

Abstract
Interfacial visible light-mediated thiol-ene photo-click reactions were developed for preparing step-growth hydrogels with multilayer structures. The effect of a non-cleavage type photoinitiator eosin-Y on visible light-mediated thiol-ene photopolymerization was first characterized using in situ photo-rheometry, gel fraction, and equilibrium swelling ratio. Next, spectrophotometric properties of eosin-Y in the presence of various relevant macromer species were evaluated using UV/Vis spectrometry. It was determined that eosin-Y was able to re-initiate thiol-ene photo-click reaction even after light exposure. Due to its small molecular weight, most eosin-Y molecules readily leached out from the hydrogels. The diffusion of residual eosin-Y from pre-formed hydrogels was exploited for fabricating multilayer step-growth hydrogels. Interfacial hydrogel coating was formed via the same visible light-mediated gelation mechanism without adding fresh initiator. The thickness of the thiol-ene gel coating could be easily controlled by adjusting visible light exposure time, eosin-Y concentration initially loaded in the core gel, or macromer concentration in the coating solution. The major benefits of this interfacial thiol-ene coating system include its simplicity and cytocompatibility. The formation of thiol-ene hydrogels and coatings neither requires nor generates any cytotoxic components. This new gelation chemistry may have great utilities in controlled release of multiple sensitive growth factors and encapsulation of multiple cell types for tissue regeneration.
Keywords: Thiol-ene, photopolymerization, hydrogel, multilayer, visible light
INTRODUCTION
Polymeric biomaterials with multilayer structures have great potential in biomedical applications, such as construction of complex tissues,1, 2 controlled release of multiple drugs at different rates,3–8 and immunoisolation for allo- or xeno-grafts.9, 10 Many physical and chemical cross-linking methods have been developed for fabricating multilayer polymers or hydrogels. For example, polyelectrolytes with opposite charges could be self-assembled into multilayer films or membranes.4, 6, 7, 11 These layer-by-layer (LbL) approaches have been used successfully in producing films with nano-scale thickness for applications such as controlled drug delivery and cell surface coating.4, 7, 12 However, the building blocks for LbL films usually comprise positively charged polymers that are potentially cytotoxicity. Further, the LbL assembly processes are often lengthy and may not be ideal for encapsulating sensitive cells. Other disadvantages include limitations in bioconjugation and drug loading capacity, as well as instability of the physically bonded films in vivo. The utility and diversity of multilayer biomaterials would greatly benefit from a chemically cross-linking method that provides long-term material stability, simplicity in coating procedures, and diversity in bioconjugation.
An attractive method to fabricate stable multilayer polymers or hydrogels is photopolymerization. Photopolymerization offers many benefits, including rapid and mild cross-linking conditions, as well as spatial-temporal control in polymerization kinetics that permits the creation of complex material structures and functionalities.13–15 A common approach to fabricate multilayer hydrogels is to prepare a layer of gel with pendent (meth)acrylate moieties that serve as anchors for subsequent homopolymerization of (meth)acrylated monomers.16, 17 Either UV or visible light could be used to cross-link multilayer polymers, as long as appropriate initiator species are included in the subsequent monomer solutions. On the downside, current photopolymerization systems for forming hydrogels carry risks of cellular damages caused by UV light, radical species, and other cytotoxic compositions required in cross-linking reactions. Furthermore, currently available photochemistries for forming multilayer hydrogels are all based on chain-growth polymerizations that may not be ideal for some cell and protein encapsulation.18, 19
Multilayer hydrogels could be fabricated using a light-independent approach. For instance, Bowman and colleagues have developed an enzymatic coating procedure for forming multilayer hydrogels.20–22 The formation of hydrogel coating on a core gel was mediated by glucose oxidase (GOx), which reacts with its substrate glucose released from a core gel to generate hydrogen peroxide (H2O2). Hydrogen peroxide further reacts with ferrous ions (Fe2+) to generate hydroxyl radicals, which initiate chain-growth polymerization through vinyl monomers. To control thickness of the hydrogel coating, one could simply control the reaction time or adjust concentrations of various components in the monomer solutions.21, 22 However, since this polymerization method is light-independent, it also loses the benefits of photopolymerizations. Another disadvantage of this method is that a total of three initiating species (glucose oxidase, glucose, Fe2+) are required, which complicate material preparation. Finally, this enzymatic reaction produces highly cytotoxic H2O2 and requires the addition of a second enzyme, catalase, to increase the cytocompatibility of this method for cell encapsulation.20
We have recently introduced a cytocompatible visible light-mediated thiol-ene photo-click reaction scheme that utilizes only a single visible light sensitive photoinitiator (eosin-Y) to initiate rapid and controllable gelation.23 This method preserves all benefits offered by step-growth thiol-ene photopolymerizations, such as rapid and mild gelation conditions, high cytocompatibility, no oxygen inhibition, idealized network structure, and capability of post-gelation modification in the presence of encapsulated cells.13, 18, 19, 24, 25 Furthermore, this gelation method not only uses visible light (400 nm < λ < 700 nm) to initiate gel cross-linking, but also greatly simplifies conventional visible light photopolymerization procedures by eliminating the use of co-monomer species.23
In this contribution, the unique visible light-mediated thiol-ene gelation was used to form multilayer hydrogels with very simple experimental setup. We first studied the influence of eosin-Y, either freshly prepared or recovered from a pre-formed gel, on thiol-ene hydrogel cross-linking efficiency and resulting hydrogel properties. We have also developed an interfacial thiol-ene photo-click reaction to fabricate multilayer step-growth hydrogels. The multilayer gels were formed via sequential visible light exposure on precursor solutions containing only macromers (no new initiator is required). We have also explored various parameters to control coating thickness. This system offers an alternative methodology for preparing hydrogel materials with complex layered structures for controlled release and tissue engineering applications.
