

Abstract
CD74, the cell surface form of the MHC class II invariant chain, is a key inflammatory factor that is involved in various immune mediated diseases as part of the Macrophage Migration Inhibitory Factor (MIF) binding complex. However, little is known about the natural regulators of CD74 in this context. In order to study the role of the HLA-DR molecule in regulating CD74, we utilized the HLA-DRα1 domain, which was shown to bind to and downregulate CD74 on CD11b+ monocytes. We found that DRα1 directly inhibited binding of MIF to CD74 and blocked its downstream inflammatory effects in the spinal cord of mice with experimental autoimmune encephalomyelitis (EAE). Potency of the DRα1 domain could be destroyed by trypsin digestion but enhanced by addition of a peptide extension (MOG-35–55 peptide) that provided secondary structure not present in DRα1. These data suggest a conformationally-sensitive determinant on DRα1-MOG that is responsible for optimal binding to CD74 and antagonism of MIF effects, resulting in reduced axonal damage and reversal of ongoing clinical and histological signs of EAE. These results demonstrate natural antagonist activity of DRα1 for MIF that was strongly potentiated by the MOG peptide extension, resulting in a novel therapeutic, DRα1-MOG-35–55,that within the limitations of the EAE model may have the potential to treat autoimmune diseases such as multiple sclerosis.
Keywords: HLA-DRα1, CD74, Macrophage migration inhibitory factor (MIF), multiple sclerosis
Introduction
The class II invariant chain (Ii, CD74) not only chaperones peptide-loaded MHC class II molecules from intracellular compartments to the surface of antigen-presenting cells but also functions as the receptor for macrophage migration inhibitory factor (MIF) when expressed on the cell surface(1, 2). MIF engagement of CD74 leads to the recruitment and activation of CD44 and CXCR2/4 to initiate signaling pathways necessary for MAPK activation and cell motility(3–5). We recently demonstrated enhanced CD74 cell surface expression on monocytes in mice with experimental autoimmune encephalomyelitis (EAE) and subjects with multiple sclerosis (MS), which implicates its involvement in the disease course(6).
Although it has been shown that MIF binds to the CD74 extracellular domain, the actual binding site is not known. In addition, little has been reported on natural anti-inflammatory regulators that can inhibit MIF/CD74 signaling either by downregulating CD74 cell surface expression or by blocking MIF binding(7, 8). Moreover, it is not clear if interactions between the MHC class II and CD74 on the cell surface influence MIF binding and subsequent signal transduction events.
We previously reported that the DRα1 domain but not the DR2β1 domain binds immunoprecipitated CD74.In the present study, we utilized the DRα1 domain in order to study the interaction of this domain with CD74 and its effect on MIF binding and signaling and its potential use as a novel therapeutic reagent.
We report herein that the HLA class II DRα1 domain binds to human and mouse monocytes through cell surface CD74 and that this interaction inhibits MIF binding and signaling. This interaction may be potentially exploited as a treatment for MIF-dependent CNS diseases. Potency of the DRα1 domain waseliminated by trypsin digestion but enhanced by addition of a peptide extension (MOG-35–55 peptide) at the N-terminus that provided discrete secondary α-helical and β-sheet structure. These data suggest a conformationally-sensitive determinant on DRα1 whose structure is influenced by an amino-terminal MOG peptide extension. We hypothesize that this conformation is responsible for optimal binding of DRα1 to CD74 and antagonism of MIF action, which resulted in reversal of clinical and histological signs of experimental autoimmune encephalomyelitis (EAE). Moreover, because the invariable DRα1 domain is present in all human subjects and would not be recognized as immunologically foreign, treatment with DRα1 constructs would not require HLA screening prior to injection and could be universally applicable as therapy forCNS or other inflammatory diseases.
Materials and Methods
Human PBMC
Peripheral blood was obtained from Healthy Control (HC) donors that were screened for HLA genotype. The study protocol received institutional review board approval. All participants signed an informed consent. Peripheral blood mononuclear cells were obtained by centrifugation through Ficoll (Histopaque 1077, Sigma). Samples were stored in liquid nitrogen until used.
Mice
DR*1501-Tg and MBP-TCR/DR2-Tg mice were bred in-house at the Veterinary Medical Unit, Portland Veterans Affairs Medical Center and used at 8–12 weeks of age. All procedures were approved and performed according to institutional guidelines.
