

Abstract
Adenovirus (Ad) derived vectors have been widely used for short or long-term gene transfer, both for gene therapy and vaccine applications. Because of the frequent pre-existing immunity against the classically used human adenovirus type 5, canine adenovirus type 2 (CAV2) has been proposed as an alternative vector for human gene transfer. The well-characterized biology of CAV2, together with its ease of genetic manipulation, offer major advantages, notably for gene transfer into the central nervous system, or for inducing a wide range of protective immune responses, from humoral to cellular immunity. Nowadays, CAV2 represents one of the most appealing nonhuman adenovirus for use as a vaccine vector. This protocol describes a simple method to construct, produce and titer recombinant CAV2 vectors. After cloning the expression cassette of the gene of interest into a shuttle plasmid, the recombinant genomic plasmid is obtained by homologous recombination in the E. coli BJ5183 bacterial strain. The resulting genomic plasmid is then transfected into canine kidney cells expressing the complementing CAV2-E1 genes (DK-E1). A viral amplification enables the production of a large viral stock, which is purified by ultracentrifugation through cesium chloride gradients and desalted by dialysis. The resulting viral suspension routinely has a titer of over 1010 infectious particles per ml and can be directly administrated in vivo.
Keywords: Immunology, Issue 82, Canine Adenovirus, viral vector, vaccination, central nervous system, gene therapy
Introduction
Over the past decades, adenoviruses (Ads) derived vectors have proven their efficacy for gene therapy and vaccination, as well as in oncolytic virotherapy 1. Ads are nonenveloped icosahedral viruses of the Adenoviridae family, which have been isolated from mammals, birds, reptiles, amphibians and fish. Since their discovery in 1953, Ads have been intensively studied as models to study virus/cell interactions and, more recently, as vector-based gene delivery systems 2. Indeed, Ads-based vectors offer many advantages, such as their broad host range and well-characterized biology, together with the ease with which they can be genetically manipulated and amplified for large-scale production.
In order to overcome clinical difficulties related to pre-existing immunity in human populations towards vectors derived from human Ads 3, we and others began to derive vectors from nonhuman Ads 4. In addition, nonhuman adenoviral vectors appear to be more adapted for mass vaccination in veterinary medicine than the classically used adenovirus type 5 (Ad5), since they should be more compliant with safety and security requirements during the risk assessment by the national regulatory authorities. In the late 1990s, we described the first recombinant CAV2 as a nonhuman alternative to vectors derived from Ad5 5. Since then, numerous studies have confirmed the potential of CAV2 vectors for therapeutic or antigenic gene transfer (for reviews 6,7). Like other Ads, CAV2-derived vectors are stable and can be produced at high titers, which facilitates their in vivo use. They are also safe, since they are not integrative, although they allow long lasting transgene expression in vivo (>1 year) 8. Remarkably, and better than human Ads serotypes, CAV2 vectors have been shown to be highly neurotropic, with a very efficient retrograde transport in axons 9,10. This intrinsic property brought the idea of using recombinant CAV2 vectors to transduce neurons of specific brain areas that are hardly accessible to lentiviral vectors, so far commonly used for gene transfer into the central nervous system 11.
Here, we describe a simple and classical protocol to construct, produce and purify nonreplicative CAV2 vectors derived from the Manhattan strain. Recombinant CAV2 genomes are constructed by cloning the desired gene of interest (GOI) in a cassette downstream of the cytomegalovirus (CMV) early promoter, which provides a ubiquitous expression. CAV2 vectors have a cloning capacity of ~4.2 kbp, which allows expressing large cDNA. In a second step, this expression cassette is inserted by homologous recombination in place of the E1 region of the CAV2 genome, leading to a nonreplicative virus (dlE1), as described initially by Chartier et al.12 Finally, viral particles are produced upon transfection of the genomic plasmid into a CAV2-E1-expressing canine kidney cell line (DK-E1) and are serially amplified prior to concentration and purification of viral stocks. The method described here uses a two-step ultracentrifugation in cesium chloride (CsCl) gradients, which enables enrichment of infectious particles with a high final titer (usually over 1010 infectious particles per ml). The viral suspension is then desalted by chromatography through disposable Sephadex G-25 columns so that it is ultimately pure enough to be directly used in vivo.