EXPERIMENTAL SECTION
Materials
Eosin-Y disodium salt was purchased from Fisher Scientific. 4-arm poly(ethylene glycol) (PEG, 20 kDa) was purchased from JenKem Technology USA. Linear PEG (10 kDa) and all other chemicals were obtained from Sigma-Aldrich unless noted otherwise.
Synthesis of PEG macromers
PEG-tetra-norbornene (PEG4NB) and PEG-di-norbornene (PEGdNB) were synthesized using an established protocol.13, 26 Briefly, 5-norbornene-2-carboxylic acid (5-fold excess of OH group on PEG) and coupling reagent N,N′-dicyclohexylcarbodiimide (DCC, 2.5-fold excess of OH group) were added to anhydrous dichloromethane (DCM). The mixture was stirred at room temperature for 30 minutes and the resulting norbornene anhydride was filtered into an addition funnel and added to a flask containing PEG (4-arm or linear), 4-(dimethylamino) pyridine (DMAP, 0.5-fold of OH group), and pyridine (0.5-fold of OH group) dissolved in anhydrous DCM. The flask was kept in an ice bath and allowed to react overnight in dark. The product was washed with 5 % sodium bicarbonate solution twice, 2 % hydrochloric acid twice, and brine once. The product was stirred for an hour with anhydrous sodium sulfate, followed by precipitation in cold ethyl ether. 1H NMR (Bruker 500) was used to confirmed high degree of PEG functionalization (> 90 %).
Hydrogel fabrication and characterization
Step-growth thiol-ene hydrogels were formed by visible light mediated photo-click reactions between macromer PEG-norbornene and cross-linker dithiothreitol (DTT).23 A unity stoichiometric ratio of thiol to ene was used in all experiments. Photoinitiator eosin-Y (concentration: 0.1 to 2.0 mM) was added to the precursor solution and the gelation was accomplished after exposing the solution to a halogen cold light lamp (AmScope Inc.) for 4 minutes at 70,000 Lux. Gel fractions and swelling ratios were characterized as reported previously without modification.26
In situ photo-rheometry was performed at room temperature in a Bohlin CVO 100 digital rheometer equipped with a light cure cell. A macromer solution was placed on a quartz plate in the light cure cell and irradiated with visible light through a flexible light guide. Visible light was turned on 30 seconds after starting time-sweep measurement (10 % strain, 1 Hz frequency, 0.1 N normal force, and a gap size of 100 μm) using a 25 mm parallel plate geometry. The moduli in the linear viscoelastic region (LVR) were reported. Gel points (i.e., crossover time) were determined at the time when storage modulus (G′) surpassed loss modulus (G″).
Retention and recovery of eosin-Y in hydrogels
Immediately after polymerization, hydrogel discs (8 mm diameter × 1 mm height) were immersed into scintillation vials containing 2 mL of PBS (pH 7.4) at 37 °C. At specific time points, a portion of solution (200 μL) was transferred to a clear 96-well plate and fresh PBS was added to maintain a constant volume (2 mL). Absorbance (516 nm) of the collected samples was measured by a microplate reader (BioTek Synergy HT) and correlated to a standard curve generated from known concentrations of eosin-Y. Mass balance calculations were performed to determine the quantity of eosin-Y retained in the hydrogels. In a similar manner, eosin-Y was recovered from the buffer and the concentration was determined by absorbance measurement using eosin-Y solutions with known concentrations.
UV/Vis absorbance of eosin-Y containing samples
Eosin-Y and various components (i.e., DTT, PEGdNB) were dissolved in PBS (pH 7.4) and exposed to visible light for 4 minutes. Non-gelling macromers were used (e.g., PEGdNB) to prevent gelation and facilitate solution-based UV/Vis spectrometric measurements. Concentrations of the components were equivalent to those used in gelation studies. The spectra of the solutions were measured between 400 and 600 nm at 1 nm increment using a microplate reader (BioTek Synergy HT) in UV/Vis absorption mode. Prior to measurements, the solutions were diluted down to equivalent of 0.02 mM eosin-Y to ensure that the absorbance values measured were within the linear range of a standard curve generated using known eosin-Y concentrations.
