

Abstract
It has recently been proposed that post-translational modification of not only the M3–M4 linker but also the M1–M2 linker of pentameric ligand-gated ion channels modulates function in vivo. To estimate the involvement of the M1–M2 linker in gating and desensitization, we engineered a series of mutations to this linker of the human adult-muscle acetylcholine receptor (AChR), the α3β4 AChR and the homomeric α1 glycine receptor (GlyR). All tested M1–M2 linker mutations had little effect on the kinetics of deactivation or desensitization compared with the effects of mutations to the M2 α-helix or the extracellular M2–M3 linker. However, when the effects of mutations were assessed with 50 Hz trains of ∼1 ms pulses of saturating neurotransmitter, some mutations led to much more, and others to much less, peak-current depression than observed for the wild-type channels, suggesting that these mutations could affect the fidelity of fast synaptic transmission. Nevertheless, no mutation to this linker could mimic the irreversible loss of responsiveness reported to result from the oxidation of the M1–M2 linker cysteines of the α3 AChR subunit. We also replaced the M3–M4 linker of the α1 GlyR with much shorter peptides and found that none of these extensive changes affects channel deactivation strongly or reduces the marked variability in desensitization kinetics that characterizes the wild-type channel. However, we found that these large mutations to the M3–M4 linker can have pronounced effects on desensitization kinetics, supporting the notion that its post-translational modification could indeed modulate α1 GlyR behavior.
Keywords: fast perfusion, glycine receptors, ion-channel kinetics, nicotinic receptors, outside-out patches, patch-clamp
Introduction
Until recently, the M3–M4 linker of pentameric ligand-gated ion channels (pLGICs) was the only identified intracellular site for post-translational modulation of channel kinetics and expression (Swope et al., 1999). Reports within the last few years, however, have suggested that post-translational modification of the other intracellular linker (the M1–M2 linker) can also regulate channel function (Krishnaswamy and Cooper, 2012). Specifically, oxidation of the cysteine at position –4′ of the M1–M2 linker of the nicotinic acetylcholine receptor (AChR) α3 subunit (Fig. 1) was proposed to cause α3 subunit-containing AChRs to enter a long-lived refractory state upon repetitive stimulation from which the channels do not recover even after long incubations in the absence of agonist. Also, palmitoylation of the aligned cysteine of the α4 AChR subunit was found to affect the expression levels of α4β2 receptors (Amici et al., 2012). Moreover, in the glycine receptor (GlyR), point mutations to residues immediately flanking the M1–M2 linker have been shown to affect the gating efficacy and EC50 values of several agonists, as well as the kinetics of entry into desensitization (Lynch et al., 1997; Saul et al., 1999; Breitinger et al., 2001). Overall, these findings underscore the need to establish the involvement of this short intracellular linker in the conformational changes associated with gating and desensitization.
Figure 1.

The pore domain of pLGICs. A, Membrane-threading pattern of an individual subunit. Yellow represents the long N- and the short C-termini; purple represents the transmembrane α-helices; and orange represents the linkers between transmembrane α-helices. The M2 α-helix lines the pore and is flanked by the intracellular M1–M2 and the extracellular M2–M3 linkers. A red triangle represents the approximate location of the –4′ position. B, Diagram representation of the unliganded closed-channel conformation of the muscle-type AChR from Torpedo (PDB ID code 2BG9) (Unwin, 2005) rendered with VMD (Humphrey et al., 1996). For clarity, the β1 subunit is omitted. The different regions of one of the two α1 subunits are color-coded as in A. The approximate location of the –4′ position in the M1–M2 linker (red triangle) was inferred upon correction of a likely register error in the original structural model (Mnatsakanyan and Jansen, 2013). C, Sequence alignment of the wild-type subunits under study. The color code is the same as in A and B. Accession numbers are provided to the left of each sequence. A cysteine occurs at position –4′ of the human AChR subunits α2, α3, α4, α6, β2, and β4, and the human 5-HT3B subunit. Also, in the muscle AChR, a cysteine occupies position –4′ of the mouse (but not the human) δ subunit.
To investigate this problem, we analyzed the effect of mutations on channel kinetics using macroscopic current recordings. We mutated the cysteine at position –4′ of the human AChR α3 subunit to several other residues and performed mutagenesis scans of the entire M1–M2 linkers of the human AChR α1 subunit and the human GlyR α1 subunit. To mimic the kinetics of neurotransmitter–receptor interaction that occur during synaptic transmission, ACh or Gly concentration jumps were applied to outside-out patches or whole cells using a fast-perfusion setup (solution exchange time ≅100 μs, for outside-out patches). We found that the effects of M1–M2 linker mutations on channel kinetics, although small compared with the effects of M2 segment or M2–M3 linker mutations, are large enough to affect the responses to fast synaptic-like stimulation (i.e., trains of brief, 1 ms pulses delivered at high frequencies). However, none of the engineered mutations mimicked the irreversible effect reported for the oxidation of the cysteine at position –4′ of the α3 AChR subunit.
To look more generally at the involvement of the cytosolic linkers in channel kinetics, we also replaced the M3–M4 linker of the human α1 GlyR with much shorter peptides. Remarkably, these constructs exhibited kinetics of deactivation similar to those of the wild-type receptor, and they did not show any less cell-to-cell variability in the kinetics of entry into desensitization. However, when the time courses were averaged across different cells, mutations to this linker did have large effects on entry into and recovery from desensitization. Moreover, our data suggest that the fast exponential component of the current decay typically observed upon sustained application of saturating Gly reflects true receptor desensitization rather than an artifact of transient local [Cl−] changes. Overall, these results are consistent with the notion that post-translational modification of the M3–M4 linker can modulate desensitization and, plausibly, channel function in intact synapses.
Materials and Methods
cDNA clones, mutagenesis, and heterologous expression.
Complementary DNAs (cDNAs) coding the adult human-muscle AChR subunits (Uniprot accession numbers: α1, P02708; β1, P11230; δ, Q07001; ε, Q04844) were provided by L. Sivilotti (University College London, London). cDNAs coding the human AChR α3 and β4 subunits (accession numbers: α3, P32297; β4, P30926) were purchased from Open Biosystems, subcloned into pcDNA3.1, and cotransfected along with cDNA coding isoform 1 of RIC-3 (provided by W.N. Green, University of Chicago, Chicago; accession number Q7Z5B4); RIC-3 is a chaperone that appeared to increase the expression of these AChRs in our experiments, consistent with previous reports (Lansdell et al., 2005; Treinin, 2008). cDNA coding the human GlyR α1 subunit isoform b (accession number: P23415) was obtained by mutating the cDNA coding the rat ortholog (accession number: P07727; provided by M.M. Slaughter, University at Buffalo, Buffalo, NY) as previously indicated (Papke et al., 2011). Mutations were engineered using either the QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies) or standard PCR-based methods, and the presence of the desired mutations and the absence of unwanted ones were confirmed by sequencing the entire coding region of the cDNA (ACGT). The different pLGICs were heterologously expressed in transiently transfected (calcium-phosphate precipitation method) HEK-293 cells. The cells were incubated with the transfection mixture for 16–18 h, and electrophysiological recordings were performed between 24 and 54 h thereafter.
Electrophysiology.
Outside-out patches were positioned opposite a double-barreled “θ-tube” (Hilgenberg) that was used to apply agonist-concentration jumps (Jonas, 1995; Elenes et al., 2006; Papke et al., 2011). The flow of solution through the θ-tube was controlled using a pressure-driven system (ALA BPS-8; ALA Scientific Instruments), and the movement of the θ-tube was achieved using a piezo-electric arm (Burleigh-LSS-3100; discontinued) controlled by pCLAMP 9.0 software and a Digidata 1322A interface (Molecular Devices). Signals from this interface were low-pass filtered (900C, Frequency Devices) at a cutoff frequency of 150 Hz before their arrival at the piezoelectric arm to reduce ringing in the θ-tube motion. This system achieved a solution-exchange time of 97 μs for the t10–90% and 101 μs for the t90–10%, as estimated from changes in the liquid-junction potential measured with an open-tip patch pipette. Constant gravity-driven perfusion was applied to the bath during experiments to wash away ligand that would otherwise accumulate in the system. All currents were recorded at −80 mV using an Axopatch 200B amplifier (Molecular Devices) with an effective bandwidth of DC−10 kHz, and the signal was digitized at 100 kHz. Data were acquired and analyzed using pCLAMP 9.0 software. Whole-cell recordings were performed using essentially the same setup as that described above for recordings in the outside-out configuration, but the relative positioning of the θ-tube and the patch pipette was adjusted so as to optimize the perfusion of the entire cell, which remained attached to the bottom of the recording chamber during the experiments. In this configuration, the solution-exchange times were 3 ms for the t10–90% and 0.4 ms for the t90–10%, as estimated from changes in the liquid-junction potential measured with an open-tip patch pipette. Clearly, these estimates do not take into account the geometry of the cell, which is expected to slow down the solution exchange even further. All recordings from the muscle AChR and the α1 GlyR were performed in the outside-out configuration, whereas those from the α3β4 AChR were performed in the whole-cell configuration.
Solutions.
Regardless of the patch-clamp configuration used, the patch-pipette solution consisted of the following (in mm): 100 KF, 40 KCl, 1 CaCl2, 11 EGTA, and 10 HEPES/KOH, pH 7.4, unless otherwise indicated. The solutions flowing through the two barrels of the θ-tube consisted of the following (in mm): 142 KCl, 5.4 NaCl, 1.8 CaCl2, 1.7 MgCl2, and 10 HEPES/KOH, pH 7.4 with or without agonist; the concentration of the agonist was 1 mm in the case of ACh and 10 mm in the case of Gly. The gravity-fed solution perfused in the background also consisted of the following (in mm): 142 KCl, 5.4 NaCl, 1.8 CaCl2, 1.7 MgCl2, and 10 HEPES/KOH, pH 7.4.
Pulse protocols.
To estimate the kinetic parameters of deactivation, series of 25 1 ms pulses of ACh or Gly were applied at a frequency of 1 Hz to outside-out patches containing the muscle AChR or the α1 GlyR; for the α3β4 AChR, 15 200 ms pulses were delivered to whole cells at a frequency of 4 pulses per minute. In all cases, the responses to all pulses of agonist within each series were averaged, and the resulting averaged traces were fit with exponential-decay functions. To assess peak-current depression in response to repetitive stimulation, series of brief pulses (as described above) were applied at the frequencies indicated in Results. Peak-current amplitudes within a recording were normalized to the peak amplitude of the first response, and the resulting values were averaged across patches. To estimate the kinetics of entry into desensitization, long, continuous pulses of agonist (2 s for the muscle AChR; 5 s for the α3β4 AChR and the α1 GlyR) were applied, and the resulting traces were fit with exponential-decay functions. To estimate the kinetics of recovery from desensitization, paired-pulse protocols were used. An initial, “conditioning” pulse was applied for 2 s. After an interpulse interval of variable duration, a “test” pulse (lasting 100 ms for outside-out and 200 ms for whole-cell recordings) was applied. The fraction of receptors that recovered from desensitization during a given interpulse interval was calculated taking into account the current level at the end of the conditioning pulse, which in some cases was non-zero. The fraction of receptors that recovered within a given interpulse interval was determined for each patch, and the resulting values were averaged across patches, plotted as a function of the duration of the interpulse interval and fit with exponential functions. It is worth noting that this way of estimating the extent of recovery from desensitization rests on the assumption that these receptor channels desensitize only while exposed to agonist but not upon agonist washout (i.e., during deactivation). We previously found that this assumption is not strictly correct for any neurotransmitter-gated ion channel (Papke et al., 2011), and hence, the recovery values plotted in Figures 4, 6, 8, and 9 (“Fraction of receptors recovered”) represent lower bounds. For the purposes of this work (in which large effects of mutations were sought), an even more rigorous analysis of the data was deemed unnecessary.
Figure 4.

Effects of M1–M2 linker mutations on recovery from desensitization of the muscle AChR. Alanine (A), glycine (B), proline (C), and valine (D) scans were performed on the M1–M2 linker of the AChR α1 subunit, as for Figures 2 and 3. The concentration of ACh during the pairs of ligand pulses was 1 mm, and the membrane potential was held constant at −80 mV. All recordings were performed using the outside-out configuration. Axis labels are the same for all four panels. Pairs of pulses consisting of a 1 s “conditioning” pulse and a subsequent 100 ms “test” pulse, separated by intervals of variable duration, were applied to outside-out patches of membrane. Data points are the averages of values taken from several patches (between two and four). Error bars indicate the corresponding SE. For all mutants, the rate of recovery from desensitization is similar to that of the wild-type.