Protocol
1. Construction of a Nonreplicative CAV2 Genome Plasmid
Note: all plasmids described in this protocol have been described previously 7 and are available upon request.
Clone the GOI into the pShuttle plasmid, using appropriate restriction enzymes, to generate pShuttle-GOI (Figure 1A).
Digest 2 μg of pShuttle-GOI by AscI/PacI. The digestion releases the Shuttle-GOI fragment (~2.9 kbp + GOI ORF sequence) from the plasmid backbone (2.8 kbp).
Purify the Shuttle-GOI fragment after agarose gel electrophoresis, using NucleoSpin Gel and PCR Clean-up kit.
In parallel, digest 1 μg of pCAV2 genomic plasmid by SwaI. The pCAV2 plasmid contains a unique SwaI site located within the E1 region (Figure 1B). Heat-inactivate this restriction enzyme at 65 °C for 20 min.
Transform competent E. coli BJ5183 bacteria with 500 ng of purified Shuttle-GOI fragment and 100 ng of linearized pCAV2 plasmid for homologous recombination (Figure 1B). Spread on LB-Amp plates. Incubate plates at 37 °C for at least 20 hr.
Perform PCR analysis directly on bacterial colonies using EmeraldAmp master mix with forward (5'-CACGAGGCCCTTTCGTCTTCAA-3') and reverse (5'-GCGGTAGTTTATCACAGTTAAATTGC-3') primers, under the following cycling conditions: 10 min at 95 °C (for one cycle), 95 °C for 1 min; 58 °C for 1 min; 72 °C for 1 min/kbp (for 39 cycles), and finally 5 min at 72 °C. A product of 1 kbp + GOI ORF sequence is amplified from pCAV-GOI positive clones, whereas no PCR product can be detected with negative clones.
Inoculate at least two positive colonies overnight each in 5 ml LB-amp medium at 37 °C in a shaker (220 rpm).
Extract plasmid DNAs using Nucleospin plasmid kit. Transform competent E. coli DH5α bacteria with an aliquot of each DNA prep. Spread bacterial suspension on LB-Amp plates and incubate overnight at 37 °C.
Inoculate two colonies of each positive clone overnight in 3 ml LB-amp medium at 37 °C in a shaker (220 rpm). Extract plasmid DNA using Nucleospin plasmid kit.
Digest the resulting DNA and the parental pCAV2 plasmid (as a control) with NdeI. The restriction pattern of parental pCAV2 is 14.8 kbp, 9 kbp, 7.9 kbp, and 1.6 kbp. The GOI expression cassette is inserted within the 7.9 kbp fragment and contains at least one NdeI site, i.e. one in the cassette and the potential sites present in the GOI.
Select positive constructions and proceed to step 2 with at least 2 independent clones.
2. Transfection of Recombinant CAV2 Genome Plasmid into DK-E1 Cells
Note: CAV2-derived non replicative vectors are genetically modified organisms classified as Biosafety Level 2 (BSL-2), whereas their use on animal models is classified as BSL-1. Please use proper containment and waste handling measures, including personal protective equipment, and work under BSL-2 laminar flow hoods.
A day before transfection, seed DK-E1 cells on a 6-well plate. Grow cells at 37 °C and 5% CO2, in Dulbecco's Modified Eagle Medium (DMEM) with high glucose, containing 7% heat-inactivated fetal calf serum (FCS), 1 mM sodium pyruvate, and 100 U/ml penicillin/100 μg/ml streptomycin. Cells should be 70-80% confluent for transfection.
Digest 2 μg of pCAV-GOI plasmid with AscI, to release the nonviral sequence of the plasmid. Check the resulting restriction pattern by 0.8% agarose gel electrophoresis. Digestion should yield a ~31 kbp fragment containing the recombinant genome and a 2 kbp fragment corresponding to the plasmid backbone.
