

Abstract
Apolipoprotein E (apoE) is the strongest known genetic risk factor for late onset Alzheimer's disease (AD). It influences amyloid-β (Aβ) clearance and aggregation, which likely contributes in large part to its role in AD pathogenesis. We recently found that HJ6.3, a monoclonal antibody against apoE, significantly reduced Aβ plaque load when given to APPswe/PS1ΔE9 (APP/PS1) mice starting before the onset of plaque deposition. To determine whether the anti-apoE antibody HJ6.3 affects Aβ plaques, neuronal network function, and behavior in APP/PS1 mice after plaque onset, we administered HJ6.3 (10 mg/kg/week) or PBS intraperitoneally to 7-month-old APP/PS1 mice for 21 weeks. HJ6.3 mildly improved spatial learning performance in the water maze, restored resting-state functional connectivity, and modestly reduced brain Aβ plaque load. There was no effect of HJ6.3 on total plasma cholesterol or cerebral amyloid angiopathy. To investigate the underlying mechanisms of anti-apoE immunotherapy, HJ6.3 was applied to the brain cortical surface and amyloid deposition was followed over 2 weeks using in vivo imaging. Acute exposure to HJ6.3 affected the course of amyloid deposition in that it prevented the formation of new amyloid deposits, limited their growth, and was associated with occasional clearance of plaques, a process likely associated with direct binding to amyloid aggregates. Topical application of HJ6.3 for only 14 d also decreased the density of amyloid plaques assessed postmortem. Collectively, these studies suggest that anti-apoE antibodies have therapeutic potential when given before or after the onset of Aβ pathology.
Keywords: Alzheimer's, amyloid, antibody, apolipoprotein E
Introduction
Alzheimer's disease (AD) is a neurodegenerative disorder characterized by the impairment of memory and other cognitive functions, as well as the presence of extracellular amyloid plaques and intracellular neurofibrillary tangles (Holtzman et al., 2011). There is substantial evidence indicating that amyloid-β (Aβ) plays a key role in the development of AD (Hardy and Selkoe, 2002; Lemere and Masliah, 2010). Previous therapeutic strategies for AD, including Aβ immunotherapy (Schenk et al., 1999; Bard et al., 2000), have focused on decreasing Aβ accumulation or removing existing Aβ aggregates by enhancing Aβ clearance. Certain types of active and passive immunization have resulted in significant side effects such as brain edema and hemorrhages (Brody and Holtzman, 2008). Given that Aβ metabolism is regulated by Aβ-binding proteins, immunization approaches targeted against proteins that regulate Aβ metabolism could also provide another treatment avenue, potentially with fewer side effects.
The ε4 allele of apolipoprotein E (apoE) has been established as the strongest genetic risk factor for late-onset AD (Corder et al., 1993; Strittmatter et al., 1993a; Strittmatter et al., 1993b; Corder et al., 1994; Belinson and Michaelson, 2009). There is compelling evidence showing that apoE affects Aβ accumulation by modulating both Aβ aggregation (Ma et al., 1994; Wisniewski et al., 1994) and clearance (Sadowski et al., 2006; Deane et al., 2008; Jiang et al., 2008; Castellano et al., 2011) and it also colocalizes with Aβ plaques (Namba et al., 1991). Recently, we found that passive immunization of APPswe/PS1ΔE9 (APP/PS1) mice before Aβ plaque onset resulted in a strong decrease of Aβ plaques and insoluble Aβ40 and Aβ42 levels (Kim et al., 2012). However, it is unknown whether passive apoE immunotherapy that begins after the presence of amyloid deposition in the brain would have similar effects. In addition, the potential mechanisms of the anti-amyloid effects and whether apoE immunotherapy benefits brain function have not been explored.
Here, we treated 7-month-old APP/PS1 mice that had preexisting Aβ plaques with the anti-apoE monoclonal antibody HJ6.3 for 21 weeks. We assessed changes in spatial learning and memory, intrinsic functional connectivity, and Aβ pathology associated with this treatment. In addition, we examined possible side effects including hypercholesterolemia, cerebral amyloid angiopathy (CAA), and CAA-associated microhemorrhage. With the aim of understanding the mechanisms of anti-apoE immunotherapy dynamically, we performed in vivo 2-photon imaging over 2 weeks after acute HJ6.3 exposure to the cortical surface. The results suggest that HJ6.3 prevents new plaque formation, inhibits Aβ plaque growth, favors the clearance of amyloid aggregates, and improves brain function.
Materials and Methods
Chronic antibody administration and pathology assessment.
Seven-month-old female APPswe/PS1ΔE9 (APP/PS1) mice on a B6C3 background (Jankowsky et al., 2004) were injected intraperitoneally with a weekly dose of 10 mg/kg body weight of anti-apoE mouse monoclonal antibody HJ6.3 (Kim et al., 2012) or PBS for 21 weeks. HJ6.3 is an IgG2b antibody. During the last 4 weeks of treatment, behavioral performance of the mice was tested. Two days after the final injection, functional connectivity optical intrinsic signal imaging (fcOIS) was recorded. Immediately after fcOIS, blood was collected via cardiac puncture and then the mice were perfused with ice-cold PBS containing 0.3% heparin. One hemibrain was fixed in 4% paraformaldehyde for histological analysis of amyloid plaque load, CAA, CAA-associated microhemorrhages, apoE, and microglial immunoreactivity. The other hemibrain was dissected and flash-frozen on dry ice for biochemical assays for Aβ and apoE. Data in all the figures are expressed as mean ± SEM unless otherwise specified. All experimental protocols were approved by the Animal Studies Committee at Washington University, Massachusetts General Hospital Subcommittee for Research, and the Animal Care and Use Committee at Harvard Medical School.
Immunohistochemistry.
Serial coronal sections at 50 μm thickness were collected from the rostral to the caudal end of each brain hemisphere using a freezing sliding microtome (Leica). Aβ plaques were immunostained using biotinylated anti-Aβ1–13 monoclonal antibody HJ3.4B (Bero et al., 2012). ApoE was immunostained using anti-apoE HJ6.8B (generated in-house), which recognizes a different epitope of apoE than HJ6.3. Activated microglia were immunostained using rat-anti CD45 (AbD Serotec), followed by biotinylated goat anti-rat IgG secondary antibody (Life Technologies). Quantitative analysis of immunopositive staining was performed as described previously (Bero et al., 2012). Briefly, images of immunostained sections were exported with NDP viewer (Hamamatsu Photonics), converted to 8-bit grayscale using ACDSee Pro 2 software (ACD Systems), thresholded to highlight positive staining, and analyzed using ImageJ software. Three sections per mouse (bregma, −1.4 mm caudal to bregma, −2.0 mm caudal to bregma) were quantified (hippocampus and overlying cortex) and the average was used to represent each mouse. For CAA quantification, the values from individual mice were normalized according to the average value of control group. Two-tailed Student's t test was used to analyze the data.
ELISA.
Brain cortices were sequentially homogenized with cold PBS, 1% Triton-X 100, and 5 m guanidine buffer in the presence of 1× protease inhibitor mixture (Roche). The levels of Aβx-40 and apoE were measured by sandwich ELISA. For Aβx-40 ELISA, anti-Aβ35–40 HJ2 was used as a capture antibody and anti-Aβ13–18 HJ5.1-biotin (Bero et al., 2011) was used as the detecting antibody. For apoE, HJ6.4 (Kim et al., 2012) was used as the capture antibody and HJ6.1-biotin (Kim et al., 2012) as the detecting antibody. Pooled C57BL/6J plasma was used as a quantification standard for murine apoE (Kim et al., 2012). Two-tailed Student's t test was used to analyze the data.
Total plasma cholesterol.
Total plasma cholesterol was assessed using a commercial kit (catalog #439-17501; Wako Chemicals) following the manufacturer's instructions. Two-tailed Student's t test was used to analyze the data.
Staining and quantitative analysis of CAA-associated microhemorrhage.
Prussian blue staining was performed to identify microhemorrhages (Fryer et al., 2003). Briefly, 15–18 equally spaced 50 μm sections, 300 μm apart, were assessed from each mouse. Sections were mounted on Superfrost slides and dried overnight. Sections were rehydrated in PBS, followed by permeablizing in PBS–Triton X-100 (0.25%) for 10 min. After being washed twice with 0.9% NaCl for 2 min/each, sections were incubated in 2% HCl containing 2% potassium ferrocyanide for 20 min. After washing with PBS, the sections were costained with 0.025% Thioflavin S (ThioS) to confirm that any putative microhemorhages were associated with CAA. The blue puncta stained by Prussian blue were counted with a light microscope. ThioS staining was visualized under the 488 nm fluorescent emission range. Two-tailed Student's t test was used to analyze the data.
