

Definition and Introduction
Molecular Imaging uses imaging technology to demonstrate molecular and cellular events in living organisms. This is in contradistinction to traditional imaging modalities such as CT, which rely on such attributes as physical density to create images.
Molecular Imaging is very attractive to both physicians and researchers, as it allows investigation into more basic aspects of disease than traditional modalities. In both research and clinical applications, Molecular Imaging has yielded great insights into disease mechanisms, and allowed patient care to be improved.
All Molecular Imaging technologies rely on the same basic model. There is: a) a probe or imaging agent (sometimes endogenous), that b) interacts with the molecular or cellular process of interest, and c) provides signal that is usually amplified in some way, that can d) be used to create an image.
This basic model has been adapted in countless ways to investigate biological questions in nearly every area of medicine. Each application typically requires problems to be overcome centered around the fundamental areas of probe design, biological interaction and image generation, with considerable expertise in a variety of fields required to create a useful imaging system.
In this review, focusing on Molecular Neuroimaging, we will show examples of such applications in the nervous system. We will successively deal with: Receptors, Neurons, Stem Cells, Tumor Cells, Migroglia, Marker genes, Bioluminescence and Alzheimer related proteins and show examples of each application.
Receptors
Some of the earliest work in Molecular Neuroimaging was aimed at investigating dopaminergic receptors. As far back as 1973, investigators had synthesized C11-labeled dopamine, and were using it in animal evaluations to map the distributions of dopamine receptors with PET [2]. 75Br-Butyrophenone compounds were used to map out the location of dopaminergic receptors in baboons with PET [3]. Human use of 3-N-[11C] methylspiperone was first reported in 1983 [4], and an atlas of human PET images showing the distribution of dopamine receptors was published soon after [5].
Dopaminergic imaging can probe at three locations in the neurotransmitter pathway: the presynaptic neuron, the postsynaptic neuron and at intraneuronal metabolism [6]. For post-synaptic imaging [11C] raclopride has a higher selectivity and more moderate affinity for dopamine D2 receptors than dopamine itself, well suited for applications in psychiatry and neurology where diseases like addiction, schizophrenia and Parkinsonism are known to be mediated through this receptor [7].[18F]-DOPA is best suited for presynaptic and intraneuronal imaging, being the radiolabeled precursor of dopamine, thus probing for dopamine metabolism [8].
The dopaminergic system is very complex, and current knowledge recognizes multiple receptor subtypes, transporters and enzymes, many of which have been targeted for probe development using a variety of ligands and radionuclides (see Table 1. Selected dopamine radioligands and their uses). These probes support a rich research and clinical infrastructure, and allow sophisticated questions to be asked on a wide variety of diseases including drug addiction, schizophrenia, Parkinsonism and many others.
Table 1.
Selected Dopamine radioligands and their uses (Adapted from Lindsey and Gatley, Applications of Clinical Dopamine imaging, Neuroimaging Clinics of North America, Nov 2006 [52])
| Target | Relevance | Radioligand | Mechanism and use |
|---|---|---|---|
| Aromatic amino acid Decarboxylase (AADC) |
Enzyme that produces dopamine From L-DOPA |
[18F]FluoroDOPA | AADC produces fluoro-dopamine that is stored in synaptic vesicles; indicates dopaminergic function |
| D1 receptor | Involved in addiction, movement disorders, parkinsonism and schizophrenia | [11C]SCH23390 | Receptor mapping |
| D2 receptor | Involved in movement disorders, parkinsonism and schizophrenia—main target of neuroleptics | [11C]raclopride [18F]fallypride [123I] epidepride [123I]IBZM |
Receptor mapping Receptor occupancy of neuroleptics Alterations in endogenous dopamine Receptor mapping Receptor mapping Similar to [11C]raclopride |
| D3 receptor | D2-like; restricted anatomically | None as yet | -- |
| D4 receptor | D2-like; important in schizophrenia | None as yet | -- |
| D5-receptor | D1-like; | None as yet | -- |
| Dopamine Transporter (DAT) | Controls extracellular dopamine; target of cocaine and similar drugs | [11C]cocaine [11C]CFT [99mTc]TRODAT [123I]beta-CIT |
Research, occupancy Transporter mapping, Loss of dopamine terminals in parkinsonism |
| Vesicular monoamine transporter (VMAT2) | Concentrates dopamine (and other monoamine neuro-transmitters) in vesicles; target of the drug reserpine | [11C]dihydrotetra benazine | Loss of dopamine nerve terminals—may be superior to DAT radioligands |
| Monoamine oxidase A (MAOA) | Destroys dopamine after reuptake in nerve terminals | [11C]clorgyline | Enzyme mapping—selective suicide inhibitor |
| Monoamine oxidase B (MAOB) | Destroys monoamines after uptake into glial cells; marker of gliosis | [11C]L-deprenyl | Enzyme mapping—selective suicide inhibitor |
The principle of dopaminergic receptor imaging is to create a radioactive ligand that will bind specifically to the dopamine receptor in question, leaving a fraction of the injected agent bound to the receptor, while the remaining fraction is cleared away by renal clearance and diffusion (Fig. 1). This allows contrast to develop, which can be detected by means of positron emission tomography to yield an image. When receptor densities are altered by pathology, such as addiction, these changes can be visualized with PET (Fig. 2). Many other receptor systems have been similarly characterized and have imaging probes to interrogate them.