Mix digested DNA with 200 μl of Jet Prime Buffer by vortexing for 10 sec. Add 4 μl of Jet Prime and mix by vortexing for 10 sec. Spin briefly to remove droplets and incubate for 10 min at room temperature.
Add the transfection mix dropwise onto cells. Gently shake plates to evenly distribute the mix and incubate at 37 °C.
3. Propagation of Recombinant CAV2
One week after transfection, collect cells and culture medium in a 15 ml polypropylene tube. Disrupt cells by three freeze-thaw cycles (-80 °C/37 °C) and clear the lysate by centrifugation for 10 min at 1,800 x g.
Collect supernatant and store at -20 °C for subsequent virus propagation.
Infect an 80-90% confluent monolayer of DK-E1 cells grown in one well of 6-well plate with 0.5 ml of the virus-containing supernatant. Incubate at 37 °C for 1 hr, under mild agitation. Remove inoculum and replace it with 1.5 ml of complete DMEM containing 5% heat-inactivated FCS.
Incubate cells at 37 °C for 3-4 days. Cells should be monitored daily for appearance of a cytopathic effect (CPE, Figures 2A-B). If no clear CPE is visible, harvest cells after 4 days of culture and repeat steps 3.1-3.4.
When a clear CPE affects the majority of cells, usually after 2-4 viral amplification rounds, collect cells with culture medium and freeze-thaw 3x as described above. Infect 80-90% confluent DK-E1 cells grown in three 10 cm diameter tissue culture dishes, using each time 0.5 ml of virus-containing supernatant.
After 3-4 days (when CPE is complete), harvest cells from these 10 cm dishes, again disrupt them by three freeze-thaw cycles, clear the lysate by centrifugation for 10 min at 1,800 x g, collect supernatant and store at -20 °C for large-scale CAV2 amplification.
4. Large-scale CAV2 Purification
Infect 80-90% confluent monolayers of DK-E1 cells grown in forty 10 cm diameter tissue culture dishes. For each dish, use 0.1 ml of virus-containing supernatant diluted in 1 ml of complete DMEM without FCS. Incubate cells at 37 °C for 3 to 4 hr under mild agitation. Add 5 ml of complete DMEM supplemented with 5% FCS.
Incubate for 3 days. Harvest infected cells from the forty 10 cm dishes in 50 ml polypropylene tubes. Pellet cells at 1,200 x g for 10 min at 4 °C and resuspend them in 15 ml of DMEM medium.
Disrupt cells by three freeze-thaw cycles (-80 °C/37 °C). Remove cell debris by centrifuging at 1,800 x g for 10 min and store supernatant at -20 °C prior to purification of viral particles.
Prepare a discontinuous CsCl gradient in a 14 ml Ultra-Clear tube. First, pour 2 ml of CsCl solution (1.40 g/ml in phosphate buffered saline) at the bottom of the tube. Then, add slowly 2 ml of CsCl solution (1.25 g/ml in phosphate buffered saline) on top of the first solution.
Load carefully the viral supernatant on top of the CsCl gradient. Fill the tube with mineral oil up to 2-3 mm from the top. Centrifuge for 1.5 hr at 130,000 x g and 18 °C in a swinging SW40 rotor.
After centrifugation, two white bands are clearly visible at the interface between the 2 CsCl solution layers (Figure 2C). Using a 21 G needle and a syringe, collect by side puncture the lower band containing mature CAV2 particles. Try as much as possible to avoid collecting the upper band, which corresponds to empty viral particles.
Prepare a continuous CsCl gradient in a 14 ml transparent tube by mixing the collected viral particles with a CsCl solution (1.34 g/ml in phosphate buffered saline). Fill the tube with mineral oil up to 2-3 mm from the top. Centrifuge for 18 hr at 130,000 x g and 18 °C in a swinging SW40 rotor.
After centrifugation, collect the white CAV2-containing band by side puncture with a syringe as described above. Again, try to avoid collecting the remaining upper band. This should be easier in this continuous gradient, which separates the two bands better. The total volume of collected suspension should not exceed 2 ml.
Equilibrate a Sephadex G-25 PD-10 column with 30 ml of PBS.
Load the viral suspension and discard the flow-through.