Multilayer hydrogel fabrication and characterization
Immediately following polymerization, PEG4NB-DTT hydrogels (25 μL, 5 mm diameter) were placed in a circular mold (8 mm diameter mold) containing macromer solution that includes PEG4NB and DTT. In some samples, 5 % of 0.2 μm Fluoresbrite® blue microparticles (Polysciences) were added for imaging purpose. These samples were exposed to the same visible light source as described earlier. Immediately after the removal of un-polymerized macromer solution, the dual-layer gels were imaged using a fluorescence microscope (Nikon-Ti). Gel coating thickness was defined from the intercept of red and blue fluorescence to the edge of outer layer and was characterized by fluorescent image analysis from at least three samples per condition. Note that eosin-Y has intrinsic red fluorescence.
In the three-layer hydrogel experiment, all layers were formed with 10 wt% PEG4NB and DTT at a unity stoichiometric ratio. Gels were prepared in a 1 mL syringe with the tip cut off. The bottom layer was formed under visible light (70,000 Lux) for 4 minutes using 2.0 mM eosin-Y as the only photoinitiator. The macromer precursor solutions in the middle and top layers (25 μL each) contained only PEG4NB and DTT (microparticles in the top layer were added for visualization purpose). These layers were formed by sequential visible light exposure for 10 minutes each (200,000 Lux). The formation of the thick coating gel construct was achieved in a similar manner. A gel disc (2 mm diameter × 1 mm height) was pre-formed using 2 mM eosin-Y and placed in a 1 mL syringe filled with macromer solution (10 wt% PEG4NB and DTT). The set up was exposed under visible light through a gooseneck light guide (200,000 Lux) for 10 minutes.
RESULTS AND DISCUSSION
Effect of eosin-Y concentration on visible light initiated thiol-ene hydrogel cross-linking
We have recently shown that the cross-linking of step-growth thiol-norbornene hydrogels could be initiated by visible light exposure with eosin-Y as the only photoinitiator.23 The major difference between this new gelation scheme and the conventional visible light-mediated chain-growth hydrogel synthesis is that no additional co-initiator or co-monomer is needed to achieve rapid gelation. Here, this unique, simple, yet effective photopolymerization method was further explored for preparing step-growth hydrogels with multilayer structures. We first investigated the effect of initiator concentration on thiol-ene gelation kinetics. For thin samples such as those cured in the in situ photo-rheometry studies (100 μm, Fig. 1A), increasing eosin-Y concentration resulted in faster gelation rate as demonstrated by the rapid gel points shown in Fig. 1B. For example, gelation using 2.0 mM eosin-Y reached gel point within 2 seconds or 10-fold faster than gelation with 0.1 mM eosin-Y (gel point ~24 seconds). The final elastic moduli of these thin gels, however, were ranging between ~6.4 kPa to ~9.1 kPa for different eosin-Y concentration used (0.1 to 2.0 mM, Fig. 1A). As shown in Scheme 1, eosin-Y was sensitized to an exited state upon visible light exposure.27 The excited eosin-Y carbanion is capable of extracting hydrogen from proton-donating thiols, such as DTT or cysteine-bearing peptides/proteins, to generate thiyl radicals responsible for initiating thiol-ene photopolymerization and gelation.13 The process repeats in a rapid step-growth manner to form a thiol-ene hydrogels.26 Therefore, higher eosin-Y concentration leads to a higher initiation rate and faster gelation.
Figure 1.

Effect of eosin-Y concentration on (A) gelation kinetics and (B) gel points of PEG4NB-DTT hydrogels formed by visible light-mediated thiol-ene photopolymerization. (PEG4NB: 10 wt%; N = 3; Mean ± S.D.)
Scheme 1.
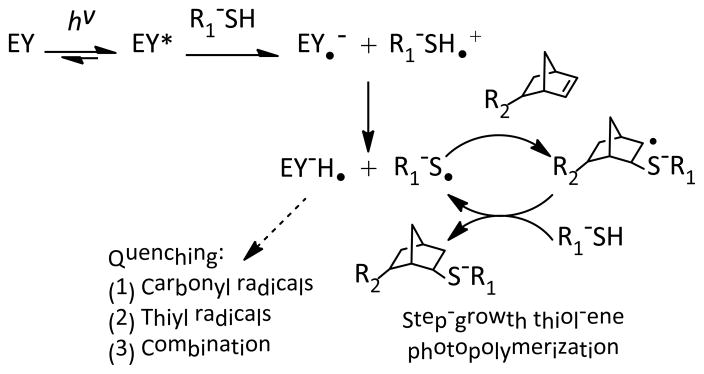
Initiation and polymerization mechanisms for visible light-mediated thiol-ene photopolymerization using eosin-Y (EY) as the sole photoinitiator. The reactions result in gel cross-linking as R1-SH and R2-norbornene represents a bi- and tetra-functional cross-linker, respectively.