Figure 1.

Line diagram showing imaging agents either binding to targets in the body, or being excreted, allowing imaging contrast to develop, as with this example of a PET imaging agent.
Figure 2.
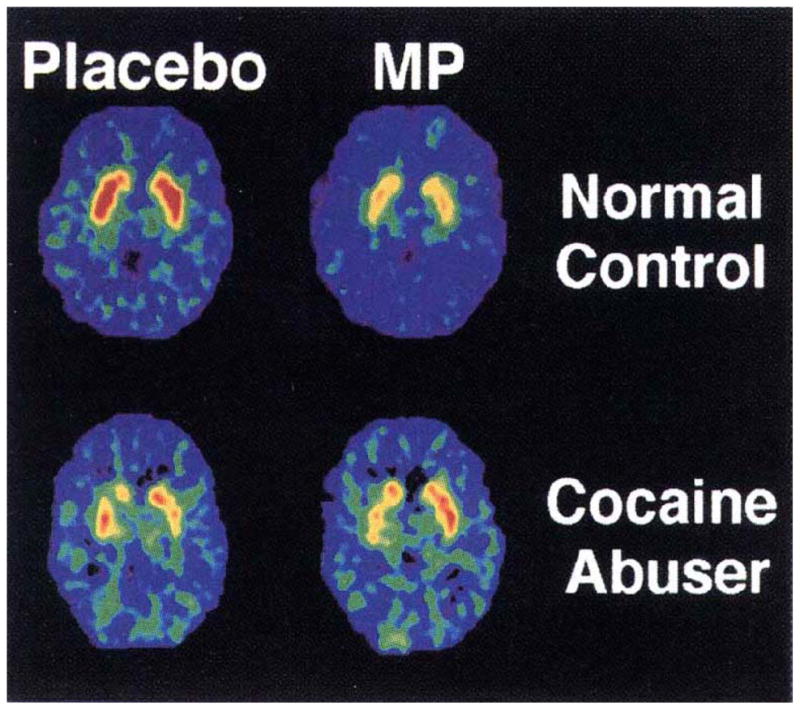
Distribution volume images of [11C] raclopride at the level of the striatum in a normal control and a detoxified cocaine-dependent subject tested after placebo (baseline) and after methylphenidate (MP) administration. Baseline binding for [11C]raclopride is reduced in the addicted subject, and changed less with an MP challenge than the normal subject, suggesting a persistent change in the brain receptor density due to prior cocaine exposure (adapted from [9], with permission).
Neurons
There is no known specific neuronal mass imaging marker, or agent that can measure and quantitate neurons only, as separate and distinct from glia and supporting cells. In areas where this is important, such as Alzheimer’s disease research, where neuronal loss is the sine qua non of the disease, substitute measures such as structural MR have been used, based on volume loss as an indirect measure of neuronal numbers (reviewed in [10]).
Radiolabeled glucose comes closest to being a neuronal marker, due to the high metabolic rate for glucose in neurons. The first radionuclide used in brain autoradiography studies was [14C]deoxyglucose, with which Sokoloff opened the study of cerebral glucose metabolism [11]. This was quickly followed by a [18F]deoxyglucose (FDG) [12] which has remained the single most important PET tracer to this day.
The mechanism of [18F] FDG brain capture is well-known. It is transported into the cell, phosphorylated and, unlike glucose, cannot be metabolized further, and remains trapped within the cytoplasm of the metabolizing cell. In other words, it allows the study of glucose transport and phosphorylation but not its subsequent metabolism. It is believed that glucose metabolism and cerebral blood flow are coupled to neuronal numbers and function, but there are still questions regarding the underlying molecular mechanisms, particularly the contributions of astrocytes and glia to FDG uptake [13]. Nevertheless, [18F] FDG PET scans are clinically routinely used to improve diagnosis of Alzheimer’s disease and distinguish it from frontotemporal dementia [14, 15]
Stem Cells
Stem cells are progenitors that can renew themselves and differentiate into progeny such as neurons, oligodendrocytes and astrocytes. Stem cells represent a therapeutic modality that could, in theory, restore neuronal tissues and functions. Because of this promise, there is intense interest in imaging the fate of these cells in transplantation experiments and trials.
One of the first techniques used was to label stem cells with HIV Tat-protein derived iron oxides. The Tat-protein carried the iron oxide nanoparticles into the intracellular space, and made the cells visible to MR imaging with T2* sequences. Cell studies [16] first demonstrated the concept, followed by animal studies [17], and a first report in humans [18]. Since then, multiple methods have evolved using different chemistries and labeling technologies to label a variety of stem cells. The discovery of combinations of clinically approved agents that could be used to label cells in a manner that might not require the extensive regulatory testing that non-conventional agents would require is appealing [19–21]. The most promising of these is the combination of Protamine and Ferumoxide (Fig. 3).
Figure 3.