Elute with fractions of 500 μl of PBS. Collect fractions 5-7, which usually contain the desalted CAV2 particles. The virus-containing fractions can easily be identified because they are opalescent. Add 150 μl of glycerol to the 1.5 ml of CAV2 suspension and store in aliquots at -80 °C.
5. Titration of CAV2 by End-point Dilution
Thaw an aliquot of purified virus on ice and perform 10-fold serial dilutions (ranging from 10-2-10-12) in serum-free DMEM.
Add 50 μl of each viral dilution in 5 wells of a 96-well plate. Add 1.5 x 104 DK-E1 cells in each well. Incubate the plate at 37 °C and 5% CO2 for 5 days.
At day 5 post-infection, monitor CPE appearance by microscopic observation (Figures 2A-B). Infectious titers are determined as median tissue culture infectious doses (TCID50), using the Reed and Muench statistical method13.
Representative Results
Production of recombinant CAV2 vectors relies on basic but important molecular biology techniques. Prior to the production of viral vectors, it is indeed necessary to construct a recombinant CAV2 genome bearing an expression cassette for the transgene. This construction involves two steps: first, the gene of interest (GOI) is cloned in a shuttle plasmid as an expression cassette (i.e. with promoter and polyadenylation signal). This functional element is flanked by two CAV2-derived genomic sequences, representing the upstream and downstream recombination regions (Figure 1A). In a second step, the expression cassette is inserted from the shuttle plasmid into the CAV2 genome, by using homologous recombination (Figure 1B). The linearization of the two DNA components will ensure a better success of this step (see sites for AscI and PacI digestion on pShuttle and SwaI on pCAV2 in Figure 1). The insertion of the GOI expression cassette will lead to the deletion of E1 in the recombinant genome.
Once the recombinant genomic plasmid has been obtained, viral particles are produced using CAV2-E1 complementing DK-E1 cells. A clear cytopathic effect, due to the large amount of viral particles produced in infected cells, can readily be visualized by brightfield microscopy (Figure 2A), or by transgene expression (example of a GFP-expressing CAV2 vector, Figure 2B). After several viral amplification rounds using fresh DK-E1 cells, viral particles are purified by ultracentrifugation on cesium chloride gradients. Concentrated viral particles appear as two opalescent bands within the cesium gradient (Figure 2C). The top (lighter) band contains empty, noninfectious particles and has to be avoided.
The resulting recombinant CAV2 vector can be used to transduce nearly all cell types, in a wide variety of species, either in vitro or in vivo. Here we show one application example: transduction of the mouse central nervous system using stereotactic injection. Since this protocol yields high-titer and pure viral stocks, injection of as little of 1 μl of PBS-diluted GFP-expressing CAV2 vector (5 x 105 TCID50) in the Dentate Gyrus (DG, Figure 3, top) leads to 40-50% neuronal transduction. Moreover, due to the ability of CAV2 to be retrogradely transported in axons, injection of 2 μl of PBS-diluted GFP-expressing CAV2 vector (2 x 106 TCID50) in the mouse striatum enables the transduction of nearly 80% of nigrostriatal neurons in the Substancia Nigra pars compacta (SNpc, Figure 3, bottom).
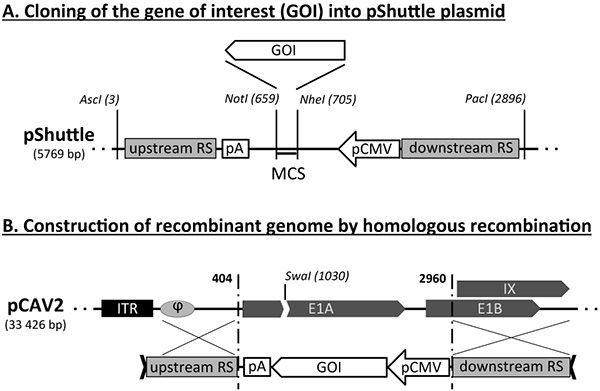 Figure 1. Schematic representation of the generation of nonreplicative CAV2 genome using homologous recombination. (A) Schematic representation of the pShuttle plasmid bearing both upstream and downstream recombination target sequences (RS) that are also shared by the CAV2 genome vector (in grey). The scheme also indicates the location of AscI and PacI restriction sites, the CMV promoter, the polyA signal, as well as of the multiple cloning site (MCS) to insert the gene of interest (GOI). (B) Schematic representation of the homologous recombination between CAV2 genomic sequence and the AscI/PacI recombination sequence of the pShuttle-GOI plasmid. The restriction site for SwaI (genome linearization) and features of the 5' end of CAV2 genome i.e. the internal terminal repeat (ITR), the encapsidation signal (φ), genes encoding the early E1A, E1B and IX proteins are also represented. The homologous recombination leads to replacement of E1A and part of E1B genes by the GOI expression cassette. Click here to view larger image.
