortical spreading depression.
Keywords: calyx of Held, FHM-1, in vivo, P/Q calcium channels, synaptic transmission
Introduction
Familial hemiplegic migraine (FHM) is a rare monogenic subtype of migraine with aura that can be considered a model for the common forms of migraine because the headache and aura features, apart from the hemiparesis, are identical (Thomsen et al., 2002). The migraine aura is due to cortical spreading depression (CSD), in which a wave of transient spike activity progresses slowly across the cortex and is followed by long-lasting neuronal suppression (Lauritzen, 1994; Somjen, 2002).
Several missense mutations in the CACNA1A gene, which encodes the pore-forming α1 subunit of voltage-gated CaV2.1 (P/Q-type) Ca2+ channels, cause FHM type 1 (FHM1; Ophoff et al., 1996; de Vries et al., 2009). Many FHM1 mutations have been subjected to molecular and biophysical analyses in heterologous cell systems (Catterall et al., 2008) with “loss-of-function” and “gain-of-function” phenotypes (for review, see Pietrobon, 2013). To study the consequences of mutated CaV2.1 channels in their neuronal environment, knock-in (KI) migraine mouse models were generated carrying either the human FHM1 R192Q (van den Maagdenberg et al., 2004) or the FHM1 S218L (van den Maagdenberg et al., 2010) mutation.
Whereas the R192Q mutation is associated with “pure” FHM, the S218L mutation leads to a particularly severe phenotype that includes cerebellar ataxia, seizures, and sometimes even fatal cerebral edema triggered by trivial head trauma (Kors et al., 2001; Chan et al., 2008; Stam et al., 2009). S218L KI mice exhibit a phenotype very similar to that in S218L patients (van den Maagdenberg et al., 2010; Gao et al., 2012) that can be explained by kinetic alterations of S218L mutant CaV2.1 channels (Tottene et al., 2005; van den Maagdenberg et al., 2010) and the occurrence of subcortical CSD (Eikermann-Haerter et al., 2011).
The principal aim of this work was to understand the physiological consequences of the S218L mutation in CaV2.1 Ca2+ channels with respect to synaptic transmission in vitro and in vivo to identify mechanisms underlying the neurobiological phenotypes of FHM and, at least to some extent, common migraine. For detailed analyses of Ca2+ influx and transmitter release in the CNS, we used the calyx of Held synapse (Forsythe, 1994; Borst et al., 1995), in which both presynaptic CaV2.1 calcium currents (IpCa) and EPSCs can be recorded. In addition, this synapse can be readily identified in vivo (Guinan and Li, 1990) and in vivo juxtacellular recordings allow the quantification of synaptic strength (Lorteije et al., 2009).
Whole-cell patch-clamp recordings of presynaptic terminals from S218L KI mice showed a strong shift of the I–V curve to more negative potentials, leading to: (1) an increase in basal [Ca2+]i, (2) increased levels of spontaneous transmitter release, (3) faster recovery from synaptic depression in the S218L KI mouse calyx, and (4) smaller action potential (AP)-elicited Ca2+ currents, but, surprisingly, (5) transmitter release that was augmented due to an increased probability of release. Therefore, we postulate that the sustained increase in basal Ca2+ induces short- and long-term changes in synaptic strength and cortical excitability, which has relevance for understanding the pathophysiology of hemiplegic and possibly other forms of migraine.
Materials and Methods
Animals and experiments.
Generation of the S218L KI mouse has been described previously (van den Maagdenberg et al., 2010). The S218L mice have been generated using a gene-targeting approach and had a 50% C57BL/6J and 50% 129Ola mixed background. Subsequently, S218L mice were backcrossed with C57BL/6J mice until the genetic background was 87.5% C57BL/6Jico and 12.5% 129Ola. From then on, heterozygous S218L mice were interbred and, from the offspring, homozygous KI and wild-type (WT) of either sex (with the same genetic background) were used for the experiments. All experiments were performed according to the European Communities Council Directive (86/609/EEC). Experiments performed in Rotterdam were approved by the animal ethics committee of the Erasmus MC. Principles of animal care were in accordance with the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET; 2003) rules for experiments performed in Argentina.
Preparation of brainstem slices.
For slice recordings, ∼50 mice between 11 and 15 d old were used in this study. Their brains were removed rapidly after decapitation and placed into an ice-cold low-Ca2+ artificial CSF (aCSF). This solution contained the following (in mm): 125 NaCl, 2.5 KCl, 3 MgSO4, 0.1 CaCl2, 1.25 NaH2PO4, 0.4 ascorbic acid, 3 myo-inositol, 2 pyruvic acid, 25 d-glucose, and 25 NaHCO3. The brainstem was glued on a Peltier-cooled chamber of a vibrating microslicer (Vibratome 3000, The Vibratome Company or Integraslice 7000, SMZ Campden Instruments). Transverse slices containing the medial nucleus of the trapezoid body (MNTB) were cut sequentially and transferred to an incubation chamber containing normal aCSF at 37°C for 30 min. After incubation, slices were allowed to return to room temperature. Slices of 200–300 μm thickness were used for both presynaptic Ca2+ currents recordings and evoked EPSC recordings. The normal aCSF had the same composition as the slicing solution except that the MgCl2 and CaCl2 concentrations were 1 and 2 mm, respectively. The pH was 7.4 when gassed with 95% O2/5% CO2.
Whole-cell patch-clamp recordings.
Slices were transferred to an experimental chamber. During recording, slices were continuously perfused with carbogenated (95% O2 and 5% CO2) aCSF maintained at room temperature (22–25°C). MNTB neurons were visualized using Nomarski contrast on a BX-50WI microscope (40×, 0.8 numerical aperture [NA] water-immersion objective; Olympus) or an Eclipse E600-FN microscope (60×, 1 NA water-immersion objective lens; Nikon). Whole-cell voltage-clamp recordings were made with patch pipettes pulled from thin-walled borosilicate glass (1155150-075-AC; Hilgenberg). Electrodes had resistances of 3.2–4.2 MΩ for presynaptic recordings and 2.9–3.2 MΩ for postsynaptic recordings when filled with internal solution. Patch solutions for presynaptic voltage-clamp recordings contained the following (in mm): 120 Cs-gluconate, 20 CsCl, 10 HEPES, 10 Na2-phosphocreatine, 10 TEA-Cl, 0.5 EGTA, 4 Mg-ATP, and 0.3 Li-GTP. The pH was adjusted to 7.2 with CsOH. Alexa Fluor 488 (40 μm; Invitrogen) was also included in the pipette solution to confirm visually that presynaptic terminals were recorded. Cs-gluconate was replaced by CsCl in the intracellular solution for postsynaptic recordings.
Whole-cell patch-clamp recordings were made using an Axopatch 200B (Molecular Devices) amplifier, a Digidata 1200 A/D converter (Molecular Devices), and pClamp 10.2 software (Molecular Devices). Data were sampled at 50 kHz and filtered at 46 kHz (low-pass Bessel filter). Series resistance values ranged between 6 and 15 MΩ for both presynaptic and postsynaptic recordings and were compensated up to 75%. Whole-cell membrane capacitance (15–25 pF) was obtained from the amplifier capacitance knob values after total compensation of cell and pipette transients generated by a 10 ms voltage step. Leak currents were subtracted online with a P/5 protocol. Calcium currents were recorded using Cs-gluconate-based solution in the presence of extracellular TTX (1 μm) and TEA-Cl (20 mm). To establish the I–V relationship of presynaptic ICa, 20 ms depolarizing pulses ranging from −70 to +20 mV (5 mV steps) were given from a holding potential of −80 mV. The average current amplitude within the first 5 ms after peak current was considered. Half-voltage activation (V1/2) was calculated by fitting the I–V curve by the following equation:
where G is conductance and k is the slope factor. Calcium current density was calculated as current over capacitance for each terminal (average Cp ∼12–13 pF). To obtain ICa activation curves, tail currents after 20 ms depolarizing pulses were normalized to the maximum peak amplitude, plotted against voltage, and fitted by a Boltzmann function. Steady-state inactivation was measured using 1.5 s conditioning step potentials applied to presynaptic terminals, followed by a 5 ms test step to the potential at the peak of the I–V curve. Currents evoked by the voltage steps were normalized, plotted against voltage, and fitted by a Boltzmann function.
APs were measured in whole-cell configuration under current-clamp mode as described by Inchauspe et al. (2010). Patch solutions for current-clamp recordings contained the following (in mm): 110 K-gluconate, 30 KCl, 10 HEPES, 10 Na-phosphocreatine, 0.2 EGTA, 2 MgATP, 0.5 LiGTP, and 1 MgCl2. Only cells that had membrane resting potential between −60 and −75 mV were selected for recording. APs in the calyx of Held were elicited by injecting depolarizing step current pulses of 1–2 nA during 0.25 ms. Averaged calyx AP was ∼110 mV, half-width ∼0.44 ms, rise time (10–90%) ∼0.33 ms, and decay time ∼0.44 ms. In addition, AP in layer 2/3 pyramidal cells (PCs) was recorded under the same experimental conditions. PC APs had a mean rise time of ∼0.53 ms, half-width of ∼1.98 ms, decay time of ∼3.1 ms, and amplitude of ∼90 mV.
mEPSCs were recorded continuously for at least three separate periods of 1 min. mEPSC amplitude and frequency were analyzed using Clampfit 10.3. EPSCs were evoked by stimulating the globular bushy cell axons in the trapezoid body at the midline using a bipolar platinum stimulating electrode and an isolated stimulator (0.1 ms duration and 50–200 μA amplitude). Strychnine (1 μm) was added to the aCSF to block inhibitory glycinergic synaptic responses. In experiments in which high-frequency trains were given to study short-term plasticity (see Figs. 4, 5), the competitive AMPAR antagonist kynurenic acid (1 mm) was added to the aCSF to minimize the impact of postsynaptic receptor desensitization, resulting in an average inhibition of ∼50% of the EPSC amplitude. Slices pretreated with EGTA-AM (200 μm) were incubated for 30 min at room temperature and then a washout for at least 30 min was performed before recording. Data analysis was done by Clampfit 10.3 software. All slice experiments were done at room temperature.
Figure 4.