DRα1 and DRα1-MOG cloning, production and purification
The DRα1 domain cloning, production and purification has been described previously (9). DRα1-MOG was built using the α1 domain construct as a template. The human MOG-35–55 peptide DNA encoding sequence was attached to the N-terminus of the DRα1 domain with a linker-thrombin-linker intervening element. The single chain gene was cloned into the NcoI and XhoI restriction sites of the pET21d(+) vector (Novagen) and transformed into E. coli BL21(DE3) expression host (Stratagene). For protein production, 4 L of Luria-Bertani medium, supplemented with 50 µg/ml of carbenicillin, were inoculated with a starting OD600 of 0.05. IPTG was added when the culture reached 0.7 OD600. Cultures were allowed to grow for 4 hours and then ice-chilled before harvesting at 7000 rpm for 6 min. After centrifugation, the pellet was resuspended in lysis buffer (50 mM Tris, 5 mM EDTA, 300 mM NaCl, pH 8), treated with lysozyme (1 ml at 10 mg/ml) for 30 min at room temperature and lysed in ice by sonication in a Branson Sonifier 450 apparatus with pulses of 1 min and pauses of 5 min. The disrupted suspension was pelleted at 7000 rpm for 6 min and the paste was resuspended in 1% Triton X-100 in lysis buffer to remove lipids and other hydrophobic contaminants. Detergent was removed by resuspending the pellet lysis buffer followed by sonication as described earlier. This was repeated 3 more times. The final pellet was solubilized in Buffer A (20mM ethanolamine, 6M Urea, pH 10) overnight at 4°C and then spun down at 40,000 g to remove particulate material. This lysate was filtered through a 0.22 µm filter twice, loaded onto a Mono-Q anion-exchange 50 ml column at a flow rate of 2 ml/min attached to an AKTA FPLC (GE Healthcare). After washing the column until no eluting material was detected at 280 nm, proteins were eluted by applying a step-wise gradient of 2 M NaCl in Buffer A. The eluate was collected in fractions of 8ml and after electrophoretic analysis those containing the target protein were pooled together. This pooled material was concentrated with a 3kDa MWCO membrane (Millipore), filtered twice through an 0.22 µm membrane and then loaded onto a Superdex 75, 16/60 size exclusion column (GE Healthcare). The loaded protein was eluted with Buffer C (20mM ethanolamine, 4 mM NaCl, 6 M Urea, pH 10) at a flow rate of 1 ml/min, collected into fractions of 1 ml and finally dialyzed against 20 mM Tris, pH 8.5 for refolding. After refolding, the DRα1-MOG protein was concentrated to 10 mg/ml, snap-frozen and stored in 1 ml aliquots at −80 °C until use.
Binding of DRα1 to immunopurified CD74 and H2-M (mouse equivalent of HLA-DM)
CD74 and H2M were serially captured from a CHAPS lysate of DR*1501-Tg mouse splenocytes, using 2 µg of rat anti-mouse CD74 (clone ln-1) and rat-anti-mouse H-2M (clone 2E5A) monoclonal antibodies (BD Pharmingen) adsorbed to Protein L Beads. DRα1 was labeled with fluorescent chromophore Alexa-488. A488-DRα1 (either un-digested or digested with trypsin) binding to CD74 or H2-M-conjugated beads was evaluated by quantification of A488-DRα1 eluted from the binding reaction as analyzed by Tris-Tricine peptide gel electrophoresis and visualized by fluorescent scanning at 488 nm with a Molecular Imager × (Biorad).
Microscopy imaging of DRα1 binding
Two million CD11b+ cells were negatively isolated from DR*1501/GFP-Tg mice by Mouse Monocyte Enrichment Kit (StemCell Technologies) and treated with 10 µg/ml DRα1 tagged with Alexa-546.The images were acquired on a high resolution wide-field Core DV system (Applied Precision™) utilizing an Olympus IX71 inverted microscope with a proprietary XYZ stage enclosed in a controlled environment chamber, transmitted-light differential interference contrast (DIC), and a solid state module for fluorescence. A Nikon Coolsnap ES2 HQ was used to acquire images as optical axis with a 60× (numerical aperture, 1.42) Plan-Apo N objective in 2 colors, FITC and TRITC. The pixel size was 0.10704 microns. The images were deconvolved with the appropriate OTF (optical transfer function) using an iterative algorithm of 10 iterations. Histograms were optimized for the most positive image and applied to all the other images for consistency before saving the images as 24 bit merged TIFF.