Figure 1. Schematic representation of the generation of nonreplicative CAV2 genome using homologous recombination. (A) Schematic representation of the pShuttle plasmid bearing both upstream and downstream recombination target sequences (RS) that are also shared by the CAV2 genome vector (in grey). The scheme also indicates the location of AscI and PacI restriction sites, the CMV promoter, the polyA signal, as well as of the multiple cloning site (MCS) to insert the gene of interest (GOI). (B) Schematic representation of the homologous recombination between CAV2 genomic sequence and the AscI/PacI recombination sequence of the pShuttle-GOI plasmid. The restriction site for SwaI (genome linearization) and features of the 5' end of CAV2 genome i.e. the internal terminal repeat (ITR), the encapsidation signal (φ), genes encoding the early E1A, E1B and IX proteins are also represented. The homologous recombination leads to replacement of E1A and part of E1B genes by the GOI expression cassette. Click here to view larger image.
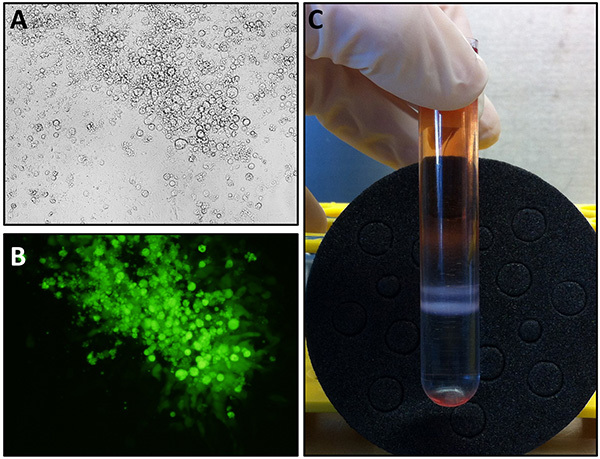 Figure 2. Representative results of CAV2 production steps. (A-B) DK-E1 cells were observed using brightfield (A) or fluorescence (B), in case of a GFP-expressing CAV2 vector, microscopy (100X). During the first amplification steps or for virus titration, individual foci of cytopathic effect are observed. (C) CAV2-containing bands are clearly visible at the end of the discontinuous CsCl gradient centrifugation, with the upper band corresponding to empty particles and the lower band to mature recombinant CAV2. Click here to view larger image.
Figure 2. Representative results of CAV2 production steps. (A-B) DK-E1 cells were observed using brightfield (A) or fluorescence (B), in case of a GFP-expressing CAV2 vector, microscopy (100X). During the first amplification steps or for virus titration, individual foci of cytopathic effect are observed. (C) CAV2-containing bands are clearly visible at the end of the discontinuous CsCl gradient centrifugation, with the upper band corresponding to empty particles and the lower band to mature recombinant CAV2. Click here to view larger image.