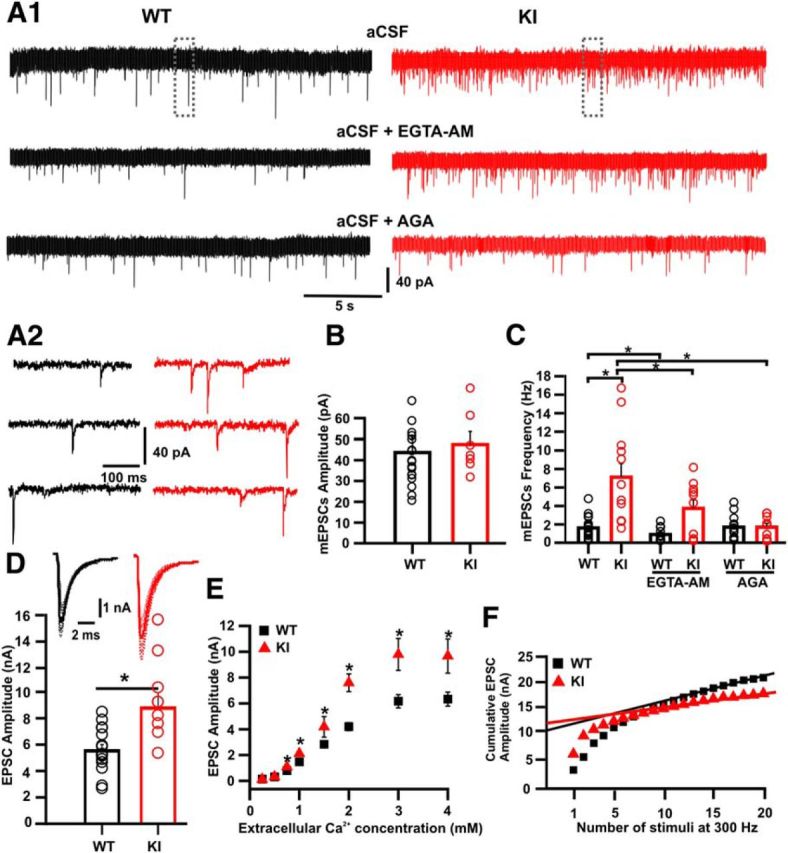
Enhanced synaptic transmission in the S218L KI calyx of Held. A1, Representative traces of mEPSCs recorded in the calyx of Held synapse in the presence of TTX (1 μm) in aCSF (top) after preincubation with EGTA-AM (middle) or in the presence of agatoxin (200 nm; bottom). A2, Boxed areas in A1 shown at higher time resolution. B, The mEPSC amplitude was similar in both genotypes (WT: 47.3 ± 5.8 pA, n = 21 and KI: 49.4 ± 5.1 pA, n = 14); however, the frequency of spontaneous events (C) was significant higher in the KI compared with WT (WT: 1.6 ± 0.3 Hz vs KI: 7.1 ± 1.5 Hz; Student's t test, p = 0.00002). After 30 min of preincubation with EGTA-AM (200 μm), the frequency was reduced in both WT and KI animals (WT: 0.9 ± 0.2 Hz, n = 13; t test, p = 0.02 vs KI: 3.73 ± 0.9 Hz, n = 10; Student's t test; p = 0.038). Spontaneous release was unaffected by ω-agatoxin-IVA (200 nm) in the WT, but was strongly reduced in KI synapses, reaching WT values (WT: 1.7 ± 0.3 Hz, n = 14; t test, p = 0.4 vs KI: 1.7 ± 0.4 Hz, n = 9; t test, p = 0.003). D, Representative traces of EPSCs (top) showing larger amplitudes of evoked synaptic currents in KI (bottom; KI: 8.8 ± 0.5 nA, n = 8 and WT: 5.5 ± 0.5 nA, n = 15; t test, p = 0.002) at an external calcium concentration of 2 mm. E, Average EPSC size was significantly larger in KI principal neurons at extracellular calcium concentrations of 0.75 mm and higher (*p = 0.002, Student's t test). F, Cumulative EPSC amplitudes during a 300 Hz train for a representative KI (red) or WT (black) whole-cell recording. Solid lines indicate the respective line fits of the last eight data points. The intersection of these lines with the ordinate provides an estimate for the cumulative EPSC evoked by the RRP (Schneggenburger et al., 1999). The relative size of the first EPSC provides an estimate of the Pr of vesicles in the RRP, which was 0.32 ± 0.03 and 0.54 ± 0.05 for WT and KI, respectively (Student's t test; p = 0.004).
Figure 5.
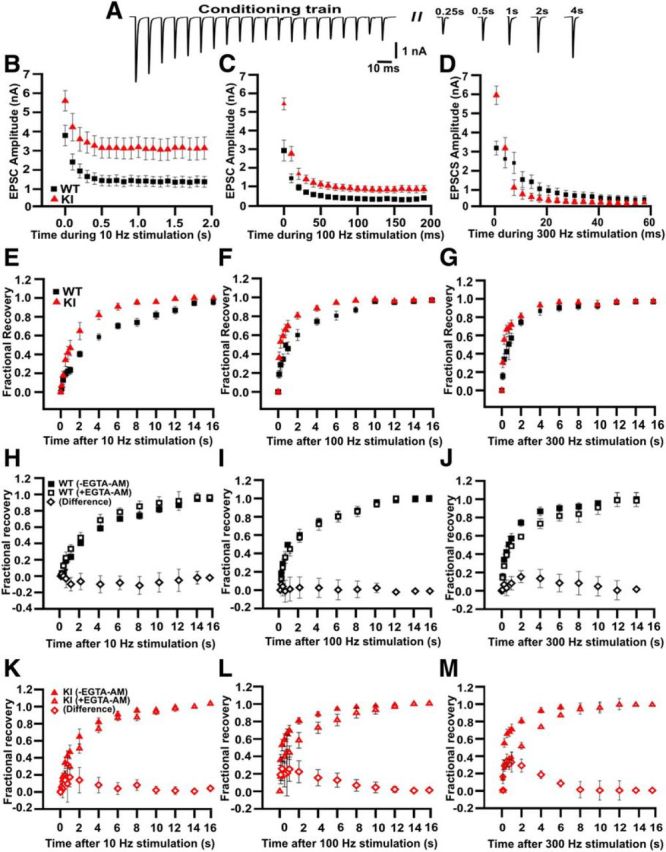
Faster recovery of EPSCs from STD in KI. A, Representative traces showing depression of EPSC amplitudes during 0.2 s stimulation at 100 Hz and recovery at different time delays after the train. B–D, Time course of EPSC depression during a 10 Hz (B), 100 Hz (C), or 300 Hz (D) stimulus train. At 10 Hz, the amplitudes of the EPSCs at the end of the train were 32.2 ± 2.7% (n = 17) and 54.4 ± 3.1% (n = 7) of the first pulse for WT and KI, respectively. At 100 Hz, the magnitude of depression was larger in WT (WT: 10.7 ± 2.2%, n = 14 vs KI: 16.7 ± 2.2%, n = 9; Student's t test, p < 0.05). At 300 Hz, the depression was larger in the KI (WT: 14.4 ± 1.1%, n = 8 vs KI: 5.1 ± 0.1%, n = 5; Student's t test, p = 0.02). E–G, Time course of recovery from synaptic depression was measured by eliciting a single test EPSC at increasing time intervals after the conditioning train. Recovery following 10 Hz trains (E) was fitted with a single exponential decay function with time constants τWT = 4.04 ± 0.26 s (n = 13) and τKI = 2.17 ± 0.7 s (n = 7), t test, p < 0.001). After a 100 Hz train (F), recovery in WT was fitted with a single exponential function (τ = 2.0 ± 0.4 s, n = 1 3), but recovery in KI needed the sum of two exponential functions for an appropriate fit (τKI,f: 94 ± 5 ms [52%], τKI,s: 2.2 ± 0.4 s [48%]). Recovery from STD after 300 Hz stimuli usually followed a double exponential time course, but in this case, no differences were found between the genotypes (τWT,f = 182 ± 45 ms [36%] and τWT,s = 2.8 ± 0.5 s [64%]; τKI,f = 141 ± 17 ms [60%] and τKI,f = 2.77 ± 0.45 s [40%]). H–M, Time course of recovery from synaptic depression before and after EGTA-AM incubation. Filled symbols represent control data (without EGTA-AM) and empty symbols represent preincubated with EGTA-AM data. The subtraction between data in the presence and absence of EGTA-AM are shown as diamonds. Recovery time course after 10 Hz were similar for both genotypes (WT: 3.9 ± 1.2 s; n = 4 and KI: 2.9 ± 0.5 s; n = 4; Student's t test, p = 0.26; after 100 Hz, WT: 2.8 ± 0.9 s; n = 5 and KI: 2.7 ± 0.9 s; n = 4; Student's t test, p = 0.47).
Ca2+ imaging.
Resting presynaptic Ca2+ concentrations were measured using the high-affinity Ca2+ indicator fura-2 (Invitrogen). In these experiments, fura-2 replaced EGTA and K+ replaced Cs+ in the presynaptic solution described in Whole-cell patch-clamp recordings section. To minimize the wash-out of endogenous Ca2+ buffers, fura-2 was loaded during a brief whole-cell recording with a pipette containing 100 μm fura-2 (Müller et al., 2007). The comparison of fluorescence intensity at the isosbestic wavelength (355 nm) after these brief recordings with the intensity reached during continuous whole-cell recordings indicated that a concentration of ∼25 μm was reached in the former.
The Ca2+-imaging system consisted of a monochromator (Polychrome V; Till Photonics) to excite fura-2 at 340 and 380 nm, a CCD camera (iXon+; Andor), and an upright microscope (BX-51WI; Olympus). Calyx terminals were visualized by a 60× objective (0.9 NA; Olympus). Camera control and data acquisition were done by Cell R software (Olympus). The illumination protocol consisted of 15 fluorescence ratio images (F340/F380) with an exposure time of 50 ms per image taken 2–3 min after loading. We used ImageJ software to perform the offline analysis. The whole fluorescent calyx was analyzed and the background fluorescence was subtracted. The ratio was converted to concentration ([Ca2+]i) using a calibration curve (Kochubey and Schneggenburger, 2010). For this propose, we made an in vitro calibration curve in small glass pipettes (rectangle capillaries of 50 μm; Vitrocom) filled with K-gluconate internal solution and fura-2, in which the free Ca2+ concentration was buffered with EGTA. The calibration comprised measurements at seven different free Ca2+ concentrations to determine the three calibration constants: Rmin, Rmax and Keff. The intracellular free Ca2+ concentration was calculated from the measured R (340/380) according to the following equation (Grynkiewicz et al., 1985):
The dissociation constant Keff was 293 nm at 25°C.
Surgical approach for in vivo recordings.
After brief exposure to isoflurane, the young-adult WT and KI mice (postnatal day 34 [P34]–P46; average, 41.2 ± 1.3 d old) were injected intraperitoneally with a ketamine–xylazine mixture (65 and 10 mg/kg, respectively). Under anesthesia, animals were supine positioned and a tracheotomy was performed. The animals were ventilated mechanically (MiniVent 845; Harvard Apparatus) with oxygen, with a frequency of ∼150/min and a stroke volume of 7 μl/g body weight. The right MNTB was accessed via ventral surgery as described previously using the right anterior inferior cerebellar artery and basilar artery as landmarks to locate the MNTB (Rodríguez-Contreras et al., 2008; Lorteije et al., 2009). Both the dura and pia were removed to expose the brain surface before recording. Ringer's solution containing the following (in mm): 135 NaCl, 5.4 KCl, 1 MgCl2, 1.8 CaCl2, 5 HEPES, pH 7.2, was applied to keep the brain surface moist. During the surgery and recording, the rectal body temperature was kept at 37°C using a homeothermic blanket (FHC).
In vivo electrophysiology and auditory stimulation.
In vivo juxtacellular (loose-patch) recordings from principal neurons were made as described previously (Lorteije et al., 2009). Thick-walled borosilicate glass micropipettes with filament were filled with Ringer's solution. The recording pipette penetrated the brain surface with a positive pressure of 300 mbar. The pressure was reduced to ∼30 mbar after passing the brain surface and no pressure was applied when recording started. Data were acquired with a MultiClamp 700B patch-clamp amplifier and pCLAMP 9.2 software (MDS Analytical Technologies). Data were filtered at 10 kHz (eight-pole Bessel filter) and sampled at 50 kHz with a 16-bit A/D converter (1322A; Digi-Data). Closed-field sound stimulation was presented as described previously (Tan and Borst, 2007). A speaker probe was inserted into the left ear canal and was stabilized with silicone elastomer. Frequency response areas were measured as described previously (Tan and Borst, 2007). A two-noise-burst stimulation protocol was designed in MATLAB Version R2008a and generated by Tucker Davis Technologies hardware (system 3, RX6 processor, PA5.1 attenuator, ED1 electrostatic driver, EC1 electrostatic speaker). It consisted of a 200 ms silent period, followed by two 400 ms bursts of wide-band noise (bandwidth 2–40 kHz; 80 dB SPL) that were presented at 6 different intervals (40, 80, 160, 320, 640, and 1280 ms), followed by a silent period for a total sweep duration of 4 s. Each interval was repeated 5 times and the total length of the stimulation protocol was 2 min. Sound intensities were calibrated as described previously (Tan and Borst, 2007). Experiments were performed in a single-walled sound-attenuated chamber (Gretch-Ken Industries).