Induction of EAE in DR2-Tg and MBP-TCR/DR2-Tg mice
Mice were screened by flow cytometry for the expression of the HLA and the TCR transgenes (10). HLA-DR2-positive male and female mice between 8 and 12 weeks of age were immunized s.c. at four sites on the flanks with 0.2ml of an emulsion of 200 g immunogenic peptide and complete Freund’s adjuvant containing 400 g of heat-killed Mycobacterium tuberculosis H37RA (10)(Difco, Detroit, MI). In addition, mice were given Pertussis toxin (Ptx) from List Biological Laboratories (Campbell, CA) on days 0 and 2 post-immunization (75 ng and 200 ng per mouse respectively). Immunized mice were assessed daily for clinical signs of EAE on a 6 point scale of combined hind limb and forelimb paralysis scores. For hind limb scores: 0 = normal; 0.5 = limp tail or mild hind limb weakness (i.e., a mouse cannot resist inversion after a 90° turn of the base of the tail); 1 = limp tail and mild hind limb weakness; 2 = limp tail and moderate hind limb weakness (i.e., an inability of the mouse to rapidly right itself after inversion); 3 = limp tail and moderately severe hind limb weakness (i.e., inability of the mouse to right itself after inversion and clear tilting of hind quarters to either side while walking); 4 = limp tail and severe hind limb weakness (hind feet can move but drag more frequently than face forward); 5 = limp tail and paraplegia (no movement of hind limbs). Front limb paralysis scores are either 0.5 for clear restriction in normal movement or 1 for complete forelimb paralysis. The combined score is the sum of the hind limb score and the forelimb score. Rarely, there is mortality of HLA-DR2 mice with severe EAE and in these cases, mice are scored as a 6 for the remainder of the experiment. Mean EAE scores and standard deviations for mice grouped according to initiation of RTL or vehicle treatment were calculated for each day and summed for the entire experiment (Cumulative Disease Index, CDI, represents total disease load). Daily mean scores were analyzed by a two-tailed Mann Whitney U test for nonparametric comparisons between Vehicle and RTL treatment groups. Mean CDIs were analyzed by a one way ANOVA with Tukey post-test, and a nonparametric one way Kruskal-Wallis ANOVA with Dunn’s multiple comparisons post-test to confirm significance between all groups.
DRα1 or DRα1-MOG-35–55 treatment of EAE in DR2-Tg mice
DRα1constructs were injected s.c. daily for 5 days at indicated doses to treat EAE induced in transgenic mice and clinical signs were scored as described above.
Flow cytometry
Four-color (fluoresceine isothiocyanate (FITC), phycoerythrin, (PE), propidium iodide (PI), allophycocyanin, (APC) fluorescence flow cytometry analyses were performed to determine the phenotypes of cells following standard antibody staining procedures. At indicated time points, cardiac blood was collected in EDTA followed by perfusion of the mice with 1× PBS. For splenocytes, single cell suspensions of spleens from vehicle and DRα1 treatment groups were prepared by homogenizing the tissue through a fine mesh screen. Blood cells were pelleted after lysis of red cells followed by washing twice with RPMI. Mononuclear cells from the spinal cord were isolated by Percoll gradient centrifugation as described (11). Cells from spleen, blood and spinal cord were resuspended in staining medium (5% BSA, 1× PBS and 0.02% sodium azide) for FACS staining. All antibodies were purchased from BD PharMingen (San Diego, CA), eBioscience (San Diego, CA) or (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) unless otherwise indicated. Cells were stained with a combination of fluorescent labeled antibodies to CD11b, CD74 and CD45. For assessing DRα1 binding, one million cells were incubated in RPMI with 5 µg DRα1-Alexa-488 for one hour at 37 C. All incubations were followed directly by a 30 minute incubation at 4 C with anti-CD11b APC (BD PharMingen., San Diego, CA), After staining, cells were washed with staining medium and analyzed immediately with a FACS Calibur using FCS express (Los Angeles, CA) software. Data represent 10,000 live gated monocytes. Absolute numbers of cells were calculated from live-gated spinal cord cells.
Binding of recombinant human MIF (rhMIF) to immunopurified CD74
rhMIF was prepared from an E. coli expression system and purified free of endotoxin by methods described previously (12). rhMIF was labeled with AlexaFluor488 (InVitrogen) following the manufacturer’s instructions. CD74 complexes were immune-adsorbed from a CHAPS-solubilized lysate from splenocytes from DR*1501-Tg mice as described elsewhere. Briefly, after lysis, splenocytes were centrifuged at 14,000 rpm to remove debris and the supernatant was mixed with Beads/Protein L-adsorbed anti-CD74 overnight at 4°C. Immune complexes were washed extensively and the CD74 complexes were left adsorbed to the beads. To assess the effect of the DRα1 on rhMIF/CD74 binding, 10 pmol of rhMIF-A488 or 10 pmol of rhMIF-A488 plus 0.3 nmol or 1 nmol of DRα1 or DR2β1 were bound to ln-1 immunoadsorbed CD74 for 16h at 4°C in 0.01% CHAPS/TEN (50mM Tris, 2mM EDTA, 150 mM NaCl, pH 7.4) buffer. Proteins were eluted by incubating the beads with 2% SDS-electrophoresis sample buffer at 98°C for 7 to 8 min and the material was separated by electrophoresis in a 10–20% SDS-PAGE. Before blotting to the PVDF membrane, the gel was scanned for AlexaFluor488 to locate and quantify bound rhMIF. The PVDF membrane was blocked with 3% BSA/PBS containing 0.05% Tween 20 and probed with ln-1 mAb conjugated to FITC for 4 to 5 h at 4°C. The membrane was scanned for FITC and CD74 bands of expected MW were localized. DRα1 bands were localized in the membrane after Coomassie blue staining of the gels.