Cell labeling with Iron oxide nanoparticles. Iron oxide colloid (Ferumoxide) is complexed to protamine, and then incubated with relevant cells, usually a stem cell population. The Protamine-Ferumoxide complex is readily endocytosed by the cells, and internalized in lysosomes. Eventually these will be broken down, but this process takes weeks, and will allow MR imaging to demonstrate the distribution of the transplanted cells in vivo (adapted from [23], with permission).
The complex regulatory environment in the US has so far delayed the broad application of stem cell labeling in clinical trials and practice. Nonetheless, the preclinical data are accumulating [22], and we remain hopeful that this technology will soon be available to US patients also. Early data from China has shown promise in the use of this technology in human patients (Fig 4).
Figure 4.

Human use of iron oxide labeled stem cells. This patient has traumatic brain injury (2A), treated with stereotactic injection of autologous iron oxide labeled stem cells, indicated by dark signal on T2* MR images (2B, marked with an asterisk, dark arrows mark injury related susceptibility). On subsequent images done days to weeks afterwards (2C–F) the dark signal, thought to represent labeled stem cells, migrate towards the lesion in a semicircular expansile front. A patient injected with non-labeled stem cells (2G–L) did not show these changes (adapted from [18], with permission).
Tumor Cells
The high metabolism of normal brain for glucose renders FDG tumor imaging in the brain relatively insensitive to tumor, because of a lack of imaging contrast between two FDG-avid tissues. There is persistent need for a tumor seeking agent that would be useful in the diagnosis and treatment of patients with CNS tumors. Most current imaging technologies rely on less specific attributes such as MR signal changes related to water content and blood-brain barrier disruption to help delineate tumors.
An interesting agent is being developed out of scorpion venom [24]. Purifying the crude venom from the scorpion Leiurus quinquestriatus yields the active compound, a small, basic, 36 amino acid peptide known as chlorotoxin (Fig. 5). Studies on scorpion venom neurotoxicity had suggested that this agent binds and inhibits chloride channels. It was also found that chlorotoxin shows a high specificity for binding gliomas and tumors of neuroectodermal origin, including more than 250 human biopsy samples [25]. While initially this was explained on the basis of the known affinity of the agent for chloride channels, eventually this explanation was not satisfactory, and current thinking revolves around binding to matrix metalloproteases secreted by gliomas. The mechanism of the observed specificity is by no means certain and remains under investigation [26]. Knowing that it does bind tumor though, has led to chlorotoxin’s chemical properties being optimized to allow improved tumor imaging and targeted therapy [27]. Clinical trials have been initiated with [131I] radiolabeled versions of chlorotoxin by administering the agent into the resection cavity following glioma surgery [28]. It was found that chlorotoxin SPECT boundaries were closer to MR T2-weighted tumor boundaries than conventional gadolinium enhanced borders. A targeted therapy trial with higher [131I] dosing was subsequently reported with good safety results [29]. The most exciting areas being explored are optical labeling techniques with near infra-red fluorescent probes that might allow surgeons to use fluorescent operating microscopes to better delineate tumor margins.
Figure 5.
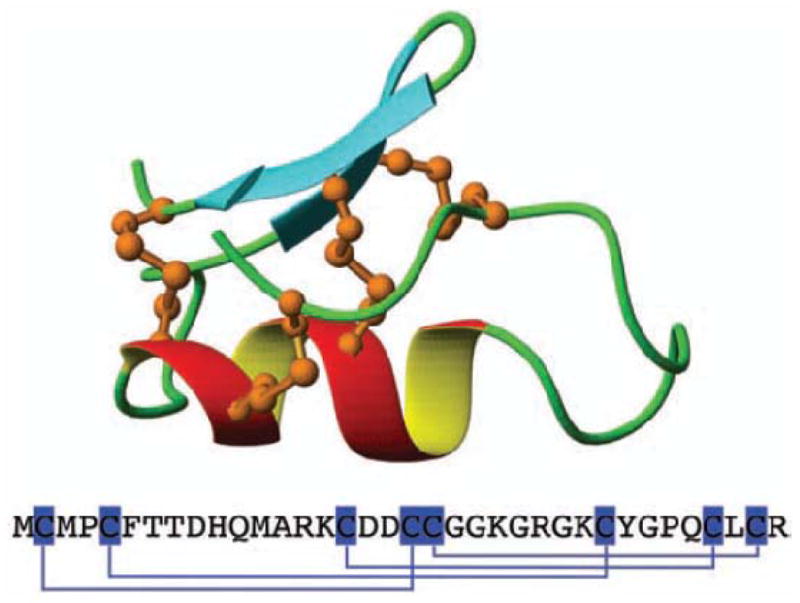
The chemical structure and amino acid structure of Chlorotoxin. A central alpha helix is connected by three disulfide bridges (orange) to an antiparallel beta-sheet. A fourth disulfide bridge links the N-terminal cysteine to the rest of the molecule (adapted from [26], with permission)
The need for tumor diagnostics in the central nervous system has led to the investigation of amino acid based tracers, with the two main contenders being [11C] methionine (MET) and [18F] ethyl-tyrosine (FET). [11C]Methionine is chemically identical to methionine, the first amino acid synthesized into every protein chain in the body, and is highly effective as a marker for protein synthesis. Cancer synthesizes a lot of protein to support fast growth, while background brain tissues have much lower protein synthesis rates. This leads to a favorable tumor to background contrast ratio (Fig. 6). The clinical implementation of [11C] compounds is very difficult due to the 20 minute half-life of the compound, limiting clinical availability to research centers with on-site cyclotrons. FET, being [18F] based has a longer half-life and the agent can be distributed through commercial channels. There have been promising studies using FET PET in combination with MR findings to derive prognostic information on patients with low grade gliomas, in order to select those at risk for progression for earlier and more aggressive therapy [30].