Morphology of the calyx.
Afferent fibers to the calyx of Held were electroporated with Alexa Fluor 594-labeled dextrans (10,000 MW; Invitrogen) at the midline in vivo in young adult mice (P30–P60) as described previously (Rodríguez-Contreras et al., 2008). One hour later, the animal was perfused and the brainstem was sliced into 40-μm-thick sections. The nuclei of the cells were stained with SYTOX Blue (1:1000; Invitrogen). A laser scanning confocal microscope (LSM 700; Zeiss) equipped with krypton-argon and helium-neon lasers was used to acquire high-resolution z-stack images (1 μm steps; 60×, NA 1.4) of randomly selected calyces of Held with a signal-to-noise ratio >20 using optimized laser power, detector gain, and pinhole diameter settings. Signal-to-noise ratio was defined as the ratio between the difference between the mean intensity of the calyx and the background divided by the SD of the background; it was, on average, 41 ± 4 (range 21–77). The number of swellings per calyx was counted on 3D-rendered (Volocity 4.2; Improvision) images of calyces with adjusted contrast and brightness. To measure surface area and volume of the calyces using the region-of-interest function in Volocity, images of calyx terminals were binary thresholded using the built-in thresholding function of ImageJ 1.46 (isodata algorithm).
Analysis of electrophysiological data.
In vivo juxtacellular data were analyzed using custom procedures in IGOR (WaveMetrics) within the NeuroMatic environment (version 2.00, kindly provided by Jason Rothman, University College–London). The maximum rate of rise of the extracellularly recorded EPSP component of the complex extracellular waveform (eEPSP′) was used as a measure for the strength of synaptic transmission. We found this measure to be more robust than the maximum amplitude of the extracellularly recorded EPSP, which was in some cells hard to delineate from the AP component of the complex extracellular waveform.
Recovery from sound-evoked STD was fitted on a dataset consisting of the last event during the sound and all events after the sound. Fitting with a simple model for short-term plasticity (Varela et al., 1997) was performed as described previously (Crins et al., 2011). In its simplest incarnation, a single depression state parameter decreases at each event with a fraction called the depletion factor (comparable to the release probability of the terminal) and recovers continuously with a single time constant. Synaptic transmission is then equal to the product of the depression state parameter and the transmission strength in the absence of short-term plasticity.
Data presentation and fitting analysis.
Activation curves were obtained from tail current amplitudes recorded at the holding potential after depolarizing pulses, and were fitted to the followoing Boltzmann equation:
Activation time constants (τ-on) were obtained by fitting the time course of activation of presynaptic Ca2+ currents evoked by depolarizing voltage steps, whereas the time constant of deactivation was measured by fitting the decaying phase of tail currents. Both time courses were fitted by a single exponential function. Statistical differences were evaluated using SigmaPlot 10. Average data are expressed and plotted as mean ± SEM. Statistical significance was determined using repeated-measures (RM)-ANOVA and Student's t test.
Drugs and reagents.
Chemicals were purchased from Sigma-Aldrich, Merck, or Tocris Bioscience.
Results
Presynaptic calcium currents in S218L KI mice
Because of the various gain-of-function phenomena in transgenic mouse models of hemiplegic migraine (van den Maagdenberg et al., 2004; Tottene et al., 2009; van den Maagdenberg et al., 2010), we studied the effect of the FHM1 S218L CaV2.1 mutation on the pharmacological and biophysical properties of IpCa currents in voltage-clamp recordings from the calyx of Held.
As expected from previous experiments (Iwasaki and Takahashi, 1998; Inchauspe et al., 2004; Fedchyshyn and Wang, 2005), the P/Q-type (CaV2.1) specific blocker ω-agatoxin-IVA (200 nm) blocked at least 90% of the current in WT mice (n = 3; Fig. 1B). In the KI mice, IpCa was inhibited to the same extent by the toxin (92.3 ± 2.3%; n = 3; Fig. 1B), indicating that there is no upregulation of other calcium channels and that P/Q-type channels are the only functional channels expressed at the calyx of Held in KI mice >11 d old.
Figure 1.
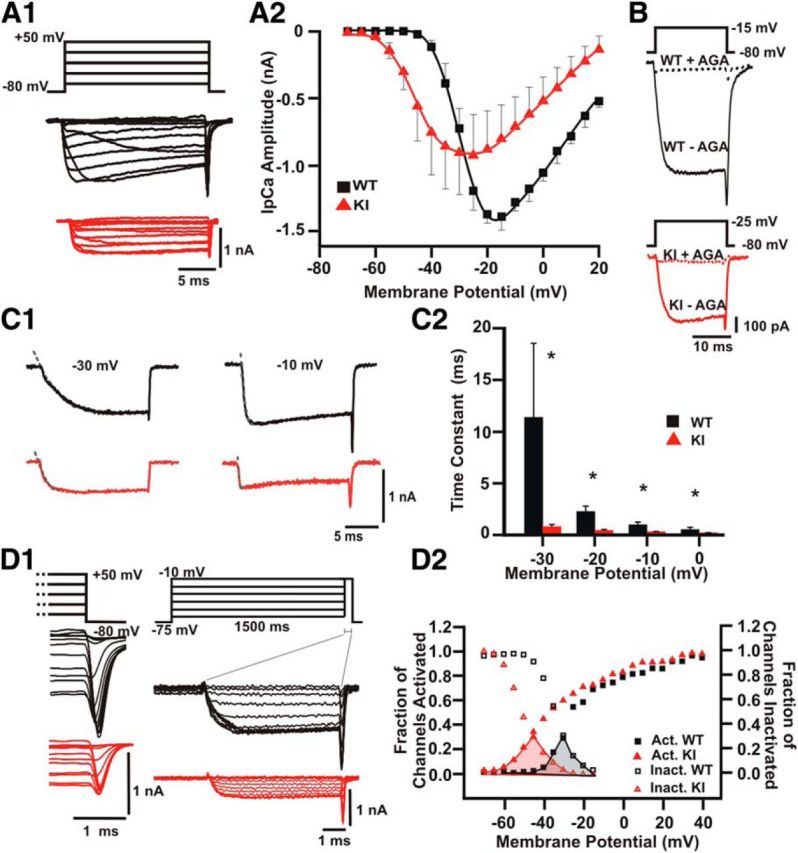
CaV2.1 (P/Q-type) calcium channels activate at more negative membrane potentials in the S218L KI mouse calyx of Held. A1, Left, Representative traces of IpCa (bottom) evoked by 20 ms depolarizing voltage steps (top) from a holding potential of −80 mV to potentials ranging from −70 to +20 mV (in 5 mV steps). A2, I–V for IpCa in the calyx of Held. Symbols are experimental data and solid lines represent cubic spline interpolation. B, Representative traces of IpCa blocked by agatoxin (200 nm) in WT (top) and KI (bottom). The blocking effect was tested at the peak of the I–V curve (i.e., −15 mV for WT and −25 mV for KI). C1, Representative examples of the Ca2+ currents evoked by steps to −30 and −10 mV for WT (black) and KI (red). Broken gray lines represent the fit of the activation time course with a single exponential function. C2, Average activation time constants of IpCa between −30 and 0 mV are smaller in the S218L mouse model. D1, Tail currents elicited after repolarization to −80 mV from different membrane potentials (left). IpCa evoked by a 5 ms voltage step to the peak of the I–V curve after applying 1.5 s conditioning prepulses to voltages from −75 to −10 mV (5 mV steps; right). D2, Normalized activation and inactivation curves for WT (black) and KI (red). The KI “window” (shaded area below the curves) is clearly shifted to the left, suggesting activation of S218L CaV2.1 Ca2+ channels already close to the resting potential. IpCa activated at more negative potentials in S218L (WT: V1/2=-23.1 ± 1.7 mV and KI: −36.7 ± 6.4 mV) with different slope factors (WT: 7.6 ± 1.2 and KI: 12.3 ± 3.2). Half-inactivation voltages were V1/2 = −34.4 ± 1.4 mV for WT (n = 9) and −50.3 ± 3.0 mV for KI (n = 4, Student's t test, p = 0.0005). The slope factor is significantly larger in WT (−8.81 ± 0.21) than in KI (−5.29 ± 0.53) animals (Student's t test, p = 0.002).
We next examined the I–V relationship of IpCa using an internal pipette solution based on Cs-gluconate (Fig. 1A1). Two important features were observed in the KI model. First, the activation of calcium currents was shifted to more negative potentials and currents were smaller (Fig. 1A2). The activation threshold of IpCa was typically at −40 mV for WT and at −60 mV in the KI, with an apparent reversal potential at +40 mV in both genotypes. Second, the peak of the I–V curve shifted to more negative potentials in the KI (WT: −17 ± 4 mV, n = 10; KI: −26 ± 7 mV, n = 5). Both I–V relationships were significantly different (RM-ANOVA, F(2,184) = 8.476; p < 0.001). The V1/2 obtained by fitting the I–V curve with Equation 1 showed a comparable shift toward hyperpolarized potentials (WT V1/2 = −29.9 ± 1.6 mV and KI: −41.1 ± 3.3 mV, p = 0.002). Furthermore, activation curves obtained from the peak amplitudes of tail currents showed a 13.6 mV shift toward hyperpolarized potentials in KI compared with WT mice (WT: V1/2= −23.1 ± 1.7 mV and KI: −36.7 ± 6.4 mV, p = 0.013). Therefore, both I–V and activation curves from S218L KI presynaptic terminals were significantly different compared with WT.
In addition, the KI IpCa activated more rapidly between voltages of −30 and +0 mV (Fig. 1C1,C2). Mean absolute IpCa to peak was 1328 ± 101 pA for WT and 923 ± 204 pA for KI (Student's t test, p = 0.04), resulting in a smaller mean IpCa density in the KI (WT: −104 ± 7 pA/pF, KI: −70 ± 16 pA/pF; Student's t test, p = 0.03; Fig. 1A2). In an independent set of experiments, IpCa was studied using an internal pipette solution based on CsCl. Again, the current density was smaller in KI mice, with the I–V curve showing a similar shift toward hyperpolarized membrane potentials (data not shown). Figure 1D2 displays WT and KI Ca2+ window currents obtained by joining the corresponding activation and inactivation curves (Fig. 1D1). The KI window current was shifted closer to the resting potential, suggesting that a larger population of CaV2.1 Ca2+ channels can be activated close to the resting potential. We conclude that the S218L KI mutation affects several biophysical properties of presynaptic Ca2+ currents, with the shift of the activation curve toward the resting membrane potential as the most conspicuous change.