While more initiators in the precursor solution accelerated initiation rate in thin hydrogel samples, higher eosin-Y concentration negatively affected network cross-linking efficiency in bulky hydrogels under the same gelling conditions (i.e., polymerization time and light intensity). This phenomenon was especially prominent for thicker gels. We fabricated thiol-ene hydrogels with two thicknesses (1 and 3 mm) using different eosin-Y concentrations (0.1 to 2.0 mM, polymerized for 4 minutes) and characterized gel fractions and equilibrium swelling ratios. As shown in Fig. 2A, while thinner gels (e.g., 1 mm) had high gel fraction (>95 %) regardless of eosin-Y concentrations, thicker gels (e.g., 3 mm) prepared with 2.0 mM eosin-Y had significantly lower gel fractions (72.5 ± 0.7 %) compared to gels prepared with 0.1 mM eosin-Y (90.5 ± 3.3 %). In general, gels with lower gel fractions reached higher equilibrium swelling ratios due to lower cross-linking efficiencies (Fig. 2B).26 Interestingly, gels prepared with 1 mm thickness also exhibited higher swelling ratios at high eosin-Y concentrations (i.e., 1.5 and 2.0 mM), even though the dependency with eosin-Y concentration was less prominent compared to thicker gels. At higher eosin-Y concentrations, there is a higher tendency of eosin-Y quenching and termination that results in pendent polymer chains (Scheme 1). While this type of network non-ideality did not cause reduction in gel fraction, it effectively decreases network cross-linking density that leads to higher gel swelling. For example, although all samples having a thickness of 1 mm have a similar gel fraction, the effects of light attenuation and eosin-Y quenching resulted in increased swelling ratio at high eosin-Y concentrations (e.g., 1.5 and 2.0 mM, Fig. 2).
Figure 2.

Effect of gel thickness and eosin-Y concentration on (A) gel fraction and (B) equilibrium swelling ratio of PEG4NB-DTT hydrogels formed by visible light-mediated thiol-ene photopolymerization for 4 minutes. (PEG4NB: 10 wt%; N = 3; Mean ± S.D.)
Comparing data presented in Fig. 1 and Fig. 2, it is clear that while eosin-Y serves as a visible light photoinitiator for thiol-ene reactions, it may also hinder network cross-linking, especially at a higher concentration or when it was used to form thicker gels. The lower gelation efficiency at a higher eosin-Y concentration was believed to be a result of a higher degree of light attenuation in thicker samples caused by the red eosin-Y. Light attenuation may result in slight network heterogeneity following cross-linking. However, due to the nature of orthogonal step-growth thiol-ene chemistry, these thiol-ene hydrogels will inherently be more homogeneous than all other chain-growth photopolymerized hydrogels. If desired, other parameters (e.g., light intensity and macromer concentrations) could be tuned to improve network cross-linking at higher eosin-Y concentration.23
Eosin-Y retention in thiol-ene hydrogels
As shown in Fig. 3A, gels cross-linked using higher eosin-Y concentrations appeared redder, even after extended (48 hours) incubation in PBS to leach out the residual dye. In addition, we found that a significant amount of eosin-Y became permanently sequestered in the gels in an eosin-Y concentration dependent manner (Fig. 3B). For example, after 48 hours incubation in PBS, roughly 26 nmol (or 25%) of eosin-Y retained in hydrogels fabricated using 2.0 mM eosin-Y. The sequestration of eosin-Y in these hydrogels was more noticeable in gels cross-linked with higher eosin-Y concentrations. The use of higher eosin-Y concentrations (2.0 mM), however, did not affect the viability of encapsulated human mesenchymal stem cells (hMSCs) as shown in our previous studies.23
Figure 3.

(A) Photographs of thiol-ene hydrogels formed by visible light-mediated, eosin-Y initiated thiol-ene photopolymerizations before (left) and after (right) swelling for 48 hours (eosin-Y concentration from left to right: 0.1, 0.5, 1.0, 1.5, 2.0 mM). (B) Effect of eosin-Y concentration on its retention in thiol-ene hydrogels. (PEG4NB: 10 wt%; N = 3; Mean ± S.D.)
The sequestration of eosin-Y in PEG hydrogels (mesh size: ~20 nm for 10 wt% PEG4NB-DTT hydrogels) was unexpected because it was unlikely for eosin-Y (MW ~692 Da) to be physically ‘trapped’ within the highly swollen and permeable gels. Based on the principles of radical polymerization, one potential explanation for the sequestration of eosin-Y is the quenching or termination reaction of protonated and radical-bearing eosin-Y (EY-H•) due to reactions with live carbonyl radicals on cross-linked PEG-norbornene or thiyl radicals on pendent DTT (Scheme 1). A second possibility for the retention/sequestration of eosin-Y in the hydrogels was the existence of binding affinity between eosin-Y and these PEG hydrogels. To test the hypothesis of potential binding affinity between eosin-Y and thiol-ene hydrogel, UV/Vis absorbance spectra of eosin-Y before and after visible light exposure were assessed in the presence of different relevant components (i.e., PEGdNB, DTT, or both) separately. The results shown in Fig. 4A indicate that visible light exposure did not significantly affect the absorbance signature of eosin-Y (both maximum absorbance and peak wavelength). On the other hand, the inclusion of DTT not only decreased ~52 % of maximum absorbance, but also caused a slight shifting of eosin-Y peak wavelength from 517 nm to 505 nm (Fig. 4B), suggesting the occurrence of photochemical reaction between DTT and eosin-Y. Interestingly, a shifting of peak wavelength from 516 nm to 522 nm was observed in the presence of PEGdNB with minimal change in maximum absorbance (Fig. 4C). No spectrophotometric difference was found when PEGdNB was replaced with hydroxyl-terminated PEG (data not shown). Further, visible light exposure in the presence of eosin-Y, PEGdNB, and DTT (i.e., with thiol-ene reactions) resulted in a higher degree of reduction in eosin-Y maximum absorbance (~64 %), while the peak wavelength shifted from 522 nm to 510 nm (Fig. 4D), a phenomenon similar to that shown in Fig. 4B. The shifting in peak wavelength from 516 nm to 522 nm without chemical reaction suggests binding affinity between eosin-Y and PEG (potentially due to hydrogen bonds). On the other hand, visible light exposure in the presence of chemically reactive species (e.g., DTT and PEGdNB) caused a shifting of peak wavelength back to a lower value (i.e., 12 nm difference). This phenomenon implies a change in eosin-Y molecular structure, potentially due to reactions between excited eosin-Y and the reactive macromer species (e.g., adduct to PEG hydrogel network).