Figure 6.
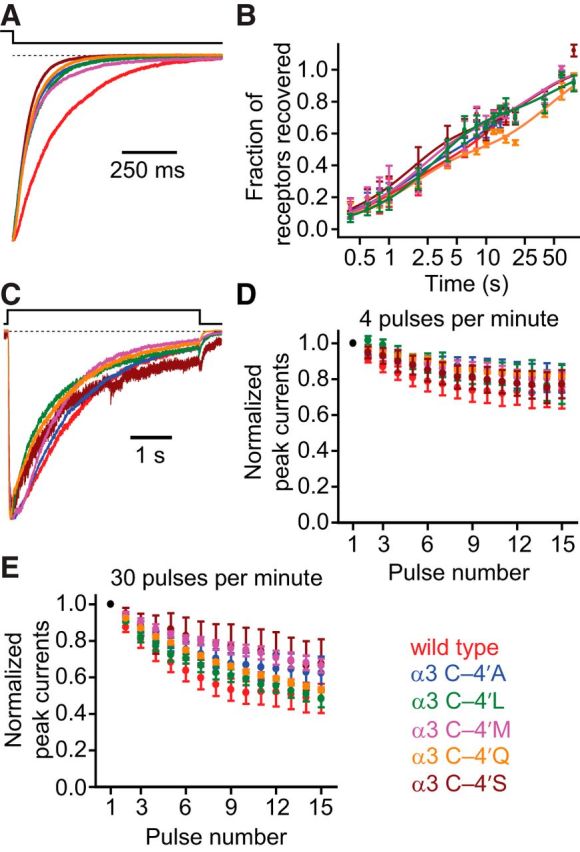
Effects of mutations to the M1–M2 linker cysteine of the α3 AChR subunit on the behavior of α3β4 receptors. A, Deactivation time courses. B, Recovery from desensitization. C, Entry into desensitization. D, E, Responses to low-frequency trains of ACh pulses. The concentration of ACh during the ligand pulses was 1 mm, and the membrane potential was held constant at −80 mV. All recordings were performed using the whole-cell configuration; the solution-exchange time in this configuration was on the order of a few milliseconds, and hence, it was too long for higher-frequency trains of briefer ligand pulses to be applied reliably. Error bars indicate SE. A, C, Dashed line indicates the current baseline. D, E, Data points indicate the average values (normalized to the peak-current response to the first pulse of ACh; black) obtained from several patches (between 3 and 12). Error bars indicate corresponding SE. Overall, point mutations to the native cysteine at position –4′ of the α3 AChR subunit tended to have very little effect on channel kinetics, and they clearly failed to evoke the irreversible loss of responsiveness to ACh reported to result from the oxidation of the native side chain (Campanucci et al., 2008, 2010; Krishnaswamy and Cooper, 2012).
Figure 8.

Effects of M1–M2 linker mutations on recovery from desensitization and high-frequency train responses of the α1 GlyR. Alanine and threonine scans were performed on the M1–M2 linker of the α1 GlyR, as for Figure 7. The concentration of Gly during the ligand pulses was 10 mm, and the membrane potential was held constant at −80 mV. All recordings were performed using the outside-out configuration. Alanine-scan (A) and threonine-scan (B) recovery from desensitization data. Axis labels are the same for both panels. Plots were generated in the same way as for Figure 4. Some of the mutations increased the number of exponential components required to obtain a good fit to the plotted data points. Alanine-scan (C) and threonine-scan (D) responses to high-frequency (50 Hz) trains of 1 ms pulses. Axis labels are the same for both panels, and the color code is the same as in A and B, respectively. Plots were generated in the same way as for Figure 5. Most M1–M2 linker mutants exhibited train responses similar to that of the wild-type. One exception was the P–2′A mutant, which exhibited very little peak-response depression even at a frequency as high as 50 Hz.
Figure 9.

Effects of M3–M4 linker mutations on the kinetic properties of the α1 GlyR. A, Sequence alignment of the wild-type and M3–M4 linker mutant α1 GlyRs under study. The “//” in the wild-type α1 GlyR and H311S sequences indicates a break in the sequence meant only to increase the clarity of the alignment. B, Time courses of deactivation. Recordings from the GlyRM3M4 311H construct were most often best fit with a single exponential component, unlike recordings from all other constructs discussed here (Table 4); the representative trace decays to a zero-current level faster than those of all other constructs because it does not exhibit the slow component of decay. C, Time courses of entry into desensitization. Of all the tested constructs, the GlyRM3M4 311H desensitized the fastest. Addition of the K24′A mutation (which, when engineered on the wild-type background, slowed down desensitization to a large degree; Fig. 7F) to this construct slowed down desensitization as well, but the time course of the GlyRM3M4 311H + K24′A channel remained faster than that of the K24′A single-point mutant. Thus, we conclude that the large deletion of the M3–M4 linker engineered in the GlyRM3M4 311H construct has a comparatively large effect on entry into desensitization. The GlyRM3M4 8xAla construct, on the other hand, desensitized the slowest. D, Time courses of recovery from desensitization. Data were gathered and analyzed using the same methods as for Figure 4. Most constructs recovered at a rate similar to that of the wild-type α1 GlyR (Table 4). The data points corresponding to the GlyRM3M4 311S, GlyRM3M4 311A, and GlyRM3M4 311H constructs and H311S point mutant nearly “fall” on top of each other, making them difficult to distinguish. The GlyRM3M4 8xAla exhibited markedly faster recovery than the wild-type (Table 4). B, C, Dashed line indicates the current baseline. B–D, The concentration of Gly during the ligand pulses was 10 mm, and the membrane potential was held constant at −80 mV. All recordings were performed using the outside-out configuration.
Time constants and decay half-times.
The time courses of deactivation, entry into desensitization, and recovery from desensitization were fit with exponential functions. The number of exponential components that best fit these data was determined visually and was found to vary among constructs and even from patch to patch for some constructs. This variability made it difficult to assess the relative effects of mutations on channel kinetics because it is not clear how to compare the parameters of, for example, a mono-exponential fit with those of a double-exponential fit. In some cases, when comparing the effects of mutations on the kinetics of deactivation or entry into desensitization, we used decay half-times; these were calculated from the parameters of the fit as the time it takes for the current to decay half-way between its peak value and its projected value at time infinity. Of course, decay half-times are a qualitative measure, and they cannot capture the behavior of complex time courses in which different exponential components of the fit have vastly different time constants (e.g., the desensitization time courses of the α1 GlyR). We deemed half-times to be useful to qualitatively compare mono-exponential fits to double-exponential fits in cases in which the time constants of the two components of the double-exponential fit were within an order of magnitude of each other (i.e., cases in which the time courses were relatively simple). In the tables, we only report the time constants of decay and their relative amplitudes because these numbers reflect kinetic behavior more accurately than do the decay half-times.
Results
The extent to which the intracellular M1–M2 linker (Fig. 1) of pLGICs is involved in channel gating and desensitization is a largely unexplored topic. Motivation to undertake a systematic study of this region was augmented by the recently proposed notion that the M1–M2 linker of the AChR α3 subunit is a post-translational modulatory site directly involved in the pathophysiology of diabetic neuropathy (Campanucci et al., 2010; Krishnaswamy and Cooper, 2012). Thus far, it is the M3–M4 linker that has been thought of as the primary intracellular modulatory hub for channel function (Swope et al., 1999); hence, the idea that the M1–M2 linker could also serve such a role seemed most intriguing to us.
Muscle-AChR mutagenesis: deactivation
Because of its high expression levels in HEK-293 cells and its negligible rundown in outside-out patches, the adult muscle AChR seemed to be an appropriate starting point for a systematic mutational study of the M1–M2 linker. The overall functional and structural similarities between this receptor and the much less tractable AChR formed by α3 and β4 subunits (Papke et al., 2011) appeared to justify this decision. Alanine, glycine, proline, and valine scans were performed on the M1–M2 linker of the human α1 subunit, between Pro 236 (corresponding to position –7′; Fig. 1C) and Gly 240 (–2′; a pore-facing position in the first intracellular turn of the M2 α-helix). Because the stoichiometry of the muscle AChR is (α1)2β1δε, each mutation is expected to be present twice in the heteropentameric complex. To quantify the effect of each mutation, we estimated the kinetics of deactivation, entry into desensitization and recovery from desensitization using fast ligand-perfusion methods, as described in detail in Materials and Methods. To the best of our knowledge, in all previous electrophysiological studies of the intracellular linkers of pLGICs, the activating ligands have been applied and washed out much more slowly and following ill-defined time courses, which complicates the interpretation of the recorded currents. Representative deactivation traces are shown in Figure 2 for the subset of mutant constructs that expressed at large enough densities to allow the recording of macroscopic currents in the outside-out configuration; the corresponding time courses were usually best fit with mono-exponential decay functions, and the estimated time constants are listed in Table 1. Compared with that of the adult wild-type, the rate of deactivation seemed to be similar in four mutants (T–6′V, D–5′A, D–5′P, S–4′G), faster in five (T–6′A, T–6′G, D–5′G, D–5′V, G–2′A), and slower in the other two (T–6′P, S–4′A). In addition to amino acid substitutions, we also engineered a construct with the D–5′ residue removed from the linker (“D–5′del”; Table 1), and another with an alanine inserted between amino acids D–5′ and S–4′ (“A–4′ins”; Table 1). A–4′ins exhibited no apparent difference in deactivation kinetics compared with the wild-type AChR, whereas the deletion of D–5′ caused the rate of deactivation to speed up by a factor of 1.7 ± 0.4 (Table 1).
Figure 2.

Effects of M1–M2 linker mutations on deactivation of the muscle AChR. Alanine (A), glycine (B), proline (C), and valine (D) scans were performed on the M1–M2 linker of the AChR α1 subunit, from position -7′ to position -2′ (Fig. 1). The concentration of ACh during the 1 ms ligand pulses was 1 mm, and the membrane potential was held constant at −80 mV. All recordings were performed using the outside-out configuration. The displayed (normalized) traces illustrate the average behavior of each construct (Table 1). E, Comparison of deactivation time courses of M1–M2 linker, M2 segment, and M2–M3 linker mutants. In all panels, a dashed line indicates the current baseline.
Table 1.