To investigate the IpCa triggered by physiological stimuli, the presynaptic terminal was depolarized with action potential waveforms (APWs) previously recorded during current-clamp recordings. IpCa amplitude and total Ca2+ charge evoked by a typical calyx APW (with half-width <0.5 ms) were significantly reduced in KI compared with WT mice (WT: 1.6 ± 0.2 nA, 0.73 ± 0.10 pC, n = 9; KI: 0.9 ± 0.2 nA, 0.44 ± 0.11 pC; n = 5; Student's t test; p = 0.04; Fig. 2A). However, no difference in the total Ca2+ charge was observed between WT and KI when APWs with longer duration and lower amplitude were applied (WT: 1.1 ± 0.2 pC; KI: 0.97 ± 0.25 pC; Student's t test; p = 0.3; Fig. 2B). The increase in IpCa charge evoked by a cortical pyramidal cell-like APW was also larger in KI (increase 115.4 ± 18.8%) than in WT (increase 48.4 ± 15.5%) mice (Student's t test; p = 0.007). Therefore, a differential response in terms of Ca2+ current generation was observed depending on the width of the APW. These results suggest that the shape of the APW is relevant to the gain-of-function in S218L KI mice, as reported previously for the R192Q KI model (Inchauspe et al., 2010).
Figure 2.

Reduced Ca2+ entry and facilitation in KI calyx of Held. A, Left, Presynaptic Ca2+ currents evoked by APWs obtained from calyx recordings are smaller in KI (red) than in WT (black). Right, Total Ca2+ entry was significantly smaller in the KI mice. B, As in A, except that APWs were recorded in pyramidal cells. Total Ca2+ entry did not differ significantly between KI and WT. C, Representative IpCa traces (bottom) generated by 100 Hz trains of APWs (top). The first and last IpCa are superimposed (right) for WT (black) and KI (red). D, Normalized current amplitudes during 100 Hz trains of APWs. The IpCa facilitation observed in WT (steady-state increase 20 ± 3%, n = 8) was significantly smaller in the S218L model (steady-state increase 7 ± 2%, n = 5; t test, p = 0.0002). E, As in D, except that the frequency was 300 Hz. Facilitation was also reduced in the KI (steady-state increase 18 ± 4%, n = 8 and 2 ± 4%, n = 5 for WT and KI, respectively; t test, p = 0.009).
The IpCa of the calyx of Held displays Ca2+-dependent facilitation (Borst and Sakmann, 1998; Cuttle et al., 1998; Forsythe et al., 1998). We analyzed IpCa facilitation during 100 Hz (200 ms) or 300 Hz (67 ms) AP trains (Fig. 2C) in S218L KI mice. Facilitation at the end of a 100 Hz AP train (Fig. 2D) was significantly smaller in KI mice (20.0 ± 2.5%, n = 8 for WT vs 7 ± 2%, n = 5 for KI; Student's t test; p = 0.002). The same behavior was present at 300 Hz (WT: 18 ± 4%, n = 8 and KI: 2.0 ± 0.4%, n = 5; Student's t test; p = 0.009; Fig. 2E).
Increased basal calcium concentration in the calyx of Held of S218L KI mice
The leftward shift in the CaV2.1 channel activation was large enough to result in a larger number of calcium channels already open at the resting membrane potential, possibly leading to an increased basal Ca2+ concentration. To test this, we preloaded calyces of Held during a brief whole-cell recording (Müller et al., 2007) with the high-affinity calcium indicator fura-2 (100 μm in the pipette; Fig. 3A). On average, basal [Ca2+]i was almost three times higher in in calyces from KI than from WT mice (WT: 121 ± 11 nm, n = 6 and KI: 295 ± 87 nm; n = 8; t test; p = 0.03; Fig. 3B), which is consistent with the idea that the leftward shift in Ca2+ channel activation increased basal Ca2+ influx at resting membrane potentials.
Figure 3.

Increased basal Ca2+ concentration in the S218L KI calyx of Held. A. Fluorescence images at an excitation wavelength of 340 nm (left) and 380 nm (right) of calyces of Held that were preloaded with fura-2 during a brief whole-cell recording from KI (top) or WT (bottom) animals. B, Ca2+ concentration at resting membrane potential was on average >2-fold higher in KI than in WT (WT: 121 ± 11 nm and KI: 295 ± 87 nm; t test, p = 0.03).
Increased miniature EPSCs at the S218L KI calyx of Held
We next tested the consequence of the increased basal calcium concentration on spontaneous transmitter release. mEPSCs were recorded in the presence of TTX (1 μm; Fig. 4A1-2). Whereas no difference was observed in their mean amplitudes (WT: −47.3 ± 5.8 pA; n = 21 and KI: −49.4 ± 5.1 pA; n = 14 cells; Fig. 4B), the frequency of mEPSCs was drastically increased in KI (WT: 1.6 ± 0.3 Hz and KI: 7.1 ± 2.1 Hz; Student's t test; p < 0.0001; Fig. 4C). To test the contribution of the increased basal Ca2+ concentration to this difference, slices were preincubated with the membrane-permeable, high-affinity calcium buffer EGTA-AM (200 μm) for 30 min. The mean frequency of mEPSCs was significantly lower in both WT and KI synapses of treated slices than of untreated slices (WT: 0.89 ± 0.2 Hz, n = 13; Student's t test, p = 0.02; KI: 3.73 ± 0.9 Hz, n = 10; Student's t test, p = 0.04) indicating that spontaneous release depends on the resting intracellular Ca2+ concentration of the calyx of Held (Awatramani et al., 2005; Habets and Borst, 2005; Korogod et al., 2005; Lou et al., 2005). To further investigate the contribution of Ca2+ influx through mutated CaV2.1 channels on the rate of spontaneous release, mEPSC frequency was measured in the presence of the CaV2.1 (P/Q-type) specific channel blocker ω-agatoxin-IVA (200 nm). Spontaneous release was unaffected in WT, but ω-agatoxin-IVA strongly reduced spontaneous rates at KI synapses to the level of WT (WT: 1.7 ± 0.3 Hz, n = 14; Student's t test, p = 0.4 vs KI: 1.7 ± 0.4 Hz, n = 9; Student's t test, p = 0.003), indicating that the increased mEPSC frequency in the KI depends on influx via mutated CaV2.1 channels at the resting potential.
Synaptic transmission strength is increased at the S218L KI calyx of Held
To test the functional consequence of the changes in the presynaptic calcium currents on evoked release, we recorded EPSCs in principal neurons of the MNTB that were evoked by afferent stimulation using a bipolar electrode placed at the midline. Both WT and KI mice showed large, unitary EPSCs, which were independent of stimulus intensity above threshold (Fig. 4D). Contrary to expectations from the IpCa measurements described in the first paragraph of the Results, the mean EPSC amplitude was larger in KI (8.7 ± 1.0 nA, n = 8) than in WT mice (5.5 ± 0.5 nA, n = 15; Student's t test; p = 0.002). We considered the possibility that other types of Ca2+ channels may be compensating in the KI mouse (Urbano et al., 2003). However, consistent with its effect on presynaptic Ca2+ currents (Fig. 1A2), ω-agatoxin-IVA abolished evoked quantal release both in WT and KI calyx of Held (data not shown), indicating absence of evidence for the upregulation of other calcium channels, which is consistent with earlier analyses in cerebellar granule cells of S218L mice (van den Maagdenberg et al., 2010) and R192Q mice (van den Maagdenberg et al., 2004).
Significantly increased transmitter release was observed in the KI at extracellular calcium concentrations ranging from 0.75 to 4 mm (Fig. 4E). For any presynaptic terminal, the number of vesicles released is given by the product of the number of vesicles available for release (readily releasable pool, RRP) and the probability of release (Pr) of these vesicles. The latter can be evaluated by estimating the fraction of the RRP released by a single AP (Schneggenburger et al., 1999). We measured the cumulative sum of EPSC amplitudes during a train of 20 stimuli at 300 Hz in WT and S218L KI mice. The last eight data points were fitted by linear regression and back-extrapolated to time zero (Fig. 4F). The zero time intersect gives an estimate of the size of the readily releasable pool of synaptic vesicles (N) multiplied by the mean quantal amplitude (q). The Pr can be estimated by dividing the mean amplitude of the first EPSC (for each cell) in the train by the Nq value. The Nq values obtained for WT and KI were 10.4 ± 2.5 nA (n = 8) and 11.20 ± 1.6 nA (n = 5) respectively. This resulted in a Pr of 0.32 ± 0.03 for WT and 0.54 ± 0.05 for KI (Student's t test; p = 0.004). We conclude that, in agreement with the increased EPSC amplitudes and mEPSC frequencies, Pr was larger in the KI.
To study the frequency dependence of synaptic transmission, evoked release was triggered by trains of low-frequency (10 Hz) or high-frequency (100 and 300 Hz) stimulation. The amplitude of the 10 Hz evoked EPSCs after 2 s stimulation depressed to 32 ± 3% (n = 17) and 55 ± 3% (n = 7) of the first EPSC for WT and KI, respectively (Student's t test, p = 0.0007). Decay time constants were τWT = 147 ± 22 ms and τKI = 164 ± 42 ms (Student's t test, p = 0.3; Fig. 5B). During 0.2 s stimulation at 100 Hz, EPSCs of WT depressed more than KI (10.7 ± 2.2%, n = 14 for WT and 16.7 ± 2.2%, n = 9 for KI; Student's t test, p = 0.001; Fig. 5C), with similar decay time constants (τWT = 11.5 ± 1.5 ms and τKI = 12.3 ± 1.6 ms, Student's t test, p = 0.35). Interestingly, this behavior was inverted at 300 Hz stimulation with larger depression in KI than WT (14.4 ± 0.1%, n = 8 for WT and 5.1 ± 0.1%, n = 5 for KI; Student's t test, p = 0.02). The rate of decay was faster in KI than in WT, with decay time constants τKI = 4.25 ± 0.65 ms and τWT = 13.2 ± 1.8 ms, respectively (Student's t test, p = 0.009; Fig. 5D).
The efficacy of synaptic transmission during repetitive stimulation is also determined by the rate of recovery from synaptic depression, which depends on the refilling rate of the RRP. The time course of recovery from synaptic depression was studied by eliciting a single test EPSC at intervals following a conditioning train. The fraction of recovery was calculated as follows:
where I1 and Iss are the amplitudes of the first and last EPSCs in the train, respectively, and Itest is the amplitude of the test EPSC. The fractional recovery could be fitted by a single exponential for the WT and KI mice after either a 10 or 100 Hz conditioning train. Recovery was faster in KI synapses after a 10 Hz train, with a time constant of τKI = 2.2 ± 0.7 s (n = 7) compared with τWT = 4.0 ± 0.3 s in WT (n = 13, p = 0.01; Fig. 5E). After 100 Hz stimulation, the recovery time constant was much faster in the KI mice, with the best fit being a double-exponential function (WT: 2.0 ± 0.4 s; KI: 94 ± 5 ms [52%], and 2.2 ± 0.4 s [48%]; Fig. 5F). Recovery from STD after 300 Hz trains also followed a double exponential time course (Fig. 5G), but in this case, no differences were found between both genotypes (WT: 182 ± 45 ms [36%] and 2.8 ± 0.5 s [64%]; KI: 141 ± 17 ms [60%] and 2.77 ± 0.45 s [40%]). The lack of a difference in the recovery at these high frequencies is consistent with the idea that, at 300 Hz, the difference in the basal Ca2+ concentration and possible Ca2+ influx during recovery between WT and KI made a relatively small impact compared with the large buildup of Ca2+ during such a high-frequency train.