Figure 4.

UV/Vis spectra of eosin-Y before (solid line) and after (dashed line) visible light exposure for 4 minutes in the presence of different components: (A) eosin-Y only; (B) eosin-Y and DTT; (C) eosin-Y and PEGdNB; (D) eosin-Y, PEGdNB, and DTT. Wavelengths indicated in each figure represent the peak absorbance before (top) and after (bottom) light exposure. (E) UV/Vis spectra of freshly prepared (solid line) and recovered (dashed line) eosin-Y. The wavelength of the peak absorbance for both samples was at 516 nm. Eosin-Y concentration in all measurements: 0.02 mM (N = 3).
Formation of thiol-ene hydrogels using released eosin-Y
As described earlier, excess eosin-Y was released into the buffer solution during hydrogel swelling. From results shown in Fig. 4, it is clear that eosin-Y, even after light exposure, still absorbs light in the visible light range. Since eosin-Y is not a cleavage type photoinitiator, we hypothesized that the excess eosin-Y could be used to initiate additional thiol-ene photo-click reactions. To demonstrate this feasibility, the spectrophotometric property of eosin-Y recovered from a pre-formed hydrogels was evaluated and compared to freshly prepared eosin-Y at an equivalent concentration (0.02 mM). The absorbance spectrum of the recovered eosin-Y overlaps with freshly prepared eosin-Y and still peaked at 516 nm (Fig. 4E), suggesting that eosin-Y could be re-excited for sequential cross-linking reactions. It is important to note that in Fig. 4B and Fig. 4D, the measurements were conducted in the presence of reactive species while the samples used in Fig. 4E were recovered eosin-Y from a cross-linked PEG gels. When recovered eosin-Y (at 0.1 mM) was used to initiate visible light-mediated thiol-ene gel cross-linking, the gel point was roughly 10 seconds slower compared to gelation using fresh eosin-Y (Fig. 5). Furthermore, a significantly lower elastic modulus (~3.6 and ~6.4 kPa for gels prepared using recovered and fresh eosin-Y, respectively) after 240 seconds of light exposure (Fig. 5) and a lower gel fraction was obtained (92% and 99% for 1 mm thick gels prepared with recovered and fresh eosin-Y, respectively). While the recovered eosin-Y does not have the same initiation capacity compared to freshly prepared eosin-Y, it is important to note that the eosin-Y concentration was kept low at 0.1 mM and a higher concentration may be used to compensate the slightly lower initiation capacity. Nonetheless, recovered eosin-Y retained the ability to re-initiate thiol-ene photopolymerization to fabricate hydrogels or conjugate biomolecules.
Figure 5.

Evolution of elastic (G′) and viscous (G′) moduli during in situ gelation of PEG4NB-DTT using fresh or recovered eosin-Y at 0.1 mM as photoinitiator (PEG4NB: 10 wt%, N = 3). Error bars were neglected for clarity.
Interfacial thiol-ene photopolymerization
Based on the previous experimental results, we hypothesized that the residual and released eosin-Y from a pre-formed gel could be utilized to fabricate multilayer hydrogels (Fig. 6A). To prove this, we first prepared a thiol-ene hydrogel disc by visible light-mediated gelation using 2 mM eosin-Y as the photoinitiator. Following cross-linking, the gel was immediately immersed in a second macromer solution containing only PEG4NB and DTT (blue microparticles were added for imaging purpose). The construct was exposed to the same visible light source and a thin layer of thiol-ene hydrogel coating was formed surrounding the core gel (Fig. 6A). From the fluorescence intensities of the hydrogel layers shown in Fig. 6B, the gel coating contained a pure red layer (from residual eosin-Y), a pure blue layer (from incorporated microparticles), and an intermediate purple zone. This partial overlap in fluorescence intensity profiles suggests the existence of molecular association between the two layers. Although we could not rule out the possibility of image artifacts, a similar phenomenon was observed in the GOx-mediated multilayer coating system developed by Johnson et al.21 The thiol-ene coating was found to extend beyond the purple zone (~130 μm in thickness, Fig. 6B), indicating that cross-linking of the coating was not limited to the diffusional distance of eosin-Y. It was possible that the small thiyl radical-bearing DTT (MW: 154.25 Da) diffused rapidly away from the core gel and caused thiol-ene reactions and cross-linking in areas beyond the diffusional distance of eosin-Y. In addition, small DTT molecules could easily penetrate the core gel and react with the unreacted norbornene moiety, thus increasing the bonding between layers upon light exposure.
