

Abstract
Background
Recent studies among males have reported a genotype-environment interaction (G × E) in which low-activity alleles at the monoamine oxidase A (MAOA) locus conferred greater sensitivity to the effects of childhood adversity on risk for conduct disorder (CD). So far, few studies of females have controlled for gene-environment correlation or used females heterozygous for this X-linked gene.
Method
Logistic regression analysis of a sample of 721 females ages 8-17 years from the longitudinal Virginia Twin Study of Adolescent Behavioral Development (VTSABD) assessed the additive effects of MAOA genotypes on risk for CD, together with the main effect of childhood adversity and parental antisocial personality disorder (ASP), as well as the interaction of MAOA with childhood adversity on risk for CD.
Results
A significant main effect of genotype on risk for CD was detected, where low-activity MAOA imparted the greatest risk to CD in girls while controlling for the significant effects of maternal ASP and childhood adversity. Significant G × E with weak effect was detected when environmental exposure was untransformed, indicating a higher sensitivity to childhood adversity in the presence of the high-activity MAOA allele. The interaction was no longer statistically significant after applying a ridit transformation to reflect the sample sizes exposed at each level of childhood adversity.
Conclusions
The main effect of MAOA on risk for CD in females, its absence in males and directional difference of interaction is suggestive of genotype-sex interaction. As the effect of G × E on risk for CD was weak, its inclusion is not justified.
Keywords: Childhood adversity, conduct disorder, gender differences, gene-environment interaction, MAOA
Introduction
The prevalence of conduct disorder (CD) is consistently higher in males than females (Simonoff et al. 1997; Steiner, 1997; Eley et al. 1999; Farrington & Loeber, 2000; Loeber et al. 2000; Moffitt et al. 2001a; Jacobson et al. 2002; Maughan et al. 2004) despite similarities in exposure to environmental risk factors, co-morbidity patterns and age of onset (Herrera & McCloskey, 2001; Moffitt et al. 2001a; Ilomaki et al. 2006). However, when stratified by a genetic risk factor, gender differences may arise in the patterns of exposure to risk factors. Thus, the study of gene-environment interaction (G × E) for CD by gender could elucidate the etiology of gender differences for this outcome.
Environmental risk factors of CD
Childhood adversity is defined as exposure to inter-parental violence, inconsistent parenting and parental neglect in order to capture the common features of salient risk factors of a difficult home environment, which are important to understanding the development of CD and to allow comparability between previous studies of G × E (Caspi et al. 2002; Foley et al. 2004). Exposure to inter-parental violence presents household aggression as a normative part of family relationships (Osofsky, 1995) and may be imitated, giving rise to difficulty in social adjustment outside the home (Dodge, 1986; Fergusson & Horwood, 1998). Males and females have been reported to be equally exposed to inter-parental violence (Moffitt et al. 2001b), although it is unclear whether this risk factor functions similarly in the development of CD in both groups (Herrera & McCloskey, 2001; Becker & McCloskey, 2002; Kinsfogel & Grych, 2004).
Inconsistent parenting is thought to contribute to CD risk by failing to restrain a child’s impulse towards deviance and antisocial behaviors (Gottfredson & Hirschi, 1990). Although rarely studied, no significant gender differences in inconsistent parenting have been reported with respect to CD (Moffitt et al. 2001b).
Parental neglect, defined as ‘the harming of a child through lack of care or supervision’ (Burgess & Conger, 1978), has been most commonly observed as a risk factor (Bassarath, 2001). It is hypothesized to manifest a lack of parental control over a child’s exposure to social risk factors outside of the home, such as deviant peers (Scaramella et al. 2002). Among juvenile delinquents, males and females were found to be equally as likely to be exposed to emotional neglect, although females were more likely to have experienced physical neglect than boys (McCabe et al. 2002).
Genetic risk for CD
Reports of gender differences in the magnitude of genetic and environmental influences in twins have been mixed. Several studies report significant gender differences in the heritability of CD and antisocial behavior in general (Graham & Stevenson, 1985; Eley et al. 1999; Jacobson et al. 2002; Hudziak et al. 2003). By contrast, other studies have reported no gender differences in additive genetic and shared environment effects for CD (Eaves et al. 1997), adult antisocial behavior (Slutske et al. 1997; Rhee & Waldman, 2002) or CD symptoms (Gelhorn et al. 2005). Results from adoption studies are also varied with respect to gender differences of genetic and environmental effects. Some have reported that the same genetic factors are responsible for antisocial behaviors in both males and females (Cadoret & Cain, 1980), whereas others suggest greater genetic effects in female CD compared with males (Langbehn et al. 1998).
Monoamine oxidase A (MAOA, EC 1.4.3.4) is responsible for the degradation of biogenic amines, including the neurotransmitters epinephrine, nor-epinephrine, dopamine and serotonin. MAOA is localized to Xp11.4-Xp11.3. A nonsense mutation in humans causes a truncation of the protein, resulting in the loss of MAOA activity (Brunner et al. 1993). Males with this mutation have engaged in impulsive/aggressive behaviors including rape, arson and assault (Brunner et al. 1993). Similarly, a mutation in transgenic mice yields a non-functioning enzyme that is also associated with increased aggressiveness and injury among male mice and their cage-mates (Cases et al. 1995). The MAOA promoter region contains a variable number tandem repeat (VNTR) polymorphism with suggested effects on mRNA transcription efficiency and may be associated with antisocial behavior (Craig, 2005).
The combined effects of genotype and environment on CD
Gene-environment interaction will be detected at the statistical level when, at the functional level, genetic differences are observed in sensitivity to an environment (Mather & Jinks, 1982). Human studies have reported G × E in the development of CD and antisocial behavior in adoption (Cadoret et al. 1995) and twin studies (Jaffee et al. 2005) in the absence of measured genotypes. Among a community sample of singletons, Caspi et al. (2002) reported findings consistent with the presence of a G × E that exacerbated risk for antisocial behavior when boys with a low-activity MAOA allele were exposed to household maltreatment, defined as maternal rejection, inconsistent presence and identification of any particular primary caregiver, harsh discipline, physical abuse, and sexual abuse among males. The finding in males has been replicated (Foley et al. 2004; Nilsson et al. 2005; Kim-Cohen et al. 2006), although non-replication has also been reported (Haberstick et al. 2005; Young et al. 2006).
Caspi et al. (2002) have also reported findings consistent with the presence of a G × E that is associated with an increased risk for CD in females. However, heterozygous females, comprising 46% of the sample, were not included because of concerns surrounding the perceived inability to estimate genotypic effects as a result of X-inactivation. The loss of such a large portion of the sample is anticipated to result in a loss of power to detect a significant genetic effect on CD diagnosis as well as an incomplete understanding of the etiology of CD in females. Thus, the inclusion and appropriate treatment of data from heterozygous females is necessary in the analysis of the effect of MAOA and G × E on risk for CD. Another study reported evidence of a significant G × E where females with the high-activity MAOA allele were at increased risk for criminal behavior when exposed to psychosocial risk, as defined by the type of housing (multi-family versus owned single family) and experiencing childhood sexual abuse (Sjöberg et al. 2007).