One hour locomotor activity and sensorimotor battery.
Locomotor activity was evaluated in HJ6.3-treated (n = 19) and PBS-treated (n = 18) mice over a 1 h period using transparent (47.6 × 25.4 × 20.6 cm high) polystyrene enclosures and computerized photobeam instrumentation, as described previously (Wozniak et al., 2004). Variables pertaining to general ambulatory activity (total ambulations) and exploratory behaviors (vertical rearings) were analyzed. All mice were also evaluated on a battery of sensorimotor tests to assess balance (ledge and platform), strength (inverted screen), coordination (pole and inclined screens), and initiation of movement (walking initiation) using previously published procedures (Wang et al., 2002; Wozniak et al., 2004). One-way ANOVA models were used to analyze differences between groups.
Morris water maze navigation.
Spatial learning and memory capabilities of the APP/PS1 mice were assessed in the Morris water maze (MWM) after 4–5 months of treatment with HJ6.3 or PBS at 11–12 months of age using a computerized tracking system (ANY-maze; Stoelting) that involved procedures similar to those previously described (Wozniak et al., 2004). The protocol included conducting cued (visible platform), place (submerged and hidden platform), and probe (platform removed) trials in a round pool (118 cm inner diameter) of opaque water. Mice were first trained on the cued condition (11 months of age) to determine whether nonassociative factors were likely to affect acquisition performance during subsequent place trials. Unlike our typical water maze procedures, in which mice experience 2 consecutive days of cued trials, the mice in the present study received 4 trials per day (60 s maximum) for 5 consecutive days in an effort to provide additional training to compensate for potentially poor swimming abilities in the APP/PS1 mice. The cued trials involved moving the platform to a different location for each trial using a 30 min intertrial interval (ITI) in the presence of very few distal spatial cues to limit spatial learning. Three days after completing the cued trials, the mice were trained on the “place” condition to learn the location of a submerged platform. The place trials were conducted in the presence of several salient distal cues positioned around the room to facilitate association of the spatial cues with the submerged platform, which remained in the same location for all trials. During the place trials, the mice received 2 blocks of 2 consecutive trials (60 s maximum for a trial; 30 s ITI spent on platform), with each block being separated by ∼2 h and each mouse being released from a different quadrant for each trial. The place trials data were analyzed over 5 blocks of trials (4 trials/block) in which each block represented the performance level for each of 5 consecutive days. Escape path length, latency, and swimming speeds were analyzed as performance variables for the cued and place trials. A probe trial was administered ∼1 h after completion of the place trials on the fifth day of testing to evaluate retention of the platform location. During the 60 s probe trial, the escape platform was removed and a mouse was placed in the quadrant diagonally opposite from the previous platform location. Times spent in the pool quadrants and the number of crossings made over the platform location (platform crossings) were recorded. Repeated-measures ANOVA (rmANOVA) models containing one between-subjects variable (treatment) and one within-subjects (repeated measures) variable (e.g., blocks of trials) were typically used to analyze the learning and memory data. The Huynh–Feldt adjustment of α levels was used for all within-subjects effects containing more than two levels to protect against violations of sphericity/compound symmetry assumptions underlying rmANOVA models. Pairwise comparisons were conducted after relevant, significant overall rmANOVA effects, which were subjected to Bonferroni's correction when appropriate.
fcOIS.
Following a previously published fcOIS protocol (White et al., 2011), mice were anesthetized with ketamine-xylazine (108 mg/kg ketamine, 17 mg/kg xylazine) and were placed on a rectally monitored heating pad maintained at 37°C (mTCII; Cell Microcontrols) with the head secured in a stereotactic frame. A midline incision was made, the scalp reflected, and the intact skull was kept moist with mineral oil. Sequential illumination was provided at four wavelengths by a ring of light-emitting diodes (LEDs) placed ∼10 cm above the head (for details, see White et al., 2011). Diffuse reflected light was detected by a cooled, frame-transfer EMCCD camera (iXon 897; Andor Technologies). The LED ring and camera were computer controlled and operated at a full frame rate of 30 Hz. The field of view covered the majority of the convexity of the cerebral cortex (∼1 cm2). Data from all mice were subjected to an initial quality check before spectroscopic analysis. Data runs (5 min) in which temporal variation in reflected light level intensity exceeded 1% for any wavelength were excluded from further analysis. This preliminary analysis resulted in 15–45 min of data per mouse. For each pixel, the differential light intensity at each wavelength was converted to differential absorption. The multiwavelength absorption coefficient data were then converted to hemoglobin concentration changes for each pixel and time point. The subsequent fc analysis used the spatiotemporal ΔHbO2 data (White et al., 2009). Data were filtered to the functional connectivity band (0.009–0.08 Hz) following previous human fMRI algorithms (Fox et al., 2005), resampled from 30 to 1 Hz, and a brain mask was created containing all pixels labeled as brain. Time traces of all brain pixels were averaged to create a global brain signal. This global signal was regressed from each pixel's time trace to remove global sources of variance, as is common in human fMRI algorithms. All image sequences and brain masks were affine transformed to a common atlas space (Franklin and Paxinos, 1996) determined by the coordinates of olfactory bulb and cerebrum, bregma, and lambda. The anterior–posterior stretch was set equal to the medial–lateral stretch. To ensure that all comparisons were performed only on the brain area visualized in all mice, the intersection of each brain mask was calculated and made symmetric. Seed placement was determined a priori using an anatomical atlas (Franklin and Paxinos, 1996). Four seed locations were positioned at coordinates corresponding to left and right motor, somatosensory, retrosplenial, and visual cortices. Seed time traces were calculated by averaging time traces within 0.25 mm (30 pixels) of a seed locus and functional connectivity maps were created by correlating each seed time trace against those in every brain pixel. Individual functional connectivity maps were averaged within each cohort to generate consensus maps for each cohort. Regional bilateral correlations were determined by correlating the seed-based time trace in each functional area with that of its contralateral homolog, thus producing six bilateral correlation values for each mouse. Pearson r values were converted to Fisher Z measures using: Z(r) = arctanh (r) for all group comparisons. To further examine bilateral connectivity across the brain, every pixel in the brain was used as a seed center and its time trace was correlated with its contralateral homolog. Individual bilateral maps were averaged within each cohort to generate consensus bilateral connectivity maps. To calculate the number (degree) of functional connections of a given pixel (node), the full pixel-to-pixel correlation matrix was computed for each mouse over the entire brain. Any node having a correlation coefficient, z(r), that was ≥0.25 was considered a connection and given a value of 1; those with a correlation coefficient <0.25 were given a value of 0. Summing over the rows of this binarized matrix produces an unweighted measure of node degree for all candidate regions. Statistical significance was determined by one-tailed t test if two groups were compared. When variance differed significantly between groups, Welch's t test was used. One-way ANOVA followed by Tukey's post hoc test for multiple comparisons were used when more than two groups were compared.
Cranial window implantation, topical application of antibodies, and multiphoton imaging.
APP/PS1 mice were anesthetized with isoflurane (1.5%) and a 4 mm piece of skull was drilled to expose the cortical surface as described previously (Spires et al., 2005). After removing the dura, 50 μl of each antibody stock (1.0 mg/ml) was topically applied before cementing the cranial window with a 5 mm glass coverslip. For a subset of experiments, both anti-HA and anti-apoE HJ6.3 antibodies were labeled with DyLight594 fluorophore following the manufacturer's instructions (Thermo Scientific). A wax ring was built along the border of the window to create a wall of water for the objective during imaging. Each mouse received an intraperitoneal injection of methoxy-XO4 (5 mg/kg) 24 h before surgery, a fluorescent compound that crosses the blood–brain barrier (BBB) and binds to plaques (Bacskai et al., 2002). Texas red or fluorescein dextran (70,000 Da molecular weight; 12.5 mg/ml in sterile PBS; Invitrogen) was also injected into a lateral tail vein to provide a fluorescent angiogram so that the shape of the vasculature would be used as a landmark to follow the exact same fields of view over time. Mice were imaged right after surgery and 14 d thereafter. The density of plaque in each field of view was quantified using ImageJ by reporting the total number of amyloid deposits per volume of cortex imaged. Each cortical volume was defined starting from surface of the brain to the last slice in which an amyloid deposit could be detected. In vivo imaging data summarizing the amount of “stable,” “absent,” and “new” plaques were organized in a contingency table and analyzed using a χ2 test. The size of amyloid deposits was evaluated by measuring their cross-sectional area from the maximal intensity after 2D projection. We calculated the ratio between the size of each plaque just after surgery (t = 0) and 2 weeks (t = 14 d) after topical application of each antibody. After the in vivo imaging on t = 14 d, the mice were killed for postmortem analysis of pathology. Data in all figures are expressed as mean ± SEM unless otherwise specified.