Figure 6.
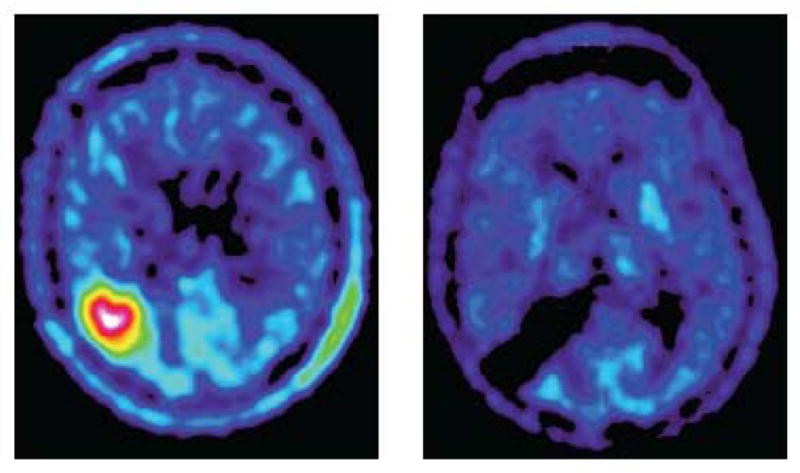
Methionine PET of a patient with an oligodendroglioma before (left) and after (right) surgery at 12 months. There is no residual. Note the good tumor to background contrast (adapted from [33], with permission).
There are numerous other PET agents under development such as Fluoro-L-Thymidine (FLT) designed to serve as probes for DNA synthesis, and [18F]choline [31] and [18F]acetate [32] to target fatty acid metabolism.
Microglia
Microglia are the resident immunocytes of the brain, a large population of cells that normally are quiescent observers. Microglia get activated in a range of pathologies, and are the starting point for most immune and inflammatory cascades involving the brain. The triggers for activation are many, and include chemical signals indicating neuronal damage, infection and inflammatory mediators.
When activated, microglia undergo dramatic changes; morphologically they change from a branchlike rarified morphology to a swollen amoeboid shape, become mobile and migrate to injured brain tissue and become phagocytic. These physical changes are accompanied by equally dramatic molecular changes, one of the more interesting of which is the expression of Translocator Protein 18 kDa (TSPO), also known as the Peripheral Benzodiazepine Receptor (PBR). This protein is located in the outer membrane of mitochondria, and functions as a part of the transportation machinery connecting the mitochondrial contents to the cytosol. It is thought to translocate cholesterol from the outer mitochondrial membrane to the inner mitochondrial membrane where cholesterol is converted to steroid derivatives by the cytochrome P450 enzyme system. It is thought to be arranged in a 5-member ring structure to mediate a channel through the membrane. During microglial activation, the TSPO/PBR protein is overexpressed, first in microglia, but also in astrocytes. The excess protein, and its tight coupling to neuroinflammation is what makes it a good target for molecular imaging [34].
The prototypical agent for imaging activated microglia is a PET analog of benzodiazepine that binds TSPO, known as PK11195. It is a [11C] agent, and thus requires on-site synthesis and a cyclotron. Imaging with this agent in Alzheimer’s disease has revealed a substantial neuroinflammatory component in some cases of the disease [35]. This has also been shown in a variety of neurologic diseases including amyotrophic lateral sclerosis [36] and multiple systems atrophy [37] There are also fluorinated analogs of PK11195 under development to allow wider use of the agent. Agents with increased specificity for microglia versus astrocytes are also under investigation [10].
Interpreting results from microglial agents is complex, and dependent on: a) the inflammatory component of the disease and b) the phase of the disease process when imaging occurs. Many diseases though have an inflammatory component at some stage, making microglial activation and its visualization by imaging an important subject for investigation.
Marker Genes
Marker gene systems are genetically encoded systems that allow a cell population to be imaged. These systems require genetic manipulation of the cells, typically by expressing a foreign protein that can be probed with a molecular imaging agent. The big advantage of such systems is that almost any cell population can be transfected, and that progeny cells will retain the marker, unlike conventional labels that dilute out as cells divide and age. The big disadvantage is that genetically manipulating a cell for human use is considered a potentially very impactful manipulation, leading to high regulatory burdens and relatively slow clinical progress compared to the large body of preclinical data available.