A statistically significant interaction may be due to either gene-environment correlation (rGE), G × E or their combination because unresolved rGE may con-found the detection of G × E. rGE occurs because parents and their children share their genes and home environments. Therefore, genetic differences between parents may contribute to environmental risk in their offspring. One taxonomy differentiates between passive, active and evocative forms of rGE (Plomin et al. 1977; Scarr & McCartney, 1983). Passive rGE is defined as children receiving genotypes that are correlated with their family environment. Evocative rGE refers to a situation where the child’s genotype and behavior elicit parental, familial or teacher responses such as neglect. Active rGE refers to individuals who seek out environments that correspond to their genetically influenced traits. rGE may play a role in CD development because (1) diagnosis of parental antisocial personality disorder (ASP) is associated with neglect of children in the household (APA, 1994), (2) ASP is a heritable disorder (Lyons et al. 1995), and (3) passive (Ge et al. 1996; Meyer et al. 2000) and evocative (O’Connor et al. 1998; Riggins-Caspers et al. 2003) rGE have been identified in the etiology of CD.
Gene-environment correlation for antisocial behavior, particularly evocative rGE, has been reported in adoption studies of CD (Ge et al. 1996; O’Connor et al. 1998). Furthermore, twin studies have reported strong additive genetic and familial effects (as measured by family adaptability) on risk for CD and evidence consistent with the presence of a passive rGE that is associated with risk for CD using an extended twin design (Meyer et al. 2000).
Recent studies using the MAOA genotype to detect G × E in CD among females have not addressed the effect of X-inactivation as a result of inconsistency in determining whether this locus is subject to X-inactivation and whether inactivation of this locus was non-random (Benjamin et al. 2000; Carrel & Willard, 2005; Fraga et al. 2005; Nordquist & Oreland, 2006; Pinsonneault et al. 2006). Consequently, our understanding of the effect of this X-linked marker on CD risk in females and any gender differences that may arise from G × E is limited. Additionally, prior studies in females have not controlled for the effects of rGE, restricting the ability to address the role of G × E in the etiology of CD in this population. Therefore, to understand the role of an X-linked marker on risk for CD and any influence it may have on gender differences in CD, females with homozygous and heterozygous MAOA genotypes were used to test for the main effects of MAOA and childhood adversity as well as any effects associated with G × E in the presence of passive rGE, defined by parental ASP, as risk factors for CD.
Method
Study population
This study is based on 721 female individual participants and their parents from the Virginia Twin Study of Adolescent and Behavioral Development (VTSABD). The eligible sample comprises twin subjects for whom data on MAOA genotype, maternal ASP and exposure to childhood adversity were available. Informed consent was obtained in writing from parents and assent was obtained from the juvenile twin subjects.
The history of this sample, including ascertainment and data collection, has been described in detail elsewhere (Meyer et al. 1996; Hewitt et al. 1997). In brief, the 1412 twin families (2824 children) who were interviewed and used for the subsequent analyses are members of a sequential cohort and were followed prospectively at approximately 15-month intervals over four waves of data collection (Maes et al. 2007). Twins and their parents were ascertained through the Virginia public and private school systems in 1987 and 1988. The first wave of data collection took place between March 1990 and March 1992 and twins in this cohort ranged in age from 8-17 years. As the study progressed, twins over the age of 17 were considered too old for inclusion and were aged out of the sample.
Assessments
Diagnosis of CD
All samples consisted of data on individual twins registered in the VTSABD on a previous 3-month history of CD at any of the four waves as assessed with the Child and Adolescent Psychiatric Assessment (CAPA) – Child and Parent Version (Angold & Costello, 2000), which is based on DSM-III-R criteria (APA, 1987). Symptoms were reported by maternal, paternal or child self-reports across all waves. A symptom was rated as being present when either of the two parents or the child endorsed an item at any wave of data collection. This rating algorithm is referred to as the ‘symptom-or’ rule and is advantageous because it uses responses from multiple informants rather than relying on a single respondent (Simonoff et al. 1997). A CD diagnosis was assigned if subjects had three or more symptoms of CD.
Measurement of childhood adversity
Three measures of negative family environment associated with CD indexed childhood adversity, specifically parental neglect, exposure to inter-parental violence and inconsistent parental discipline. Parental neglect was assessed by parent report and used three items to determine a lack of care severe enough to be recognized by individuals outside the home, including notification from others on the lack of general care for the children, illness due to insufficient parental care and failure to seek medical attention for the children when such care was necessary. Exposure to inter-parental violence was measured by child report and used two items to determine whether parents make physical contact (i.e. pushing, shoving or hitting) with one another during disagreements. Inconsistent parental discipline was obtained by child report to determine whether each parent maintained consistent responses to child rule-breaking. Responses to the binary items were summed and used as a scale ranging from 0 to 7.
Parental ASP symptoms
Maternal and paternal ASP symptoms were measured separately as the sum of the following seven binary items obtained by personal interview with mothers: inconsistent work behavior, failure to conform to social norms and laws, irritability/aggression or involvement in fighting or assault, failure to honor financial obligations, impulsivity, recklessness in the safety of self or others, and no long-term (>1 year) monogamous romantic relationships. Responses to the binary items were summed to create an ordinal score ranging from 0 to 7.
DNA extraction and MAOA genotyping
DNA was obtained from buccal cells using a cytology brush for collection. DNA was isolated using the InstaGene Matrix kit protocol for cell lysis absorption (Bio-Rad Laboratories, Hercules, CA, USA). Genotyping of the MAOA promoter polymorphism used samples with a working concentration of 5-20 ng/ml. Primer sequences were used as described previously (Sabol et al. 1998), specifically MAO APT1 labeled with the FAM-6 fluorophore (5′-ACAGCCTGACCGTG-GAGAAG-3′) and MAO APB1 (5′-GAACGGACGCTCCATTCGGA-3′). Polymerase chain reaction (PCR) amplification of the MAOA promoter region VNTR was performed in 96-well microtitre plates, using a 10-ml volume containing 50-200 ng of genomic DNA, 10 × PCR buffer (Invitrogen), 0.3 mM 2′-deoxynucleoside 5′-triphosphate (Invitrogen), 50 mM magnesium chloride, 0.3 mmol each of forward and reverse primer, and 0.5 U Platinum Taq DNA polymerase (Invitrogen). Cycling reactions were performed on a PTC-225 DNA engine (MJ Research Inc., Waltham, MA, USA) with a 3-min initial denaturation at 95 °C, followed by 35 cycles at 95 °C for 3 min, 62 °C for 1 min, 72 °C for 1.5 min, and concluding with a final extension at 72 °C for 8 min.
Classification of MAOA activity was assigned to each allele resulting from previous work in the efficiency of transcription activity of the MAOA gene promoter as low activity for the 3- and 5-repeat alleles and high activity for the 3.5- and 4-repeat alleles (Sabol et al. 1998). Products were analyzed using an SCE-9610 capillary sequencer (Spectrumedix, State College, PA, USA), ROX-labeled GX-500 size standard and Genospectrum v. 2.6 DNA fragment analysis software (SpectruMedix).
Data analysis
Determination of study sample representativeness
The subsample of females included in this study was compared with the remaining females in VTSABD who were not included to determine whether the subsample was representative of the VTSABD population. Childhood adversity, maternal ASP symptoms, age of entry into the study, and census-based measures of socio-economic indicators were compared using a Wilcoxon rank sum test. Frequency of CD diagnosis was compared using the x2 test.