Kinetic parameters of M1–M2 linker mutants of the muscle AChRa
| α1-subunit mutation | τdeactivation (ms) | τdeactivation ratio (mutant/wild type) | τentry into desensitization (2 components) (ms) | τentry into desensitization (1 component) (ms) | τrecovery from desensitization (ms) |
|---|---|---|---|---|---|
| Wild type | 2.61 ± 0.11 | — | 36.4 ± 5.2 (0.571 ± 0.043) | 22.3 ± 3.4 | 402 ± 46 (0.752 ± 0.061) |
| 243 ± 65 | (21/43) | 3530 ± 1020 | |||
| T–6′A | 1.38 ± 0.24 | 0.53 ± 0.21 | 24.8 ± 7.5 (0.588 ± 0.138) | 26.3 ± 6.9 | 549 ± 31 (0.756 ± 0.029) |
| 124 ± 41 | (7/12) | 5710 ± 760 | |||
| T–6′G | 0.75 ± 0.06 | 0.29 ± 0.12 | — | 10.9 ± 2.0 | 398 ± 41 (0.782 ± 0.050) |
| (5/5) | 4520 ± 1310 | ||||
| T–6′P | 5.19 ± 0.22 | 1.99 ± 0.25 | 12.0 ± 0.6 (0.522 ± 0.060) | 15.2 ± 1.6 | 2540 ± 170 |
| 31.1 ± 4.1 | (2/4) | ||||
| T–6′V | 2.34 ± 0.23 | 0.90 ± 0.23 | — | 13.4 ± 3.6 | 1030 ± 440 (0.492 ± 0.262) |
| (5/5) | 4210 ± 1480 | ||||
| D–5′A | 3.20 ± 0.62 | 1.23 ± 0.33 | 48.3 ± 7.9 (0.756 ± 0.111) | 12.2 ± 2.8 | 763 ± 91 (0.782 ± 0.071) |
| 374 ± 253 | (8/10) | 6660 ± 2300 | |||
| D–5′G | 2.05 ± 0.15 | 0.79 ± 0.18 | — | 17.3 ± 3.6 | 445 ± 38 (0.849 ± 0.037) |
| (5/5) | 9500 ± 3640 | ||||
| D–5′P | 2.94 ± 0.49 | 1.13 ± 0.30 | — | 23.1 ± 3.6 | 1000 ± 140 (0.787 ± 0.091) |
| (5/5) | 7630 ± 3340 | ||||
| D–5′V | 1.74 ± 0.32 | 0.67 ± 0.24 | — | 11.7 ± 1.1 | 602 ± 92 (0.778 ± 0.091) |
| (5/5) | 4910 ± 2080 | ||||
| S–4′A | 4.13 ± 0.48 | 1.58 ± 0.31 | — | 13.2 ± 2.6 | 930 ± 80 (0.846 ± 0.057) |
| (10/10) | 8320 ± 3440 | ||||
| S–4′G | 2.23 ± 0.57 | 0.85 ± 0.31 | — | 25.8 ± 3.7 | 900 ± 56 |
| (3/3) | |||||
| G–2′A | 1.83 ± 0.19 | 0.70 ± 0.20 | 3.6 ± 2.3 (0.527 ± 0.009) | 8.99 ± 4.29 | 439 ± 62 (0.775 ± 0.067) |
| 18.5 ± 4.7 | (6/8) | 5320 ± 1970 | |||
| D–5′del | 1.52 ± 0.11 | 0.58 ± 0.16 | 9.0 (0.656) | 48.4 ± 15.0 | — |
| 37.8 | (7/8) | ||||
| A–4′ins | 2.30 ± 1.20 | 0.88 ± 0.44 | — | 14.3 ± 4.5 | — |
aAll errors are SEs. The errors associated with the ratios of deactivation time constants were calculated by error propagation. Some desensitization time courses required two exponential components to achieve a good fit, whereas others required only one; average values for both sets of time constants are presented. In the one-component data column, fractions are presented for each construct: in each case, the numerator is the number of patches from which the kinetics of entry into desensitization were best fit with a single exponential component, and the denominator is the total number of patches from which desensitization time courses were recorded. Each time course was obtained from a different patch of membrane. Because of the small current amplitudes recorded from outside-out patches containing the α1 D–5′del and α1 A–4′ins constructs, it was not possible to estimate their time constants of recovery from desensitization. —, Not applicable.
To gain a basis of comparison for the size of the effects of the described M1–M2 linker mutations, we also studied constructs bearing single-point mutations in the M2 segment or the M2–M3 linker (Table 2). Mutations to the aliphatic, lumen-facing side chains of the M2 α-helix are known to slow down the kinetics of deactivation drastically (e.g., Labarca et al., 1995; Filatov and White, 1995), supporting the widely accepted notion that this segment is integrally involved in the conformational changes associated with channel gating. Many mutations to the M2–M3 linker are also known to have large effects on the deactivation time course (Grosman et al., 2000; Jha et al., 2007; Lee et al., 2009). In our experiments, the α1 L9′A mutation (located in the middle of M2) slowed down the rate of deactivation by a factor of 27.0 ± 2.2 (comparing half-times of decay to allow a direct comparison between time courses best fit with different numbers of components, and determining the standard error by propagating the standard errors associated with these half-times; see Materials and Methods), whereas the α1 S27′I mutation (located in the M2–M3 linker) slowed it down by a factor of 7.0 ± 0.8. In contrast, none of the M1–M2 linker mutations slowed down or sped up the time course of deactivation by a factor larger than 3.5 (the factor corresponding to the effect of the α1 T–6′G mutation; Fig. 2; Table 1).
Table 2.
Kinetic parameters of M2 segment and M2–M3 linker mutants of the muscle AChRa
| α1-subunit mutation | Site of mutation | τdeactivation (ms) | τentry into desensitization (ms) |
|---|---|---|---|
| Wild type | — | 2.61 ± 0.11 (20/29) | 22.3 ± 3.4 (21/43) |
| L9′A | M2 segment | 29.5 ± 18.1 (0.472 ± 0.111) | 511 ± 115 (6/6) |
| 206 ± 68 (3/3) | |||
| L9′A + T–6′G | M2 segment + | 49.5 ± 0.6 (0.837 ± 0.070) | 518 ± 247 (5/5) |
| M1–M2 linker | 350 ± 49 (2/4) | ||
| S27′I | M2–M3 linker | 18.2 ± 3.2 (7/8) | 47.3 ± 13.1 (6/11) |
aAll errors are SEs. For some constructs, the number of exponential components required to achieve a good fit to the deactivation and/or desensitization time courses varied among patches, which complicated the direct comparison of data across different experiments. Hence, only the kinetic parameters corresponding to the most common type of fit are presented. For all data columns, fractions are presented for each construct: the numerator is the number of patches from which the time courses were best fit with the indicated number of exponential components, and the denominator is the total number of patches from which such time courses were recorded. Each time course was obtained from a different patch of membrane. —, Not applicable.
One caveat that needs to be borne in mind, however, when trying to assess the effects of mutations on channel gating and desensitization using phenomenological observables such as the deactivation time course is that these quantities may be quite insensitive. For example, for a hypothetical channel with two agonist-binding sites whose activation-deactivation behavior can be modeled by a simple, linear C ⇌ CA ⇌ CA2 ⇌ OA2 scheme and whose individual activations have a high open probability (∼1), the time constant of deactivation is approximately given by the following (Colquhoun and Hawkes, 1981; Wyllie et al., 1998):
where β2 and α2 are the opening and closing rate constants of the diliganded receptor-channel, respectively, and k– is the agonist-dissociation rate constant from the diliganded closed-channel state (i.e., the rate constant governing the CA2 → CA step). In the particular case of the wild-type muscle AChR, the ratio β2/k– is ∼1. Hence, from Equation 1, it can be seen that the deactivation time constant of the muscle AChR would be prolonged by mutations that increase the β2/k– ratio more than it would be shortened by mutations that lower this ratio. For example, whereas a mutation that increases the β2/k– ratio by a factor of, say, 10 is expected to increase the deactivation time constant by a factor of ∼5.5, a mutation that lowers this ratio by a factor of 10 is expected to lower the deactivation time constant by a factor of only ∼1.8. And whereas a mutation that increases the β2/k– ratio by a factor of 1000 is expected to increase the deactivation time constant by a factor of ∼500, a mutation that lowers this ratio by a factor of 1000 is expected to lower the deactivation time constant by a factor of only ∼2.0. Importantly, this “asymmetric” contribution of gain-of-function and loss-of-function mutations to the deactivation time course also holds for more realistic kinetic models of channel function. It follows, then, that it is not insightful to directly compare the factors by which the α1 L9′A and T–6′G mutations affect the deactivation time course because the L9′A mutation slows deactivation down and the T–6′G mutation speeds it up. Thus, to obtain a better estimate of the magnitude of the effects of these two mutations relative to each other, we tested the effect of combining the two mutations in the same construct. This double mutant exhibited deactivation kinetics similar to those of the α1 L9′A single mutant (Fig. 2E and Table 2), from which we infer (assuming that the effects of the individual mutations are additive) that mutations to the M1–M2 linker have a comparatively small impact on channel function. Also, it seems safe to conclude that the rearrangement of the muscle-AChR M1–M2 linker during gating is more limited than that undergone by the M2 α-helix or the M2–M3 linker, a notion consistent with studies of other eukaryotic members of the pLGIC superfamily (Panicker et al., 2004).
Muscle-AChR mutagenesis: desensitization
We were also interested in determining the effect of the M1–M2 linker mutations on the kinetics of desensitization. This interest was motivated in part because a naturally occurring mutation to this region in the GlyR α1 subunit has been shown to affect the time course of desensitization and lead to hyperekplexia in humans (Saul et al., 1999). To this end, we estimated the rates of entry into and recovery from desensitization for all muscle AChR M1–M2 linker mutants studied above.
The comparison of mutant and wild-type desensitization behavior of the muscle AChR is complicated by the cell-to-cell variability in its kinetics (Franke et al., 1993). Wild-type entry-into-desensitization time courses generally fell into two categories: those best fit with one exponential component (as judged visually) and those best fit with two (Table 1). Time courses that were best fit with a single exponential component had an average decay half-time of 15.4 ± 2.4 ms, whereas those best fit with two had an average of 45.8 ± 7.7 ms. Comparing these half-times of decay, the one-component time courses decayed faster than those best fit with two components by a factor of 3.0 ± 0.3. Eight of 13 M1–M2 linker mutants exhibited only the faster-type exponential component observed for the wild-type AChR; thus, the entry-into-desensitization time course of these mutants was faster (Fig. 3; Table 1). Furthermore, some of these mutations seemed to speed up the time course of entry into desensitization to rates even faster than the single exponential component of the wild-type. The most extreme effects were seen for the α1 G–2′A and α1 T–6′G constructs, which desensitized faster by factors of 2.5 ± 0.4 and 2.0 ± 0.2, respectively (only patches giving rise to mono-exponential decays were considered in this comparison).
Figure 3.
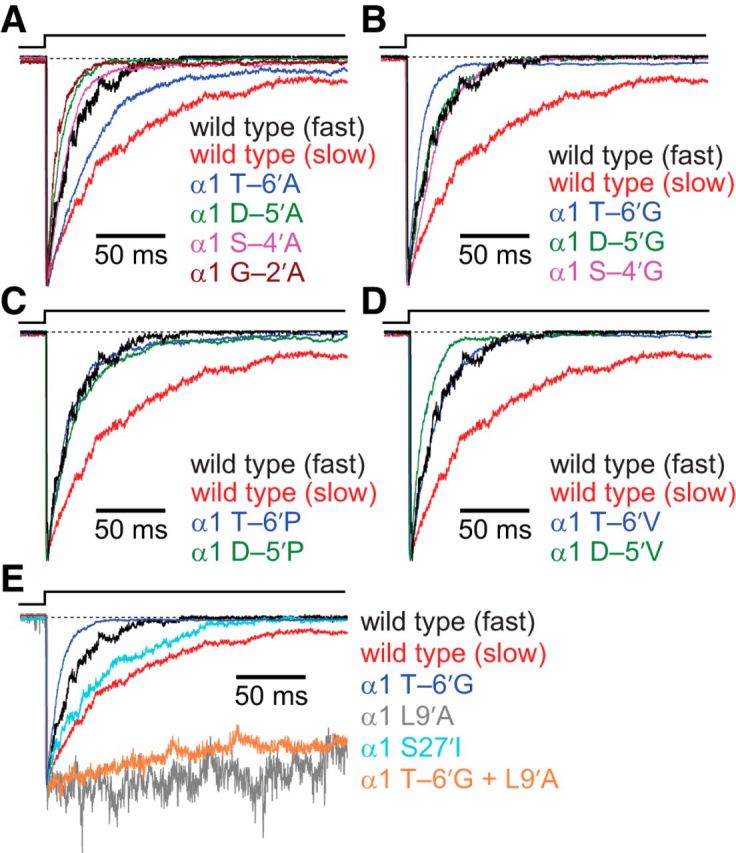
Effects of M1–M2 linker mutations on entry into desensitization of the muscle AChR. Alanine (A), glycine (B), proline (C), and valine (D) scans were performed on the M1–M2 linker of the AChR α1 subunit, as for Figure 2. The concentration of ACh during the 2 s ligand pulses was 1 mm, and the membrane potential was held constant at −80 mV. All recordings were performed using the outside-out configuration. In all panels, two traces exemplifying the fast-type (mono-exponential) and slow-type (double-exponential) time courses recorded from the wild-type channel are shown in black and red, respectively (see also Table 1). All other (normalized) traces illustrate the average behavior of each construct. Mutations to the M1–M2 linker tended to result in constructs with kinetics of entry into desensitization similar to those of the faster wild-type behavior. E, Comparison of entry into desensitization time courses of M1–M2 linker, M2 segment, and M2–M3 linker mutants. Recordings from constructs bearing the α1 L9′A mutation had small peak amplitudes, most likely because of low levels of channel expression in the patches of membrane that were successfully assayed; hence, upon normalization, these traces appear noisier. As was the case for deactivation, the α1 L9′A + T-6′G double mutant construct had a time course of entry into desensitization similar to that of the construct bearing the α1 L9′A mutation alone. In all panels, a dashed line represents the current baseline.