The enzyme Thymidine Kinase (TK) is responsible for the phosphorylation of thymidine, an essential step in the synthesis of DNA. The mammalian version of TK is very fastidious, and will only accept thymidine or very close chemical analogues of it as a substrate for phosphorylation. By contrast, the Herpes Simplex Virus (HSV) has a version of the enzyme that has far less substrate specificity. This difference was exploited by expressing the HSV version of TK (hsvTK) in genetically modified cells, and offering PET labeled thymidine-like substrates that would be accepted by hsvTK, but rejected by the mammalian enzyme. Wherever these PET analogues were phosphorylated and trapped (analogous to FDG), the enzyme had to be present.
This ingenious system was first described in glioma cell lines using a retroviral vector and probing with several agents, of which 5-iodo-2′-fluoro-2′deoxy-1-β-D-arabinofuranosyluracil (FIAU) was found to be the best [38]. This agent was used in humans to show relatively low transduction in a gene therapy trial for glioblastoma, a very important result demonstrating how imaging can be used to judge the efficacy of novel therapies [39].
Multiple further developments yielded other tracers for this marker system, culminating in 9-[4-[18F]fluoro-3-(hydroxymethyl)butyl]guanine (FHBG) [40], an agent that was subsequently FDA approved for human use.
Bioluminescent Imaging
Bioluminescent Imaging/Luciferase imaging is a non-invasive in-vivo optical imaging modality that is based on the release of light due to enzyme catalysis. The enzyme, luciferase, is a naturally occurring luminescent protein found in insects like the North American firefly (Photinus pyralis). In the firefly, luciferase breaks down its substrate D-luciferin by oxidation, and emits light (bioluminescence) in a reaction that requires the presence of Mg 2+, ATP and O2 (reviewed in [41]). This reaction is what allows the firefly to shine at night as part of its courtship rituals. When the luciferase enzyme is genetically transferred into cells or organisms of interest, it becomes a marker gene that can be detected by its light emission.
There are other examples of luminescent proteins such as those from Renilla luciferase (RLuc) from the soft coral Renilla reniformis [42] and bacterial luciferase (Lux) from marine Vibreo bacteria [43].
The major advantages of using in-vivo luciferase reporter systems include 1) high signal to noise imaging (there is almost no background light emission in mammalian biology) and 2) the relative ease and low cost imaging acquisition instrumentation available (camera technology is better developed and cheaper than gamma detection technology). Cross validation of BLI with other imaging modalities like MRI has been described for orthotropic rat glioma models and demonstrated that both MRI and BLI could be used to determine kinetics of tumor growth and therapy response over time [44]. PET studies that involve the detection of reporter genes in mouse models [45] have also been used to validate BLI as an imaging modality for in-vivo gene delivery [46]. BLI was predicted to be 100- to 1000 fold more sensitive than PET for detecting adenoviral-mediated gene transfer to liver and muscle tissue [46]. BLI is rapidly gaining strength as an optical imaging tool for studying true tumor microenvironment in genetically engineered mouse models (GEMMs), specifically involving genetically encoded in-vivo luciferase reporters (reviewed in [47]).
While bioluminescent imaging is of great importance in experimental imaging research, it unfortunately has limited human translation potential. The photons emitted are relatively quickly attenuated by tissues, and in larger organisms like humans, imaging can only be done of superficial structures, like the skin or mucosa. This physical limitation, along with the difficulties of using genetically modified cells in humans, has limited the application of bioluminescent imaging to research models.
Alzheimer related proteins
Alzheimers disease is a highly prevalent cause of dementia, with large public health consequences. The disease is characterized by the accumulation of extracellular plaques of amyloid beta (Abeta) protein and intracellular tangles of tau protein. It is widely accepted that these proteins are central to the neuronal loss that underlies this disease [48].
There has been great interest in measuring Abeta protein in living subjects for several reasons. The clinical diagnosis is challenging and sometimes wrong, frequently due to other causes of dementia; there are no laboratory tests to serve as a gold standard (definitive diagnosis is possible only on post-mortem tissues); and the disease has a very long time course, often decades.
The first agent that successfully measured Abeta protein was Pittsburgh Compound B (PIB) [1]. PIB, also known as N-methyl-[11C]2-(4-methylaminophenyl)-6-hydroxybenzothiazole, is a [11C]compound modeled on thioflavin-T, a compound used as a histological dye to stain for amyloid. The human studies (performed in Sweden) showed a striking accumulation of PIB in the brain of Alzheimer’s patients, markedly different from normal control patients (Fig. 7). This study was a huge advance in the care of Alzheimer’s patients, being the first in vivo test that could assess the Abeta status of a patient directly, giving hope that it might serve as a biomarker that could be used to assess therapies directed at Abeta protein accumulation. This would allow the monitoring of therapy provided early in the course of the disease, when outcomes might still be modifiable.
Figure 7.