Test of Hardy-Weinberg equilibrium (HWE)
Female allele frequencies were determined using a randomly selected allele from each MAOA genotype. As human males are not diploid on the X chromosome, HWE was tested in the female genotypes. Male and female allele frequencies were then tested for significant differences in distribution as a populationlevel evaluation of HWE. Male genotypes (n = 578) from the VTSABD were obtained as part of a previously reported replication study of G × E in risk for CD among males (Foley et al. 2004).
Assessment of environmental exposure
Alternate scales of childhood adversity measured as 0/1+, 0/1/2+ or 0/1/2/3+ exposures were considered. As there were no a priori expectations of the most appropriate scale for measuring environmental exposure and none of these measures offered significant improvement in the prediction of CD over scales using the full range of measure (0-5 exposures), each scale could be considered equally informative. Furthermore, a scale consisting of 0/1/2/3+ exposures increases the cell size of those exposed at the highest levels and minimizes loss of information that results from collapsing the scale. Therefore, the ordinal measure of childhood adversity using a scale of 0/1/2/3+ exposures was used. This measure was treated as a continuous variable to maintain model interpretability while attempting to address the issue of low frequency at the highest levels of exposure. Maternal ASP (measured as 0-5 symptoms) was treated in an identical manner and a scale of 0/1/2/3+ symptoms was used.
Testing for additive and dominance effects of MAOA on CD in females
In the absence of experimental data to determine the levels of MAOA expression of homozygotes and heterozygotes, it remains possible to specify their phenotypic differences by dummy coding of the homozygous differences and heterozygous deviations between genotypes at the locus (Mather & Jinks, 1982; Falconer & Mackay, 1996). Thus, the phenotype midway between the homozygous phenotypes is defined as m. The value h is used to identify the phenotypic departure of the heterozygote from m. +d and −d are the phenotypic differences of the homozygotes from the mid-point. Thus, d refers to the fixable or additive genetic variation, and h reflects the unfixable heritable variation or dominance properties. Adapting the framework to reflect a dichotomous phenotype, the contribution associated with one homozygous genotype (AA) to the phenotype can be denoted as 1, and the contribution of the other homozygous genotype (aa) is defined as −1 and the heterozygote as 0 (Fig. 1) (Fisher et al. 1932).
Fig. 1.
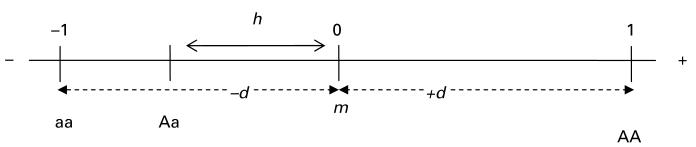
Genotypic representation of a continuous trait (adapted from Mather & Jinks, 1982).
Three possible models can be specified for the effects of the MAOA locus in females using the classical biometrical-genetic approach (Fisher, 1918; Mather & Jinks, 1982): (1) an ‘additive’ model, or one with no dominance in which the phenotypic mean of the heterozygotes is between the means of the two homozygotes; (2) a model of ‘complete dominance’ in which the heterozygote mean is the same as either of the two homozygotes; and (3) a model of ‘incomplete dominance’ in which the mean phenotype of heterozygotes resides between the two homozygotes, while differing from the mid-point. Both additive and dominance effects may interact with the environment if genotypes differ in their sensitivity to the environment.
A preliminary sequence of four logistic regression models (incomplete dominance, additive, dominance for low-activity MAOA, and dominance for high-activity MAOA) was tested to evaluate the relationship between MAOA and CD in the absence of environmental exposure. Each genetic model was compared with a null model in which only the intercept was included.
Testing for main and interaction effects of MAOA activity and environmental risks to CD
Models of decreasing complexity were fitted. First, the model of simple regression of all main effects and all combinations of two- and three-way interactions was fitted. Subsequent models removed nonsignificant parameters as measured by p values >0.05, until no significant increase in deviance resulted. The number of paternal ASP symptoms was not found to be a significant predictor of CD nor to improve model fit as either a main or interaction term and was excluded from the model. Models were compared using goodness-of-fit and parsimony, assessed by the deviance and Akaike’s Information Criterion (AIC) respectively. Lower values of AIC indicate more parsimonious models and lower values of deviance imply improved model fit (Neale & Maes, 2002).
The best-fitting model for risk of CD was selected based on (1) the lowest value of AIC, (2) non-significant differences in deviance from more complex models and (3) significant parameter estimates. Logistic regression was conducted using PROC GENMOD in SAS version 9.1.3 (SAS Institute, Cary, NC, USA). Random residual effects of twin resemblance and repeated measurement were accommodated by using the Generalized Estimating Equation (GEE) algorithm incorporated in the SAS GENMOD procedure on the simplifying assumption of constant correlation between measures within monozygotic and dizygotic twin clusters.
Environmental measures in the final model were standardized to a mean of 0 and a standard deviation of 1 to adequately establish the magnitude of main and interaction effects of final models using PROC STANDARD in SAS. A main effect is defined here as the effect of a parameter averaged across all levels of the other parameters in the model, with each parameter in the model having a mean of 0.
Results
Sample representativeness
Female twins included in this study were younger than those who were not (p<0.0001) because of older participants aging out of the study after age 18 before data collection was completed over all waves. The rates of CD were comparable between study participants (3.1%) and those not included (2.9%, p = 0.88). The average number of maternal ASP symptoms between subsample members (0.89±0.98) and those not included (0.81±0.93) did not differ significantly (p = 0.11). Individuals included in this study were similar for measured census-based indicators of socioeconomic status such as median family income (p = 0.26), rural versus urban residence (p = 0.99) and college education (p = 0.44). Thus, although the females in this study were younger than the larger sample, they represent the total sample with respect to CD prevalence and risk factor exposure.
MAOA allele frequency and test of HWE
The MAOA allele distribution (Table 1) is comparable to those in other studies (Caspi et al. 2002), with 3- and 4-repeat alleles having the highest frequencies. There were no significant differences in expected allele frequencies resulting from female genotypes (p = 0.86), nor between males and female allele frequencies (p = 0.10), and thus no significant departure from HWE.
Table 1. Monoamine oxidase A (MAOA) allele distribution.
| Repeat number |
Males |
Females |
||||
|---|---|---|---|---|---|---|
| Allele | Activity | n | (%) | n | (%) | |
| 1 | 3 | Low | 170 | (28.3) | 235 | (32.2) |
| 2 | 3.5 | High | 12 | (2.0) | 12 | (1.6) |
| 3 | 4 | High | 415 | (69.1) | 468 | (64.1) |
| 4 | 5 | Low | 2 | (0.3) | 12 | (1.6) |
| 5 | 2 | Low | 2 | (0.3) | 3 | (0.4) |
Prevalence of CD and exposure to childhood adversity and symptoms of parental ASP
There were 53 (7.4%) females with CD. Childhood adversity [x2 (df = 3) = 30.6, p<0.0001] and maternal ASP [x2 (df = 3) = 18.0, p = 0.0004] were significantly associated with CD. Paternal ASP was not significantly associated with CD [x2 (df = 3) = 4.34, p = 0.23]. The associations between parental ASP symptoms and childhood adversity as an indicator of passive rGE were assessed by Spearman correlation. Increasing maternal (r = 0.23, p<0.0001) and paternal ASP (r = 0.16, p<0.01) were significantly associated with childhood adversity, indicating a role of passive rGE through maternal ASP on risk for CD.