The influence of intracellular Ca2+ on the speed of the recovery from STD was investigated by loading the presynaptic terminal with EGTA by incubating the slices with EGTA-AM (200 μm; Wang and Kaczmarek, 1998). This treatment had no effect on recovery from STD after stimulation at 10 or 100 Hz in the WT synapse. However, it did slow the recovery after 300 Hz stimulation, as reported previously (Inchauspe et al., 2007; Inchauspe et al., 2012). In contrast, EGTA-AM treatment of the KI synapses reduced the speed of recovery from STD (Fig. 5H–M) at three different frequencies, suggesting a link between the fast recovery of STD observed in the KI synapse and the increased basal Ca2+ concentration in the KI presynaptic terminal.
S218L KI calyx of Held has normal morphology in KI
The adult calyx of Held morphology has a complex structure characterized by numerous fingers or stalks and swellings. We compared the calyceal morphology in KI and WT animals, because morphological alterations occur in the cerebellum of the S218L mouse (van den Maagdenberg et al., 2010) and short-term synaptic depression is correlated with gross morphology of the calyx terminal (Grande and Wang, 2011). Afferent fibers were fluorescently labeled in vivo by electroporation in P30–P60 animals, followed by confocal imaging of fixed brainstem slices (Fig. 6). Morphology of KI (n = 11 from 3 animals) and WT (n = 11 from 3 animals) calyces was similar. All calyces had many swellings originating from thin necks of third order branches. A previous morphological study classified the calyces in three groups based on the number of swellings; terminals in group I had <6 swellings, group II had 6–14, and group III had >15 swellings. In WT calyces, 3 of 11 calyces belonged to group II and the remainder to group III. In the KI, 4 of 11 calyces belonged to group II and the remainder to group III. Terminals with <6 swellings were not observed, which may be related to the younger age studied in Grande and Wang (2011), which was focused on P16–P19 mice. Surface area (555 ± 48 μm2 in KI vs 613 ± 45 μm2 in WT; p = 0.39) and volume of calyx terminals (1513 ± 87 μm3 in KI vs 1571 ± 77 μm3 in WT; p = 0.62) were also similar between S218L KI and WT calyces. We therefore conclude that it is unlikely that differences in the gross morphology were responsible for the functional differences observed between WT and KI terminals.
Figure 6.
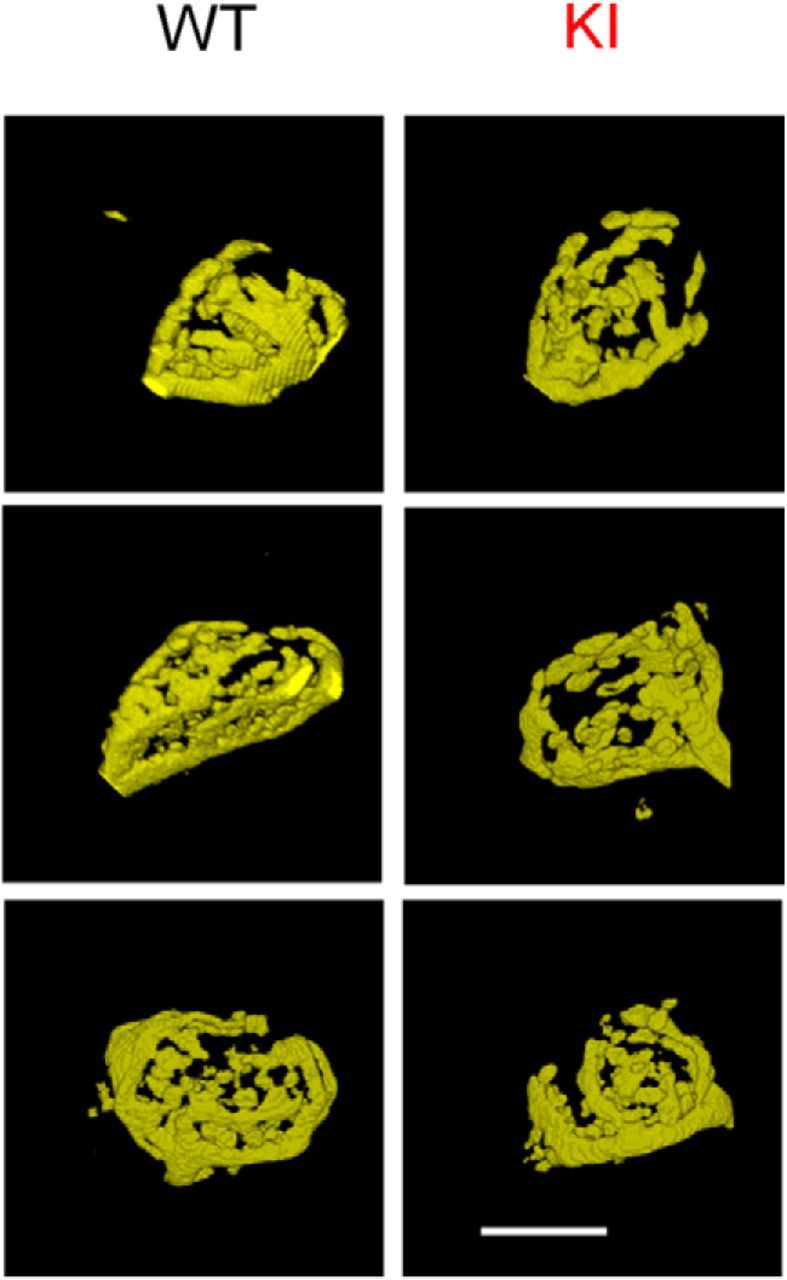
Comparison of morphology of calyx of Held from WT and S218L KI. Images of 3D-rendered representative calyces from WT (left) and S218L KI animals (right). Scale bar, 10 μm. Note that the calyx of Held has normal morphology in KI animals, suggesting that morphological differences are not responsible for the observed functional differences.
S218L KI shows more short-term depression in vivo
To test the consequence of the observed differences in slice recordings in the intact brain, we made juxtacellular recordings from the MNTB of anesthetized S218L KI and littermate WT animals. Recordings from the calyx of Held synapse are readily identified by their extracellular waveform (Guinan and Li, 1990; Lorteije et al., 2009), which had similar shape in KI and WT cells (Fig. 7A,E). Spontaneous firing in the WT animal averaged 26 ± 9 Hz (mean ± SEM; n = 11; range 0.8–102 Hz). Frequency response areas were similar in KI and WT cells (data not shown). Presentation of a 400 ms, 80 dB noise burst elicited a clear firing increase (Fig. 7B,F). During auditory stimulation, it reached a maximum frequency of 312 ± 21 Hz within the first 10 ms and decayed to 153 ± 16 Hz during the last 50 ms of the tone (Fig. 7C). Spontaneous frequency of the KI animal averaged 30 ± 7 Hz (n = 18; range 1.7–97 Hz). In S218L KI animals, the maximum firing frequency during the first 10 ms of auditory stimulation was 326 ± 18 Hz; the steady-state frequency during the last 50 ms was 175 ± 12 Hz (n = 18; Fig. 7G). Spontaneous, maximum evoked and steady-state evoked frequencies did not differ statistically between WT and KI animals (p > 0.25 in each case).
Figure 7.
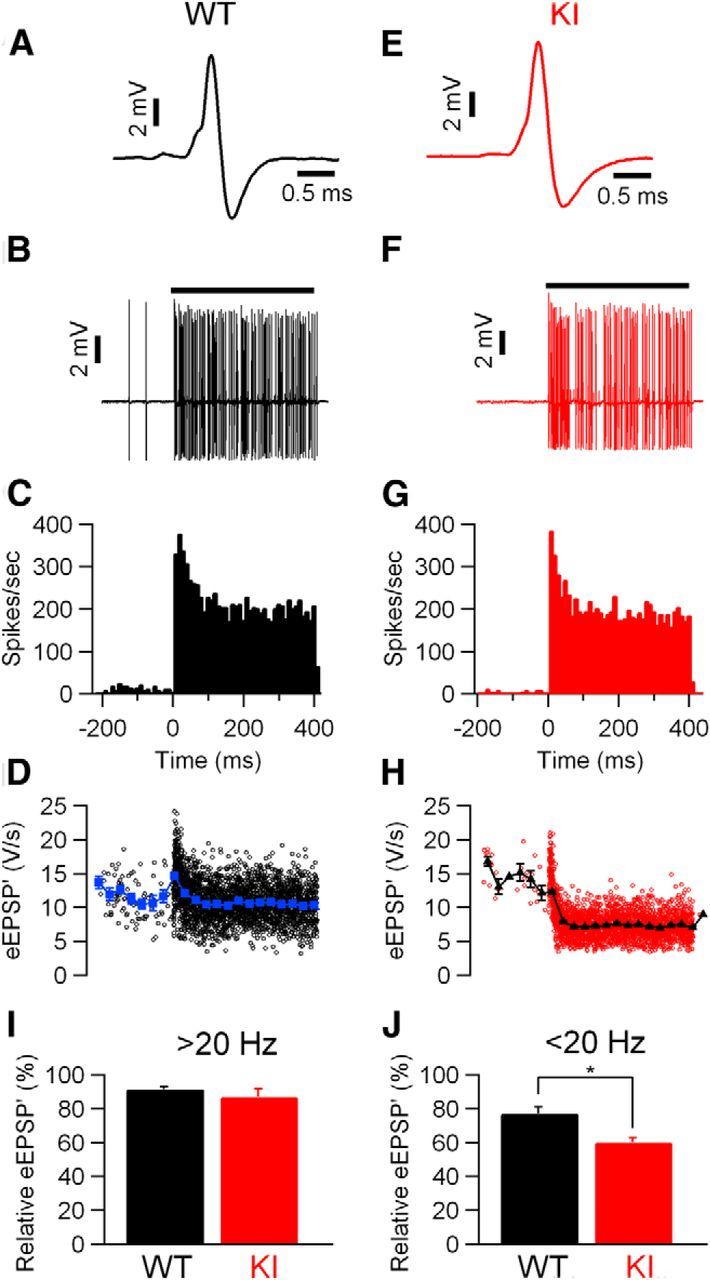
Larger sound-evoked STD in S218L KI mice. A, Complex extracellular waveform in juxtacellular recording from a principal neuron in MNTB of a WT animal. B, Increase in frequency during a 400 ms, 80 dB noise burst. Spontaneous frequency was 19 Hz. C, Peristimulus time histogram showing primary-like response to sound. D, Amplitudes of eEPSP′. Closed triangles indicate time-binned averages. Bin size 30 ms. A–D are from the same WT recording. E–H, As in A–D, respectively, except the recording was from a unit in an S218L KI animal; spontaneous frequency was 7.9 Hz. I, Comparison of relative eEPSP′ amplitudes during the last 50 ms of auditory stimulation of cells with a spontaneous frequency >20 Hz in both WT (black) and KI (red) animals. J, Comparison of relative eEPSP′ amplitudes of cells with a spontaneous frequency <20 Hz in both WT (black) and KI (red) animals. The amplitudes differed significantly (*p = 0.003).