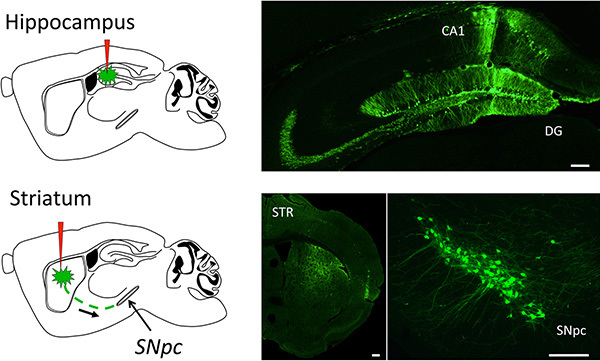 Figure 3. Representative results of CNS gene transfer using CAV2. A GFP expressing CAV2 was injected by stereotaxy in different areas of the central nervous system. The top picture shows representative results of the transduction of mouse hippocampal neurons, after stereotactic injection of 1 x 106 TCID50 in the Dentate Gyrus (GD) and 5 x 105 TCID50 in the Cornus Ammonis 1 (CA1) areas. For the bottom pictures, 2 x 106 TCID50 were stereotaxically injected in the striatum of mice and GFP staining was assayed in the striatum (STR, bottom left) or in the Substantia Nigra pars compacta (SNpc, bottom right), to demonstrate the ability of CAV2 vectors to be retrogradely transported from one CNS area to another. GFP expression was assayed 2 weeks after vector inoculation. Importantly, barely any immune reaction was detected at this time point, as assayed by IBA-1 staining for activated microglia (not shown). The white bars correspond to 100 μm. Click here to view larger image.
Figure 3. Representative results of CNS gene transfer using CAV2. A GFP expressing CAV2 was injected by stereotaxy in different areas of the central nervous system. The top picture shows representative results of the transduction of mouse hippocampal neurons, after stereotactic injection of 1 x 106 TCID50 in the Dentate Gyrus (GD) and 5 x 105 TCID50 in the Cornus Ammonis 1 (CA1) areas. For the bottom pictures, 2 x 106 TCID50 were stereotaxically injected in the striatum of mice and GFP staining was assayed in the striatum (STR, bottom left) or in the Substantia Nigra pars compacta (SNpc, bottom right), to demonstrate the ability of CAV2 vectors to be retrogradely transported from one CNS area to another. GFP expression was assayed 2 weeks after vector inoculation. Importantly, barely any immune reaction was detected at this time point, as assayed by IBA-1 staining for activated microglia (not shown). The white bars correspond to 100 μm. Click here to view larger image.
Discussion
The simple method described herein, adapted from the well-documented Ad5 technology, allows the efficient production of nonreplicative CAV2 vectors with a typical cloning capacity of ~4.2 kbp. Regarding time considerations, a recombinant CAV2 genome is usually generated in two weeks, whereas it will take an additional 5 weeks to amplify, purify and titer viral suspensions.
Although the production of the first batch of CAV2-derived vectors may appear time-consuming, it should be reminded that CAV2 production only requires the initial transfection of one well of a 6-well plate, which minimizes costs for plasmid production, transfection reagents, etc. Moreover, once the first batch of viral particles has been obtained, it can be amplified by a simple infection of the DK-E1 complementing cell line. Here, we proposed a method to purify CAV2 particles based on ultracentrifugation, a commonly available technology in many laboratories. However, purification of CAV2 virions using chromatography has also been described14. Most purified CAV2 viral suspensions have infectious titers greater than 1010 TCID50/ml. Viral particles can alternatively be titrated using quantitative real-time PCR15. CAV2 preparations are stable for years when stored in 10% glycerol (vol/vol) at -80 °C. It is therefore possible to administer these vectors directly in vivo, provided that a minimal two-fold dilution is used to avoid potential glycerol toxicity. Finally, a viral sample can be refrozen up to 2x without significant loss of infectious titer, making its use convenient for large-scale in vivo experiments. Because of its growing interest for human gene transfer, a GMP process for recombinant CAV2 has been recently developed16.
Once the transgene has been cloned into the pShuttle plasmid using conventional molecular biology techniques; a critical step of this protocol consists in obtaining the recombinant CAV2 genomic plasmid. Commercially-available, electroporation-competent E. coli BJ5183 bacteria are to our knowledge best suited for this operation, although homemade competent cells can also be used, albeit with sometimes lower transformation efficiency. The recombination process in E. coli BJ5183 bacteria, which is necessary to generate the genomic plasmid, may occasionally produce clones that will not enable viral production even if they have integrated the recombinant cassette. Thus, it is highly recommended to transfect at least two independent genomic clones to maximize chances of a successful rescue of recombinant CAV2 virions. Generation of the first viral particles after transfection of the recombinant genome in DK-E1 cells is however the most limiting step in the production of recombinant CAV2 vectors.