Prevalence of CD and exposure to environmental risk factors by MAOA genotype
Prevalence of CD was higher in females with low/low genotypes (14.6%) than either low/high (6.2%) or high/high genotypes (5.7%) [x2 (df = 2) = 8.8, p = 0.01]. Within the sample, 41.6% had a high/high MAOA genotype, 45.3% had the heterozygous genotype and 13.1% were found to have a low/low genotype.
Exposure to maternal [Spearman’s rank correlation coefficient (ρ)=−0.06, p = 0.10] or paternal ASP symptoms (ρ=−0.3, p = 0.55) was not associated with daughter’s MAOA genotype. Females with low/low genotype had increased exposure to childhood adversity, although differences were non-significant. In a test of evocative rGE, MAOA genotype did not predict exposure to childhood adversity using a linear regression approach (β = 1.8, p = 0.24), suggesting that MAOA genotype does not affect exposure to childhood adversity.
Testing for additive and dominance effects of MAOA genotype on CD in females
The most parsimonious model of the contribution of MAOA genotype to risk for CD included only additive effects (Table 2, model 2, deviance = 372.6, AIC = 374.6). MAOA was subsequently modeled in an additive fashion (low activity = 1, low/high activity = 0 and high activity=−1) because (1) dominance effects were not significant within the additive/dominance model, (2) improvement in model fit was observed for the more parsimonious additive model and (3) a model of dominance in the direction of high activity (model 4) seemed to oversimplify the effect of the heterozygotes.
Table 2. Summary of model-fitting statistics of genotypic contribution to conduct disorder in females.
| Deviance difference from null |
p values |
|||||
|---|---|---|---|---|---|---|
| Model – MAOA function | df | Deviance | AIC | Additive | Dominance | |
| Null | 0 | 378.55 | 378.55 | |||
| Additive/dominance | 2 | 7.31 | 371.24* | 375.24 | 0.01 | 0.24 |
| Additivity | 1 | 5.94 | 372.61* | 374.61 | 0.04 | |
| Dominance, low activity | 1 | 2.10 | 376.45 | 378.45 | 0.24 | |
| Dominance, high activity | 1 | 7.02 | 371.53* | 373.53 | 0.01 | |
MAOA, Monoamine oxidase A; df, degrees of freedom; AIC, Akaike’s Information Criterion.
Significant difference of deviance at p ≤ 0.05.
Testing for main and interaction effects of MAOA genotype and childhood adversity on risk for CD
The best-fitting model of risk for CD in females (Table 3, model 4, deviance = 338.57 and AIC = 336.57) included (1) MAOA considered as a genotype with additive variance in the heterozygous females, (2) childhood adversity, (3) maternal ASP symptoms, and (4) the interaction of childhood adversity and MAOA genotype.
Table 3. Summary of backwards elimination model fitting in females to predict conduct disorder.
| Model specified | p value | Deviance | AIC | Parameters | |
|---|---|---|---|---|---|
| Model 1 | 1. Childhood adversity | 0.006 | 337.45 | 351.45 | 7 |
| (full model) | 2. Maternal ASP symptoms | 0.02 | |||
| 3. MAOA | 0.13 | ||||
| 4. MAOA × maternal ASP | 0.74 | ||||
| 5. MAOA × childhood adversity | 0.10 | ||||
| 6. Maternal ASP × childhood adversity | 0.55 | ||||
| 7. Childhood adversity × maternal | 0.50 | ||||
| ASP × MAOA | |||||
| Model 2 | 1. Childhood adversity | 0.003 | 337.65 | 349.65 | 6 |
| (drop 7) | 2. Maternal ASP symptoms | 0.03 | |||
| 3. MAOA | 0.12 | ||||
| 4. MAOA × maternal ASP | 0.94 | ||||
| 5. MAOA × childhood adversity | 0.05 | ||||
| 6. Maternal ASP × childhood adversity | 0.52 | ||||
| Model 3 | 1. Childhood adversity | 0.003 | 337.65 | 347.65 | 5 |
| (drop 4) | 2. Maternal ASP symptoms | 0.02 | |||
| 3. MAOA | 0.03 | ||||
| 5. MAOA × childhood adversity | 0.06 | ||||
| 6. Maternal ASP × childhood adversity | 0.50 | ||||
| Model 4 | 1. Childhood adversity | 0.0003 | 338.57 | 346.57 | 4 |
| (drop 6) | 2. Maternal ASP symptoms | 0.006 | |||
| 3. MAOA | 0.02 | ||||
| 5. MAOA × childhood adversity | 0.05 |
AIC, Akaike’s Information Criterion; ASP, antisocial personality disorder; MAOA, Monoamine oxidase A.
Significant G × E is present when controlling for the main effects of passive rGE (maternal ASP symptoms), MAOA genotype and exposure to childhood adversity (Table 4). The significant effect of MAOA on risk for CD reflects the effect of the low/low genotype across low levels of exposure to childhood adversity. The direction of the interaction effect represents the increased risk associated with the high/high and heterozygous genotypes in the presence of higher levels of childhood adversity. The detection of this interaction occurs where the distribution of exposure to childhood adversity is the greatest, highlighting that the ability to detect this interaction is derived from the extremes of the distribution. However, very few cases reside at the highest levels of exposure (Table 5), indicating low power to detect significant G × E using this model. In an attempt to address this issue, a modified ridit transformation (Bross, 1958) was performed to adjust the measure of environmental exposure by the sample size at each level of childhood adversity. The modified ridit transformation determines a score for each category, which is defined as the percentile rank of an item in the population. Therefore, each ridit score reflects the category severity of an ordinal scale and sample size for each level and limits the variance of each level to produce a measure with a uniform distribution having a range between 0 and 1.
Table 4. Parameter estimates and odds ratios for model used to estimate conduct disorder risk in females.
| Final model | Estimate | OR | 95%CI | p value |
|---|---|---|---|---|
| MAOA | 0.46 | 1.59 | 1.03-2.47 | 0.04 |
| Childhood adversity | 0.54 | 1.72 | 1.32-2.25 | <0.0001 |
| Maternal ASP | 0.40 | 1.50 | 1.12-2.00 | 0.006 |
| Childhood adversity × MAOA |
−0.26 | 0.77 | 0.59-0.99 | 0.05 |
OR, Odds ratio; CI, confidence interval; MAOA, monoamine oxidase A; ASP, antisocial personality disorder.
Table 5. Prevalence of female conduct disorder by childhood adversity and monoamine oxidase A (MAOA) genotype.
| Level of exposure to childhood adversity |
Low MAOA |
Low/high MAOA |
High MAOA |
|||
|---|---|---|---|---|---|---|
| n/Total | % | n/Total | % | n/Total | % | |
| 0 | 7/60 | 11.7 | 8/222 | 3.6 | 6/213 | 2.8 |
| 1 | 2/13 | 15.4 | 5/44 | 11.4 | 4/41 | 9.8 |
| 2 | 5/17 | 29.4 | 6/48 | 12.5 | 2/33 | 6.1 |
| ≥3 | 0/6 | 0 | 3/12 | 25.0 | 5/12 | 41.7 |
| Total | 14/96 | 14.6 | 22/326 | 6.2 | 17/299 | 5.7 |
After ridit transformation, a significant main effect of the low-activity MAOA allele on CD remained (b = 0.46, odds ratio (OR) 1.58, 95% confidence interval (CI) 1.01-2.48, p = 0.05) and G × E was non-significant (β=−0.25, OR 0.78, 95% CI 0.57-1.07, p = 0.12). Thus, there is evidence for a weak main effect of MAOA genotype on CD in females. Furthermore, the effects of childhood adversity (β = 0.58, OR 1.78, 95% CI 1.13-1.93, p = 0.0006) and maternal ASP (β = 0.47, OR 1.60, 95% CI 0.99-1.86, p = 0.006) on CD remained significant after ridit transformation. However, significant G × E associated with CD was no longer present.