To obtain some perspective on the relative size of the effects of mutations to the M1–M2 linker on the rate of entry into desensitization, we again turned to the M2 segment and the M2–M3 linker (Fig. 3E; Table 2). Although the α1 S27′I mutation (in the M2–M3 linker) had little impact, the α1 L9′A mutation (in M2) greatly slowed down entry into desensitization, suggesting that the effects of the M1–M2 linker mutations are comparatively small. It is not surprising, then, that the desensitization kinetics of the α1 L9′A + T–6′G double mutant closely resembled those of the α1 L9′A single mutant. Because we also found recovery from desensitization to be largely unaffected by M1–M2 linker mutations (Fig. 4; Table 1), we conclude that mutations to the M1–M2 linker do not strongly affect the kinetics of conformational transitions involving the desensitized state. As is the case for deactivation, it seems that the rearrangement of the muscle-AChR M1–M2 linker during desensitization is more limited than that undergone by the M2 segment.
Muscle-AChR mutagenesis: response to synaptic-like stimulation
A physiologically more relevant way of probing the effect of a mutation on the function of a neurotransmitter-gated ion channel is to test its response to synaptic-like stimulation. To this end, we analyzed the responses of wild-type and M1–M2 linker mutant AChRs to high-frequency (50 Hz) trains of brief (1 ms) pulses of saturating ACh (1 mm). We found that, with the exception of the α1 T–6′A mutant, all mutant constructs exhibited more pronounced peak-response depression than the wild-type AChR (Fig. 5). Because desensitization can contribute to the receptor's response to repetitive stimulation (Elenes et al., 2006, 2009; Papke et al., 2011), the relatively strong peak-response depression of the mutants is consistent with their somewhat faster desensitization kinetics. It is interesting to note that, even though the M1–M2 linker mutations had relatively small effects when deactivation and desensitization were evaluated as separate phenomena, these small changes manifested as larger changes when channel kinetics were probed with more complex (and physiologically more relevant) pulse protocols.
Figure 5.
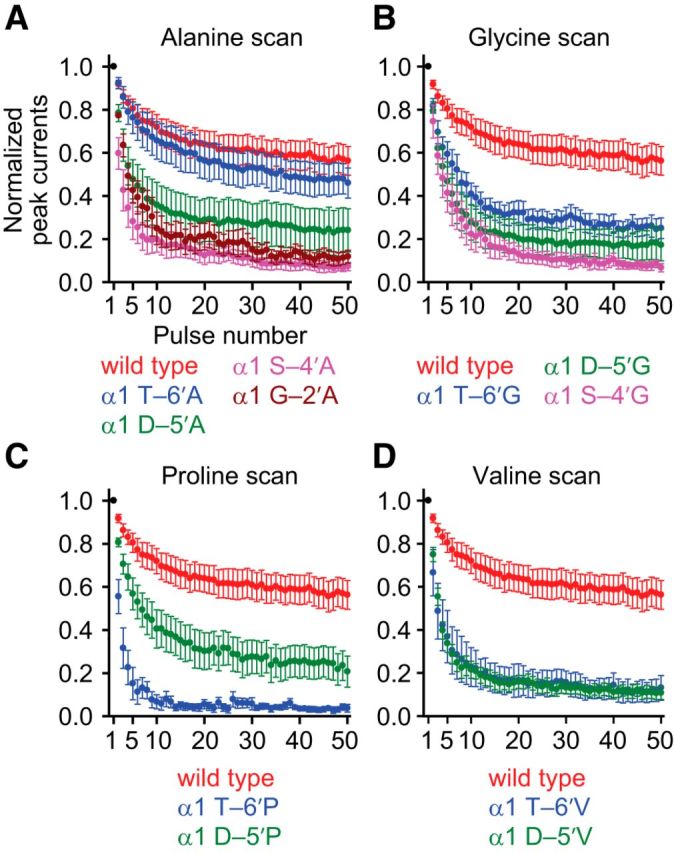
Effects of M1–M2 linker mutations on the response to repetitive stimulation of the muscle AChR. Alanine (A), glycine (B), proline (C), and valine (D) scans were performed on the M1–M2 linker of the AChR α1 subunit, as for Figures 2–4. The concentration of ACh during the 1 ms ligand pulses was 1 mm, and the membrane potential was held constant at −80 mV. All recordings were performed using the outside-out configuration. Axis labels are the same for all four panels. Data points are the average values (normalized to the peak-current response to the first pulse of ACh, in black) obtained from several patches (between 4 and 14). Error bars indicate the corresponding SE. For all four panels, the train responses exhibited peak-response depression consistent with the kinetics shown in Figures 2–4. Interestingly, some M1–M2 linker mutations have a large effect on the response to repetitive stimulation despite only having a relatively small effect on deactivation and desensitization.
α3β4 AChR mutagenesis
It has recently been proposed that the cysteine at position –4′ of the M1–M2 linker of the AChR α3 subunit (Fig. 1) serves as a site of post-translational redox modulation of channel function. Certainly, the oxidation of this cysteine was suggested to cause α3β4 AChRs to enter a refractory state upon repetitive stimulation (Campanucci et al., 2008) that was deemed to be distinct from the well-characterized desensitized state in that channels do not recover even after prolonged incubations in the absence of agonist. Furthermore, this unprecedented functional effect was proposed to be potentially critical to the pathophysiology of diabetic neuropathy (Campanucci et al., 2010; Krishnaswamy and Cooper, 2012). Importantly, this M1–M2 linker position has implications for other neuronal nicotinic AChRs, as well: mutating the residue at position –4′ of the α4 subunit from cysteine to serine was found to lower the expression levels of α4β2 receptors without affecting the potency of epibatidine or the affinity for nicotine (Amici et al., 2012).
To investigate the involvement of position –4′ in gating and desensitization of neuronal AChRs, we studied the functional effect of a series of mutations at this position of the human α3 subunit in α3β4 receptors. The stoichiometry of this AChR is not unique, with both (α3)2(β4)3 and (α3)3(β4)2 combinations being possible, although in HEK-293 cells, the (α3)3(β4)2 stoichiometry is expected to predominate (Krashia et al., 2010). Because of the rapid rundown of these channels in excised patches (Sivilotti et al., 1997; Nelson and Lindstrom, 1999; Papke et al., 2011), we resorted to using the whole-cell configuration. Although the slower rates of solution exchange that can typically be achieved in this configuration of the patch-clamp technique undermine the usefulness of these experiments when it comes to quantifying the kinetic behavior of ligand-gated ion channels, we nevertheless consider them useful for gaining qualitative insight into the effects of mutations. Because oxidation of the cysteine at position –4′ of the α3 subunit was reported to lead to the irreversible loss of channel function, we reasoned that mutating this cysteine to other amino acids could evoke a similar phenomenon. Indeed, side-chain mutations are expected to mimic, and in most cases even to exceed, the chemical changes that accompany the oxidation of the free thiol of cysteine (-SH) to the sulfenic- (-SOH), sulfinic- (-SO2H), or sulfonic-acid (-SO3H) form of the side chain.
Mutating the cysteine at position –4′ of the α3 subunit to alanine, leucine, methionine, glutamine, or serine made little to no difference to the kinetics of deactivation, recovery from desensitization or entry into desensitization of α3β4 AChRs (Fig. 6). Of most relevance here, the responses to trains of repetitive stimulation (4 or 30 pulses per minute) were also wild-type-like (Fig. 6D,E). There are two evident possibilities: one is that the oxidized thiol is involved in very specific interactions with its microenvironment that are not readily mimicked by the tested amino acid substitutions; another one is that oxidation leads to the formation of disulfide bonds between neighboring –4′ cysteines or to the formation of mixed disulfides between these cysteines and small intracellular thiol groups (such as glutathione), neither of which is expected to be mimicked by conventional amino-acid mutagenesis. More experimental work is necessary to understand the discrepancy between the lack of effect of mutations and the extreme effect of thiol oxidation.
α1 GlyR M1–M2 mutagenesis: deactivation
To determine whether mutations to the M1–M2 linker affect the kinetics of gating or desensitization in pLGICs other than the AChR, threonine and alanine scans were performed on the M1–M2 linker of the (homomeric) human α1 GlyR, between Asn 245 (–7′; Fig. 1C) and Pro 250 (–2′), and the mutant constructs were assayed using the same experimental procedures that were used for the muscle AChR. The results from these scans show that most tested mutations do not affect the rate of deactivation (Fig. 7A,B; Table 3). One notable exception is the A–3′T mutation, which (comparing decay half-times) slowed deactivation by a factor of 3.7 ± 0.4 (Fig. 7B).
Figure 7.
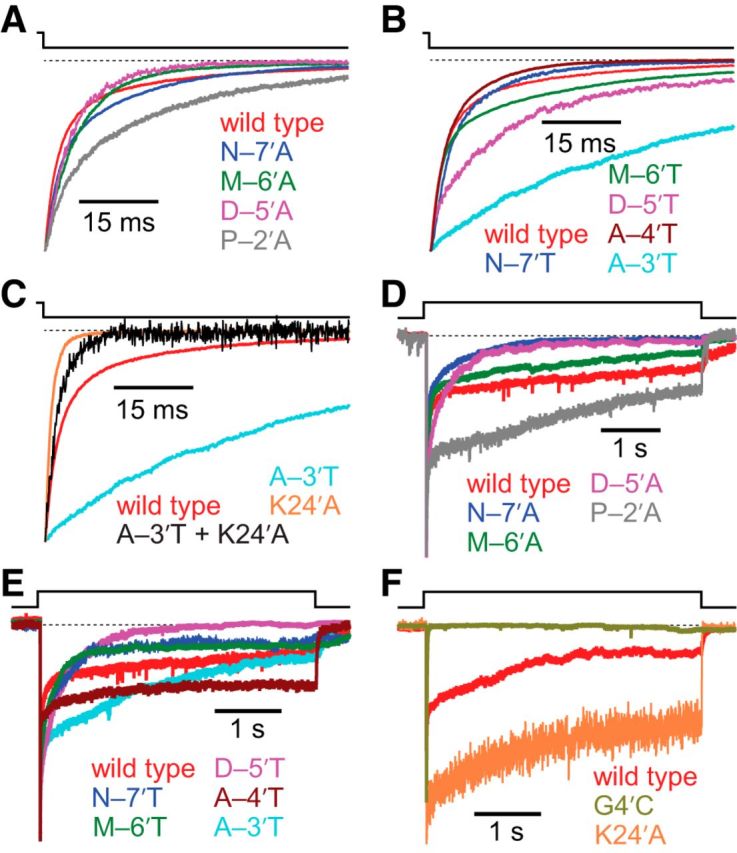
Effects of M1–M2 linker mutations on deactivation and entry into desensitization of the α1 GlyR. Alanine and threonine scans were performed on the M1–M2 linker of the α1 GlyR, from position –7′ to position –2′ (Fig. 1). The concentration of Gly during the ligand pulses was 10 mm, and the membrane potential was held constant at −80 mV. All recordings were performed using the outside-out configuration. Alanine-scan (A) and threonine-scan (B) time courses of deactivation. Most mutations to the M1–M2 linker had little to no effect on deactivation kinetics; the most notable exception was the A–3′T mutation, which slowed the deactivation time course down by a factor of 3.7 ± 0.4. C, Comparison of deactivation time courses of the A–3′T M1–M2 linker mutant and the K24′A M2–M3 linker mutant. Because the A–3′T + K24′A double mutant deactivated with a time course more similar to that of the K24′A single mutant than to that of the wild-type or the A–3′T single mutant, we infer that the loss-of-function K24′A mutation has a larger effect than the gain-of-function A–3′T mutation. Alanine-scan (D) and threonine-scan (E) time courses of entry into desensitization. The desensitization time courses of wild-type GlyRs are highly complex, typically requiring three exponential components (Table 3) to achieve a good fit; this complexity was observed for all studied M1–M2 mutant constructs, too. The time courses also exhibited considerable cell-to-cell variability, such that recordings from some cells required two or four exponential components, instead. Hence, although the data presented here exemplify the average behavior of the wild-type and mutant constructs, they do not capture the full range of responses observed. F, Desensitization time courses of wild-type, and M2 segment and M2–M3 linker mutants. A comparison with the time courses in D and E reveals that the G4′C M2 mutant desensitizes faster, and the K24′A M2–M3 linker mutant desensitizes more slowly, than any of the tested M1–M2 linker mutants. In all panels, a dashed line indicates the current baseline.
Table 3.