We used the maximum of the first derivative of the extracellular recorded EPSP (eEPSP′) of the characteristic complex waveforms (Fig. 7A,E) from the principal neurons as a measure for the strength of synaptic transmission, as described in the Materials and Methods. At sound onset, the amplitudes often showed a transient increase in both KI (14 of 18 cells) and WT (8 of 11 cells) animals. The ratio between the average amplitudes of the second sound-evoked eEPSP′ and the eEPSP′ before sound stimulation, which we used as an estimate for synaptic facilitation, was similar in KI and WT (1.10 ± 0.043, n = 18 vs 1.02 ± 0.045, n = 10, respectively; p = 0.20). The average interval between the second and the first sound-evoked eEPSP′ were similar as well between KI and WT (3.6 ± 0.3 ms vs 4.6 ± 0.7 ms, respectively; p = 0.091). Later during the sound presentation, the average eEPSP′ in both WT and KI animals often decreased to a lower level, indicating that the high firing frequencies induced STD (Fig. 7D,H). The amount of STD depended strongly on spontaneous firing frequency in both KI and WT animals. Both KI and WT cells with a spontaneous frequency >20 Hz showed little STD (87 ± 5% of control, n = 8 and 92 ± 1%, n = 3, respectively; p = 0.6; Fig. 7I), presumably because cells with a high spontaneous firing frequency are tonically depressed by the spontaneous activity (Hermann et al., 2007; Hermann et al., 2009; Lorteije et al., 2009; Wang et al., 2013). In contrast, cells with spontaneous frequency <20 Hz, on average, showed more STD. In these cells, STD was much more pronounced in KI cells than in WT cells (60 ± 3%; n = 10 vs 77 ± 4% of control; n = 8; p = 0.003; Fig. 7J).
Faster depression and faster recovery from depression in S218L KI in vivo
To further investigate the difference in STD between KI and WT mice, we estimated the time course of STD during sound stimulation. To quantify how many events it took for the synapse to reach steady-state depression, the relation between the eEPSP′ and its event number was plotted (Fig. 8A,B). The decay of the amplitudes could generally be well described by a single-exponential function, both in the presence (Fig. 8A) and absence (Fig. 8B) of synaptic facilitation. Steady state was reached more rapidly in KI than in WT (τKI: 3.6 ± 0.2 events, n = 15 vs τWT: 6.8 ± 1.4 events, n = 8; p = 0.05; Fig. 7E). Recovery from depression in cells showing >15% depression was quantified by fitting the eEPSP′ after the 400 ms noise stimulation with a single exponential function (Fig. 8C, D). Recovery varied between cells, but was overall faster in KI than in WT cells (τKI: 66 ± 16 ms, n = 8 vs τWT: 286 ± 112 ms, n = 6; p = 0.04; Fig. 8F).
Figure 8.
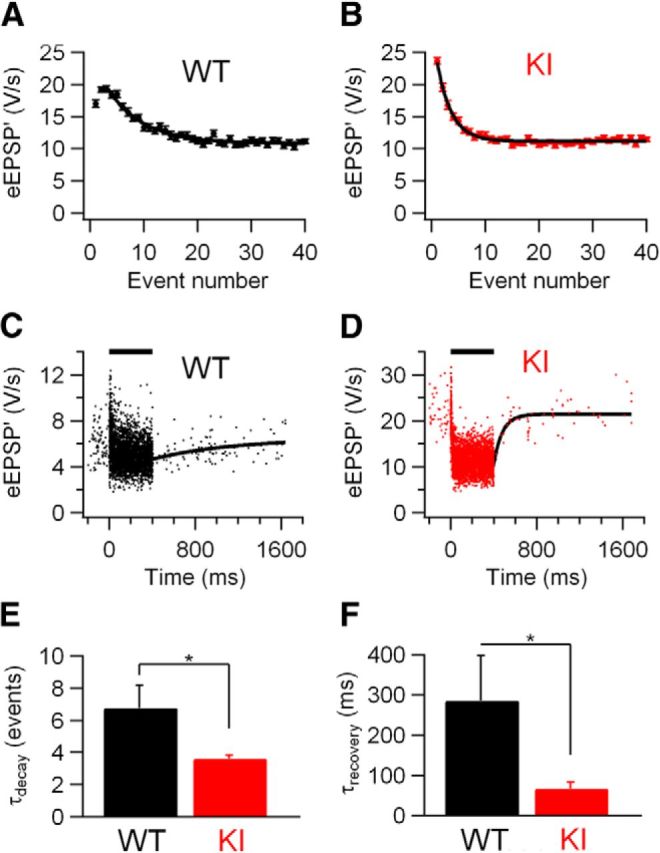
Faster kinetics of STD in S218L KI mice. A, Amplitude of eEPSP′ against sound-evoked event number from recording of WT cell. Error bars indicate SEM. Solid line is fit with single exponential function with τ = 6.7 events. B, Sound-evoked eEPSP′ from KI recording. Solid line is fit with single exponential function with τ = 2.7 events. C, Sound-evoked eEPSP′ in WT animal. Solid line is fit of recovery from sound-evoked STD with single exponential function with time constant τ = 763 ms. D, Sound-evoked eEPSP′ in KI animal. Solid line is recovery fit with τ = 81 ms. E, Comparison of results of fit of depression time course as shown in A and B for WT and KI. F, Comparison of recovery time constants as shown in C and D for WT and KI. A and C are from the same WT recording; B and D are from the same unit from an S218L KI animal.
A potential limitation of the in vivo situation is that, in contrast to slice studies discussed in the first part of the Results section each unit recorded in vivo has different firing frequencies and firing patterns, which makes it harder to exclude that the observed differences are not the consequence of subtle differences in firing. We therefore fitted the amplitudes with a simple model for short-term plasticity (Varela et al., 1997), as described in the Materials and Methods. To simplify the comparisons, we neglected any facilitation and used only a single depletion time constant. Despite the simplicity of the model, amplitudes were reasonably well described by the model fits, in agreement with an earlier study in slices (Hermann et al., 2009) and as indicated by the good match of predicted and measured steady-state depression (73 ± 4% vs 72 ± 4%). In cells with spontaneous frequency <20 Hz, the fitted depletion factor was larger in KI (0.057 ± 0.006; n = 8) than in WT (0.012 ± 0.003; n = 8) animals (p = 0.00001). The predicted time constant of recovery from depression was significantly faster in KI (91 ± 11 ms; n = 8) than in WT (459 ± 121 ms; n = 8) animals (p = 0.019), which is in agreement with the direct fits of the recovery after the sound bursts. The model explained a much larger part of the variance in the eEPSP′ amplitudes in the KI than in the WT (47.3 ± 3.7% vs 14.8 ± 4.5%, respectively) animals (p < 0.0001).
Discussion
Our results show how fundamental features of neuronal communication are influenced by a human FHM1-causing CaV2.1 Ca2+ channel mutation, S218L. At the calyx of Held, the negative shift in the current activation associated with the S218L FHM1 mutation resulted in increased activation of the current, already at the resting membrane potential, leading to a higher presynaptic Ca2+ concentration and increased Pr both in slice recordings and in vivo. Remarkably, this gain-of-function phenotype was observed despite a reduction of AP-induced presynaptic Ca2+ influx.
A hyperpolarizing shift is a common feature of FHM1 calcium channel mutations (van den Maagdenberg et al., 2004, 2010; Tottene et al., 2005, 2009; Gao et al., 2012). The S218L mutation caused a hyperpolarizing shift of ∼13 mV, which is considerably larger than the 6.5 mV reported in the disease-related R192Q in the calyx (Inchauspe et al., 2010). The larger shift of the S218L is in agreement with studies of the cloned channel expressed in a cell line (Tottene et al., 2005) and of this KI in cerebellar Purkinje or granule cells (Tottene et al., 2005; Gao et al., 2012). The S218L indeed has much more severe consequences in mice and patients than the R192Q mutation, including a neurological phenotype of cerebellar ataxia, fatal seizure, and a higher susceptibility of CSD (Kors et al., 2001; Stam et al., 2009; van den Maagdenberg et al., 2010; Eikermann-Haerter et al., 2011).
The large, hyperpolarizing shift observed with the S218L was accompanied by several other biophysical changes. Calcium-dependent facilitation of the IpCa at the calyx of Held is characterized by a small hyperpolarizing shift in their activation (Borst and Sakmann, 1998; Cuttle et al., 1998). Our results indicate that this shift was occluded by the much larger shift induced by the S218L mutation. The S218L may render Cav2.1 channels in a basally facilitated state, in agreement with experiments on cloned channels (Adams et al., 2010). In addition, because the sensor that is responsible for calcium-dependent facilitation of calcium channels has relatively high affinity for Ca2+, the increased resting Ca2+ level may also have contributed to bringing the mutated Cav2.1 channels in a facilitated state. We noticed a hyperpolarizing shift in the inactivation as well, which was similar in size as for the activation. The effects of FHM1 mutations on Ca2+ channel inactivation are notoriously variable (Pietrobon, 2012). The sensor that mediates calcium-dependent inactivation also has high affinity for Ca2+ (Lin et al., 2012), suggesting that increased calcium-dependent inactivation caused by the hyperpolarizing shift in the activation may have contributed to the hyperpolarizing shift in the inactivation. Because of the parallel shift in activation and inactivation, the so-called window-current, the steady-state current that is available at different voltages and also shifted to hyperpolarizing voltages. Several findings indicated that activation of the mutated Ca2+ channels already around the resting membrane potential plays a central role in the gain-of-function phenotype observed at the calyx of Held synapse and other synapses. There was an increased resting Ca2+ concentration, which is consistent with the reported efficacy of KCa activators in restoring cerebellar functioning of S218L KI mice (Gao et al., 2012). We observed a clearly increased mEPSC frequency, in agreement with results obtained at the neuromuscular junction end plate (Kaja et al., 2010; van den Maagdenberg et al., 2010). This increased mEPSC frequency was reduced by incubation in EGTA-AM and the difference in mEPSC frequency between WT and KI disappeared in the presence of the CaV2.1 Ca2+ channel blocker ω-agatoxin-IVA, indicating that an increased presynaptic resting Ca2+ concentration caused by increases in the conductance of the mutated CaV2.1 Ca2+ channels at rest was responsible for this increase.
The identity of the calcium sensor that is responsible for increasing spontaneous vesicle release when the presynaptic Ca2+ concentration is modestly increased, is still debated. Synaptotagmin-1/2 may contribute to this release (Xu et al., 2009), but another Ca2+-binding protein, which is not synaptotagmin-7, protein kinase C, or Doc2A/B, most likely contributes as well (Sun et al., 2007; Fioravante et al., 2011; Pang et al., 2011; Bacaj et al., 2013).
The higher resting Ca2+ concentration was presumably also responsible for the increase in the evoked EPSCs. No clear changes in the size of the RRP were observed in the KI, indicating that the increased EPSC size was caused by an increase in the Pr of vesicles in the RRP. It has been well established that, at the calyx of Held synapse, small increases in resting Ca2+ concentration can already evoke large changes in transmitter release; increases of <100 nm can already lead to a doubling of the size of the afferently evoked EPSC (Awatramani et al., 2005; Habets and Borst, 2005; Korogod et al., 2005; Lou et al., 2005). At the same time, the IpCa density was reduced, presumably as a result of a homeostatic mechanism to reduce the impact of the S218L mutation, which could include the strong leftward shift in the voltage dependence of inactivation, leading to a larger fraction of inactivated channels already at the holding potential of −80 mV or to calcium-dependent inactivation due to the increased basal Ca2+ concentration (Forsythe et al., 1998; Lin et al., 2012). IpCa total charge evoked by a single calyx of Held APW was ∼60% smaller in the S218L KI than in the WT. However, when IpCa was elicited by a longer APW (i.e., such as that from pyramidal cortical neurons), calcium currents in both genotypes were similar. Therefore, the APW could be partially involved in the gain-of-function in S218L, as described for the FHM1 R192Q mouse model (Inchauspe et al., 2010).