There was an increased prevalence of CD for the low/low MAOA genotype at all levels of exposure in females (Fig. 2). However, there were no affected individuals with the low/low MAOA genotype at the highest level of exposure (Table 5).
Fig. 2.

Prevalence of female conduct disorder by childhood adversity and monoamine oxidase A (MAOA) genotype.
Discussion
We have demonstrated that the inclusion of females heterozygous for the low- and high-activity MAOA alleles is reasonable and yields meaningful results despite the ambiguity around the issue of X-inactivation by defining both the homozygous (additive) and heterozygous (dominance) effects of MAOA. Additionally, the risk for CD associated with the heterozygous MAOA genotype is between that of the homozygous groups and resembles that of the highactivity genotype (Meyer-Lindenberg et al. 2006).
A study of X-inactivation using human-rodent somatic cell hybrids reported that MAOA among several other genes on the X chromosome escapes inactivation (Carrel & Willard, 2005). However, other studies report that MAOA is subject to random X-inactivation. Benjamin et al. (2000) reported non-skewed patterns of inactivation in genomic DNA obtained from blood samples. A study of monozygotic female twins described non-skewed inactivation in a majority (85%) of samples (Fraga et al. 2005), supporting random X-inactivation. Another study of allelic expression of a single nucleotide polymorphism in exon 6 of MAOA in human skin fibroblasts also demonstrated random monoallelic expression (Nordquist & Oreland, 2006).
A recent study reported that MAOA is subject to X-inactivation using a measure of allelic expression imbalance in human brain tissue concluded that there was no evidence for skewing in normal individuals (Pinsonneault et al. 2006). Furthermore, a recent study of functional response of MAOA genotype for amygdala and cingulate volume demonstrated the functioning of heterozygous females to be in between that of the homozygotes (Meyer-Lindenberg et al. 2006). Therefore, in the presence of inconsistency on whether MAOA is subject to X-inactivation, the inclusion of the additive effects of MAOA genotype on risk to CD is appropriate and consistent with the majority of findings reported in recent molecular and neuroscience literature.
Gender differences in risk for CD
Among females, the persistence of a modest main genetic effect (OR 1.59) of the low/low MAOA genotype while controlling for all other risk factors is striking because there was no significant effect of MAOA on CD in males of the same sample. The observation of a main genetic effect in females rather than males has been reported in twin studies of antisocial behavior. Significant additive genetic effects have been reported to account for a greater amount of variation of ASP in females as compared with males (Eley et al. 1999; Jacobson et al. 2002). However, Gelhorn et al. (2005) demonstrated equal contributions of unmeasured environmental and genetic effects across gender after controlling for prevalence differences in CD symptoms between males and females and such an approach to these data may alter these results. Ultimately, low/low MAOA genotype does not predispose a female to CD, but suggests an increased risk for CD at lower levels of childhood adversity compared with the heterozygous and high/high genotypes.
In models where the measurement of childhood adversity was untransformed, significant G × E with weak effect (OR 0.77, p = 0.05) was detected. The direction of the interaction in females differed from that of males such that the high-activity MAOA allele conferred greater risk for CD at the highest level of childhood adversity in females. Among males, risk for CD increased with increasing exposure to childhood adversity in those with the low-activity MAOA genotype (Caspi et al. 2002; Foley et al. 2004; Nilsson et al. 2005; Kim-Cohen et al. 2006). The difference in direction of interaction along with the presence of a main effect of MAOA in females and its absence in males in this sample (Foley et al. 2004) is suggestive of genotype-sex interaction and has been detected in other studies of MAOA and aggression (Meyer-Lindenberg et al. 2006; Sjöberg et al. 2007). However, there was no significant G × E associated with CD in females after transformation of the measure of childhood adversity into modified ridit scores, lending little support for the inclusion of G × E. A simulation study demonstrated how the consistent false detection of G × E might occur as a result of the treatment of measurement scale for either an outcome or the environment (Eaves, 2006). Thus, the detection of G × E in this study may be contingent on the more or less arbitrary placement of the threshold for diagnosis or environmental exposure (Eaves, 2006). Although the modified ridit transformation reflected the sample size of individuals at each level of childhood adversity, any transformation of scale often leads to the loss of significant interaction (Eaves, 2006). Therefore, although the data illustrate patterns of G × E that differ by sex, the significance of such trends should be interpreted with caution given the vulnerability of G × E to scale.
The detection of G × E depended upon the absence of CD in six individuals with the low/low MAOA genotype who were also exposed to high levels of childhood adversity. The absence of CD diagnosis in these participants reflects measurement of CD at all waves and does not indicate loss to follow-up. Furthermore, upon inspection of the items used for CD diagnosis, these individuals endorsed items reflecting covert symptoms including lying and truancy rather than overt symptoms such as interpersonal aggression. Therefore, an item response theory approach to measure CD using symptoms to reflect a latent trait rather than a diagnosis is anticipated to have better psychometric properties and improve power to detect G × E (Eaves et al. 2005).
The estimate of a passive rGE associated with risk for CD in females, as measured by the association between child CD and maternal ASP, was significant. Although maternal ASP may be associated with childhood risk to CD, its effect is apparently not mediated by childhood adversity because the effect of maternal ASP remains significant in models that include maternal ASP as a covariate. However, the genotypic differences in sensitivity to childhood adversity may relate to a general measure of family dysfunction rather than simply a specified measure of childhood adversity or maternal ASP as reported in a recent study of the interaction of family dysfunction and genetic effects in outcomes of antisocial symptoms (Button et al. 2005). As there are significant associations in this sample between paternal and maternal ASP symptoms, assortative mating for ASP among adults may result in a household with family dysfunction. Thus, it is plausible that children receive their genotypes as well as their environmental exposure from the parents in the form of family dysfunction and related social cues to manage the environment through interpersonal interactions (passive rGE). Additionally, gender differences in processing the home environment may explain the gender differences for CD symptoms. This may also explain why female CD is more likely to result from disrupted relationships with carers or peers and females are more likely to engage in interpersonal violence against family members or intimate partners (Moffitt et al. 2001a; Ehrensaft, 2005). The results from this study encourage family-centered prevention efforts interested in altering environmental exposure to childhood adversity and treating parental ASP, because no genotypic group is completely protected from the effects of household difficulty.
Further analysis of the transmission of antisocial behavior between twins and their parents is required to resolve the role of childhood adversity in the correlation between parental ASP and child CD. Larger samples would enable the characterization of profiles of covariates that differentiate between subjects who have high genetic risk yet did not manifest the disorder. However, the small number of subjects and large number of potential covariates precludes such post-hoc mining of the data for this purpose. It would be helpful to test the assumptions of X-linked inheritance on CD diagnosis to determine whether they explain the lower prevalence of CD in females compared to males.