Multiphoton imaging.
In vivo multiphoton imaging was performed using an Olympus FluoView FV1000MPE multiphoton laser-scanning system mounted on an Olympus BX61WI microscope and an Olympus 25× objective (numerical aperture = 1.05). A DeepSee Mai Tai Ti:sapphire mode-locked laser (Mai Tai; Spectra-Physics) generated two-photon excitation at 800 nm, and detectors containing three photomultiplier tubes (Hamamatsu) collected emitted light in the range of 420–460, 495–540, and 575–630 nm. Mice were placed on the microscope stage, heated using a heating pad, and feedback regulation was obtained from a rectal temperature probe (Harvard Apparatus). Z-series (2 μm steps, depth of ∼200 μm, 512 × 512 pixels) were taken to cover a large surface of the window. The laser power was measured and adjusted before each imaging and the settings of the photomultiplier tubes were maintained unchanged throughout the different imaging sessions.
Postmortem analysis after in vivo imaging.
Serial paraffin sections at 4 μm thickness or frozen sections at 40 μm thickness were immunostained with rabbit anti-human amyloidβ (N) antibody (Immuno-Biological Laboratories) and ThioS. Immunostained brain sections were scanned using a Nanozoomer slide scanner (Hamamatsu Photonics). For the postmortem analysis after in vivo imaging, images were taken (1.25×, 20×) using an Olympus BX51 epifluorescence upright microscope equipped with a CCD camera model DP70 (Olympus). For the localization analysis of HJ6.3 (see Fig. 10), images were acquired on a Zeiss LSM 510 META confocal microscope equipped with two-photon Coherent Chameleon laser. Quantitative analyses of amyloid load and plaque density were done using the BIOQUANT software (version 6.90.10; MBSR) after application of an optical threshold. This software is coordinated with the motorized stage of an upright Leica DMRB microscope equipped with a CCD camera (model DC330; DAGE-MTI). Immunostained amyloid plaques were thresholded under the 10× objective after background correction to avoid uneven lighting. Results of plaque load were analyzed using a nonparametric Mann–Whitney test because the data did not follow a normal distribution (according to the D'Agostino and Pearson test). For each staining method independently, a mixed-effect model with treatment as the fixed effect and mouse and section within mouse as the random effects was fitted to assess the differences between treated groups in the value of plaque size (with log of plaque size as the outcome) while accounting for variability between mice and section within mice. The plaque size ratios were compared between each treatment using one-way ANOVA followed by post hoc Tukey multiple comparison tests.
Figure 10.

Affinity of fluorescent-labeled DyLight 594-apoE antibody HJ6.3 to CAA and Aβ plaques. Fifty micrograms of fluorescently labeled DyLight 594-apoE antibody HJ6.3 or DyLight 594-anti-HA control antibody (1.0 mg/ml) were topically applied to the brain surface of 5- to 7-month-old APP/PS1 mice. Plaques and CAA were imaged by in vivo two-photon microscopy and immunohistochemistry with the rabbit anti-human Aβ (N) antibody (Immuno-Biological Laboratories). A, Representative photographs of in vivo imaging after 30 min of incubation with antibody, showing that only the DyLight 594-apoE antibody HJ6.3 can bind apoE in CAA (yellow arrow) and amyloid plaques (white arrow) after a single topical application to the brain. Scale bar, 100 μm. B, Representative photographs of immunohistochemistry after 2 h of incubation with antibody, showing that the DyLight 594-apoE antibody HJ6.3 can bind apoE in small, methoxy-XO4-negative amyloid plaque (yellow arrow) and in a methoxy-XO4-positive amyloid plaque (white arrow) after a single topical application to the brain. Scale bar, 50 μm.
Results
HJ6.3 decreased Aβ plaque load in the brain of APP/PS1 mice
To determine whether an anti-apoE antibody decreased Aβ deposition in APP/PS1 mice that had already developed significant Aβ deposition, we assessed APP/PS1 mice after 5 months of peripheral administration of HJ6.3 (10 mg/kg/week). We immunostained Aβ plaques using an Aβ-specific antibody HJ3.4B (Fig. 1A). In cerebral cortex immediately dorsal to the hippocampus, HJ6.3 decreased plaque load by ∼20% (Fig. 1B). In thalamus, HJ6.3 decreased plaque load by a greater amount (∼40%; Fig. 1B). In hippocampus, HJ6.3 did not significantly change plaque load compared with the PBS group (Fig. 1B).
Figure 1.

Reduction of Aβ plaque deposition by the anti-apoE antibody HJ6.3. Seven-month-old female APP/PS1 mice received 10 mg/kg weekly intraperitoneal injections of HJ6.3 or PBS for 21 weeks. A, Brain sections of the HJ6.3- or PBS-treated mice were stained with the anti-Aβ antibody HJ3.4B. Scale bar, 1 mm. B, Aβ plaque load in cortex, thalamus, and hippocampus were quantified (n = 13–17/group; *p < 0.05).
To determine whether fibrillar plaques were altered by HJ6.3, we stained brain sections with ThioS (Fig. 2A). HJ6.3 significantly reduced ThioS plaque load in cortex (Fig. 2B, top). In thalamus (Fig. 2B, middle), plaque load was reduced by ∼ 40%, whereas in hippocampus, plaque load was not changed (Fig. 2B, bottom).
Figure 2.

Attenuation of fibrillar amyloid deposition by the anti-apoE antibody HJ6.3. Seven-month-old female APP/PS1 mice received 10 mg/kg weekly intraperitoneal injections of HJ6.3 or PBS for 21 weeks until 12 months of age. A, Fibrillar amyloid plaques on brain sections of the HJ6.3- or PBS-treated mice were stained with ThioS. Scale bar, 500 μm. B, Quantification of fibrillar amyloid plaque load in cortex, thalamus, and hippocampus (n = 13–17/group; *p < 0.05; **p < 0.01).
Changes of brain and blood apoE levels resulting from HJ6.3 treatment
Because apoE is the direct target of the anti-apoE antibody HJ6.3, we investigated whether peripheral administration of HJ6.3 altered apoE levels in brain and blood. Immunostaining using an apoE antibody directed to a different apoE epitope, HJ6.8B, showed decreased apoE immunostaining in the cerebral cortex in HJ6.3-treated mice (Fig. 3A,B). Biochemical analysis of apoE in the brain tissue lysates demonstrated that apoE was decreased primarily in the 1% Triton X-100 fraction, whereas the levels in the PBS and 5 m guanidine fraction were unaltered (Fig. 4A, top). In plasma, there was no change in apoE levels due to HJ6.3 (Fig. 4B, top).
Figure 3.

Effects of the anti-apoE antibody HJ6.3 on brain apoE. Seven-month-old female APP/PS1 mice received 10 mg/kg weekly intraperitoneal injections of HJ6.3 or PBS for 21 weeks. ApoE in the cortex of the HJ6.3- or PBS-treated mice was immunostained with HJ6.8B (A) and the percentage area covered by the staining was quantified (B) (n = 15–18/group; *p < 0.05; **p < 0.01). Scale bar, 400 μm.
Figure 4.

Effects of the anti-apoE antibody HJ6.3 on Aβ and apoE levels in the brain and blood. A, The cortices of the HJ6.3- or PBS-treated mice were homogenized in PBS, followed by 1% Triton X-100 and 5 m guanidine (Guan). ApoE and Aβx-40 levels in the brain tissue lysates in the PBS, Triton X-100, and Guan fractions were measured by ELISA. The level of apoE and Aβx-40 in all fractions combined is also shown (n = 10–18/group). B, Plasma apoE and Aβx-40 levels in HJ6.3- and PBS-treated animals were measured by ELISA (n = 15–18/group; *p < 0.05; **p < 0.01).
HJ6.3 influenced Aβ levels in brain and blood
To determine whether the anti-apoE antibody HJ6.3 affects Aβ accumulation and solubility in the brain, we did a three-step sequential extraction of cortical tissue using PBS, 1% Triton X-100, and 5 m guanidine. Aβx-40 was then analyzed in each fraction (Fig. 4A). In the PBS-soluble fraction, HJ6.3 treatment had no significant effects on Aβx-40. In the Triton X-100 fraction, in which the apoE level was significantly lower in the HJ6.3-treated group, Aβx-40 was significantly reduced by HJ6.3. In the 5 m guanidine fraction, which contains the insoluble Aβ and >100× the Aβ present in the other fractions, Aβx-40 was reduced significantly. The combination of all fractions of Aβx-40 showed similar findings as the guanidine fraction because this fraction contains the majority of Aβ in the brain of plaque-bearing mice (Fig. 4A). In plasma, HJ6.3 treatment resulted in a 40–60% increase in Aβx-40 without affecting apoE levels (Fig. 4B).