Figure 6.

(A) Fluorescence micrograph and (B) intensity profile of a dual-layer hydrogel formed by sequential visible light-mediated thiol-ene photopolymerization. Red and blue fluorescence were from the eosin-Y added in the core gel and blue microparticles added in the coating macromer solution, respectively. (10 wt% PEG4NB-DTT pre-formed gel with 2.0 mM eosin-Y, 10 wt% PEG4NB-DTT and 2 minutes of secondary polymerization, 5 % blue particles, Scale bar: 200 μm)
The thiol-ene interfacial photopolymerization developed here is similar to the GOx-mediated coating system developed by Johnson and Bowman et al.20–22 and the interfacial PEG-diacrylate (PEGDA) photopolymerization system pioneered by Hubbell et al.3, 9, 28–30 The major differences between our system and the previous ones are two-fold: (1) our coating chemistry is a step-growth gelation mechanism, and (2) the macromer solution used in our thiol-ene interfacial coating procedures contained only macromers (i.e., PEG4NB and DTT) without additional co-initiator or co-monomer. Further, our controlled experiments demonstrated that this interfacial cross-linking was not due to initiator-free thiol-ene photopolymerization as gelation was not observed after prolonged light exposure (> 1 hour) without the presence of eosin-Y (data not shown).
Controlling the thickness of thiol-ene coating
The amount of eosin-Y released from these thiol-ene hydrogels was roughly proportional to its initial concentration in the polymer precursor solution (Fig. 3B). Therefore, we hypothesized that the thickness of the second gel layer could be readily controlled by increasing either the concentration of eosin-Y in the precursor solution of the core gel or the duration of the secondary visible light exposure, both of which allow eosin-Y to diffuse further away from the core gel surface. Results in Fig. 7A confirmed our hypothesis that higher eosin-Y concentration leached out from the core gel resulted in thicker hydrogel coatings. However, Fig. 7A also shows that the growth rates of coating thickness are not linear and decrease as time. For example, as we increased the coating time from 0.5 to 2 minutes with 0.5 mM eosin-Y in the core gel, the thickness of the coating increased non-linearly to roughly 220 μm (Fig. 7A). Further prolonging coating time only marginally increased the coating thickness. This behavior was likely due to a monotonic decrease in eosin-Y concentration further away from the core gel surface, and a lower eosin-Y concentration reduces the initiation rate of thiol-ene photopolymerization upon visible light exposure (Fig. 1B).
Figure 7.

(A) Effect of secondary visible light exposure time (or coating time) and eosin-Y concentration in the core gel on the thickness of thiol-ene hydrogel coating. (B) Effect of PEG4NB macromer concentration on the thickness of thiol-ene hydrogel coating (eosin-Y in the core gel: 2.0 mM; coating time: 2 min). Core gels were fabricated with 10 wt% PEG4NB. (N = 3; Mean ± S.D.)
In addition to light exposure time and eosin-Y concentration in the core gel, thickness of the thiol-ene hydrogel coating could also be controlled by the macromer concentration in the coating precursor solution. For example, increasing macromer concentration from 5 to 30 wt% of PEG4NB ([DTT] was adjusted such that [SH]/[ene] = 1) increased the thickness of the coating almost linearly from ~180 to ~1150 μm (Fig. 7B). The trend was most likely caused by acceleration in propagation rate at higher macromer (PEG4NB) concentrations (i.e., Rp = kp[S•][ene]). While further studies are required to elucidate the mechanism and successive coated gel property, the linearity of coating thickness as a function of macromer concentration provides a highly controllable way of forming gel coating with different thicknesses. Note that while DTT was used as a model cross-linker here, other bis-cysteine-containing peptides can be easily incorporated as cell-responsive linker. With excess amount of norbornene groups (i.e., higher than unity ene-to-thiol ratio), mono-cysteine-bearing or thiolated peptides/proteins can also be conjugated to render the otherwise inert PEG coating bioactive.