Apparently, the effect of the increase in resting Ca2+ concentration outweighed the reduction in AP-induced Ca2+ influx. PKC may be involved in the increase in evoked release resulting from an increased basal Ca2+ concentration (Bouhours et al., 2011; Chu et al., 2012). The increase of EPSCs is also similar to the situation during posttetanic potentiation (Habets and Borst, 2005; Korogod et al., 2005). Activation of PKC and myosin light chain kinase have been implicated in the increased release during PTP (Korogod et al., 2007; Lee et al., 2008; Fioravante et al., 2011; Pang et al., 2011). However, to what extent the synaptic phenotype observed here overlaps with what is observed in normal calyces during PTP remains to be established.
During trains, complex effects were observed on STD. During 300 Hz trains, there was increased depression, presumably because of the higher Pr, as reported at the neuromuscular junction (Kaja et al., 2010). The recovery from STD after trains of stimuli was clearly faster in the S218L KI model, as was also observed for the R192Q KI model (Inchauspe et al., 2012), whereas it is slower in Cacna1a knock-out mice (Inchauspe et al., 2007; Giugovaz-Tropper et al., 2011). The speedup of recovery was presumably responsible for the decrease in the STD during 10 or 100 Hz trains. Under normal conditions, it has been well established that there is a fast component to the recovery when stimuli are large enough to deplete almost the entire RRP (Wang and Kaczmarek, 1998). The exact mechanism underlying this fast recovery component is still debated (for review, see Neher and Sakaba, 2008), but a Ca2+-calmodulin-dependent effect on vesicle recruitment (Sakaba and Neher, 2001), which is mediated by Munc-13 (Lipstein et al., 2013), is important. It is possible that the Ca2+-calmodulin- and Munc-13-dependent increase in vesicle recruitment, which normally needs near-maximal stimuli (Lee et al., 2012; Lipstein et al., 2013), is now operating already at more modest frequencies.
Both the increased STD and the faster recovery were also observed during in vivo recordings in young adult animals, illustrating the impact of the S218L mutation on tone responses. This observation was somewhat remarkable, because our estimates for the Pr of vesicles from the RRP in slices obtained with the back extrapolation method were much higher than the analogous “depletion factor” obtained in vivo from the fits with the depression model. Several factors are responsible for this discrepancy in these Pr estimates, including the lower Ca2+ concentration in vivo (Lorteije et al., 2009), tonic depression as a result of spontaneous activity (Hermann et al., 2007, Hermann et al., 2009; Lorteije et al., 2009; Wang et al., 2013), and the use of the back-extrapolation method for estimating the RRP and Pr in slices, which tends to provide a relatively high estimate for Pr (Wang et al., 2013). However, because the evoked frequencies were similar in WT and KI animals, these considerations do not affect our conclusions with regard to the differences in synaptic transmission between the two.
We speculate that the sustained increase in the intracellular Ca2+ concentration may contribute to the permanent symptoms in S218L patients that are not observed in “pure” FHM1, such as ataxia and/or nystagmus with cerebellar atrophy (Pietrobon, 2013). The gain-of-function of transmitter release may disrupt the finely tuned balance between excitation and inhibition in neuronal circuits (Shu et al., 2003), resulting in a persistent state of hyperexcitability and thereby an increased susceptibility for CSD that is believed to initiate attacks of migraine with aura.
Footnotes
The authors declare no competing financial interests.
This work was supported by the Wellcome Trust no. 084636 (UK), UBACYT X-223, PICT BID 1728 OC.AR.PICT 2005 N 32113, FONCYT (ANPCyT) PICT BID 1728 OC.AR.PICT 2006 N 199 (Argentina; to O. D. U.) and by the Dutch Fund for Economic Structure Reinforcement (FES, 0908 ‘NeuroBasic PharmaPhenomics project’; J.G.G.B.). We thank M.E. Martin for excellent technical assistance and animal breeding, Juan D. Goutman for helpful assistance with Ca2+ imaging experiments, Francisco J. Urbano for helpful comments on this manuscript, Ype Elgersma and Steven Kushner for the use of their confocal microscope, and Freek Hoebeek for advice on breeding.
References
- Adams PJ, Rungta RL, Garcia E, van den Maagdenberg AM, MacVicar BA, Snutch TP. Contribution of calcium-dependent facilitation to synaptic plasticity revealed by migraine mutations in the P/Q-type calcium channel. Proc Natl Acad Sci U S A. 2010;107:18694–18699. doi: 10.1073/pnas.1009500107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awatramani GB, Price GD, Trussell LO. Modulation of transmitter release by presynaptic resting potential and background calcium levels. Neuron. 2005;48:109–121. doi: 10.1016/j.neuron.2005.08.038. [DOI] [PubMed] [Google Scholar]
- Bacaj T, Wu D, Yang X, Morishita W, Zhou P, Xu W, Malenka RC, Südhof TC. Synaptotagmin-1 and synaptotagmin-7 trigger synchronous and asynchronous phases of neurotransmitter release. Neuron. 2013;80:947–959. doi: 10.1016/j.neuron.2013.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JGG, Sakmann B. Facilitation of presynaptic calcium currents in the rat brainstem. J Physiol. 1998;513:149–155. doi: 10.1111/j.1469-7793.1998.149by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst JGG, Helmchen F, Sakmann B. Pre- and postsynaptic whole-cell recordings in the medial nucleus of the trapezoid body of the rat. J Physiol. 1995;489:825–840. doi: 10.1113/jphysiol.1995.sp021095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhours B, Trigo FF, Marty A. Somatic depolarization enhances GABA release in cerebellar interneurons via a calcium/protein kinase C pathway. J Neurosci. 2011;31:5804–5815. doi: 10.1523/JNEUROSCI.5127-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, Dib-Hajj S, Meisler MH, Pietrobon D. Inherited neuronal ion channelopathies: new windows on complex neurological diseases. J Neurosci. 2008;28:11768–11777. doi: 10.1523/JNEUROSCI.3901-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YC, Burgunder JM, Wilder-Smith E, Chew SE, Lam-Mok-Sing KM, Sharma V, Ong BK. Electroencephalographic changes and seizures in familial hemiplegic migraine patients with the CACNA1A gene S218L mutation. J Clin Neurosci. 2008;15:891–894. doi: 10.1016/j.jocn.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Chu Y, Fioravante D, Thanawala M, Leitges M, Regehr WG. Calcium-dependent isoforms of protein kinase C mediate glycine-induced synaptic enhancement at the calyx of Held. J Neurosci. 2012;32:13796–13804. doi: 10.1523/JNEUROSCI.2158-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crins TT, Rusu SI, Rodríguez-Contreras A, Borst JGG. Developmental changes in short-term plasticity at the rat calyx of Held synapse. J Neurosci. 2011;31:11706–11717. doi: 10.1523/JNEUROSCI.1995-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuttle MF, Tsujimoto T, Forsythe ID, Takahashi T. Facilitation of the presynaptic calcium current at an auditory synapse in rat brainstem. J Physiol. 1998;512:723–729. doi: 10.1111/j.1469-7793.1998.723bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries B, Frants RR, Ferrari MD, van den Maagdenberg AM. Molecular genetics of migraine. Hum Genet. 2009;126:115–132. doi: 10.1007/s00439-009-0684-z. [DOI] [PubMed] [Google Scholar]
- Eikermann-Haerter K, Yuzawa I, Qin T, Wang Y, Baek K, Kim YR, Hoffmann U, Dilekoz E, Waeber C, Ferrari MD, van den Maagdenberg AM, Moskowitz MA, Ayata C. Enhanced subcortical spreading depression in familial hemiplegic migraine type 1 mutant mice. J Neurosci. 2011;31:5755–5763. doi: 10.1523/JNEUROSCI.5346-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedchyshyn MJ, Wang LY. Developmental transformation of the release modality at the calyx of Held synapse. J Neurosci. 2005;25:4131–4140. doi: 10.1523/JNEUROSCI.0350-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioravante D, Chu Y, Myoga MH, Leitges M, Regehr WG. Calcium-dependent isoforms of protein kinase C mediate posttetanic potentiation at the calyx of Held. Neuron. 2011;70:1005–1019. doi: 10.1016/j.neuron.2011.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsythe ID. Direct patch recording from identified presynaptic terminals mediating glutamatergic EPSCs in the rat CNS, in vitro. J Physiol. 1994;479:381–387. doi: 10.1113/jphysiol.1994.sp020303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsythe ID, Tsujimoto T, Barnes-Davies M, Cuttle MF, Takahashi T. Inactivation of presynaptic calcium current contributes to synaptic depression at a fast central synapse. Neuron. 1998;20:797–807. doi: 10.1016/S0896-6273(00)81017-X. [DOI] [PubMed] [Google Scholar]
- Gao Z, Todorov B, Barrett CF, van Dorp S, Ferrari MD, van den Maagdenberg AM, De Zeeuw CI, Hoebeek FE. Cerebellar ataxia by enhanced CaV2.1 currents is alleviated by Ca2+-dependent K+-channel activators in Cacna1aS218L mutant mice. J Neurosci. 2012;32:15533–15546. doi: 10.1523/JNEUROSCI.2454-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giugovaz-Tropper B, González-Inchauspe C, Di Guilmi MN, Urbano FJ, Forsythe ID, Uchitel OD. P/Q-type calcium channel ablation in a mice glycinergic synapse mediated by multiple types of Ca2+ channels alters transmitter release and short term plasticity. Neuroscience. 2011;192:219–230. doi: 10.1016/j.neuroscience.2011.06.021. [DOI] [PubMed] [Google Scholar]
- Grande G, Wang LY. Morphological and functional continuum underlying heterogeneity in the spiking fidelity at the calyx of Held synapse in vitro. J Neurosci. 2011;31:13386–13399. doi: 10.1523/JNEUROSCI.0400-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Guinan JJ, Jr, Li RY. Signal processing in brainstem auditory neurons which receive giant endings (calyces of Held) in the medial nucleus of the trapezoid body of the cat. Hear Res. 1990;49:321–334. doi: 10.1016/0378-5955(90)90111-2. [DOI] [PubMed] [Google Scholar]
- Habets RL, Borst JGG. Post-tetanic potentiation in the rat calyx of Held synapse. J Physiol. 2005;564:173–187. doi: 10.1113/jphysiol.2004.079160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann J, Pecka M, von Gersdorff H, Grothe B, Klug A. Synaptic transmission at the calyx of Held under in vivo like activity levels. J Neurophysiol. 2007;98:807–820. doi: 10.1152/jn.00355.2007. [DOI] [PubMed] [Google Scholar]
- Hermann J, Grothe B, Klug A. Modeling short-term synaptic plasticity at the calyx of Held using in vivo-like stimulation patterns. J Neurophysiol. 2009;101:20–30. doi: 10.1152/jn.90243.2008. [DOI] [PubMed] [Google Scholar]