Effects of HJ6.3 on microglial activation
To determine whether HJ6.3 had an impact on microglial activation, we immunostained brain sections of the mice treated with HJ6.3 or PBS using CD45, a marker of activated microglia (Fig. 5A). In cerebral cortex, the CD45 staining load was decreased by 40% after HJ6.3 treatment (Fig. 5B).
Figure 5.

Effects of the anti-apoE antibody HJ6.3 on microglial activation. Seven-month-old female APP/PS1 mice received 10 mg/kg weekly intraperitoneal injections of HJ6.3 or PBS for 21 weeks until 12 months of age. Activated microglia in the cortex were immunostained with anti-CD45 antibody (A) and the area covered by CD45 staining was quantified (B) (n = 15–18/group; *p < 0.05). Scale bar, 400 μm.
Anti-apoE antibody HJ6.3 improves spatial learning but slows motor responses
To determine whether HJ6.3 produced functional effects in APP/PS1 mice, we assessed HJ6.3- or PBS-treated mice starting at 11 months of age on several behavioral tests 4 months after beginning treatment. We first evaluated the mice on a 1 h locomotor activity test and a battery of sensorimotor measures followed by the MWM. The rmANOVAs conducted on the cued trials data from the MWM showed that the HJ6.3- and PBS-treated groups did not differ in terms of path length (Fig. 6A) or latency (data not shown) to find the platform and escape out of the water. This suggests that the two groups exhibited similar levels of cued learning performance. However, the results of an rmANOVA revealed that the HJ6.3-treated mice swam significantly more slowly than the PBS-treated group during certain blocks within the cued trials (treatment effect: F(1,35) = 6.99, p = 0.012; treatment × blocks of trials interaction: F(4,140) = 3.37, p = 0.025). Subsequent pairwise comparisons showed that significant differences were found for block 3 (p = 0.004) and large differences were also observed on blocks 2 (p = 0.037), 4 (p = 0.014), and 5 (p = 0.015). Together, these results suggest that nonassociative influences (e.g., sensorimotor, visual, or motivational disturbances) did not affect the performance of the HJ6.3-treated mice relative to the PBS-treated controls in terms of simple cued learning. Due to differences in swimming speeds, we used path length to evaluate acquisition performance in the place trials of the water maze.
Figure 6.

HJ6.3 treatment improved spatial learning performance in the MWM. Seven month-old female APP/PS1 mice received 10 mg/kg weekly intraperitoneal injections of HJ6.3 or PBS for 21 weeks until 12 months of age. During the last 4 weeks of treatment, the performance of HJ6.3-treated (n = 19) and PBS-treated (n = 18) mice in the MWM task was tested. A, The HJ6.3- and PBS-treated mice did not differ in performance with regard to path length during the cued trials. B, However, the HJ6.3-treated mice exhibited significantly improved performance in terms of showing decreased path length during the place (spatial learning) trials compared with the PBS-treated group (treatment effect: *p = 0.035), with differences being greatest during block 3 (†p = 0.019). C, PBS-treated mice did not show a spatial bias for the target quadrant during the probe trial because they did not spend significantly more time in that quadrant compared with the times in each of the other quadrants. In contrast, the HJ6.3-treated mice did show a spatial bias for the target quadrant because they spent significantly more time in the target quadrant compared with the times spent in each of the other pool quadrants (*p < 0.014).
In contrast to the cued trials results, an rmANOVA of the path length data from the place (spatial learning) trials showed that the HJ6.3-treated mice had significantly shorter path lengths to the submerged platform across trials (Fig. 6B) compared with the PBS-treated control mice (treatment effect: F(1,35) = 4.79, p = 0.035), with differences being greatest during block 3 (p = 0.019). In addition, similar to the results from the cued trials, the HJ6.3-treated mice swam significantly more slowly than the PBS-treated controls during certain blocks within the place trials (treatment effect: F(1,35) = 13.51, p = 0.0008; treatment × blocks of trials interaction: F(4,140) = 4.01, p = 0.005), with pairwise comparisons showing that significant differences occurred during blocks 2–5 (p < 0.001). Analysis of the probe trial data indicated that the groups did not differ in terms of platform crossings (data not shown) or time spent in the target quadrant. The HJ6.3-treated mice did, however, show a spatial bias for the target quadrant (Fig. 6C) in that they spent significantly more time in the target quadrant relative to the times they spent in each of the other quadrants (p < 0.014), whereas the PBS-treated group did not show a spatial bias for the target quadrant. Overall, these results suggest that HJ6.3 improved spatial learning in APP/PS1 mice.
HJ6.3 improves brain functional connectivity in APP/PS1 mice
To determine whether HJ6.3 antibody treatment restored disrupted functional connectivity in the mouse brain, we compared functional connectivity patterns (Fig. 7A) in APP/PS1 mice treated with HJ6.3 and PBS using fcOIS (White et al., 2011; Bero et al., 2012). We have shown previously that fcOIS is a sensitive imaging modality capable of measuring functional connectivity between brain regions in healthy mice (White et al., 2011; Bero et al., 2012) and that there is functional disruption in APP/PS1 mice after Aβ deposition (White et al., 2011; Bero et al., 2012). APP/PS1 mice treated with HJ6.3 exhibited improved bilateral functional correlation strength in visual cortex with trends toward improvement in motor and somatosensory cortices (Fig. 7A,B). To more closely assess the effects of HJ6.3 on global bilateral functional connectivity, full-field bilateral functional connectivity maps were created for every mouse and averaged within each cohort (Fig. 7C). Compared with PBS-treated mice, bilateral functional connectivity in the HJ6.3 group appeared to be more globally restored and to resemble that of healthy, wild-type mice (Fig. 7C), as described previously (White et al., 2011; Bero et al., 2012). Although bilateral connectivity magnitude provides a useful metric of homotopic interhemispheric connectivity, this is only a limited measure of the rich connectivity structure otherwise observed over the mouse brain. To examine more closely the effects of HJ6.3 on the topology of functional connectivity, we analyzed the number of functional connections (node degree) in the four brain regions investigated (Fig. 7D). From this analysis, we observed significant increases in the node degree of motor, somatosensory, retrosplenial, and visual cortices compared with the PBS-treated group.
Figure 7.

HJ6.3 treatment improved brain functional connectivity assessed by fcOIS. Seven-month-old female APP/PS1 mice received 10 mg/kg weekly intraperitoneal injections of HJ6.3 or PBS for 21 weeks. At the age of 12 months, fcOIS was performed to assess the resting state functional connectivity of the brain before euthanizing the mice. A, Composite, group-averaged, functional correlation maps of motor, somatosensory, retrosplenial, and visual cortices in HJ6.3-treated (n = 15) and PBS-treated (n = 14) APP/PS1 mice. Black circles denote seed position. B, Regional bilateral functional correlation of PBS-treated (blue) and HJ6.3-treated (red) APP/PS1 mice. C, Consensus bilateral functional connectivity maps generated for HJ6.3 and PBS treated APP/PS1 mice and age-matched untreated wild-type B6C3 mice. The maps for age-matched wild-type B6C3 mice were regenerated from the data reported in Bero et al. (2012). D, Node degree in motor, somatosensory, retrosplenial, and visual cortices (*p < 0.05; **p < 0.01).
Lack of potential side effects on cholesterol, CAA, and CAA-associated microhemorrhage
Being a key apolipoprotein in certain lipoprotein complexes, apoE plays an important role in plasma lipid metabolism. Given that the plasma cholesterol level of apoE-deficient mice is fivefold higher than that of wild-type mice, a potential side effect of apoE passive immunotherapy is that the apoE antibodies could elevate the cholesterol level by lowering plasma apoE levels or blocking apoE function. We found that injection of the anti-apoE antibody HJ6.3 did not affect plasma cholesterol (Fig. 8A).
Figure 8.

No effects of HJ6.3 on plasma cholesterol, CAA, or CAA-associated microhemorrhage. Seven-month-old APP/PS1 mice received 10 mg/kg weekly intraperitoneal injections of HJ6.3 or PBS for 21 weeks until 12 months of age. A, Total plasma cholesterol levels. B, CAA on the surface of the cortex was identified by anti-Aβ antibody HJ3.4B immunostaining and the area covered by CAA was quantified. C, CAA-associated microhemorrhage was detected by Prussian blue and ThioS costaining (n = 13–18/group).