Fabrication of multilayer hydrogels
We further conducted proof-of-principle studies to explore the utility of this new interfacial gelation scheme on forming multilayer hydrogels. As shown in Fig. 8A, we synthesized a three-layer hydrogel construct using sequential visible light mediated thiol-ene photopolymerizations. To fabricate this simple multilayer hydrogel, we only added eosin-Y in the bottom layer. The formation of the middle and top layers was due to the diffusion of eosin-Y from the immediate adjacent layer. Although no additional initiator was added in the middle and top layers, the thickness of each layer after 10 minutes visible light exposure was similar (~2 mm). This was due to the use of a fixed pre-polymer solution volume (25 μL) in each layer and a three-fold higher light intensity (200,000 Lux) that resulted in complete gelation. Controlled experiments also showed that the formation of multilayer gels was not due to initiator-free polymerization as gelation did not occur (after 60 minutes light exposure) without the presence of eosin-Y. In addition, Fig. 8B shows the formation of a thick gel coating (~ 5 mm (dia.) × 6 mm (h) cylinder) from a thin hydrogel disk (2 mm (dia.) × 1 mm (h)). This coating was formed without the inclusion of additional initiator in the coating macromer solution. More importantly, light attenuation was not a significant issue in forming this thick construct because there was no eosin-Y in the precursor solution and the gel cross-linking reaction was initiated from the surface of the core gel. The results of Fig. 8A and 8B show that the formation of the multi-layer construct was not solely due to eosin-Y diffusion but surface-mediated polymerization. In the previous study (Fig. 7), eosin-Y diffusion might have played a more significant role resulting in limited growth in gel thickness. In Fig. 8, surface-mediated polymerization might have played a more significant role in allowing the growing of subsequent gel layers beyond the diffusional distant of eosin-Y. Although long exposure time and higher light intensity (200,000 Lux) were used in forming these thick multilayer hydrogels, the thickness could also be easily controlled via manipulating coating time, macromer concentration, and eosin-Y content as shown in Fig. 7. Due to the step-growth nature of the reactions, we believe that the properties in each layer will remain similar. However, from the view point of constructing biomimetic tissues with multi-layer constructs, it is actually beneficial to have layer of gels with different properties. Future studies will focus on generation and characterization of multilayer gel structures with different material properties (e.g., crosslinking density, reaction kinetics, etc.).
Figure 8.

(A) Photograph of a three-layer thiol-ene hydrogel formed from sequential visible light-mediated thiol-ene photopolymerization. PEG4NB macromer concentration was 10 wt% in each layer. Eosin-Y concentration in the bottom layer was 2.0 mM. 5 wt% of blue microparticles was added in the top layer for visualization purpose. (B) Photograph of an example small gel disc (left, 2 mm Dia. x 1 mm H) used to fabricate a thick gel coating (right, 6 mm Dia. x 6 mm H). (Note: gel in the left curled up due to partial drying)
While conventional visible light-mediated photopolymerization techniques could be used to fabricate multilayer hydrogels,16, 31, 32 additional initiating species and co-monomers are required to generate sufficient radicals for initiating cross-linking. Furthermore, prior visible light gelation systems were all based on chain-growth photopolymerization that has been shown detrimental to sensitive cells and growth factors.18, 19 Chain-growth polymerized gels also have heterogeneous network structures,29 which may not be ideal for releasing therapeutically-relevant molecules. The unique visible light-mediated interfacial thiol-ene gel coating system is comparable to the glucose oxidase (GOx) mediated gel coating system reported by Bowman and colleagues.20–22 The GOx-mediated gel coating system, however, uses multiple initiator components (i.e., GOx, glucose, and ferrous ion) to initiate polymerization reactions. Because the process produces highly cytotoxic hydrogen peroxide (H2O2), a second enzyme, catalase, has to be included in the macromer solution to prevent cellular damage. Another disadvantage of GOx-mediated polymer coating system is that GOx, being a bulky enzyme (Rh ~43 Å), has a tendency of being trapped within the growing polymer layer. The presence of this additional component in the gel coating may cause unwanted complications. The interfacial thiol-ene gelation scheme presented here overcomes many disadvantages associated with other multilayer hydrogel systems and additional work is underway to exploit the utilities of this gel coating system in tissue engineering applications.
CONCLUSION
In conclusion, we have developed a simple yet effective visible light-mediated interfacial thiol-ene photopolymerization scheme that can be used to create multilayer gel structures for biomedical applications. In addition to characterizing the effects of eosin-Y concentration on gel cross-linking efficiency, we also verified that part of the eosin-Y retained its ability to re-initiate thiol-ene photo-crosslinking. Utilizing this unique property, we have further designed step-growth hydrogels with multilayer structures and a wide range of thicknesses (from tens of microns to a few millimeters). No additional initiator is required in the preparation of hydrogel coating or multilayer gel, given that sufficient eosin-Y is available from the core or adjacent hydrogel layers. This complete step-growth multilayer hydrogel system does not require and does not generate any cytotoxic components. Therefore, these multilayer gels may serve as a highly cytocompatible platform for creating complex multifunctional tissue substitutes.
Acknowledgments
This project was supported by the Department of Biomedical Engineering at IUPUI, a Pilot & Feasibility grant from the Indiana Diabetes Research Center at IU School of Medicine, and NIH/NIBIB (R21EB013717). The authors thank Dr. Chang Seok Ki for his technical assistance.