- Inchauspe CG, Martini FJ, Forsythe ID, Uchitel OD. Functional compensation of P/Q by N-type channels blocks short-term plasticity at the calyx of Held presynaptic terminal. J Neurosci. 2004;24:10379–10383. doi: 10.1523/JNEUROSCI.2104-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inchauspe CG, Forsythe ID, Uchitel OD. Changes in synaptic transmission properties due to the expression of N-type calcium channels at the calyx of Held synapse of mice lacking P/Q-type calcium channels. J Physiol. 2007;584:835–851. doi: 10.1113/jphysiol.2007.139683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inchauspe CG, Urbano FJ, Di Guilmi MN, Forsythe ID, Ferrari MD, van den Maagdenberg AM, Uchitel OD. Gain of function in FHM-1 Ca(V)2.1 knock-in mice is related to the shape of the action potential. J Neurophysiol. 2010;104:291–299. doi: 10.1152/jn.00034.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inchauspe CG, Urbano FJ, Di Guilmi MN, Ferrari MD, van den Maagdenberg AM, Forsythe ID, Uchitel OD. Presynaptic CaV2.1 calcium channels carrying familial hemiplegic migraine mutation R192Q allow faster recovery from synaptic depression in mouse calyx of Held. J Neurophysiol. 2012;108:2967–2976. doi: 10.1152/jn.01183.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki S, Takahashi T. Developmental changes in calcium channel types mediating synaptic transmission in rat auditory brainstem. J Physiol. 1998;509:419–423. doi: 10.1111/j.1469-7793.1998.419bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaja S, Van de Ven RC, Broos LA, Frants RR, Ferrari MD, Van den Maagdenberg AM, Plomp JJ. Severe and progressive neurotransmitter release aberrations in familial hemiplegic migraine type 1 Cacna1a S218L knock-in mice. J Neurophysiol. 2010;104:1445–1455. doi: 10.1152/jn.00012.2010. [DOI] [PubMed] [Google Scholar]
- Kochubey O, Schneggenburger R. Ca2+ uncaging in nerve terminals. In: Konnerth A, Helmchen F, editors. Optical imaging in neuroscience: a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 2010. pp. 151–160. [Google Scholar]
- Korogod N, Lou X, Schneggenburger R. Presynaptic Ca2+ requirements and developmental regulation of posttetanic potentiation at the calyx of Held. J Neurosci. 2005;25:5127–5137. doi: 10.1523/JNEUROSCI.1295-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korogod N, Lou X, Schneggenburger R. Posttetanic potentiation critically depends on an enhanced Ca2+ sensitivity of vesicle fusion mediated by presynaptic PKC. Proc Natl Acad Sci U S A. 2007;104:15923–15928. doi: 10.1073/pnas.0704603104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kors EE, Terwindt GM, Vermeulen FL, Fitzsimons RB, Jardine PE, Heywood P, Love S, van den Maagdenberg AM, Haan J, Frants RR, Ferrari MD. Delayed cerebral edema and fatal coma after minor head trauma: role of the CACNA1A calcium channel subunit gene and relationship with familial hemiplegic migraine. Ann Neurol. 2001;49:753–760. doi: 10.1002/ana.1031. [DOI] [PubMed] [Google Scholar]
- Lauritzen M. Pathophysiology of the migraine aura. The spreading depression theory. Brain. 1994;117:199–210. doi: 10.1093/brain/117.1.199. [DOI] [PubMed] [Google Scholar]
- Lee JS, Kim MH, Ho WK, Lee SH. Presynaptic release probability and readily releasable pool size are regulated by two independent mechanisms during posttetanic potentiation at the calyx of Held synapse. J Neurosci. 2008;28:7945–7953. doi: 10.1523/JNEUROSCI.2165-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Ho WK, Lee SH. Actin-dependent rapid recruitment of reluctant synaptic vesicles into a fast-releasing vesicle pool. Proc Natl Acad Sci U S A. 2012;109:E765–E774. doi: 10.1073/pnas.1114072109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KH, Erazo-Fischer E, Taschenberger H. Similar intracellular Ca2+ requirements for inactivation and facilitation of voltage-gated Ca2+ channels in a glutamatergic mammalian nerve terminal. J Neurosci. 2012;32:1261–1272. doi: 10.1523/JNEUROSCI.3838-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipstein N, Sakaba T, Cooper BH, Lin KH, Strenzke N, Ashery U, Rhee JS, Taschenberger H, Neher E, Brose N. Dynamic control of synaptic vesicle replenishment and short-term plasticity by Ca2+-calmodulin-Munc13–1 signaling. Neuron. 2013;79:82–96. doi: 10.1016/j.neuron.2013.05.011. [DOI] [PubMed] [Google Scholar]
- Lorteije JA, Rusu SI, Kushmerick C, Borst JGG. Reliability and precision of the mouse calyx of Held synapse. J Neurosci. 2009;29:13770–13784. doi: 10.1523/JNEUROSCI.3285-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou X, Scheuss V, Schneggenburger R. Allosteric modulation of the presynaptic Ca2+ sensor for vesicle fusion. Nature. 2005;435:497–501. doi: 10.1038/nature03568. [DOI] [PubMed] [Google Scholar]
- Müller M, Felmy F, Schwaller B, Schneggenburger R. Parvalbumin is a mobile presynaptic Ca2+ buffer in the calyx of Held that accelerates the decay of Ca2+ and short-term facilitation. J Neurosci. 2007;27:2261–2271. doi: 10.1523/JNEUROSCI.5582-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E, Sakaba T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron. 2008;59:861–872. doi: 10.1016/j.neuron.2008.08.019. [DOI] [PubMed] [Google Scholar]
- Ophoff RA, Terwindt GM, Vergouwe MN, van Eijk R, Oefner PJ, Hoffman SM, Lamerdin JE, Mohrenweiser HW, Bulman DE, Ferrari M, Haan J, Lindhout D, van Ommen GJ, Hofker MH, Ferrari MD, Frants RR. Familial hemiplegic migraine and episodic ataxia type-2 are caused by mutations in the Ca2+ channel gene CACNL1A4. Cell. 1996;87:543–552. doi: 10.1016/S0092-8674(00)81373-2. [DOI] [PubMed] [Google Scholar]
- Pang ZP, Bacaj T, Yang X, Zhou P, Xu W, Südhof TC. Doc2 supports spontaneous synaptic transmission by a Ca2+-independent mechanism. Neuron. 2011;70:244–251. doi: 10.1016/j.neuron.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietrobon D. Calcium channels and migraine. Biochim Biophys Acta. 2013;1828:1655–1665. doi: 10.1016/j.bbamem.2012.11.012. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Contreras A, van Hoeve JS, Habets RL, Locher H, Borst JGG. Dynamic development of the calyx of Held synapse. Proc Natl Acad Sci U S A. 2008;105:5603–5608. doi: 10.1073/pnas.0801395105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaba T, Neher E. Calmodulin mediates rapid recruitment of fast-releasing synaptic vesicles at a calyx-type synapse. Neuron. 2001;32:1119–1131. doi: 10.1016/S0896-6273(01)00543-8. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron. 1999;23:399–409. doi: 10.1016/S0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Shu Y, Hasenstaub A, McCormick DA. Turning on and off recurrent balanced cortical activity. Nature. 2003;423:288–293. doi: 10.1038/nature01616. [DOI] [PubMed] [Google Scholar]
- Somjen GG. Ion regulation in the brain: implications for pathophysiology. Neuroscientist. 2002;8:254–267. doi: 10.1177/1073858402008003011. [DOI] [PubMed] [Google Scholar]
- Stam AH, Luijckx GJ, Poll-Thé BT, Ginjaar IB, Frants RR, Haan J, Ferrari MD, Terwindt GM, van den Maagdenberg AM. Early seizures and cerebral oedema after trivial head trauma associated with the CACNA1A S218L mutation. J Neurol Neurosurg Psychiatry. 2009;80:1125–1129. doi: 10.1136/jnnp.2009.177279. [DOI] [PubMed] [Google Scholar]
- Sun J, Pang ZP, Qin D, Fahim AT, Adachi R, Südhof TC. A dual-Ca2+-sensor model for neurotransmitter release in a central synapse. Nature. 2007;450:676–682. doi: 10.1038/nature06308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan ML, Borst JGG. Comparison of responses of neurons in the mouse inferior colliculus to current injections, tones of different durations, and sinusoidal amplitude-modulated tones. J Neurophysiol. 2007;98:454–466. doi: 10.1152/jn.00174.2007. [DOI] [PubMed] [Google Scholar]
- Thomsen LL, Eriksen MK, Roemer SF, Andersen I, Olesen J, Russell MB. A population-based study of familial hemiplegic migraine suggests revised diagnostic criteria. Brain. 2002;125:1379–1391. doi: 10.1093/brain/awf132. [DOI] [PubMed] [Google Scholar]
- Tottene A, Pivotto F, Fellin T, Cesetti T, van den Maagdenberg AM, Pietrobon D. Specific kinetic alterations of human CaV2.1 calcium channels produced by mutation S218L causing familial hemiplegic migraine and delayed cerebral edema and coma after minor head trauma. J Biol Chem. 2005;280:17678–17686. doi: 10.1074/jbc.M501110200. [DOI] [PubMed] [Google Scholar]
- Tottene A, Conti R, Fabbro A, Vecchia D, Shapovalova M, Santello M, van den Maagdenberg AM, Ferrari MD, Pietrobon D. Enhanced excitatory transmission at cortical synapses as the basis for facilitated spreading depression in Ca(v)2.1 knockin migraine mice. Neuron. 2009;61:762–773. doi: 10.1016/j.neuron.2009.01.027. [DOI] [PubMed] [Google Scholar]
- Urbano FJ, Piedras-Rentería ES, Jun K, Shin HS, Uchitel OD, Tsien RW. Altered properties of quantal neurotransmitter release at endplates of mice lacking P/Q-type Ca2+ channels. Proc Natl Acad Sci U S A. 2003;100:3491–3496. doi: 10.1073/pnas.0437991100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Maagdenberg AM, Pietrobon D, Pizzorusso T, Kaja S, Broos LA, Cesetti T, van de Ven RC, Tottene A, van der Kaa J, Plomp JJ, Frants RR, Ferrari MD. A Cacna1a knockin migraine mouse model with increased susceptibility to cortical spreading depression. Neuron. 2004;41:701–710. doi: 10.1016/S0896-6273(04)00085-6. [DOI] [PubMed] [Google Scholar]
- van den Maagdenberg AM, Pizzorusso T, Kaja S, Terpolilli N, Shapovalova M, Hoebeek FE, Barrett CF, Gherardini L, van de Ven RC, Todorov B, Broos LA, Tottene A, Gao Z, Fodor M, De Zeeuw CI, Frants RR, Plesnila N, Plomp JJ, Pietrobon D, Ferrari MD. High cortical spreading depression susceptibility and migraine-associated symptoms in Ca(v)2.1 S218L mice. Ann Neurol. 2010;67:85–98. doi: 10.1002/ana.21815. [DOI] [PubMed] [Google Scholar]
- Varela JA, Sen K, Gibson J, Fost J, Abbott LF, Nelson SB. A quantitative description of short-term plasticity at excitatory synapses in layer 2/3 of rat primary visual cortex. J Neurosci. 1997;17:7926–7940. doi: 10.1523/JNEUROSCI.17-20-07926.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LY, Kaczmarek LK. High-frequency firing helps replenish the readily releasable pool of synaptic vesicles. Nature. 1998;394:384–388. doi: 10.1038/28645. [DOI] [PubMed] [Google Scholar]
- Wang T, Rusu SI, Hruskova B, Turecek R, Borst JGG. Modulation of synaptic depression of the calyx of Held synapse by GABAB receptors and spontaneous activity. J Physiol. 2013;591:4877–4894. doi: 10.1113/jphysiol.2013.256875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Pang ZP, Shin OH, Südhof TC. Synaptotagmin-1 functions as a Ca2+ sensor for spontaneous release. Nat Neurosci. 2009;12:759–766. doi: 10.1038/nn.2320. [DOI] [PMC free article] [PubMed] [Google Scholar]