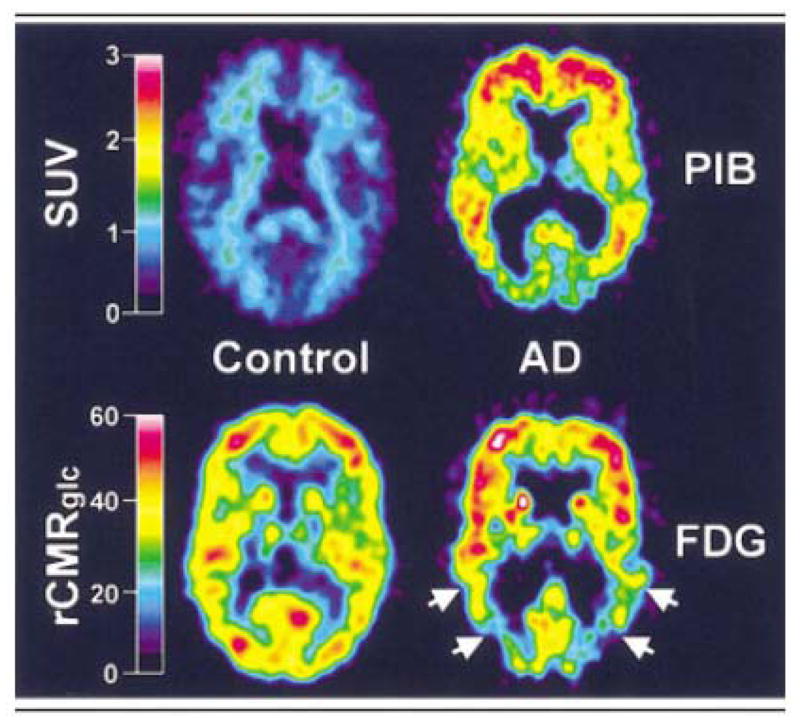
A normal control (left panels) and Alzheimer patient (right panels) are compared with PIB imaging (top panels) and FDG imaging parametric maps (bottom panels). There is marked retention of PIB in the brain of the Alzheimer patient compared to the control. The changes on the FDG image, while still apparent, are much less striking (taken from [1]).
PiB agent, however, is a [11C] agent, with a short half-life, requiring an on-site cyclotron and individual synthesis. This limits applications to major research centers, and provided the impetus for the development of fluorinated analogs. Currently, three 18F-labeled tracers are under development and being investigated in clinical trials by pharmaceutical companies: flutemetamol, florbetapir, and florbetaben[49].
Research is also ongoing into alternative targets, such as tau protein [50] and alphasynuclein [51]. It is very likely that we will have an extensive armamentarium of imaging compounds to dissect the molecular pathology of Alzheimer’s disease.
Conclusion
Molecular imaging is a fascinating and dynamic diagnostic counterpart to molecular medicine. Finding ways to interrogate molecular and cellular targets of interest is likely to vastly increase our diagnostic abilities, and help us to guide therapies to the benefit of our patients.
Acknowledgments
We thank David Bier from medical graphics for designing Figure 1 and technically managing referenced figures. We thank Anita Vallier for administrative assistance with the manuscript.
Supported in part by: NINDS R01NS070742
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Klunk WE, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Annals of neurology. 2004;55(3):306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 2.Fowler JS, et al. Synthesis and preliminary evaluation in animals of carrier-free 11C-1-dopamine hydrochloride: X. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 1973;14(11):867–9. [PubMed] [Google Scholar]
- 3.Moerlein SM, et al. Evaluation of 75Br-labelled butyrophenone neuroleptics for imaging cerebral dopaminergic receptor areas using positron emission tomography. European journal of nuclear medicine. 1986;12(4):211–6. doi: 10.1007/BF00256924. [DOI] [PubMed] [Google Scholar]
- 4.Wagner HN, Jr, et al. Imaging dopamine receptors in the human brain by positron tomography. Science. 1983;221(4617):1264–6. doi: 10.1126/science.6604315. [DOI] [PubMed] [Google Scholar]
- 5.Inoue Y, et al. Atlas of dopamine receptor images (PET) of the human brain. Journal of computer assisted tomography. 1985;9(1):129–40. doi: 10.1097/00004728-198501000-00024. [DOI] [PubMed] [Google Scholar]
- 6.Heiss WD, Herholz K. Brain receptor imaging. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2006;47(2):302–12. [PubMed] [Google Scholar]
- 7.Cumming P, et al. The competition between endogenous dopamine and radioligands for specific binding to dopamine receptors. Annals of the New York Academy of Sciences. 2002;965:440–50. doi: 10.1111/j.1749-6632.2002.tb04185.x. [DOI] [PubMed] [Google Scholar]
- 8.Elsinga PH, Hatano K, Ishiwata K. PET tracers for imaging of the dopaminergic system. Current medicinal chemistry. 2006;13(18):2139–53. doi: 10.2174/092986706777935258. [DOI] [PubMed] [Google Scholar]
- 9.Volkow ND, et al. Decreased striatal dopaminergic responsiveness in detoxified cocaine-dependent subjects. Nature. 1997;386(6627):830–3. doi: 10.1038/386830a0. [DOI] [PubMed] [Google Scholar]
- 10.Reiman EM, Jagust WJ. Brain imaging in the study of Alzheimer’s disease. NeuroImage. 2012;61(2):505–16. doi: 10.1016/j.neuroimage.2011.11.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sokoloff L, et al. The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. Journal of neurochemistry. 1977;28(5):897–916. doi: 10.1111/j.1471-4159.1977.tb10649.x. [DOI] [PubMed] [Google Scholar]
- 12.Reivich M, et al. Measurement of local cerebral glucose metabolism in man with 18F-2-fluoro-2-deoxy-d-glucose. Acta neurologica Scandinavica Supplementum. 1977;64:190–1. [PubMed] [Google Scholar]
- 13.Pellerin L, et al. Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia. 2007;55(12):1251–62. doi: 10.1002/glia.20528. [DOI] [PubMed] [Google Scholar]
- 14.Ishii K. Clinical application of positron emission tomography for diagnosis of dementia. Annals of nuclear medicine. 2002;16(8):515–25. doi: 10.1007/BF02988628. [DOI] [PubMed] [Google Scholar]