References
- 1.Karpiak JV, Ner Y, Almutairi A. Adv Mater. 2012;24:1466–1470. doi: 10.1002/adma.201103501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu ZS, Calvert P. Adv Mater. 2000;12:288–291. [Google Scholar]
- 3.An YJ, Hubbell JA. J Control Release. 2000;64:205–215. doi: 10.1016/s0168-3659(99)00143-1. [DOI] [PubMed] [Google Scholar]
- 4.Antipov AA, Sukhorukov GB, Donath E, Mohwald H. J Physical Chem B. 2001;105:2281–2284. [Google Scholar]
- 5.Zhang JT, Chua LS, Lynn DM. Langmuir. 2004;20:8015–8021. doi: 10.1021/la048888i. [DOI] [PubMed] [Google Scholar]
- 6.Wood KC, Boedicker JQ, Lynn DM, Hammond PT. Langmuir. 2005;21:1603–1609. doi: 10.1021/la0476480. [DOI] [PubMed] [Google Scholar]
- 7.Mehrotra S, Lynam D, Maloney R, Pawelec KM, Tuszynski MH, Lee I, Chan C, Sakamoto J. Adv Funct Mater. 2010;20:247–258. doi: 10.1002/adfm.200901172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen XY, Wu W, Guo ZZ, Xin JY, Li JS. Biomaterials. 2011;32:1759–1766. doi: 10.1016/j.biomaterials.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 9.Cruise GM, Hegre OD, Lamberti FV, Hager SR, Hill R, Scharp DS, Hubbell JA. Cell Transplantation. 1999;8:293–306. doi: 10.1177/096368979900800310. [DOI] [PubMed] [Google Scholar]
- 10.Hume PS, Bowman CN, Anseth KS. Biomaterials. 2011;32:6204–6212. doi: 10.1016/j.biomaterials.2011.04.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang X, Chen H, Zhang HY. Chem Commun. 2007:1395–1405. doi: 10.1039/b615590a. [DOI] [PubMed] [Google Scholar]
- 12.Tang ZY, Wang Y, Podsiadlo P, Kotov NA. Adv Mater. 2006;18:3203–3224. [Google Scholar]
- 13.Fairbanks BD, Schwartz MP, Halevi AE, Nuttelman CR, Bowman CN, Anseth KS. Adv Mater. 2009;21:5005–5010. doi: 10.1002/adma.200901808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lin CC, Anseth KS. Pharm Res-dordr. 2009;26:631–643. doi: 10.1007/s11095-008-9801-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nguyen KT, West JL. Biomaterials. 2002;23:4307–4314. doi: 10.1016/s0142-9612(02)00175-8. [DOI] [PubMed] [Google Scholar]
- 16.Kizilel S, Sawardecker E, Teymour F, Perez-Luna VH. Biomaterials. 2006;27:1209–1215. doi: 10.1016/j.biomaterials.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 17.Cuchiara MP, Allen ACB, Chen TM, Miller JS, West JL. Biomaterials. 2010;31:5491–5497. doi: 10.1016/j.biomaterials.2010.03.031. [DOI] [PubMed] [Google Scholar]
- 18.Lin CC, Raza A, Shih H. Biomaterials. 2011;32:9685–9695. doi: 10.1016/j.biomaterials.2011.08.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McCall JD, Anseth KS. Biomacromolecules. 2012;13:2410–2417. doi: 10.1021/bm300671s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnson LM, Fairbanks BD, Anseth KS, Bowman CN. Biomacromolecules. 2009;10:3114–3121. doi: 10.1021/bm900846m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Johnson LM, DeForest CA, Pendurti A, Anseth KS, Bowman CN. Acs Appl Mater Inter. 2010;2:1963–1972. doi: 10.1021/am100275n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shenoy R, Bowman CN. Biomaterials. 2012;33:6909–6914. doi: 10.1016/j.biomaterials.2012.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shih H, Lin CC. Macromol Rapid Comm. 2012 doi: 10.1002/marc.201200605. (In press) [DOI] [PubMed] [Google Scholar]
- 24.Hoyle CE, Bowman CN. Angew Chem Int Edit. 2010;49:1540–1573. doi: 10.1002/anie.200903924. [DOI] [PubMed] [Google Scholar]
- 25.Anderson SB, Lin CC, Kuntzler DV, Anseth KS. Biomaterials. 2011;32:3564–3574. doi: 10.1016/j.biomaterials.2011.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shih H, Lin CC. Biomacromolecules. 2012;13:2003–2012. doi: 10.1021/bm300752j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grotzinger C, Burget D, Jacques P, Fouassier JP. Polymer. 2003;44:3671–3677. [Google Scholar]
- 28.Sawhney AS, Pathak CP, Hubbell JA. Biomaterials. 1993;14:1008–1016. doi: 10.1016/0142-9612(93)90194-7. [DOI] [PubMed] [Google Scholar]
- 29.Cruise GM, Hegre OD, Scharp DS, Hubbell JA. Biotechnol Bioeng. 1998;57:655–665. doi: 10.1002/(sici)1097-0290(19980320)57:6<655::aid-bit3>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 30.Cruise GM, Scharp DS, Hubbell JA. Biomaterials. 1998;19:1287–1294. doi: 10.1016/s0142-9612(98)00025-8. [DOI] [PubMed] [Google Scholar]
- 31.Papavasiliou G, Songprawat P, Perez-Luna V, Hammes E, Morris M, Chiu YC, Brey E. Tissue Eng Part C-Methods. 2008;14:129–140. doi: 10.1089/ten.tec.2007.0355. [DOI] [PubMed] [Google Scholar]
- 32.Kizilel S, Perez-Luna VH, Teymour F. Langmuir. 2004;20:8652–8658. doi: 10.1021/la0496744. [DOI] [PubMed] [Google Scholar]