In previous studies of active and passive immunization involving the generation of certain anti-Aβ antibodies, an increase of CAA and CAA-associated microhemorrhage has been observed (Pfeifer et al., 2002). To determine whether passive immunotherapy targeting apoE results in similar side effects, we assessed CAA and CAA-associated microhemorrhage. The CAA present on the cortical surface stained with Aβ-specific antibody HJ3.4B was quantified. HJ6.3 did not alter the relative area covered by CAA compared with the PBS-treated group (Fig. 8B). The amount of CAA-associated microhemorrhage in APP/PS1 mice at 12 months of age was low and we did not detect any increase in CAA-associated microhemorrhage associated with HJ6.3 administration (Fig. 8C).
Topical application of the anti-apoE HJ6.3 dynamically alters amyloid deposition over 2 weeks
To further assess early dynamic changes in amyloid deposition associated with apoE immunotherapy, HJ6.3 or IgG subtype-matched anti-HA (i.e., irrelevant epitope) control antibodies were topically applied to the surface of brain and in vivo two-photon microscopy was used to monitor Aβ plaques the day of surgery (t = 0) and 14 d after antibody treatment (t = 14 d). After 2 weeks of incubation, the same fields of view were imaged again and amyloid plaques were classified as “absent” when they could not be found again, “stable” when they were imaged again, and “new” for newly formed deposits over the 2 weeks of incubation. Although most deposits were stable over time, direct application of HJ6.3 reduced the amount of newly formed plaques (“new” plaques) and was associated with clearance of some amyloid deposits (“absent” plaques; Fig. 9A,B).
Figure 9.

In vivo imaging of Aβ plaques after topical application of the anti-apoE antibody HJ6.3. Fifty microliters of the anti-apoE antibody HJ6.3 or anti-HA control antibody (1.0 mg/ml) were topically applied to the brain surface of 5- to 7-month-old APP/PS1 mice. Plaques were monitored through the cranial window using two-photon microscopy the day of surgery and 14 d after treatment, following the exact same fields of view over time. A, Representative in vivo images of amyloid plaques the day of surgery (t = 0) and 14 d (t = 14 d) after topical application of control (anti-HA) or the anti-apoE antibody HJ6.3. Yellow and white arrows respectively show “absent” and “new” plaques. Scale bar, 100 μm. B, Contingency table summarizing the number of “absent,” “stable,” and “new” plaques 2 weeks after topical application of the anti-apoE antibody HJ6.3 and anti-HA antibodies (n = 7/group; p < 0.0001, χ2 test).
To further establish the connection between the presence of anti-apoE antibody and changes in amyloid deposition, fluorescent-labeled DyLight 594-HJ6.3 antibody was applied to the surface of the brain. We confirmed that the anti-apoE antibody HJ6.3 can bind directly to intraparenchymal amyloid deposits and to CAA (Fig. 10A). Some Aβ plaques, especially those located deeper within the cortical parenchyma, were not stained with DyLight 594-HJ6.3 antibody, likely due to limitations in diffusion over the several hours of observation. To determine the type of plaques that were bound by DyLight 594-HJ6.3, brain sections were immunostained with anti-Aβ antibody after the application of the DyLight 594-conjugated antibody. Immunohistochemistry showed that DyLight 594-HJ6.3 can also bind small, methoxy-XO4-negative nonfibrillar plaques, as well as methoxy-XO4-positive fibrillar plaques (Fig. 10B). Interestingly, we observed that 91% of the “absent” plaques that could not be detected after 14 d were previously labeled with HJ6.3 (Fig. 11A,B). At the same time, 56% of “stable” plaques were also bound by HJ6.3, but the ratio of plaque size of these deposits decreased between 0 and 14 d, a change that was not observed with nonlabeled plaques or with the anti-HA control antibody (Fig. 11C,D). We therefore concluded that topical application of anti-apoE antibody HJ6.3 acutely modified the course of amyloid deposition over 2 weeks, leading to the clearance of a small percentage of amyloid plaques while leading to shrinkage of others, a process associated with a direct interaction between the anti-apoE antibody and apoE present within Aβ deposits.
Figure 11.

In vivo dynamic measurement of Aβ plaques after treatment with HJ6.3. Fifty microliters of fluorescently labeled DyLight 594-apoE antibody HJ6.3 or DyLight 594-anti-HA control antibody (1.0 mg/ml) were topically applied to the brain surface of 5- to 7-month-old APP/PS1 mice. Plaques were monitored by two-photon microscopy. A, Representative in vivo images of the fluorescently labeled DyLight 594-HJ6.3 and methoxy-XO4 the day of application (t = 0) or 14 d after (t = 14 d), showing that the plaque that got cleared (yellow arrow) was previously labeled by DyLight 594-HJ6.3, as opposed to nonlabeled plaque (white arrow). Scale bar, 100 μm. B, Contingency table summarizing the number of “absent,” “stable,” and “new” plaques according to their initial binding with anti-apoE HJ6.3 and anti-HA antibodies. C, Representative in vivo images of the fluorescently labeled DyLight 594-HJ6.3 and methoxy-XO4 the day of application (t = 0) or 14 d after (t = 14 d), showing that the size of HJ6.3-labeled plaque decreased after topical application of labeled HJ6.3 (yellow arrow), as opposed to nonlabeled plaque (indicated by white arrow). Scale bar, 100 μm. D, Graph bar indicating the size ratio between day 0 and day 14 after topical application of the control antibody or the anti-apoE antibody HJ6.3 (n = 3/group; *p < 0.05, ***p < 0.001 for one-way ANOVA followed by post hoc Tukey test and p < 0.0001 for χ2 test).
Acute exposure with HJ6.3 also affects nonfibrillar Aβ plaques
As mentioned previously, acute exposure with HJ6.3 for 2 weeks significantly altered the dynamic of amyloidosis, a phenomenon associated with the direct binding of anti-apoE antibody to apoE-bound amyloid deposits. We then aimed at determining whether these changes could also be observed by postmortem immunohistological analysis. Amyloid load and amyloid plaque density were evaluated 15 d after topical application with the anti-apoE HJ6.3 or anti-HA antibodies (Fig. 12A). The stereological analysis specifically focused on the amyloid deposits present within the cortical area underneath the cranial window. To correct for interindividual variability, the results were normalized for each mouse for the amount of plaques in the hippocampus, a deeper cerebral area that was not in contact with the antibodies. The plaque number per square millimeter after HJ6.3 topical application was significantly lower by 40% compared with anti-HA control antibody (Fig. 12C); however, the plaque load per square millimeter was not changed significantly (Fig. 12B). This was likely because the overall effect on plaques was modest and the plaques most affected have the smallest cross-sectional area and thus likely have a proportionately small impact on the measure of amyloid burden. Surprisingly, ThioS staining showed no differences in either plaque load or plaque number per square millimeter (data not shown) between HJ6.3 and anti-HA control antibody. In addition, whereas the distribution of plaque size tended to show that HJ6.3 topical application preferentially affected smaller deposits when assessed by immunohistological staining (p < 0.075; Fig. 12D), no such trend was observed with the ThioS staining (data not shown). These results may indicate that, whereas small changes were observed on dense-core plaques by in vivo two-photon imaging, acute exposure with anti-apoE antibody primarily decreased the density of less fibrillar small amyloid deposits.
Figure 12.