Subsequent viral amplification steps are usually not limiting. We routinely produce viral stocks using forty 10 cm dishes of infected cells, but the final titer will not differ significantly if starting with 30 or 60 dishes. Once cells have been infected for viral production (step 4.1), we usually wait for 3 days before harvesting them. It is however highly recommended to daily monitor the infection progress to ensure that cells will be harvested at the onset of infection and not too late. Indeed, if infection proceeds faster than expected, a large cytopathic effect may lead to the release of the newly synthesized virions in the culture medium. Therefore, it is preferable to collect cells even earlier than suggested in the protocol, rather than losing most viral particles because of a generalized cytopathic effect. In any event, if cells appear to be dying less than 36 hr postinfection, it is likely that this is not due to CAV2 infection. In this case, it is recommended to restart the viral preparation from the beginning. We also highly recommend working with freshly thawed cells.
After ultracentrifugation through cesium gradients, we described the procedure to collect the white band containing mature virions, by performing a side puncture of the tube. In our hands, we believe that this represents the best way to collect the maximum of viral particles in a minimal volume, which is highly preferable for all future steps, either the second ultracentrifugation or dialysis. However, it is also possible to collect the band using a 1 ml pipette tip, after having removed as much of as possible the upper part of the gradient, without exceeding a volume of 1 ml per band. Once a viral suspension has been produced, presence of the transgene within the viral genome can be verified using PCR analysis directly on the viral stock, after proteinase K digestion to disassemble viral capsids. In addition, transgene expression can be sought after transduction of any susceptible cell type by immunofluorescence, qRT-PCR or western blotting analysis.
As illustrated in Figure 3, CAV2-derived vectors exhibit a remarkable ability to efficiently transduce neurons of the CNS, and can be efficiently transported from one brain area to another by retrograde axonal transport. These properties support the potential interest of CAV2 vectors for gene therapy of the CNS, as well as for the targeted expression of any transgene in selected areas of animal brains. Adenoviral vectors are also increasingly being used to silence gene expression using shRNA17,18. Combined with the specificities of CAV2 tropism, it is therefore tempting to speculate that CAV2-derived vectors could be used to drive shRNA expression in specific brain areas, more efficiently than human serotypes, in order to downregulate targeted neuronal genes. One can also use recombinant CAV2 as vaccination vectors to induce a strong immunity against any antigenic transgene, upon intramuscular or oral delivery, as widely documented during the past decade19. Finally, human Adenovirus derived vectors have also been successfully used in oncotherapy. The increased safety offered by the use of a nonhuman adenovirus for gene therapy, further stresses the potential of CAV2-derived vectors as a new tool to explore antitumoral cell therapy.
Disclosures
Authors have nothing to disclose.
Acknowledgments
This work was supported financially by grants from INSERM, the CNRS, the Fondation pour la Recherche Médicale and the Agence Nationale pour la Recherche (ANR- 10-Blanc-1322) to D.G-D; The European Commission's Seventh Framework Program to B.K.; M.S. was supported by a postdoctoral fellowship from FRM (programme "Equipe FRM 2009"); C.B. was supported by a postdoctoral fellowship from UE, under grant agreement number 245266 (Orbivac).