- 15.Ishii K, et al. Cerebral glucose metabolism in patients with frontotemporal dementia. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 1998;39(11):1875–8. [PubMed] [Google Scholar]
- 16.Weissleder R, et al. Magnetically labeled cells can be detected by MR imaging. Journal of magnetic resonance imaging : JMRI. 1997;7(1):258–63. doi: 10.1002/jmri.1880070140. [DOI] [PubMed] [Google Scholar]
- 17.Lewin M, et al. Tat peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nature biotechnology. 2000;18(4):410–4. doi: 10.1038/74464. [DOI] [PubMed] [Google Scholar]
- 18.Zhu J, Zhou L, XingWu F. Tracking neural stem cells in patients with brain trauma. The New England journal of medicine. 2006;355(22):2376–8. doi: 10.1056/NEJMc055304. [DOI] [PubMed] [Google Scholar]
- 19.Frank JA, et al. Clinically applicable labeling of mammalian and stem cells by combining superparamagnetic iron oxides and transfection agents. Radiology. 2003;228(2):480–7. doi: 10.1148/radiol.2281020638. [DOI] [PubMed] [Google Scholar]
- 20.Frank JA, et al. Magnetic intracellular labeling of mammalian cells by combining (FDA-approved) superparamagnetic iron oxide MR contrast agents and commonly used transfection agents. Academic radiology. 2002;9(Suppl 2):S484–7. doi: 10.1016/s1076-6332(03)80271-4. [DOI] [PubMed] [Google Scholar]
- 21.Arbab AS, et al. Efficient magnetic cell labeling with protamine sulfate complexed to ferumoxides for cellular MRI. Blood. 2004;104(4):1217–23. doi: 10.1182/blood-2004-02-0655. [DOI] [PubMed] [Google Scholar]
- 22.Gutova M, et al. Magnetic Resonance Imaging Tracking of Ferumoxytol-Labeled Human Neural Stem Cells: Studies Leading to Clinical Use. Stem cells translational medicine. 2013;2(10):766–775. doi: 10.5966/sctm.2013-0049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jasmin, et al. Labeling stem cells with superparamagnetic iron oxide nanoparticles: analysis of the labeling efficacy by microscopy and magnetic resonance imaging. Methods in molecular biology. 2012;906:239–52. doi: 10.1007/978-1-61779-953-2_18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Veiseh M, et al. Tumor paint: a chlorotoxin:Cy5.5 bioconjugate for intraoperative visualization of cancer foci. Cancer research. 2007;67(14):6882–8. doi: 10.1158/0008-5472.CAN-06-3948. [DOI] [PubMed] [Google Scholar]
- 25.Lyons SA, O’Neal J, Sontheimer H. Chlorotoxin, a scorpion-derived peptide, specifically binds to gliomas and tumors of neuroectodermal origin. Glia. 2002;39(2):162–73. doi: 10.1002/glia.10083. [DOI] [PubMed] [Google Scholar]
- 26.Stroud MR, Hansen SJ, Olson JM. In vivo bio-imaging using chlorotoxin-based conjugates. Current pharmaceutical design. 2011;17(38):4362–71. doi: 10.2174/138161211798999375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akcan M, et al. Chemical re-engineering of chlorotoxin improves bioconjugation properties for tumor imaging and targeted therapy. Journal of medicinal chemistry. 2011;54(3):782–7. doi: 10.1021/jm101018r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hockaday DC, et al. Imaging glioma extent with 131I-TM-601. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2005;46(4):580–6. [PubMed] [Google Scholar]
- 29.Mamelak AN, et al. Phase I single-dose study of intracavitary-administered iodine-131-TM-601 in adults with recurrent high-grade glioma. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2006;24(22):3644–50. doi: 10.1200/JCO.2005.05.4569. [DOI] [PubMed] [Google Scholar]
- 30.Floeth FW, et al. Prognostic value of O-(2-18F-fluoroethyl)-L-tyrosine PET and MRI in low-grade glioma. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2007;48(4):519–27. doi: 10.2967/jnumed.106.037895. [DOI] [PubMed] [Google Scholar]
- 31.Lam WW, et al. Promising role of [18F] fluorocholine PET/CT vs [18F] fluorodeoxyglucose PET/CT in primary brain tumors-early experience. Clinical neurology and neurosurgery. 2011;113(2):156–61. doi: 10.1016/j.clineuro.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 32.Nishii R, et al. Pharmacokinetics, metabolism, biodistribution, radiation dosimetry, and toxicology of (18)F-fluoroacetate ((18)F-FACE) in non-human primates. Molecular imaging and biology : MIB : the official publication of the Academy of Molecular Imaging. 2012;14(2):213–24. doi: 10.1007/s11307-011-0485-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smits A, Baumert BG. The Clinical Value of PET with Amino Acid Tracers for Gliomas WHO Grade II. International journal of molecular imaging. 2011;2011:372509. doi: 10.1155/2011/372509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trapani A, et al. Targeting of the Translocator Protein 18 kDa (TSPO): A Valuable Approach for Nuclear and Optical Imaging of Activated Microglia. Bioconjugate chemistry. 2013;24(9):1415–28. doi: 10.1021/bc300666f. [DOI] [PubMed] [Google Scholar]