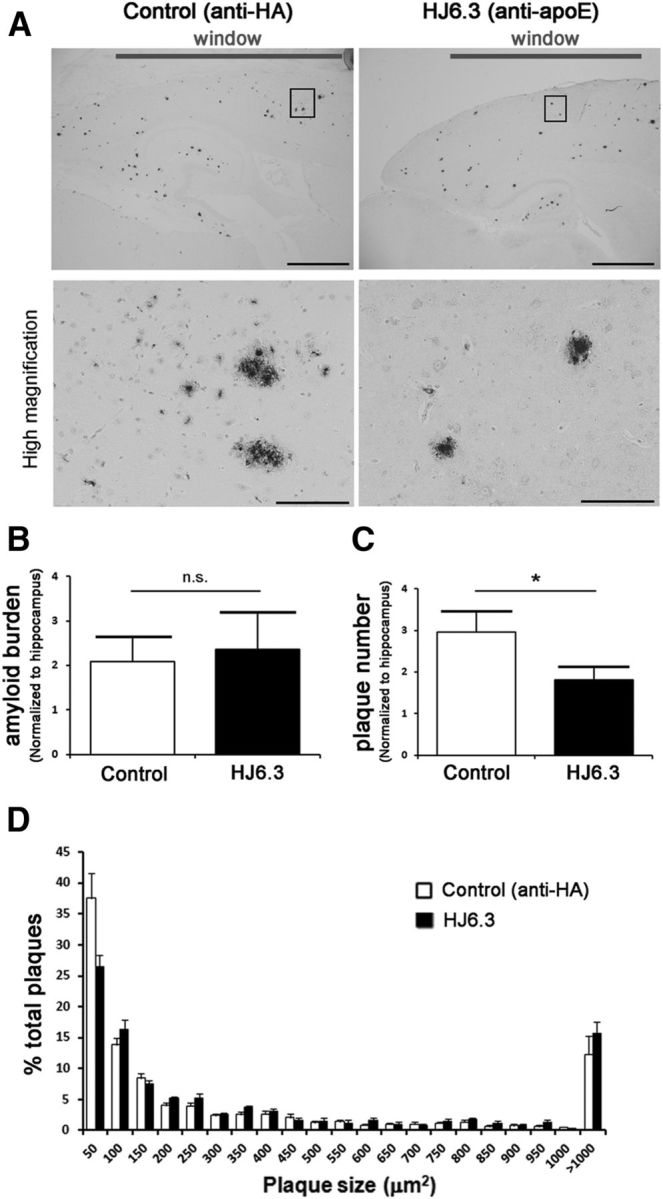
Reduction of the number of amyloid plaques after acute exposure with the anti-apoE antibody HJ6.3. Fifty microliters of the anti-apoE antibody HJ6.3 or anti-HA control antibody (1.0 mg/ml) were topically applied to the brain surface of 5- to 7-month-old APP/PS1 mice. A, Fifteen days after application, amyloid plaques were stained with the rabbit anti-human Aβ (N) antibody (Immuno-Biological Laboratories). Scale bars: top, 2 mm; bottom, 100 μm. B, C, Bar graphs summarizing the amyloid load (B) and plaque number (C) quantified in the cortical region under the cranial window, normalized to the amount of plaques within the hippocampus. D, Graph bar representing the plaque size distribution between the application of the control antibody and the anti-apoE antibody HJ6.3 (n = 9/group; *p < 0.05, nonparametric Mann–Whitney test).
Discussion
In the present study, we report that peripheral weekly administration of apoE antibodies into mice with preexisting Aβ plaques attenuates plaque load and insoluble Aβ without causing significant side effects such as hypercholesterolemia, CAA, or CAA-associated microhemorrhage. ApoE levels in the brain were decreased by antibody treatment and Aβ levels in the blood were increased. Importantly, anti-apoE antibody treatment improved spatial learning performance during place trials in the MWM and enhanced the resting-state bilateral functional connectivity in different cortical regions. In vivo two-photon microscopic imaging demonstrated that HJ6.3 bound to Aβ plaques and cleared some of them or led to their shrinkage, particularly those containing apoE. Together, the data suggest that further development of anti-apoE antibodies may be warranted as a potential therapeutic strategy.
One of the major mechanisms for removal of the plaques in response to some anti-Aβ antibodies is microglial-mediated phagocytosis (Bard et al., 2000; Demattos et al., 2012). For apoE passive immunotherapy, we previously observed an increase of reactive microglia with a 14 d short-term passive immunization with HJ6.3 (Kim et al., 2012), suggesting the involvement of microglia in the antibody-mediated Aβ removal. In the present study, 5 months of weekly administration of the anti-apoE antibody HJ6.3 decreased microglial activity. This time-dependent change in microglial activation agrees with the pattern observed with Aβ passive immunization, in which short-term Aβ antibody administration with certain anti-Aβ antibodies increases reactive microglia whereas long-term administration decreases reactive microglia (Wilcock et al., 2003; Wilcock et al., 2004b). The decrease of microglial activation associated with long-term immunization is likely a consequence of reduced plaque load and associated microgliosis (Liu et al., 2001). Although our present study does not rule out the possibility that anti-apoE antibody-mediated phagocytosis plays a role in the clearance of Aβ colocalized with apoE in the plaques, it may suggest that apoE antibody could suppress Aβ pathology through additional mechanisms via modifying apoE levels or function. ApoE plays an important role in amyloidosis progression (Dodart et al., 2005; Hudry et al., 2013). Previous work has shown that apoE colocalizes with newly formed Aβ deposits and that apoE4 carriers show more apoE containing newly formed plaques than individuals with other apoE genotypes, suggesting the involvement of apoE in the initiation of plaque formation (Thal et al., 2005). In the chronic treatment experiment of the present study, HJ6.3 was more effective at decreasing Aβ deposits in thalamus than in cortex and hippocampus, likely due to the fact that thalamus has fewer initial preexisting plaques than cortex and hippocampus. Furthermore, the fact that chronic but not acute application of anti-apoE antibody decreased fibrillar plaques may also suggest that the major effect of the anti-apoE antibody was to suppress amyloidosis progression, as opposed to removing preexisting plaques.
The in vivo two-photon microscopy experiments provided direct evidence that the anti-apoE antibody prevented the formation of new plaques and decreased the size of existing plaques upon binding. This was particularly true for plaques containing apoE. Collectively, our data suggest that the anti-apoE antibody HJ6.3 is able to suppress further Aβ deposition and to enhance the clearance or shrinkage of a small percentage of plaques after antibody application if plaques are bound by the HJ6.3 antibody. Importantly, when peripherally administrated into mice with preexisting Aβ deposition, the anti-apoE antibody HJ6.3 was able to improve spatial learning performance and functional connectivity in addition to its effects on Aβ pathology.
The role of apoE in AD pathogenesis has been studied extensively, but there is still debate regarding whether increasing or decreasing apoE is the best strategy in regard to AD therapeutics (Ashford, 2002; Teter et al., 2002). LXR agonists have been shown to decrease Aβ deposition in the brain while modestly increasing apoE expression (Koldamova et al., 2005; Zelcer et al., 2007; Fitz et al., 2010). However, these agents also strongly increase apoE lipidation by increasing ABCA1 and affecting other inflammatory mechanisms. Conversely, blocking the interaction between apoE and Aβ (Sadowski et al., 2006) and decreasing the apoE level by overexpressing low-density lipoprotein receptor (Kim et al., 2009) or haploinsufficiency of apoE (Bales et al., 1999; Holtzman et al., 2000; Kim et al., 2011; Bien-Ly et al., 2012) results in decreased amyloidosis. The anti-apoE antibody in the present study did not alter plasma apoE levels while decreasing apoE level in the brain, as detected by immunostaining associated with plaques, providing evidence that lowering Aβ-associated apoE in the brain plays a beneficial role in Aβ-amyloid-related pathogenesis. Interestingly, using biochemical analysis after anti-apoE antibody treatment, apoE levels decreased mainly in the Triton X-100 fraction of the cortical tissue extract. It is unlikely that the change of apoE in the Triton X-100 fraction is only a consequence of the reduction of coaggregated Aβ in the Triton X-100 fraction because, if this were the case, we would have expected apoE to decrease in the guanidine fraction where the Aβ levels changed as well. In fact, the apoE level in the guanidine fraction was not changed. Triton X-100 is a nonionic detergent that is widely used to extract molecules and lipids from lipid-containing structures such as cell membranes. Our observation suggests the possibility that the anti-apoE antibody HJ6.3 targeted apoE associated with brain lipids and suppressed amyloidosis.
Interestingly, the anti-apoE antibody HJ6.3 increased plasma Aβ levels without affecting plasma apoE. For certain Aβ antibodies, such as m266, increases of peripheral Aβ has been observed because antibody prevented Aβ from being degraded via binding to it and potentially increasing net Aβ efflux out of the brain (DeMattos et al., 2002; Yamada et al., 2009). If a similar mechanism occurred in the present study, then apoE antibody could stabilize apoE as well as Aβ associated with apoE. However, we found that the plasma apoE level did not increase along with Aβ levels upon binding with antibodies. Therefore, the increase of plasma Aβ could be mediated through another mechanism that is due to a central or peripheral effect. Expression of apoE3 and apoE4 in the mouse brain results in diminished efflux of Aβ into the blood compared with apoE2 (Hudry et al., 2013). Overexpression of an apoE receptor, a low-density lipoprotein receptor, lowers the brain apoE level and enhances the rate of brain-to-blood transportation of Aβ (Castellano et al., 2012). In the present study, HJ6.3 may be enhancing Aβ transport across the BBB or enhancing its CSF/ISF to plasma flow via decreasing the apoE level or sequestering apoE.