References
- Kaufmann JK, Nettelbeck DM. Virus chimeras for gene therapy, vaccination, and oncolysis: adenoviruses and beyond. Trends Mol. Med. 2012;18:365–376. doi: 10.1016/j.molmed.2012.04.008. [DOI] [PubMed] [Google Scholar]
- Curr Topics Microbiol. Immunol. 2010;343:195–224. doi: 10.1007/82_2010_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zak DE, et al. Merck Ad5/HIV induces broad innate immune activation that predicts CD8(+) T-cell responses but is attenuated by preexisting Ad5 immunity. Proc. Natl. Acad. Sci. U.S.A. 2012;109:3503–3512. doi: 10.1073/pnas.1208972109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paillard F. Advantages of non-human adenoviruses versus human adenoviruses. Hum. Gene Ther. 1997;8:2007–2009. doi: 10.1089/hum.1997.8.17-2007. [DOI] [PubMed] [Google Scholar]
- Ackermann A, Guelzow T, Staeheli P, Schneider U, Heimrich B. Visualizing viral dissemination in the mouse nervous system, using a green fluorescent protein-expressing Borna disease virus vector. J. Virol. 2010;84:5438–5442. doi: 10.1128/JVI.00098-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bru T, Salinas S, Kremer EJ. An update on canine adenovirus type 2 and its vectors. Viruses. 2010;2:2134–2153. doi: 10.3390/v2092134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouet-Cararo C, et al. Canine adenoviruses elicit both humoral and cell-mediated immune responses against rabies following immunisation of sheep. Vaccine. 2011;29:1304–1310. doi: 10.1016/j.vaccine.2010.11.068. [DOI] [PubMed] [Google Scholar]
- Soudais C, Skander N, Kremer EJ. Long-term in vivo transduction of neurons throughout the rat CNS using novel helper-dependent CAV-2 vectors. FASEB J. 2004;18:391–393. doi: 10.1096/fj.03-0438fje. [DOI] [PubMed] [Google Scholar]
- Deinhardt K, et al. Rab5 and Rab7 control endocytic sorting along the axonal retrograde transport pathway. Neuron. 2006;52:293–305. doi: 10.1016/j.neuron.2006.08.018. [DOI] [PubMed] [Google Scholar]
- Soudais C, Laplace-Builhe C, Kissa K, Kremer EJ. Preferential transduction of neurons by canine adenovirus vectors and their efficient retrograde transport in vivo. FASEB J. 2001;15:2283–2285. doi: 10.1096/fj.01-0321fje. [DOI] [PubMed] [Google Scholar]
- Peltekian E, Garcia L, Danos O. Neurotropism and retrograde axonal transport of a canine adenoviral vector: a tool for targeting key structures undergoing neurodegenerative processes. Mol. Ther. 2002;5:25–32. doi: 10.1006/mthe.2001.0517. [DOI] [PubMed] [Google Scholar]
- Chartier C, et al. Efficient generation of recombinant adenovirus vectors by homologous recombination in Escherichia coli. J. Virol. 1996;70:4805–4810. doi: 10.1128/jvi.70.7.4805-4810.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed LJ, Muench H. A simple method of estimating fifty percent endpoints. Am. J. Hyg. 1938;27:493–497. [Google Scholar]
- Segura MM, Puig M, Monfar M, Chillon M. Chromatography purification of canine adenoviral vectors. Human Gene Ther. Methods. 2012;23:182–197. doi: 10.1089/hgtb.2012.058. [DOI] [PubMed] [Google Scholar]
- Segura MM, et al. A real-time PCR assay for quantification of canine adenoviral vectors. Journal of virological. 2010;163:129–136. doi: 10.1016/j.jviromet.2009.09.010. [DOI] [PubMed] [Google Scholar]
- Fernandes P, et al. Bioprocess development for canine adenovirus type 2 vectors. Gene Ther. 2012. [DOI] [PubMed]
- Rinne A, Littwitz C, Bender K, Kienitz MC, Pott L. Adenovirus-mediated delivery of short hairpin RNA (shRNA) mediates efficient gene silencing in terminally differentiated cardiac myocytes. Methods Mol. Biol. 2009;515:107–123. doi: 10.1007/978-1-59745-559-6_7. [DOI] [PubMed] [Google Scholar]
- Huang B, Kochanek S. Adenovirus-mediated silencing of huntingtin expression by shRNA. Hum. Gene Ther. 2005;16:618–626. doi: 10.1089/hum.2005.16.618. [DOI] [PubMed] [Google Scholar]
- Tordo N, et al. Canine adenovirus based rabies vaccines. Dev. Biol. 2008;131:467–476. [PubMed] [Google Scholar]