These results should be evaluated in the light of the following limitations. First, we were unable to estimate risk for CD among females with low MAOA activity also experiencing three or more exposures to childhood adversity because of a lack of observations (Fig. 2). Consequently, G × E may have been initially overestimated and after transformation was lost. The variable strength of G × E highlights the issue of scale in our measurement and treatment of environmental exposure towards the detection of G × E and genetic effects in humans, reinforcing the need for better measures of environmental risk for psychiatric disorders. Second, this study is a cross-sectional analysis of longitudinal data from four waves of data. Age was not included as a covariate and these results therefore reflect the risk for CD associated with childhood adversity, maternal ASP and MAOA throughout adolescence. Third, these analyses treated CD as a categorical outcome and ignored the additional information that might be reflected by using indices of severity such as symptom counts or by differentiating subtypes such as aggressive and non-aggressive behaviors. Fourth, the occurrence of X-inactivation in females has resulted in little attention to differences in enzyme function for different MAOA genotypes in females. Finally, all participants were Caucasian and the results may not generalize to populations of differing ethnicities and cultural norms.
Acknowledgements
This work was supported by National Institutes of Health Grants MH-45268 and MH-068521 and NIMH Training Grant MH-20030. We acknowledge the contribution of the VTSABD, now part of the Mid-Atlantic Twin Registry (MATR), for ascertainment of subjects for this study. We thank Dr Dawn Thiselton for her helpful comments on earlier versions of this manuscript.
Footnotes
Declaration of Interest
None.
References
- Angold A, Costello EJ. The Child and Adolescent Psychiatric Assessment (CAPA) Journal of the American Academy of Child and Adolescent Psychiatry. 2000;39:39–48. doi: 10.1097/00004583-200001000-00015. [DOI] [PubMed] [Google Scholar]
- APA . Diagnostic and Statistical Manual of Mental Disorders. 3rd edn American Psychiatric Association; Washington, DC: 1987. [Google Scholar]
- APA . Diagnostic and Statistical Manual of Mental Disorders. 4th edn American Psychiatric Association; Washington, DC: 1994. [Google Scholar]
- Bassarath L. Conduct disorder: a biopsychosocial review. Canadian Journal of Psychiatry. 2001;46:609–616. doi: 10.1177/070674370104600704. [DOI] [PubMed] [Google Scholar]
- Becker KB, McCloskey LA. Attention and conduct problems in children exposed to family violence. American Journal of Orthopsychiatry. 2002;72:83–91. doi: 10.1037//0002-9432.72.1.83. [DOI] [PubMed] [Google Scholar]
- Benjamin D, Van Bakel I, Craig IW. A novel expression based approach for assessing the inactivation status of human X-linked genes. European Journal of Human Genetics. 2000;8:103–108. doi: 10.1038/sj.ejhg.5200427. [DOI] [PubMed] [Google Scholar]
- Bross IDJ. How to use ridit analysis. Biometrics. 1958;14:18–38. [Google Scholar]
- Brunner HG, Nelen M, Breakefield XO, Ropers HH, van Oost BA. Abnormal behavior associated with a point mutation in the structural gene for monoamine oxidase A. Science. 1993;262:578–580. doi: 10.1126/science.8211186. [DOI] [PubMed] [Google Scholar]
- Burgess DL, Conger RD. Family interaction in abusive, neglectful and normal families. Child Development. 1978;49:1163–1173. [PubMed] [Google Scholar]
- Button TMM, Scourfield J, Martin N, Purcell S, McGuffin P. Family dysfunction interacts with genes in the causation of antisocial symptoms. Behavior Genetics. 2005;35:115–120. doi: 10.1007/s10519-004-0826-y. [DOI] [PubMed] [Google Scholar]
- Cadoret RJ, Cain C. Sex differences in predictors of antisocial behavior in adoptees. Archives of General Psychiatry. 1980;37:1171–1175. doi: 10.1001/archpsyc.1980.01780230089013. [DOI] [PubMed] [Google Scholar]
- Cadoret RJ, Yates WR, Troughton E, Woodworth G, Stewart MA. Genetic-environmental interaction in the genesis of aggressivity and conduct disorders. Archives of General Psychiatry. 1995;52:916–924. doi: 10.1001/archpsyc.1995.03950230030006. [DOI] [PubMed] [Google Scholar]
- Carrel L, Willard HF. X-inactivation profile reveals extensive variability in X-linked gene expression in females. Nature. 2005;434:400–404. doi: 10.1038/nature03479. [DOI] [PubMed] [Google Scholar]
- Cases O, Seif I, Grimsby J, Gaspar P, Chen K, Puournin S, Müeller U, Aguet M, Babinet C, Chen Shih J, De Maeyer E. Aggressive behavior and altered amounts of brain serotonin and norepinephrine in mice lacking MAOA. Science. 1995;268:1763–1766. doi: 10.1126/science.7792602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi A, McClay J, Moffitt TE, Mill J, Martin J, Craig IW, Taylor A, Poulton R. Role of genotype in the cycle of violence in maltreated children. Science. 2002;297:851–854. doi: 10.1126/science.1072290. [DOI] [PubMed] [Google Scholar]
- Craig IW. The role of monoamine oxidase A, MAOA, in the aetiology of antisocial behaviour: the importance of gene-environment interactions. Novartis Foundation Symposium. 2005;268:227–237. doi: 10.1002/0470010703.ch16. discussion 237-253. [DOI] [PubMed] [Google Scholar]
- Dodge KA. A social-information processing model of social competence in children. In: Perlmutter M, editor. Minnesota Symposium in Child Psychology. Vol. 18. Erlbaum; Hillsdale, NJ: 1986. pp. 77–125. [Google Scholar]
- Eaves L, Erkanli A, Silberg J, Angold A, Maes H, Foley DL. Application of Baysian inference using Gibbs sampling to item-response theory modeling of multi-symptom genetic data. Behavior Genetics. 2005;35:765–780. doi: 10.1007/s10519-005-7284-z. [DOI] [PubMed] [Google Scholar]
- Eaves LJ. Genotype × environment interaction in psychopathology: fact or artifact? Twin Research and Human Genetics. 2006;9:1–8. doi: 10.1375/183242706776403073. [DOI] [PubMed] [Google Scholar]
- Eaves LJ, Silberg JL, Meyer JM, Maes HH, Simonoff E, Pickles A, Rutter M, Neale MC, Reynolds CA, Erikson MT, Heath AC, Loeber R, Truett KR, Hewitt JK. Genetics and developmental psychopathology: 2. The main effects of genes and environment on behavioral problems in the Virginia Twin Study of Adolescent Behavioral Development. Journal of Child Psychology and Psychiatry. 1997;38:965–980. doi: 10.1111/j.1469-7610.1997.tb01614.x. [DOI] [PubMed] [Google Scholar]
- Ehrensaft MK. Interpersonal relationships and sex differences in the development of conduct problems. Clinical Child and Family Psychology Review. 2005;8:39–63. doi: 10.1007/s10567-005-2341-y. [DOI] [PubMed] [Google Scholar]