Kinetic parameters of M1–M2 linker mutants of the α1 GlyRa
| Mutation | τdeactivation (ms) | τentry into desensitization (ms) | τrecovery from desensitization (s) |
|---|---|---|---|
| Wild type | 3.95 ± 0.48 (0.544 ± 0.036) | 4.92 ± 1.08 (0.448 ± 0.044) | 2.80 ± 0.27 |
| 28.8 ± 3.5 | 100 ± 22 (0.274 ± 0.045) | ||
| (21/29) | 1,380 ± 420 (0.278 ± 0.030) | ||
| (15/38) | |||
| N–7′A | 4.17 ± 1.37 (0.475 ± 0.079) | 4.92 ± 2.73 (0.353 ± 0.098) | 1.90 ± 0.17 (0.845 ± 0.072) |
| 21.8 ± 5.5 | 45.3 ± 13.1 (0.337 ± 0.133) | 13.5 ± 7.6 | |
| (6/6) | 609 ± 165 (0.310 ± 0.090) | ||
| (4/6) | |||
| N–7′T | 3.09 ± 0.68 (0.689 ± 0.061) | 3.59 ± 0.83 (0.298 ± 0.055) | 1.42 ± 0.23 (0.748 ± 0.148) |
| 12.8 ± 2.6 | 43.4 ± 18.9 (0.293 ± 0.090) | 5.98 ± 2.53 | |
| (6/6) | 1,140 ± 470 (0.409 ± 0.096) | ||
| (5/7) | |||
| M–6′A | 4.96 ± 0.26 (0.701 ± 0.035) | 4.98 ± 1.21 (0.248 ± 0.042) | 6.41 ± 2.40 |
| 15.2 ± 1.3 | 54.2 ± 14.9 (0.336 ± 0.084) | ||
| (3/3) | 962 ± 299 (0.416 ± 0.082) | ||
| (6/7) | |||
| M–6′T | 1.30 ± 0.37 (0.596 ± 0.193) | 3.30 ± 2.44 (0.239 ± 0.117) | 1.15 ± 0.16 (0.692 ± 0.100) |
| 15.7 ± 5.9 | 33.3 ± 3.9 (0.172 ± 0.005) | 5.94 ± 1.57 | |
| (3/4) | 369 ± 77 (0.589 ± 0.112) | ||
| (2/5) | |||
| D–5′A | 3.35 ± 0.49 (0.614 ± 0.037) | 21.7 ± 10.8 (0.491 ± 0.113) | 2.58 ± 0.30 |
| 12.1 ± 1.6 | 265 ± 128 | ||
| (3/6) | (3/6) | ||
| D–5′T | 8.81 ± 3.58 (0.613 ± 0.126) | 17.5 ± 10.5 (0.193 ± 0.036) | 1.71 ± 0.14 |
| 40.7 ± 16.2 | 238 ± 30 (0.382 ± 0.026) | ||
| (4/5) | 878 ± 8 (0.425 ± 0.048) | ||
| (3/6) | |||
| A–4′T | 2.77 ± 0.90 (0.581 ± 0.065) | 6.25 ± 2.5 (0.194 ± 0.062) | 0.102 ± 0.012 (0.518 ± 0.026) |
| 9.78 ± 1.86 | 69.1 ± 20.1 (0.306 ± 0.117) | 3.62 ± 0.46 | |
| (6/6) | 1,440 ± 750 (0.500 ± 0.081) | ||
| (3/6) | |||
| A–3′T | 12.8 ± 3.0 (0.255 ± 0.072) | 3.42 ± 0.56 (0.346 ± 0.100) | 0.114 ± 0.020 (0.495 ± 0.046) |
| 50.5 ± 4.1 | 36.7 ± 6.1 (0.148 ± 0.026) | 2.40 ± 0.47 | |
| (6/9) | 3,840 ± 2,600 (0.506 ± 0.079) | ||
| (4/8) | |||
| P–2′A | 3.70 ± 0.52 (0.307 ± 0.042) | 59.0 ± 20.6 (0.247 ± 0.035) | 0.247 ± 0.052 (0.441 ± 0.040) |
| 35.7 ± 6.7 | 6,010 ± 1,430 | 6.52 ± 0.83 | |
| (5/7) | (6/9) |
aAll errors are SEs. The fractions in parentheses have the same meaning as in Table 2. Each time course was obtained from a different patch of membrane.
To get a basis of comparison for the size of this effect, we engineered an M2 segment mutation (G4′C) and an M2–M3 linker mutation (K24′A). Both mutations had clear effects on the kinetics of deactivation (Table 4): comparing half-times of decay, the G4′C mutation sped up deactivation by a factor of 5.9 ± 1.5, whereas the K24′A mutations sped it up by a factor of 7.2 ± 1.7. Thus, the effects of the tested M2 α-helix and M2–M3 linker mutations seem to be larger than that of the A–3′T mutation. However, because both of these mutations sped up deactivation and the A–3′T mutation slowed it down, a direct comparison of the magnitudes of these effects is not insightful. To address this problem, we engineered a double mutant construct containing both the A–3′T and K24′A mutations. Because this construct had deactivation kinetics closely resembling those of the K24′A alone (Fig. 7C; Table 4), and assuming that the effects of the individual mutations are additive, we conclude that the magnitude of the effect of the A–3′T on deactivation is comparatively small. Consequently, as is the case for the AChR, the changes experienced in the microenvironment around the M1–M2 linker of the α1 GlyR during deactivation are expected to be small compared with those around the M2 segment or the M2–M3 linker.
Table 4.
Kinetic parameters of other mutants of the α1 GlyRa
| Construct | τdeactivation (ms) | τentry into desensitization (ms) | τrecovery from desensitization (s) |
|---|---|---|---|
| Wild type | 3.95 ± 0.48 (0.544 ± 0.036) | 4.92 ± 1.08 (0.448 ± 0.044) | 2.80 ± 0.27 |
| 28.8 ± 3.5 | 100 ± 22 (0.274 ± 0.045) | ||
| (21/29) | 1380 ± 420 (0.278 ± 0.030) | ||
| (15/38) | |||
| G4′C | 1.66 ± 0.37 | 11.1 ± 3.6 | — |
| (5/9) | (5/9) | ||
| K24′A | 1.36 ± 0.22 | 2320 ± 693 | 0.281 ± 0.038 |
| (14/14) | C = 0.41 ± 0.05 | ||
| (9/15) | |||
| K24′A + | 3.3 ± 0.42 | 1650 ± 940 (0.498 ± 0.046) | 0.227 ± 0.046 |
| A–3′T | (7/7) | 21,700 ± 8700 (0.278 ± 0.053) | |
| C = 0.22 ± 0.08 | |||
| (4/10) | |||
| GlyRM3M4 311S | 4.91 ± 0.64 (0.451 ± 0.036) | 9.34 ± 2.24 (0.321 ± 0.087) | 2.05 ± 0.23 |
| 28.0 ± 3.3 | 165 ± 44 (0.328 ± 0.091) | ||
| (16/18) | 1130 ± 390 (0.352 ± 0.076) | ||
| (8/20) | |||
| GlyRM3M4 311A | 4.28 ± 0.79 (0.394 ± 0.046) | 7.52 ± 1.84 (0.293 ± 0.048) | 2.03 ± 0.12 |
| 23.8 ± 1.8 | 86.8 ± 21.4 (0.374 ± 0.050) | ||
| (12/15) | 492 ± 128 (0.334 ± 0.073) | ||
| (8/17) | |||
| GlyRM3M4 311H | 7.07 ± 0.73 | 7.24 ± 1.18 (0.406 ± 0.031) | 2.02 ± 0.23 |
| (9/18) | 68.7 ± 9.2 | ||
| (10/19) | |||
| H311S | 5.08 ± 1.34 (0.515 ± 0.063) | 7.12 ± 3.06 (0.286 ± 0.035) | 1.80 ± 0.14 |
| 17.2 ± 3.1 | 45.8 ± 16.0 (0.335 ± 0.021) | ||
| (6/9) | 240 ± 78 (0.334 ± 0.073) | ||
| (5/9) | |||
| GlyRM3M4 8xAla | 16.0 ± 5.56 (0.686 ± 0.037) | 28.0 ± 17.7 (0.592 ± 0.0075) | 0.702 ± 0.076 |
| 96.4 ± 27.9 | 166 ± 83 (0.181 ± 0.025) | ||
| (6/13) | 8210 ± 4570 (0.227 ± 0.065) (6/10) | ||
| GlyRM3M4 311H + K24′A | 1.09 ± 0.24 | 109 ± 22 (0.565 ± 0.105) | 0.164 ± 0.030 |
| (4/6) | 2490 ± 1540 | ||
| (5/6) | |||
| GlyRM3M4 8xAla + G4′C | 1.08 ± 0.18 | 2.10 ± 0.25 | — |
| (7/7) | (8/8) |
aAll errors are SEs. The fractions in parentheses have the same meaning as in Tables 2 and 3. Unlike all other tested mutants, the two constructs containing the GlyR α1 K24′A mutation exhibited desensitization time courses that seemed to reach a non-zero current level at steady state, as judged from the channel's sustained currents at the end of 1 minute applications of saturating Gly. Steady-state current levels were normalized to their time courses' peak amplitude values to allow averaging across patches; these average values are presented in the table as “C.” The α1 GlyR G4′C mutant exhibited an unusually fast, and apparently irreversible, rundown in the outside-out configuration. As a result, it was not possible to determine its rate of recovery from desensitization. Because of its poor functional expression, it was also not possible to quantify the rate of recovery from desensitization of the α1 GlyRM3M4 8xAla + G4′C construct. Each time course was obtained from a different patch of membrane. —, Not applicable.
α1 GlyR M1–M2 mutagenesis: desensitization and train responses
Comparison of desensitization kinetics across mutants was complicated by the complexity of the time courses, which typically required three exponential components to achieve a good fit (Fig. 7D,E). Therefore, in this case, it would not be sensible to use a simplifying metric, such as the half-time of decay or the weighted time constant, to make comparisons among mutants; too much detail would be lost. Thus, for the wild-type and mutants, we report in full detail the time constants and associated relative amplitudes averaged across patches (Table 3), and we do not attempt to compare these time courses using simplified empirical parameters.
Although most mutations in the scan had no apparent effect on desensitization, there were a few exceptions. The P–2′A mutation seemed to eliminate the fastest component from most patches, causing slower desensitization (Fig. 7D; Table 3), whereas the N–7′A and M–6′T mutations seemed to significantly speed up the slowest component, leading to faster desensitization (Fig. 7D,E; Table 3). The D–5′A mutant seemed to combine these two effects: the fastest component of desensitization was eliminated, and the slowest component was faster than that of the wild-type.
To gain insight into the relative size of these effects, we turned again to the G4′C and K24′A mutants. The G4′C mutation sped up desensitization, essentially leaving only the fast component (Fig. 7F; Table 4). The K24′A mutation was unusual in that it slowed down desensitization to a large extent while also speeding up deactivation greatly (Fig. 7C,F; Table 4). The effects of the K24′A and G4′C mutations demonstrate that mutations to the M2 segment and M2–M3 linker can have much more extreme effects on desensitization than any of the tested M1–M2 linker mutations. We also found that recovery from desensitization is largely unaffected by mutations in this intracellular linker (Fig. 8A,B; Table 3). Thus, as is the case for the conformational rearrangements involved in deactivation, these results imply that the M1–M2 linker is unlikely to undergo large changes in microenvironment during the transitions to and from the desensitized state.
To estimate the effects of the M1–M2 linker mutations on the response of the α1 GlyR to physiological stimulation, we also exposed the wild-type and mutant receptors to 50 Hz trains of 1 ms pulses of 10 mm Gly. In most cases, M1–M2 linker mutants displayed a wild-type-like behavior (Fig. 8C,D). One notable exception, however, is the P–2′A mutant, which showed much less peak-current depression over the course of the series of pulses (Fig. 8C); this reduced depression can be explained by the slower time course of desensitization of this construct (Fig. 7D). Thus, the P–2′A mutation provides another example of the notion that, even though a mutation might not have a large effect on the kinetics of deactivation and desensitization, it can still have a sizable impact on the response to high-frequency, synaptic-like stimulation.