Increased CAA and CAA-associated microhemorrhage (Pfeifer et al., 2002; Wilcock et al., 2004a) are side effects associated with previous passive Aβ immunotherapy, especially with those Aβ antibodies that target aggregated forms of Aβ if the effector function of the antibody is intact (Pfeifer et al., 2002; Wilcock et al., 2004a; Racke et al., 2005). The mechanism of the anti-Aβ antibody-induced increase of CAA associated microhemorrhage is not entirely clear. The anti-apoE antibody HJ6.3 did not cause a detectable increase of CAA or CAA-associated microhemorrhages. This may be due to the anti-apoE antibody enhancing both apoE and Aβ clearance, but via a different mechanism from certain Aβ antibodies that favor Aβ clearance but may enhance Aβ transport along the interstitial fluid surrounding arterioles where CAA forms. Although apoE is known to play a key role in CAA development (Holtzman et al., 2000; Fryer et al., 2003), therapeutic antibodies targeting apoE may induce fewer or no side effects associated with CAA. The somewhat modest effect of the anti-apoE antibody HJ6.3 on established CNS amyloid levels may also contribute to the lack of CAA and CAA-associated brain microhemorrhages. In addition, the APP/PS1 mice that we used in these studies had a relatively small amount of CAA. In future studies, it will be important to compare some of the differences in anti-Aβ-associated versus anti-apoE-associated effects of CAA and CAA-associated brain hemorrhage in another mouse model. In addition, it will be important to assess the effects of anti-apoE antibodies in animals that express human apoE isoforms. Such experiments may lead to a better understanding of the involvement and consequences of both apoE and Aβ in plaque and CAA formation and aid in treatment development.
Footnotes
This work was supported by the BrightFocus Foundation (Grant A2013037F to F.L.), AstraZeneca (D.M.H.), the Cure Alzheimer's Fund (D.M.H.), the Dreyfoos Program (B.T.H. and E.H.), the Uehara Memorial Foundation (postdoctoral fellowship to Y.H.), and the National Institutes of Health (Grant R37 AG13956 to D.M.H.).
D.M.H. and Washington University have ownership interests in C2N Diagnostics. D.M.H. is on the scientific advisory board of C2N Diagnostics, consults for AstraZeneca, Genentech, and Eli Lilly, and his laboratory receives research grants from AstraZeneca and Eli Lilly. The remaining authors declare no competing financial interests.
References
- Ashford JW. ApoE4: is it the absence of good or the presence of bad? J Alzheimers Dis. 2002;4:141–143. doi: 10.3233/jad-2002-4303. [DOI] [PubMed] [Google Scholar]
- Bacskai BJ, Klunk WE, Mathis CA, Hyman BT. Imaging amyloid-beta deposits in vivo. J Cereb Blood Flow Metab. 2002;22:1035–1041. doi: 10.1097/00004647-200209000-00001. [DOI] [PubMed] [Google Scholar]
- Bales KR, Verina T, Cummins DJ, Du Y, Dodel RC, Saura J, Fishman CE, DeLong CA, Piccardo P, Petegnief V, Ghetti B, Paul SM. Apolipoprotein E is essential for amyloid deposition in the APP(V717F) transgenic mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 1999;96:15233–15238. doi: 10.1073/pnas.96.26.15233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, et al. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- Belinson H, Michaelson DM. Pathological synergism between amyloid-beta and apolipoprotein E4–the most prevalent yet understudied genetic risk factor for Alzheimer's disease. J Alzheimers Dis. 2009;17:469–481. doi: 10.3233/JAD-2009-1065. [DOI] [PubMed] [Google Scholar]
- Bero AW, Yan P, Roh JH, Cirrito JR, Stewart FR, Raichle ME, Lee JM, Holtzman DM. Neuronal activity regulates the regional vulnerability to amyloid-beta deposition. Nat Neurosci. 2011;14:750–756. doi: 10.1038/nn.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bero AW, Bauer AQ, Stewart FR, White BR, Cirrito JR, Raichle ME, Culver JP, Holtzman DM. Bidirectional relationship between functional connectivity and amyloid-beta deposition in mouse brain. J Neurosci. 2012;32:4334–4340. doi: 10.1523/JNEUROSCI.5845-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bien-Ly N, Gillespie AK, Walker D, Yoon SY, Huang Y. Reducing human apolipoprotein E levels attenuates age-dependent Abeta accumulation in mutant human amyloid precursor protein transgenic mice. J Neurosci. 2012;32:4803–4811. doi: 10.1523/JNEUROSCI.0033-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody DL, Holtzman DM. Active and passive immunotherapy for neurodegenerative disorders. Annu Rev Neurosci. 2008;31:175–193. doi: 10.1146/annurev.neuro.31.060407.125529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, Goate AM, Bales KR, Paul SM, Bateman RJ, Holtzman DM. Human apoE isoforms differentially regulate brain amyloid-beta peptide clearance. Sci Transl Med. 2011;3:89ra57. doi: 10.1126/scitranslmed.3002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano JM, Deane R, Gottesdiener AJ, Verghese PB, Stewart FR, West T, Paoletti AC, Kasper TR, DeMattos RB, Zlokovic BV, Holtzman DM. Low-density lipoprotein receptor overexpression enhances the rate of brain-to-blood Abeta clearance in a mouse model of beta-amyloidosis. Proc Natl Acad Sci U S A. 2012;109:15502–15507. doi: 10.1073/pnas.1206446109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921–923. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC, Jr, Rimmler JB, Locke PA, Conneally PM, Schmader KE, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. 1994;7:180–184. doi: 10.1038/ng0694-180. [DOI] [PubMed] [Google Scholar]
- Deane R, Sagare A, Hamm K, Parisi M, Lane S, Finn MB, Holtzman DM, Zlokovic BV. apoE isoform-specific disruption of amyloid beta peptide clearance from mouse brain. J Clin Invest. 2008;118:4002–4013. doi: 10.1172/JCI36663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMattos RB, Bales KR, Cummins DJ, Paul SM, Holtzman DM. Brain to plasma amyloid-beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer's disease. Science. 2002;295:2264–2267. doi: 10.1126/science.1067568. [DOI] [PubMed] [Google Scholar]
- Demattos RB, Lu J, Tang Y, Racke MM, Delong CA, Tzaferis JA, Hole JT, Forster BM, McDonnell PC, Liu F, Kinley RD, Jordan WH, Hutton ML. A plaque-specific antibody clears existing beta-amyloid plaques in Alzheimer's disease mice. Neuron. 2012;76:908–920. doi: 10.1016/j.neuron.2012.10.029. [DOI] [PubMed] [Google Scholar]
- Dodart JC, Marr RA, Koistinaho M, Gregersen BM, Malkani S, Verma IM, Paul SM. Gene delivery of human apolipoprotein E alters brain Abeta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2005;102:1211–1216. doi: 10.1073/pnas.0409072102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitz NF, Cronican A, Pham T, Fogg A, Fauq AH, Chapman R, Lefterov I, Koldamova R. Liver X receptor agonist treatment ameliorates amyloid pathology and memory deficits caused by high-fat diet in APP23 mice. J Neurosci. 2010;30:6862–6872. doi: 10.1523/JNEUROSCI.1051-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox MD, Snyder AZ, Vincent JL, Corbetta M, Van Essen DC, Raichle ME. The human brain is intrinsically organized into dynamic, anticorrelated functional networks. Proc Natl Acad Sci U S A. 2005;102:9673–9678. doi: 10.1073/pnas.0504136102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin KB, Paxinos G. The mouse brain in stereotaxic coordinates. Ed 1. San Diego: Academic; 1996. [Google Scholar]
- Fryer JD, Taylor JW, DeMattos RB, Bales KR, Paul SM, Parsadanian M, Holtzman DM. Apolipoprotein E markedly facilitates age-dependent cerebral amyloid angiopathy and spontaneous hemorrhage in amyloid precursor protein transgenic mice. J Neurosci. 2003;23:7889–7896. doi: 10.1523/JNEUROSCI.23-21-07889.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Holtzman DM, Fagan AM, Mackey B, Tenkova T, Sartorius L, Paul SM, Bales K, Ashe KH, Irizarry MC, Hyman BT. Apolipoprotein E facilitates neuritic and cerebrovascular plaque formation in an Alzheimer's disease model. Ann Neurol. 2000;47:739–747. [PubMed] [Google Scholar]