- Eley TC, Lichtenstein P, Stevenson J. Sex differences in the etiology of aggressive and non-aggressive antisocial behavior: results from two twin studies. Child Development. 1999;70:155–168. doi: 10.1111/1467-8624.00012. [DOI] [PubMed] [Google Scholar]
- Falconer DS, Mackay TFC. Introduction to Quantitative Genetics. 4th edn Longan; New York: 1996. [Google Scholar]
- Farrington DP, Loeber R. Epidemiology of juvenile violence. Child and Adolescent Psychiatric Clinics of North America. 2000;9:733–748. [PubMed] [Google Scholar]
- Fergusson DM, Horwood LJ. Exposure to interparental violence in childhood and psychosocial adjustment in young adulthood. Child Abuse and Neglect. 1998;22:339–357. doi: 10.1016/s0145-2134(98)00004-0. [DOI] [PubMed] [Google Scholar]
- Fisher RA. The correlation between relatives on the supposition of Mendelian inheritance. Transactions of the Royal Society of Edinburgh. 1918;52:399–433. [Google Scholar]
- Fisher RA, Immer FR, Tedin O. The genetical interpretation of statistics of the third degree in the study of quantitative inheritance. Genetics. 1932;17:107–124. doi: 10.1093/genetics/17.2.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley DL, Eaves LJ, Wormley B, Silberg JL, Maes HH, Kuhn J, Riley B. Childhood adversity, monoamine oxidase A genotype, and risk for conduct disorder. Archives of General Psychiatry. 2004;61:738–744. doi: 10.1001/archpsyc.61.7.738. [DOI] [PubMed] [Google Scholar]
- Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J, Boix-Chornet M, Sanchez-Aguilera A, Ling C, Carlsson E, Poulsen P, Vaag A, Stephan Z, Spector TD, Wu YZ, Plass C, Esteller M. Epigenetic differences arise during the lifetime of monozygotic twins. Proceedings of the National Academy of Sciences USA. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X, Conger RD, Cadoret RJ, Neiderhiser JM, Yates WR, Troughton E, Stewart MA. The developmental interface between nature and nurture: a mutual influence model of child antisocial behavior and parent behaviors. Developmental Psychology. 1996;32:574–589. [Google Scholar]
- Gelhorn HL, Stallings MC, Young SE, Corley RP, Rhee SH, Hewitt JK. Genetic and environmental influences on conduct disorder: symptom, domain and full-scale analyses. Journal of Child Psychology and Psychiatry. 2005;46:580–591. doi: 10.1111/j.1469-7610.2004.00373.x. [DOI] [PubMed] [Google Scholar]
- Gottfredson M, Hirschi T. A General Theory of Crime. Stanford University Press; Stanford, CA: 1990. [Google Scholar]
- Graham P, Stevenson J. A twin study of genetic influences on behavioral deviance. Journal of the American Academy of Child Psychiatry. 1985;24:33–41. doi: 10.1016/s0002-7138(09)60407-6. [DOI] [PubMed] [Google Scholar]
- Haberstick BC, Lessem JM, Hopfer CJ, Smolen A, Ehringer MA, Timberlake D, Hewitt J. Monoamine oxidase A (MAOA) and antisocial behaviors in the presence of childhood and adolescent maltreatment. American Journal of Medical Genetics. Part B, Neuropsychiatric Genetics. 2005;135:59–64. doi: 10.1002/ajmg.b.30176. [DOI] [PubMed] [Google Scholar]
- Herrera VM, McCloskey LA. Gender differences in the risk for delinquency among youth exposed to family violence. Child Abuse and Neglect. 2001;25:1037–1051. doi: 10.1016/s0145-2134(01)00255-1. [DOI] [PubMed] [Google Scholar]
- Hewitt JK, Silberg JL, Rutter M, Simonoff E, Meyer JM, Maes H, Pickles A, Neale MC, Loeber R, Erickson MT, Kendler KS, Heath AC, Truett KR, Reynolds CA, Eaves LJ. Genetics and developmental psychopathology: 1. Phenotypic assessment in the Virginia Twin Study of Adolescent Behavioral Development. Journal of Child Psychology and Psychiatry. 1997;38:943–963. doi: 10.1111/j.1469-7610.1997.tb01613.x. [DOI] [PubMed] [Google Scholar]
- Hudziak JJ, van Beijsterveldt CE, Bartels M, Rietveld MJ, Rettew DC, Derks EM, Boomsma DI. Individual differences in aggression: genetic analyses by age, gender, and informant in 3-, 7-, and 10-year-old Dutch twins. Behavior Genetics. 2003;33:575–589. doi: 10.1023/a:1025782918793. [DOI] [PubMed] [Google Scholar]
- Ilomaki E, Viilo K, Hakko H, Marttunen M, Makikyro T, Rasanen P. Familial risks, conduct disorder and violence: a Finnish study of 278 adolescent boys and girls. European Journal of Child and Adolescent Psychiatry. 2006;15:46–51. doi: 10.1007/s00787-006-0507-x. [DOI] [PubMed] [Google Scholar]
- Jacobson KC, Prescott CA, Kendler KS. Sex differences in the genetic and environmental influences on the development of antisocial behavior. Development and Psychopathology. 2002;14:395–416. doi: 10.1017/s0954579402002110. [DOI] [PubMed] [Google Scholar]
- Jaffee SR, Caspi A, Moffitt TE, Dodge KA, Rutter M, Taylor A, Tully LA. Nature × nurture: genetic vulnerabilities interact with physical maltreatment to promote conduct problems. Development and Psychopathology. 2005;17:67–84. doi: 10.1017/s0954579405050042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim-Cohen J, Caspi A, Taylor A, Williams B, Newcombe R, Craig IW, Moffitt TE. MAOA, maltreatment, and gene-environment interaction predicting children’s mental health: new evidence and a meta-analysis. Molecular Psychiatry. 2006;11:903–913. doi: 10.1038/sj.mp.4001851. [DOI] [PubMed] [Google Scholar]
- Kinsfogel KM, Grych JH. Interparental conflict and adolescent dating relationships: integrating cognitive, emotional, and peer influences. Journal of Family Psychology. 2004;18:505–515. doi: 10.1037/0893-3200.18.3.505. [DOI] [PubMed] [Google Scholar]
- Langbehn DR, Cadoret RJ, Yates WR, Troughton EP, Stewart MA. Distinct contributions of conduct and oppositional defiant symptoms to adult antisocial behavior: evidence from an adoption study. Archives of General Psychiatry. 1998;55:821–829. doi: 10.1001/archpsyc.55.9.821. [DOI] [PubMed] [Google Scholar]
- Loeber R, Burke JD, Lahey BB, Winters A, Zera M. Oppositional defiant and conduct disorder: a review of the past 10 years, part I. Journal of the American Academy of Child and Adolescent Psychiatry. 2000;39:1468–1484. doi: 10.1097/00004583-200012000-00007. [DOI] [PubMed] [Google Scholar]
- Lyons MJ, True WR, Eisen SA, Goldberg J, Meyer J, Faraone SV, Eaves LJ, Tsuang MT. Differential heritability of adult and juvenile antisocial traits. Archives of General Psychiatry. 1995;52:906–915. doi: 10.1001/archpsyc.1995.03950230020005. [DOI] [PubMed] [Google Scholar]
- Maes H, Silberg J, Neale MC, Eaves LJ. Genetic and cultural transmission of antisocial behavior: an extended twin parent model. Twin Research and Human Genetics. 2007;10:136–150. doi: 10.1375/twin.10.1.136. [DOI] [PubMed] [Google Scholar]