α1 GlyR M3–M4 mutagenesis: deactivation
Mutations to the M3–M4 linker of the AChR, the γ-aminobutyric-acid type-A receptor (GABAAR), the serotonin receptor (5HT3R), and the α3 homomeric GlyR have been reported to affect the kinetics of gating and desensitization (Swope et al., 1999; Breitinger et al., 2002, 2009; Baptista-Hon et al., 2013), but quantitative insight into the magnitude of these effects has been largely lacking. Encouraged by the finding that pLGICs containing much shorter M3–M4 linkers, as a result of naturally occurring variations (Bocquet et al., 2009; Gonzalez-Gutierrez and Grosman, 2010, 2012) or engineered mutations (Jansen et al., 2008; McKinnon et al., 2012), display all the functional properties of a full-length eukaryotic channel (i.e., they activate, deactivate, desensitize, and recover from desensitization), we set out to study the effect of major changes in the amino acid sequence of this intracellular linker. Although our efforts to study the M3–M4 linker of the muscle AChR have thus far failed (we found the levels of functional expression to be extremely low), we have succeeded in generating a number of drastic M3–M4 linker mutants of the α1 GlyR (Fig. 9A; Table 4).
We began by replacing a 75-residue stretch of amino acids of the GlyR α1 subunit (starting with H311 and ending with K385) with a heptapeptide (SQPARAA) consisting of two residues of the M3–M4 linker (SQ) and the first five residues of the M4 α-helix (PARAA) of the bacterial homolog GLIC (Tasneem et al., 2005), as originally done for the 5-HT3A and GABAC receptors (Jansen et al., 2008), and subsequently, for the α1 GlyR (Moroni et al., 2011); hereafter, we refer to this construct as “GlyRM3M4 311S” (the naming scheme for all studied M3–M4 constructs is presented in Fig. 9A). We do not know what structure this heptapeptide adopts when spliced into the sequence of the α1 GlyR, nor do we know whether the PARAA stretch becomes the new N-terminal end of the M4 α-helix or it remains in the cytosol as part of a cytosolic linker. These uncertainties notwithstanding, we chose this heptapeptide because its presence in place of the much longer, native M3–M4 linker yields functional channels (Moroni et al., 2011). Because the GlyRM3M4 311S construct contains a predicted phosphorylation site (S311, www.cbs.dtu.dk/services/NetPhos/) (Blom et al., 1999) that could plausibly impact channel kinetics, we also engineered constructs with mutations to this residue (GlyRM3M4 311A, GlyRM3M4 311H, and the single-point mutant H311S on the wild-type background; Fig. 9A) and a construct with an alanine octapeptide replacing most of the linker (GlyRM3M4 8xAla; Fig. 9A).
The GlyRM3M4 311S and GlyRM3M4 311A constructs exhibited slightly slower kinetics of deactivation than those of the wild-type: time courses of both mutants had two components of decay, with the slower component being slightly more prominent than that of the wild-type (Fig. 9B; Table 4). The GlyRM3M4 311H construct most often only required a one-component exponential function to achieve a good fit of the deactivation time course compared with the two components required for the wild-type. Comparison of deactivation half-times showed that the GlyRM3M4 311H mutant decays faster than the wild-type by a factor of 1.4 ± 0.3. It is remarkable that the deactivation time course is sensitive to the residue at position 311 despite its relative insensitivity to the substitution of a hexapeptide for the 72-residue stretch between position 311 and the M4 α-helix; perhaps, position 311 is close enough to the C-terminal end of the M3 α-helix to directly impact its orientation in the membrane. To further investigate this possibility, we engineered a point mutation at this position while leaving the rest of the linker intact (H311S; Fig. 9A; Table 4). This construct deactivated with a time course similar to that of the wild-type (Fig. 9B), suggesting that the effect of this position on channel function depends on whether the M3–M4 linker is truncated. Finally, we also measured the deactivation kinetics of the GlyRM3M4 8xAla mutant and found that (comparing half-times of decay) the deactivation time course of this construct is slower than that of the wild-type by a factor of 2.3 ± 0.4 (Fig. 9B; Table 4). To assess the magnitude of this effect, we engineered the G4′C mutation (which we found to speed up deactivation when introduced in the wild-type background) on the background of the GlyRM3M4 8xAla construct and found that the average deactivation time constant of this multiple mutant is similar to that of the G4′C single mutant (Table 4). We conclude that the effect of the extensive mutations to the M3–M4 linker on deactivation is small compared with the effect of the G4′C single-residue mutation.
α1 GlyR M3–M4 mutagenesis: desensitization
Consistent with the notion that post-translational modification of the M3–M4 linker can impact desensitization kinetics (Swope et al., 1999), we found that most mutations to this linker have an effect on the time course of entry into desensitization and that some of these effects are large (Fig. 9C). In most cases, it was the time constant and the relative amplitude of the slowest component that were affected (Table 4). Although the desensitization time courses of the GlyRM3M4 311S construct were usually similar to those of the wild-type, the desensitization time courses of the GlyRM3M4 311A and the H311S single mutant constructs had significantly faster slow components of desensitization. The effect of the GlyRM3M4 311H mutation is even more striking in that it appeared to completely eliminate the slowest component of desensitization from most patches (Table 4). Visually, it is clear that all three constructs (GlyRM3M4 311A, GlyRM3M4 311H, and H311S) desensitized faster than the wild-type (Fig. 9C).
To gain a basis of comparison for the extent to which desensitization was sped up in the GlyRM3M4 311H construct, we estimated the degree to which this effect was offset by the K24′A mutation, a mutation that (when engineered on the background of the wild-type) slows down desensitization greatly (Fig. 7F; Table 4). The GlyRM3M4 311H + K24′A channel exhibited desensitization kinetics intermediate between those of the K24′A single mutant and those of the GlyRM3M4 311H. Although, because of the complexity of the desensitization time course, it is unclear how one would determine which effect is larger, it is clear that the effect of the GlyRM3M4 311H is large enough to revert most of the effect of the K24′A mutation: the GlyRM3M4 311H + K24′A channel desensitizes “completely” (i.e., the currents attain a zero level at equilibrium), and the values of the two time constants required to fit the time courses are similar to those of the two slower components required to fit the wild-type's (Fig. 9C; Table 4). We conclude that the effect of the GlyRM3M4 311H on desensitization kinetics is large.
In contrast to the mutants described above, the GlyRM3M4 8xAla construct desensitized more slowly than the wild-type, with the slowest component of decay also being the most affected (Fig. 9C; Table 4). To assess the magnitude of this effect, we engineered the G4′C mutation (which we found to speed up desensitization when introduced in the wild-type background; Fig. 7F) on the background of the GlyRM3M4 8xAla construct and found that this multiple mutant desensitizes even faster than the G4′C single mutant (Table 4). Thus, as was the case for deactivation, the kinetics of desensitization of the GlyRM3M4 8xAla + G4′C construct were closer to those of the G4′C single-residue mutant than to those of the extensively mutated GlyRM3M4 8xAla channel. Because of its low functional expression levels, we could not assess the kinetics of recovery from desensitization of the GlyRM3M4 8xAla + G4′C construct (Fig. 9D).
α1 GlyR: cell-to-cell variability of desensitization time courses
As previously reported (Papke et al., 2011), there are multiple levels of complexity in the desensitization time courses of the α1 GlyR. Certainly, the number of exponential components necessary to achieve a good fit, as well as the values of the time constants and their relative amplitudes, change from cell to cell (Fig. 10A). Even a cursory inspection of the recordings reveals that, in some patches, entry into desensitization of the wild-type α1 GlyR was multiphasic, with a rapid decay to a non-zero value from which further decay proceeded very slowly (Fig. 10A, red trace), whereas in other patches desensitization was as fast as that of the muscle AChR (Fig. 10A, green trace). Like others (Gentet and Clements, 2002), we suspected that this wide range of behaviors might be the result of cell-to-cell variability in the extent of post-translational modifications to the (long) intracellular M3–M4 linker, and we wondered whether its removal would eliminate the variability in desensitization kinetics.
Figure 10.

Heterogeneity of α1 GlyR desensitization kinetics. A, Desensitization time courses recorded from three different patches of membrane containing the wild-type α1 GlyR. The traces in blue and green required two exponential components to achieve a good fit. However, the slower time constant of the exponential decay fitted to the trace in blue was an order of magnitude slower than that required to fit the time course in green. The time course in red required four exponential components to achieve a good fit. The differences between the displayed traces are indicative of the typical cell-to-cell variation observed with these receptors. In all cases, a fast-decaying component (4.92 ± 1.08 ms; Table 4) that would probably be missed in whole-cell recordings (even if a rapid, θ-tube-type perfusion of ligand were used) is present. B, Desensitization time courses recorded from three different patches of membrane containing the GlyRM3M4 311S construct. The trace in red required two exponential components to achieve a good fit, and the traces in green and blue required three. C, Desensitization time courses recorded from three different patches of membrane containing the GlyRM3M4 8xAla construct. The traces in green and red required three exponential components to achieve a good fit, whereas the trace in blue required four. For all three panels, the concentration of Gly during the ligand pulses was 10 mm, and the membrane potential was held constant at −80 mV. All recordings were performed using the outside-out configuration. In all panels, a dashed line indicates the current baseline. To allow for an easier visual comparison of the time courses across patches, all current traces were normalized to their peak values.
The time courses of entry into desensitization recorded from the deletion mutant α1 GlyRM3M4 311S were as variable as those recorded from the wild-type channel (Fig. 10B). More specifically, of a total of 20 outside-out patches of membrane containing this mutant construct, the recordings obtained from three patches required one exponential component to achieve a good fit; those from seven patches required two components; those from eight patches required three components; and those from two patches required four components. It could be argued that the phosphorylation state of the serine at position 311 (a likely phosphorylation site; www.cbs.dtu.dk/services/NetPhos/) (Blom et al., 1999) underlies the observed kinetic variability, but the GlyR M3M4 8xAla construct (which lacks any such intracellular consensus sequences) appeared similarly variable (Fig. 10C). Indeed, time courses recorded from this construct required two exponential components in two patches, three in six patches, and four in two patches. We infer that the cell-to-cell variability in the time course of entry into desensitization must stem from variability in regions of the protein other than this large intracellular domain.
The fast exponential component of α1 GlyR desensitization
One of the most enigmatic features of the desensitization time course of the α1 GlyR is its fastest exponential component, the time constant and relative amplitude of which (in our experiments) were as fast as 0.49 ms and as high as 70%, respectively (a similar fast component can also be frequently seen in desensitization time courses of GABAARs but not in those of the cation-selective muscle AChR) (e.g., Papke et al., 2011). Several ideas have been put forward to account for the occurrence of this rapid decay in the currents, ranging from the notion that it represents an artifact caused by the transient depletion/accumulation of Cl− in the tip of the patch pipette (which would cause a shift of the Cl− equilibrium potential and an ensuing decrease of the single-channel current) (Karlsson et al., 2011) to the proposal that it represents a genuine property of the desensitization phenomenon that becomes more prevalent at high receptor densities through, perhaps the formation of channel clusters (Legendre et al., 2002). A number of observations that we made during our experiments support the idea that this fast-decaying component reflects true desensitization. First, the concentration of Cl− in the pipette was high: 42 mm for all traces shown in Figures 7D–F, 9C, and 10, and 154 mm for the trace shown in Figure 11A. The fact that inward currents recorded in the outside-out configuration displayed a fast-decaying component even in the presence of these high Cl− concentrations in the pipette runs counter to the notion that transient Cl− depletion underlies this phenomenon; increasing the concentration of Cl− in the pipette would reduce the effect of the local depletion of Cl− (the recorded currents were carried by Cl− flowing out of the pipette). Second, the size of the peak current did not dictate the relative amplitude of the fast component, as would be expected from a simple relationship between the size of the inward currents and the degree of Cl− depletion in the pipette. This lack of correspondence is shown with two example traces in Figure 11B: the peak-current value corresponding to the blue trace is 530 pA, and its fastest component (τ = 1.01 ± 0.05 ms) accounts for 53% of the total decay, whereas the peak-current value of the red trace is nearly twice as large (930 nA), and its fastest component (τ = 1.28 ± 0.08 ms) accounts for only 15% of the total decay. Third, a mutation (K24′A, in the M2–M3 linker) eliminated the fast component from the desensitization time course (Fig. 7F), which leads us to speculate that the M2–M3 linker might hold the structural key to this fast phase of desensitization. Fourth, using patch-pipettes of the same overall shape and dimensions, and the same intracellular and extracellular solutions, we have never observed such a fast-decaying component in desensitization time courses recorded from the (cation-selective) muscle AChR (Papke et al., 2011).
Figure 11.