- 35.Cagnin A, et al. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358(9280):461–7. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- 36.Turner MR, et al. Evidence of widespread cerebral microglial activation in amyotrophic lateral sclerosis: an [11C](R)-PK11195 positron emission tomography study. Neurobiology of disease. 2004;15(3):601–9. doi: 10.1016/j.nbd.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 37.Gerhard A, et al. [11C](R)-PK11195 PET imaging of microglial activation in multiple system atrophy. Neurology. 2003;61(5):686–9. doi: 10.1212/01.wnl.0000078192.95645.e6. [DOI] [PubMed] [Google Scholar]
- 38.Tjuvajev JG, et al. Imaging the expression of transfected genes in vivo. Cancer research. 1995;55(24):6126–32. [PubMed] [Google Scholar]
- 39.Jacobs A, et al. Positron-emission tomography of vector-mediated gene expression in gene therapy for gliomas. Lancet. 2001;358(9283):727–9. doi: 10.1016/s0140-6736(01)05904-9. [DOI] [PubMed] [Google Scholar]
- 40.Yaghoubi S, et al. Human pharmacokinetic and dosimetry studies of [(18)F]FHBG: a reporter probe for imaging herpes simplex virus type-1 thymidine kinase reporter gene expression. Journal of nuclear medicine : official publication, Society of Nuclear Medicine. 2001;42(8):1225–34. [PubMed] [Google Scholar]
- 41.Contag CH, Bachmann MH. Advances in in vivo bioluminescence imaging of gene expression. Annu Rev Biomed Eng. 2002;4:235–60. doi: 10.1146/annurev.bioeng.4.111901.093336. [DOI] [PubMed] [Google Scholar]
- 42.Lorenz WW, et al. Isolation and expression of a cDNA encoding Renilla reniformis luciferase. Proc Natl Acad Sci U S A. 1991;88(10):4438–42. doi: 10.1073/pnas.88.10.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meighen EA. Molecular biology of bacterial bioluminescence. Microbiol Rev. 1991;55(1):123–42. doi: 10.1128/mr.55.1.123-142.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rehemtulla A, et al. Rapid and quantitative assessment of cancer treatment response using in vivo bioluminescence imaging. Neoplasia. 2000;2(6):491–5. doi: 10.1038/sj.neo.7900121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tjuvajev JG, et al. Imaging herpes virus thymidine kinase gene transfer and expression by positron emission tomography. Cancer Res. 1998;58(19):4333–41. [PubMed] [Google Scholar]
- 46.Wu JC, et al. Noninvasive optical imaging of firefly luciferase reporter gene expression in skeletal muscles of living mice. Mol Ther. 2001;4(4):297–306. doi: 10.1006/mthe.2001.0460. [DOI] [PubMed] [Google Scholar]
- 47.Kocher B, Piwnica-Worms D. Illuminating cancer systems with genetically engineered mouse models and coupled luciferase reporters in vivo. Cancer Discov. 2013;3(6):616–29. doi: 10.1158/2159-8290.CD-12-0503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bignante EA, et al. Amyloid beta precursor protein as a molecular target for amyloid beta-induced neuronal degeneration in Alzheimer’s disease. Neurobiology of aging. 2013;34(11):2525–37. doi: 10.1016/j.neurobiolaging.2013.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Herholz K, Ebmeier K. Clinical amyloid imaging in Alzheimer’s disease. Lancet neurology. 2011;10(7):667–70. doi: 10.1016/S1474-4422(11)70123-5. [DOI] [PubMed] [Google Scholar]
- 50.Fodero-Tavoletti MT, et al. 18F-THK523: a novel in vivo tau imaging ligand for Alzheimer’s disease. Brain : a journal of neurology. 2011;134(Pt 4):1089–100. doi: 10.1093/brain/awr038. [DOI] [PubMed] [Google Scholar]
- 51.Kikuchi A, et al. In vivo visualization of alpha-synuclein deposition by carbon-11-labelled 2-[2-(2-dimethylaminothiazol-5-yl)ethenyl]-6-[2-(fluoro)ethoxy]benzoxazole positron emission tomography in multiple system atrophy. Brain : a journal of neurology. 2010;133(Pt 6):1772–8. doi: 10.1093/brain/awq091. [DOI] [PubMed] [Google Scholar]
- 52.Lindsey KP, Gatley SJ. Applications of clinical dopamine imaging. Neuroimaging clinics of North America. 2006;16(4):553–73. vii–viii. doi: 10.1016/j.nic.2006.06.003. [DOI] [PubMed] [Google Scholar]