- Mather K, Jinks JL. Biometrical Genetics. Chapman & Hall; London: 1982. [Google Scholar]
- Maughan B, Rowe R, Messer J, Goodman R, Meltzer H. Conduct disorder and oppositional defiant disorder in a national sample: developmental epidemiology. Journal of Child Psychology and Psychiatry. 2004;45:609–621. doi: 10.1111/j.1469-7610.2004.00250.x. [DOI] [PubMed] [Google Scholar]
- McCabe KM, Lansing AE, Garland A, Hough R. Gender differences in psychopathology, functional impairment, and familial risk factors among adjudicated delinquents. Journal of the American Academy of Child and Adolescent Psychiatry. 2002;41:860–867. doi: 10.1097/00004583-200207000-00020. [DOI] [PubMed] [Google Scholar]
- Meyer JM, Rutter M, Silberg JL, Maes HH, Simonoff E, Shillady LL, Pickles A, Hewitt JK, Eaves LJ. Familial aggregation for conduct disorder symptomatology: the role of genes, marital discord and family adaptability. Psychological Medicine. 2000;30:759–774. doi: 10.1017/s0033291799002408. [DOI] [PubMed] [Google Scholar]
- Meyer JM, Silberg JL, Simonoff E, Kendler KS, Hewitt JK. The Virginia Twin-Family Study of Adolescent Behavioral Development: assessing sample biases in demographic correlates of psychopathology. Psychological Medicine. 1996;26:1119–1133. doi: 10.1017/s0033291700035844. [DOI] [PubMed] [Google Scholar]
- Meyer-Lindenberg A, Buckholtz JW, Kolachana B, Hariri AR, Pezawas L, Blasi G, Wabnitz A, Honea R, Verchiniski B, Callicott JH, Egan M, Mattay V, Weinberger DR. Neural mechanisms of genetic risk for impulsivity and violence in humans. Proceedings of the National Academy of Sciences USA. 2006;103:6269–6274. doi: 10.1073/pnas.0511311103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffitt TE, Caspi A, Rutter M, Silva PA. Sex Differences in Antisocial Behavior. Cambridge University Press; Cambridge, UK: 2001a. [Google Scholar]
- Moffitt TE, Caspi A, Rutter M, Silva PA. Sex Differences in Antisocial Behavior: Conduct Disorder, Delinquency, and Violence in the Dunedin Longitudinal Study. Cambridge University Press; Cambridge, UK: 2001b. Sex effects in risk predictors for antisocial behavior: are males exposed to more risk factors for antisocial behavior? pp. 109–122. [Google Scholar]
- Neale MC, Maes HM. Methodology for Genetic Studies of Twins and Families. Kluwer; Dordrecht, The Netherlands: 2002. [Google Scholar]
- Nilsson KW, Sjöberg RL, Damberg M, Leppert J, Öhrvik J, Alm PO, Lindstrom L, Oreland L. Role of monoamine oxidase A genotype and psychosocial factors in male adolescent criminal activity. Biological Psychiatry. 2005;59:121–127. doi: 10.1016/j.biopsych.2005.06.024. [DOI] [PubMed] [Google Scholar]
- Nordquist N, Oreland L. Monoallelic expression of MAOA in skin fibroblasts. Biochemical and Biophysical Research Communications. 2006;348:763–767. doi: 10.1016/j.bbrc.2006.07.131. [DOI] [PubMed] [Google Scholar]
- O’Connor TG, Deater-Deckard K, Fulker D, Rutter M, Plomin R. Genotype-environment correlations in late childhood and early adolescence: antisocial behavioral problems and coercive parenting. Developmental Psychology. 1998;34:970–981. doi: 10.1037//0012-1649.34.5.970. [DOI] [PubMed] [Google Scholar]
- Osofsky JD. Children who witness domestic violence: the invisible victims. SRCD Social Policy Report. 1995;9:1–16. [Google Scholar]
- Pinsonneault JK, Papp AC, Sadee W. Allelic mRNA expression of X-linked monoamine oxidase A (MAOA) in human brain: dissection of epigenetic and genetic factors. Human Molecular Genetics. 2006;15:2636–2649. doi: 10.1093/hmg/ddl192. [DOI] [PubMed] [Google Scholar]
- Plomin R, DeFries JC, Loehlin JC. Genotype-environment interaction and correlation in the analysis of human behavior. Psychological Bulletin. 1977;84:309–322. [PubMed] [Google Scholar]
- Rhee SH, Waldman ID. Genetic and environmental influences on antisocial behavior: a meta-analysis of twin and adoption studies. Psychological Bulletin. 2002;128:490–529. [PubMed] [Google Scholar]
- Riggins-Caspers KM, adoret RJ, Knutson JF, Langbehn D. Biology-environment interaction and evocative biology-environment correlation: contributions of harsh discipline and parental psychopathology to problem adolescent behaviors. Behavior Genetics. 2003;33:205–220. doi: 10.1023/a:1023434206261. [DOI] [PubMed] [Google Scholar]
- Sabol SZ, Hu S, Hamer D. A functional polymorphism in the monoamine oxidase A gene promoter. Human Genetics. 1998;103:273–279. doi: 10.1007/s004390050816. [DOI] [PubMed] [Google Scholar]
- Scaramella LV, Conger RD, Spoth R, Simons RL. Evaluation of a social contextual model of delinquency: a cross-study replication. Child Development. 2002;73:175–195. doi: 10.1111/1467-8624.00399. [DOI] [PubMed] [Google Scholar]
- Scarr S, McCartney K. How people make their own environments: a theory of genotype-environment effects. Child Development. 1983;54:424–435. doi: 10.1111/j.1467-8624.1983.tb03884.x. [DOI] [PubMed] [Google Scholar]
- Simonoff E, Pickles A, Meyer JM, Silberg JL, Maes HH, Loeber R, Rutter M, Hewitt JK, Eaves LJ. The Virginia Twin Study of Adolescent Behavioral Development. Influences of age, sex, and impairment on rates of disorder. Archives of General Psychiatry. 1997;54:801–808. doi: 10.1001/archpsyc.1997.01830210039004. [DOI] [PubMed] [Google Scholar]
- Sjöberg RL, Nilsson KW, Wargelius HL, Leppert J, Lindstrom L, Oreland L. Adolescent girls and criminal activity: role of MAOA-LPR genotype and psychosocial factors. American Journal of Medical Genetics, Part B. Neuropsychiatric Genetics. 2007;144:159–164. doi: 10.1002/ajmg.b.30360. [DOI] [PubMed] [Google Scholar]
- Slutske WS, Heath AC, Dinwiddie SH, Madden PA, Bucholz KK, Dunne MP, Statham DJ, Martin NG. Modeling genetic and environmental influences in the etiology of conduct disorder: a study of 2,682 adult twin pairs. Journal of Abnormal Psychology. 1997;106:266–279. doi: 10.1037//0021-843x.106.2.266. [DOI] [PubMed] [Google Scholar]
- Steiner H. Practice parameters for the assessment and treatment of children and adolescents with conduct disorder. Journal of the American Academy of Child and Adolescent Psychiatry. 1997;36:S122–S139. doi: 10.1097/00004583-199710001-00008. [DOI] [PubMed] [Google Scholar]
- Young SE, Smolen A, Hewitt JK, Haberstick BC, Stallings MC, Corley RP, Crowley TJ. Interaction between MAO-A genotype and maltreatment in the risk for conduct disorder: failure to confirm in adolescent patients. American Journal of Psychiatry. 2006;163:1019–1025. doi: 10.1176/ajp.2006.163.6.1019. [DOI] [PubMed] [Google Scholar]