The fast component of entry into desensitization of the α1-GlyR. A, Current decay in the presence of ∼150 mm KCl in the pipette. The fast exponential component in this outside-out patch had a time constant of 3.67 ms and accounted for 62% of the total decay. Thus, even at this high concentration of intrapipette Cl−, the fast component is a conspicuous feature of the decay time course. B, Two example traces of entry into desensitization recorded from two different outside-out patches. As was the case for all other α1-GlyR traces shown in this work (with the exception of the trace in A), the intrapipette Cl− concentration was 42 mm. As indicated in the text, although the peak-current amplitude of the trace in red was larger, the relative amplitude of the faster exponential component in this trace was smaller than that of the trace in blue. It is unlikely, then, that the fast exponential component observed in A and B reflects a decrease in the intrapipette Cl− concentration during the recording of Cl−-carried inward currents. This notion is reinforced by the identification of a mutation in the M2–M3 linker that eliminates this fast component (Fig. 7F). The concentration of Gly during the ligand pulses was 10 mm, and the membrane potential was held constant at −80 mV. In both panels, a dashed line indicates the current baseline.
Discussion
Recent developments in the pLGIC field (Campanucci et al., 2008, 2010; Jansen et al., 2008) have renewed the interest in two understudied regions of these receptor channels: the intracellular M1–M2 and M3–M4 linkers. Here, we examined the involvement of these domains in channel function using mutagenesis, macroscopic-current recordings, a variety of ligand-pulse protocols, and a method for the fast application and washout of ligand. As could be expected, we found that some mutations affect function more than others, but we were surprised to realize that the channels' multiexponential responses were often complex enough (Tables 1–4) that the effects of mutations could not be compared and classified as being “small,” “large,” or anything in between in an objective manner. Thus, in an attempt to provide a more intuitive measure of the effects of mutations than can be obtained from the comparison of time constants and amplitudes of multiexponential fits, we also engineered specific mutations in the M2 α-helix and the M2–M3 linker known to have extreme effects on the kinetics of gating and desensitization.
In the case of the M1–M2 linker, we found that the effects of mutations were invariably small relative to the effects of mutations to the M2 α-helix or the M2–M3 linker for the human muscle AChR (Figs. 2 and 3), the human α3β4 AChR (Fig. 6), and the human α1 GlyR (Fig. 7). For example, the effect of the M1–M2 linker α1 GlyR A–3′T mutation (which clearly slowed down the deactivation time course) was overwhelmed by the addition of the M2–M3 linker K24′A mutation (which sped it up), such that the double mutant behaved essentially like the construct with the K24′A mutation alone (Fig. 7C). We conclude that, compared with the M2 segment or the M2–M3 linker, the M1–M2 linker of these three receptors is unlikely to undergo large changes in conformation during gating or desensitization.
Regarding the α3β4 AChR, it could be argued that our failure to observe functional changes with mutations to the cysteine at position –4′ of the α3-subunit's M1–M2 linker may be the result of the slow perfusion that can typically be achieved in the whole-cell configuration (the behavior of these AChRs could not be studied in the outside-out configuration because the channel activity runs down upon patch excision). However, although a slow perfusion may certainly conceal minor effects (such as those observed for the muscle AChR), the irreversible loss of responsiveness upon repetitive stimulation reported for the α3β4 AChR under conditions of elevated oxidative stress (Campanucci et al., 2008, 2010; Krishnaswamy and Cooper, 2012) is far from subtle and would have been detected clearly in our whole-cell recordings. Undoubtedly, the chemical aspects of the mechanism that links the cysteine at position –4′ of the α3 AChR subunit, the increased production of reactive oxygen species, and the impairment of channel function remain to be elucidated; clearly, mutations do not seem to recapitulate the effect of cysteine oxidation. In this context, it is perhaps interesting to note that mutation of the cysteine at position –4′ of the α4 AChR subunit to serine also failed to affect the function of α4β2 receptors (Amici et al., 2012).
As for the M3–M4 linker of the α1 GlyR, we found that none of the engineered mutations had a large effect on the deactivation kinetics compared with the tested M2 segment or M2–M3 linker point mutations (Table 4). This finding is consistent with observations made in a study of another α1 GlyR M3–M4 linker deletion (Moroni et al., 2011). It is remarkable that deleting such a large stretch of amino acids leaves the kinetics of deactivation largely unaffected, especially considering that more conservative mutations to the linker in the (closely related) α3 GlyRs have been shown to have a large effect on gating (Breitinger et al., 2009). Hence, although our mutagenesis study suggests that deactivation is relatively insensitive to even extensive changes to the M3–M4 linker amino-acid sequence, there remains the possibility that this result depends on the member of the pLGIC superfamily under consideration. Mutations to this linker did, however, affect the kinetics of entry into desensitization to a degree that is comparable with the effect of mutations in the M2–M3 linker. This result supports the notion that the M3–M4 linker may act as a site of post-translational modulation of desensitization kinetics.
In addition to probing channel kinetics with the application of “simple” pulse protocols, we also applied trains of brief pulses of saturating neurotransmitter at 50 Hz, a physiologically relevant frequency. Intriguingly, we found that mutations that would have been judged to have little effect on function on the basis of the channel's responses to deactivation, entry-into-desensitization, and recovery-from-desensitization protocols, had a profound impact on the responses to 50 Hz repetitive stimulation (Figs. 5 and 8C). Most muscle-AChR M1–M2 linker mutations, for example, led to substantially more peak-current depression than was seen with the wild-type channel, very likely because of the increased rate of entry into desensitization of these mutants. Although this increase was generally modest, the higher probability of the channels entering the desensitized state (both within and between ligand pulses) (Papke et al., 2011) adds up throughout the high-frequency trains of neurotransmitter pulses leading to the progressive decline in peak amplitude. Collectively, these findings show that the response to physiological stimulation may be sensitive to small changes in channel kinetics and thus suggest that repetitive synaptic-like trains of ligand pulses should be routinely applied in mutagenesis studies of neurotransmitter-gated ion channels.
One of the most striking observations of this work is the large cell-to-cell variability in the time courses of entry into desensitization, especially for the α1 GlyR, and one of the plausible mechanisms underlying this phenomenon is variable channel phosphorylation. For the muscle AChR, it has been shown that the phosphorylation of a number of serines (Safran et al., 1987; Schroeder et al., 1991; Paradiso and Brehm, 1998) and a tyrosine (Hopfield et al., 1988; Wagner et al., 1991) in the M3–M4 linker of the δ subunit, as well as a tyrosine in the M3–M4 linker of the β1 subunit (Wagner et al., 1991), alters the kinetics of desensitization. Similarly, phosphorylation of native and recombinant GlyRs has been found to affect the kinetics of gating and desensitization (Agopyan et al., 1993; Vaello et al., 1994; Gentet and Clements, 2002), and a phosphorylation site has been identified in the M3–M4 linker of this receptor (Ruiz-Gómez et al., 1991). However, our results indicate that, although mutations to the M3–M4 linker may affect the kinetics of desensitization, they do not affect the extent to which the time courses of desensitization vary from cell to cell, even when the mutations involve extensive deletions that eliminate known and predicted phosphorylation sites (Fig. 10). We speculate that naturally occurring variations in the lipid composition (Fong and McNamee, 1986; daCosta and Baenziger, 2009; Nothdurfter et al., 2010) or perhaps even in the extent of N-glycosylation (Sumikawa and Miledi, 1989; Gehle et al., 1997; Dellisanti et al., 2007) might contribute to the observed variability. Speculating even further, it is also conceivable that pLGICs interact with other protein partners (Jones et al., 2010) and that these interactions affect the receptors' kinetic behavior in a manner that differs from cell to cell.
We also presented results that argue for the notion that the fast component of the exponential decay of the α1-GlyR inward currents during exposure to saturating Gly represents true desensitization rather than a mere decrease in the single-channel current amplitude because of the local depletion of intrapipette Cl−. Furthermore, it has been suggested that GlyR clustering increases the relative amplitude of this fast component (Legendre et al., 2002). Thus, to the extent that GlyRs form clusters in intact synapses, this fast component is likely to occur in vivo and, hence, to be physiologically relevant. On the other hand, the reported lack of a single-channel correlate for this sharp component in steady-state, cell-attached recordings (Pitt et al., 2008) is curious and makes us wonder whether the different ligand-application protocols, stationary in the cell-attached configuration versus pulse-like in the outside-out experiments, may have anything to do with this observation. Undoubtedly, further studies will be required to unveil the mechanistic origin and physiological implications of this elusive aspect of channel desensitization in GlyRs.
Footnotes
This work was supported by National Institutes of Health Grant R01-NS042169 to C.G. and Grant T32GM008276 to D.P., the Britta L. and Charles J. Wolfe Award for Diabetes Research (to D.P.), and the University of Illinois College of Medicine Craig Summer Research Fellowship (to D.P.). We thank G. Gonzalez-Gutierrez for adapting our fast-perfusion setup to perform whole-cell recordings; S. Elenes (Universidad de Colima, Colima, Mexico) for critical advice on fast-perfusion experiments; and A. Holmstrom, C. Johnson, M. Pasquini, J. Pizarek, and M. Rigby for technical assistance.
The authors declare no competing financial interests.