- Holtzman DM, Morris JC, Goate AM. Alzheimer's disease: the challenge of the second century. Sci Transl Med. 2011;3:77sr1. doi: 10.1126/scitranslmed.3002369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudry E, Dashkoff J, Roe AD, Takeda S, Koffie RM, Hashimoto T, Scheel M, Spires-Jones T, Arbel-Ornath M, Betensky R, Davidson BL, Hyman BT. Gene transfer of human apoe isoforms results in differential modulation of amyloid deposition and neurotoxicity in mouse brain. Sci Transl Med. 2013;5:212ra161. doi: 10.1126/scitranslmed.3007000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowsky JL, Fadale DJ, Anderson J, Xu GM, Gonzales V, Jenkins NA, Copeland NG, Lee MK, Younkin LH, Wagner SL, Younkin SG, Borchelt DR. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum Mol Genet. 2004;13:159–170. doi: 10.1093/hmg/ddh019. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Lee CY, Mandrekar S, Wilkinson B, Cramer P, Zelcer N, Mann K, Lamb B, Willson TM, Collins JL, Richardson JC, Smith JD, Comery TA, Riddell D, Holtzman DM, Tontonoz P, Landreth GE. ApoE promotes the proteolytic degradation of Abeta. Neuron. 2008;58:681–693. doi: 10.1016/j.neuron.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Castellano JM, Jiang H, Basak JM, Parsadanian M, Pham V, Mason SM, Paul SM, Holtzman DM. Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular A beta clearance. Neuron. 2009;64:632–644. doi: 10.1016/j.neuron.2009.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Jiang H, Park S, Eltorai AE, Stewart FR, Yoon H, Basak JM, Finn MB, Holtzman DM. Haploinsufficiency of human APOE reduces amyloid deposition in a mouse model of amyloid-beta amyloidosis. J Neurosci. 2011;31:18007–18012. doi: 10.1523/JNEUROSCI.3773-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Eltorai AE, Jiang H, Liao F, Verghese PB, Kim J, Stewart FR, Basak JM, Holtzman DM. Anti-apoE immunotherapy inhibits amyloid accumulation in a transgenic mouse model of Abeta amyloidosis. J Exp Med. 2012;209:2149–2156. doi: 10.1084/jem.20121274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koldamova RP, Lefterov IM, Staufenbiel M, Wolfe D, Huang S, Glorioso JC, Walter M, Roth MG, Lazo JS. The liver X receptor ligand T0901317 decreases amyloid beta production in vitro and in a mouse model of Alzheimer's disease. J Biol Chem. 2005;280:4079–4088. doi: 10.1074/jbc.M411420200. [DOI] [PubMed] [Google Scholar]
- Lemere CA, Masliah E. Can Alzheimer disease be prevented by amyloid-beta immunotherapy? Nat Rev Neurol. 2010;6:108–119. doi: 10.1038/nrneurol.2009.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Wang K, Gao HM, Mandavilli B, Wang JY, Hong JS. Molecular consequences of activated microglia in the brain: overactivation induces apoptosis. J Neurochem. 2001;77:182–189. doi: 10.1046/j.1471-4159.2001.t01-1-00216.x. [DOI] [PubMed] [Google Scholar]
- Ma J, Yee A, Brewer HB, Jr, Das S, Potter H. Amyloid-associated proteins alpha 1-antichymotrypsin and apolipoprotein E promote assembly of Alzheimer beta-protein into filaments. Nature. 1994;372:92–94. doi: 10.1038/372092a0. [DOI] [PubMed] [Google Scholar]
- Namba Y, Tomonaga M, Kawasaki H, Otomo E, Ikeda K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer's disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991;541:163–166. doi: 10.1016/0006-8993(91)91092-F. [DOI] [PubMed] [Google Scholar]
- Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, Mathews PM, Jucker M. Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science. 2002;298:1379. doi: 10.1126/science.1078259. [DOI] [PubMed] [Google Scholar]
- Racke MM, Boone LI, Hepburn DL, Parsadainian M, Bryan MT, Ness DK, Piroozi KS, Jordan WH, Brown DD, Hoffman WP, Holtzman DM, Bales KR, Gitter BD, May PC, Paul SM, DeMattos RB. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhage in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyloid beta. J Neurosci. 2005;25:629–636. doi: 10.1523/JNEUROSCI.4337-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski MJ, Pankiewicz J, Scholtzova H, Mehta PD, Prelli F, Quartermain D, Wisniewski T. Blocking the apolipoprotein E/amyloid-beta interaction as a potential therapeutic approach for Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:18787–18792. doi: 10.1073/pnas.0604011103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, et al. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Spires TL, Meyer-Luehmann M, Stern EA, McLean PJ, Skoch J, Nguyen PT, Bacskai BJ, Hyman BT. Dendritic spine abnormalities in amyloid precursor protein transgenic mice demonstrated by gene transfer and intravital multiphoton microscopy. J Neurosci. 2005;25:7278–7287. doi: 10.1523/JNEUROSCI.1879-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Saunders AM, Schmechel D, Pericak-Vance M, Enghild J, Salvesen GS, Roses AD. Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A. 1993a;90:1977–1981. doi: 10.1073/pnas.90.5.1977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, Schmechel D, Saunders AM, Goldgaber D, Roses AD. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci U S A. 1993b;90:8098–8102. doi: 10.1073/pnas.90.17.8098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teter B, Raber J, Nathan B, Crutcher KA. The presence of apoE4, not the absence of apoE3, contributes to AD pathology. J Alzheimers Dis. 2002;4:155–163. doi: 10.3233/jad-2002-4305. [DOI] [PubMed] [Google Scholar]
- Thal DR, Capetillo-Zarate E, Schultz C, Rüb U, Saido TC, Yamaguchi H, Haass C, Griffin WS, Del Tredici K, Braak H, Ghebremedhin E. Apolipoprotein E co-localizes with newly formed amyloid beta-protein (Abeta) deposits lacking immunoreactivity against N-terminal epitopes of Abeta in a genotype-dependent manner. Acta Neuropathol. 2005;110:459–471. doi: 10.1007/s00401-005-1053-1. [DOI] [PubMed] [Google Scholar]
- Wang Q, Bardgett ME, Wong M, Wozniak DF, Lou J, McNeil BD, Chen C, Nardi A, Reid DC, Yamada K, Ornitz DM. Ataxia and paroxysmal dyskinesia in mice lacking axonally transported FGF14. Neuron. 2002;35:25–38. doi: 10.1016/S0896-6273(02)00744-4. [DOI] [PubMed] [Google Scholar]
- White BR, Snyder AZ, Cohen AL, Petersen SE, Raichle ME, Schlaggar BL, Culver JP. Resting-state functional connectivity in the human brain revealed with diffuse optical tomography. Neuroimage. 2009;47:148–156. doi: 10.1016/j.neuroimage.2009.03.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White BR, Bauer AQ, Snyder AZ, Schlaggar BL, Lee JM, Culver JP. Imaging of functional connectivity in the mouse brain. PLoS One. 2011;6:e16322. doi: 10.1371/journal.pone.0016322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, DiCarlo G, Henderson D, Jackson J, Clarke K, Ugen KE, Gordon MN, Morgan D. Intracranially administered anti-Abeta antibodies reduce beta-amyloid deposition by mechanisms both independent of and associated with microglial activation. J Neurosci. 2003;23:3745–3751. doi: 10.1523/JNEUROSCI.23-09-03745.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, Morgan D. Passive immunotherapy against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004a;1:24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Rojiani A, Rosenthal A, Levkowitz G, Subbarao S, Alamed J, Wilson D, Wilson N, Freeman MJ, Gordon MN, Morgan D. Passive amyloid immunotherapy clears amyloid and transiently activates microglia in a transgenic mouse model of amyloid deposition. J Neurosci. 2004b;24:6144–6151. doi: 10.1523/JNEUROSCI.1090-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski T, Castaño EM, Golabek A, Vogel T, Frangione B. Acceleration of Alzheimer's fibril formation by apolipoprotein E in vitro. Am J Pathol. 1994;145:1030–1035. [PMC free article] [PubMed] [Google Scholar]
- Wozniak DF, Hartman RE, Boyle MP, Vogt SK, Brooks AR, Tenkova T, Young C, Olney JW, Muglia LJ. Apoptotic neurodegeneration induced by ethanol in neonatal mice is associated with profound learning/memory deficits in juveniles followed by progressive functional recovery in adults. Neurobiol Dis. 2004;17:403–414. doi: 10.1016/j.nbd.2004.08.006. [DOI] [PubMed] [Google Scholar]
- Yamada K, Yabuki C, Seubert P, Schenk D, Hori Y, Ohtsuki S, Terasaki T, Hashimoto T, Iwatsubo T. Abeta immunotherapy: intracerebral sequestration of Abeta by an anti-Abeta monoclonal antibody 266 with high affinity to soluble Abeta. J Neurosci. 2009;29:11393–11398. doi: 10.1523/JNEUROSCI.2021-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelcer N, Khanlou N, Clare R, Jiang Q, Reed-Geaghan EG, Landreth GE, Vinters HV, Tontonoz P. Attenuation of neuroinflammation and Alzheimer's disease pathology by liver x receptors. Proc Natl Acad Sci U S A. 2007;104:10601–10606. doi: 10.1073/pnas.0701096104. [DOI] [PMC free article] [PubMed] [Google Scholar]