References
- Agopyan N, Tokutomi N, Akaike N. Protein kinase A-mediated phosphorylation reduces only the fast desensitizing glycine current in acutely dissociated ventromedial hypothalamic neurons. Neuroscience. 1993;56:605–615. doi: 10.1016/0306-4522(93)90360-R. [DOI] [PubMed] [Google Scholar]
- Amici SA, McKay SB, Wells GB, Robson JI, Nasir M, Ponath G, Anand R. A highly conserved cytoplasmic cysteine residue in the alpha4 nicotinic acetylcholine receptor is palmitoylated and regulates protein expression. J Biol Chem. 2012;287:23119–23127. doi: 10.1074/jbc.M111.328294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baptista-Hon DT, Deeb TZ, Lambert JJ, Peters JA, Hales TG. The minimum M3–M4 loop length of neurotransmitter-activated pentameric receptors is critical for the structural integrity of cytoplasmic portals. J Biol Chem. 2013;288:21558–21568. doi: 10.1074/jbc.M113.481689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;294:1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- Bocquet N, Nury H, Baaden M, Le Poupon C, Changeux JP, Delarue M, Corringer PJ. X-ray structure of a pentameric ligand-gated ion channel in an apparently open conformation. Nature. 2009;457:111–114. doi: 10.1038/nature07462. [DOI] [PubMed] [Google Scholar]
- Breitinger HG, Villmann C, Becker K, Becker CM. Opposing effects of molecular volume and charge at the hyperekplexia site α1 (P250) govern glycine receptor activation and desensitization. J Biol Chem. 2001;276:29657–29663. doi: 10.1074/jbc.M100446200. [DOI] [PubMed] [Google Scholar]
- Breitinger HG, Villmann C, Rennert J, Ballhausen D, Becker CM. Hydroxylated residues influence desensitization behaviour of recombinant α3 glycine receptor channels. J Neurochem. 2002;83:30–36. doi: 10.1046/j.1471-4159.2002.01109.x. [DOI] [PubMed] [Google Scholar]
- Breitinger HG, Villmann C, Melzer N, Rennert J, Breitinger U, Schwarzinger S, Becker CM. Novel regulatory site within the TM3–4 loop of human recombinant α3 glycine receptors determines channel gating and domain structure. J Biol Chem. 2009;284:28624–28633. doi: 10.1074/jbc.M109.043174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanucci VA, Krishnaswamy A, Cooper E. Mitochondrial reactive oxygen species inactivate neuronal nicotinic acetylcholine receptors and induce long-term depression of fast nicotinic synaptic transmission. J Neurosci. 2008;28:1733–1744. doi: 10.1523/JNEUROSCI.5130-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanucci V, Krishnaswamy A, Cooper E. Diabetes depresses synaptic transmission in sympathetic ganglia by inactivating nAChRs through a conserved intracellular cysteine residue. Neuron. 2010;66:827–834. doi: 10.1016/j.neuron.2010.06.010. [DOI] [PubMed] [Google Scholar]
- Colquhoun D, Hawkes AG. On the stochastic properties of single ion channels. Proc R Soc Lond B Biol Sci. 1981;211:205–235. doi: 10.1098/rspb.1981.0003. [DOI] [PubMed] [Google Scholar]
- daCosta CJ, Baenziger JE. A lipid-dependent uncoupled conformation of the acetylcholine receptor. J Biol Chem. 2009;284:17819–17825. doi: 10.1074/jbc.M900030200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellisanti CD, Yao Y, Stroud JC, Wang ZZ, Chen L. Crystal structure of the extracellular domain of nAChR α1 bound to α-bungarotoxin at 1.94 Å resolution. Nat Neurosci. 2007;10:953–962. doi: 10.1038/nn1942. [DOI] [PubMed] [Google Scholar]
- Elenes S, Ni Y, Cymes GD, Grosman C. Desensitization contributes to the synaptic response of gain-of-function mutants of the muscle nicotinic receptor. J Gen Physiol. 2006;128:615–627. doi: 10.1085/jgp.200609570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elenes S, Decker M, Cymes GD, Grosman C. Decremental response to high-frequency trains of acetylcholine pulses but unaltered Ca2+ fractional currents in a panel of “slow-channel syndrome” nicotinic receptor mutants. J Gen Physiol. 2009;133:151–169. doi: 10.1085/jgp.200810089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filatov GN, White MM. The role of conserved leucines in the M2 domain of the acetylcholine receptor in channel gating. Mol Pharmacol. 1995;48:379–384. [PubMed] [Google Scholar]
- Fong TM, McNamee MG. Correlation between acetylcholine receptor function and structural properties of membranes. Biochemistry. 1986;25:830–840. doi: 10.1021/bi00352a015. [DOI] [PubMed] [Google Scholar]
- Franke C, Parnas H, Hovav G, Dudel J. A molecular scheme for the reaction between acetylcholine and nicotinic channels. Biophys J. 1993;64:339–356. doi: 10.1016/S0006-3495(93)81374-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehle VM, Walcott EC, Nishizaki T, Sumikawa K. N-glycosylation at the conserved sites ensures the expression of properly folded functional ACh receptors. Brain Res Mol Brain Res. 1997;45:219–229. doi: 10.1016/S0169-328X(96)00256-2. [DOI] [PubMed] [Google Scholar]
- Gentet LJ, Clements JD. Binding site stoichiometry and the effects of phosphorylation on human α1 homomeric glycine receptors. J Physiol. 2002;544:97–106. doi: 10.1113/jphysiol.2001.015321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Gutierrez G, Grosman C. Bridging the gap between structural models of nicotinic receptor superfamily ion channels and their corresponding functional states. J Mol Biol. 2010;403:693–705. doi: 10.1016/j.jmb.2010.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Gutierrez G, Lukk T, Agarwal V, Papke D, Nair SK, Grosman C. Mutations that stabilize the open state of the Erwinia chrisanthemi ligand-gated ion channel fail to change the conformation of the pore domain in crystals. Proc Natl Acad Sci U S A. 2012;109:6331–6336. doi: 10.1073/pnas.1119268109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosman C, Salamone FN, Sine SM, Auerbach A. The extracellular linker of muscle acetylcholine receptor channels is a gating control element. J Gen Physiol. 2000;116:327–340. doi: 10.1085/jgp.116.3.327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfield JF, Tank DW, Greengard P, Huganir RL. Functional modulation of the nicotinic acetylcholine receptor by tyrosine phosphorylation. Nature. 1988;336:677–680. doi: 10.1038/336677a0. [DOI] [PubMed] [Google Scholar]
- Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- Jansen M, Bali M, Akabas MH. Modular design of Cys-loop ligand-gated ion channels: functional 5-HT3 and GABA ρ1 receptors lacking the large cytoplasmic M3M4 loop. J Gen Physiol. 2008;131:137–146. doi: 10.1085/jgp.200709896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha A, Cadugan DJ, Purohit P, Auerbach A. Acetylcholine receptor gating at extracellular transmembrane domain interface: the Cys-loop and M2–M3 linker. J Gen Physiol. 2007;130:547–558. doi: 10.1085/jgp.200709856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas P. Fast application of agonists to isolated membrane patches. In: Sakmann B, Neher E, editors. Single-channel recording. New York: Plenum; 1995. pp. 231–243. [Google Scholar]
- Jones AK, Buckingham SD, Sattelle DB. Proteins interacting with nicotinic acetylcholine receptors: expanding functional and therapeutic horizons. Trends Pharmacol Sci. 2010;31:455–462. doi: 10.1016/j.tips.2010.07.001. [DOI] [PubMed] [Google Scholar]
- Karlsson U, Druzin M, Johansson S. Cl− concentration changes and desensitization of GABAA and glycine receptors. J Gen Physiol. 2011;138:609–626. doi: 10.1085/jgp.201110674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krashia P, Moroni M, Broadbent S, Hofmann G, Kracun S, Beato M, Groot-Kormelink PJ, Sivilotti LG. Human α3β4 neuronal nicotinic receptors show different stoichiometry if they are expressed in Xenopus oocytes or mammalian HEK293 cells. PLoS One. 2010;5:e13611. doi: 10.1371/journal.pone.0013611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnaswamy A, Cooper E. Reactive oxygen species inactivate neuronal nicotinic acetylcholine receptors through a highly conserved cysteine near the intracellular mouth of the channel: implications for diseases that involve oxidative stress. J Physiol. 2012;590:39–47. doi: 10.1113/jphysiol.2011.214007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labarca C, Nowak MW, Zhang H, Tang L, Deshpande P, Lester HA. Channel gating governed symmetrically by conserved leucine residues in the M2 domain of nicotinic receptors. Nature. 1995;376:514–516. doi: 10.1038/376514a0. [DOI] [PubMed] [Google Scholar]
- Lansdell SJ, Gee VJ, Harkness PC, Doward AI, Baker ER, Gibb AJ, Millar NS. RIC-3 enhances functional expression of multiple nicotinic acetylcholine receptor subtypes in mammalian cells. Mol Pharmacol. 2005;68:1431–1438. doi: 10.1124/mol.105.017459. [DOI] [PubMed] [Google Scholar]
- Lee WY, Free CR, Sine SM. Binding to gating transduction in nicotinic receptors: Cys-loop energetically couples to pre-M1 and M2–M3 regions. J Neurosci. 2009;29:3189–3199. doi: 10.1523/JNEUROSCI.6185-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre P, Muller E, Badiu CI, Meier J, Vannier C, Triller A. Desensitization of homomeric alpha1 glycine receptor increases with receptor density. Mol Pharmacol. 2002;62:817–827. doi: 10.1124/mol.62.4.817. [DOI] [PubMed] [Google Scholar]
- Lynch JW, Rajendra S, Pierce KD, Handford CA, Barry PH, Schofield PR. Identification of intracellular and extracellular domains mediating signal transduction in the inhibitory glycine receptor chloride channel. EMBO J. 1997;16:110–120. doi: 10.1093/emboj/16.1.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon NK, Bali M, Akabas MH. Length and amino acid sequence of peptides substituted for the 5-HT3A receptor M3M4 loop may affect channel expression and desensitization. PLoS One. 2012;7:e35563. doi: 10.1371/journal.pone.0035563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mnatsakanyan N, Jansen M. Experimental determination of the vertical alignment between the second and third transmembrane segments of the muscle nicotinic acetylcholine receptors. J Neurochem. 2013;125:843–854. doi: 10.1111/jnc.12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroni M, Biro I, Giugliano M, Vijayan R, Biggin PC, Beato M, Sivilotti LG. Chloride ions in the pore of glycine and GABA channels shape the time course and voltage dependence of agonist currents. J Neurosci. 2011;31:14095–14106. doi: 10.1523/JNEUROSCI.1985-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson ME, Lindstrom J. Single channel properties of human α3 AChRs: impact of β2 β4 and α5 subunits. J Physiol. 1999;516:657–678. doi: 10.1111/j.1469-7793.1999.0657u.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nothdurfter C, Tanasic S, Di Benedetto B, Rammes G, Wagner EM, Kirmeier T, Ganal V, Kessler JS, Rein T, Holsboer F, Rupprecht R. Impact of lipid raft integrity on 5-HT3 receptor function and its modulation by antidepressants. Neuropsychopharmacology. 2010;35:1510–1519. doi: 10.1038/npp.2010.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panicker S, Cruz H, Arrabit C, Suen KF, Slesinger PA. Minimal structural rearrangement of the cytoplasmic pore during activation of the 5-HT3A receptor. J Biol Chem. 2004;279:28149–28158. doi: 10.1074/jbc.M403545200. [DOI] [PubMed] [Google Scholar]
- Papke D, Gonzalez-Gutierrez G, Grosman C. Desensitization of neurotransmitter-gated ion channels during high-frequency stimulation: a comparative study of Cys-loop, AMPA and purinergic receptors. J Physiol. 2011;589:1571–1585. doi: 10.1113/jphysiol.2010.203315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradiso K, Brehm P. Long-term desensitization of nicotinic acetylcholine receptors is regulated via protein kinase A-mediated phosphorylation. J Neurosci. 1998;18:9227–9237. doi: 10.1523/JNEUROSCI.18-22-09227.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt SJ, Sivilotti LG, Beato M. High intracellular chloride slows the decay of glycinergic currents. J Neurosci. 2008;28:11454–11467. doi: 10.1523/JNEUROSCI.3890-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Gómez A, Vaello ML, Valdivieso F, Mayor F., Jr Phosphorylation of the 48-kDa subunit of the glycine receptor by protein kinase C. J Biol Chem. 1991;266:559–566. [PubMed] [Google Scholar]
- Safran A, Sagi-Eisenberg R, Neumann D, Fuchs S. Phosphorylation of the acetylcholine receptor by protein kinase C and identification of the phosphorylation site within the receptor δ subunit. J Biol Chem. 1987;262:10506–10510. [PubMed] [Google Scholar]
- Saul B, Kuner T, Sobetzko D, Brune W, Hanefeld F, Meinck HM, Becker CM. Novel GLRA1 missense mutation (P250T) in dominant hyperekplexia defines and intracellular determinant of glycine receptor channel gating. J Neurosci. 1999;19:869–877. doi: 10.1523/JNEUROSCI.19-03-00869.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder W, Meyer HE, Buchner K, Bayer H, Hucho F. Phophorylation sites of the nicotinic acetylcholine receptor: a novel site detected in position δS362. Biochemistry. 1991;30:3583–3588. doi: 10.1021/bi00228a032. [DOI] [PubMed] [Google Scholar]
- Sivilotti LG, McNeil DK, Lewis TM, Nassar MA, Schoepfer R, Colquhoun D. Recombinant nicotinic receptors, expressed in Xenopus oocytes, do not resemble native rat sympathetic ganglion receptors in single-channel behavior. J Physiol. 1997;500:123–138. doi: 10.1113/jphysiol.1997.sp022004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumikawa K, Miledi R. Assembly and N-glycosylation of all ACh receptor subunits are required for their efficient insertion into plasma membranes. Brain Res Mol Brain Res. 1989;5:183–192. doi: 10.1016/0169-328X(89)90034-X. [DOI] [PubMed] [Google Scholar]
- Swope SL, Moss SJ, Raymond LA, Huganir RL. Regulation of ligand-gated ion channels by protein phosphorylation. Adv Second Messenger Phosphoprotein Res. 1999;33:49–78. doi: 10.1016/S1040-7952(99)80005-6. [DOI] [PubMed] [Google Scholar]
- Tasneem A, Iyer LM, Jakobsson E, Aravind L. Identification of the prokaryotic ligand-gated ion channels and their implications for the mechanism and origins of animal Cys-loop ion channels. Genome Biol. 2005;6:R4. doi: 10.1186/gb-2004-6-1-r4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treinin M. RIC-3 and nicotinic acetylcholine receptors: biogenesis, properties, and diversity. Biotechnol J. 2008;3:1539–1547. doi: 10.1002/biot.200800179. [DOI] [PubMed] [Google Scholar]
- Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4Å resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Vaello ML, Ruiz-Gómez A, Lerma J, Mayor F., Jr Modulation of inhibitory glycine receptors by phosphorylation by protein kinase C and cAMP-dependent protein kinase. J Biol Chem. 1994;269:2002–2008. [PubMed] [Google Scholar]
- Wagner K, Edson K, Heginbotham L, Post M, Huganir RL, Czernik AJ. Determination of the tyrosine phosphorylation sites of the nicotinic acetylcholine receptor. J Biol Chem. 1991;266:23784–23789. [PubMed] [Google Scholar]
- Wyllie DJ, Béhé P, Colquhoun D. Single-channel activations and concentration jumps: comparison of recombinant NR1a/NR2A and NR1a/NR2D NMDA receptors. J Physiol. 1998;510:1–18. doi: 10.1111/j.1469-7793.1998.001bz.x. [DOI] [PMC free article] [PubMed] [Google Scholar]