Electrophoresis and western blot
Samples were analyzed by reducing and non-reducing Laemli SDS-PAG electrophoresis and when necessary, blotted onto PVDF at 400mA for 45 minutes. Membranes then were blocked with 3% BSA in PBS and 0.05% Tween 20 either for 3 h or overnight before probing with the appropriate antibody.
Circular dichroism (CD) spectrometry
Before CD analysis MOG-35–55, DRα1, DRα1-MOG-35–55, DR2β1 and the MOG-35–55 peptide were dialyzed in 20 mM Tris buffer pH 8.5 and adjusted to a concentration of 1 mg/ml. Proteins were greater than 95% pure as assessed by SDS PAGE under non-reducing conditions. Far UV (180–260nm) spectra of the samples were run on an AVIV Model 215 circular dichroism spectropolarimeter at 0.5 or 1nm nm intervals using an 0.1 cm light path with a 3 seconds average time in triplicate at 25°C. For deconvolution, the blank (20mM Tris, pH 8.5) average readings were subtracted from the samples average readings and plotted as molar ellipticity
Histology
Mice were deeply anesthetized with isoflurane, heparinized and perfused with 100 ml of 4% paraformaldehyde in 0.1 M sodium phosphate buffer (pH 7.4), and fixed at 4°C for 24 hr. The spinal cords were dissected from the spinal columns and sectioned 1–2 mm in length from cervical, thoracic and lumbar cords. Tissues were placed in 0.1 M sodium phosphate buffer (PH 7.4), immersed fixed with 5% glutaraldehyde in 0.1 M sodium phosphate buffer (pH 7.4) for three days at 4° C, post-fixed with 1% osmium tetroxide (in 0.1 M phosphate buffer) for 3.5 hr, dehydrated in ethanol, and embedded in plastic. Semi-thin sections (0.5 um) were stained with toluidine blue and photographed using a light microscope.
CD4+ and CD11b+ cell co-cultures
Spleens were collected from MBP-TCR/DR2-Tg mice 9 days after immunization (as described before) (13) with MBP-85–99/CFA/Ptx. CD4+ and CD11b+ cells were negatively isolated (StemCell Technologies). Cell purity was >85%. Carboxy fluorescein succinimidyl ester (CFSE) labeled MBP-85–99-specific CD4+ T cells were co-cultured with CD11b+ cells at a 1:2 ratio (for CD11b+) in 96 well round-bottom plates for 3 days at 37°C in the presence of MBP-85–99 and/or DRα1 or DRα1-MOG-35–55. Following incubation, cells were harvested, washed and stained with fluorescent-labeled anti-CD3 to study T-cell proliferation concomitant with CFSE dilution.
Statistical analysis
Statistical analysis was performed using the Mann Whitney U- test and Student’s t-test, unless mentioned otherwise. Values of p<0.05 were considered significant.
Results
DRα1 binds to CD74 and H2-M
We recently have demonstrated that the DRα1 but not the DR2β1 domain binds to immunopurified CD74 (9, 14). Experimental and crystallographic studies have shown that MHC class II interacts with invariant chain (CD74) and H2-M (HLA-DM in humans) in different subcellular compartments and they have shown that the contact region is mainly located in the MHC α1 domain (15–17). Thus, the DRα1 domain was tested for binding activity to these membrane-anchored proteins using in vitro assays with immunoprecipitated CD74 and H2-M from DR*1501-Tg mouse splenocytes. As shown in Figure 1a, the recombinant DRα1 domain bound to both CD74 and H2-M. Digestion of DRα1 with trypsin yielded fragments that could still bind to H2-M but not to CD74, suggesting different structural requirements for binding of the DRα1 fragments to each of these molecules. These data suggest that the DRα1 constructs mimic the binding properties of the full length HLA-DR molecule and provide a binding interface for both CD74 and HLA-DM.
Figure 1. Binding of DRα1 to immunopurified CD74 and H2-M molecules andto CD11b+ monocytes.
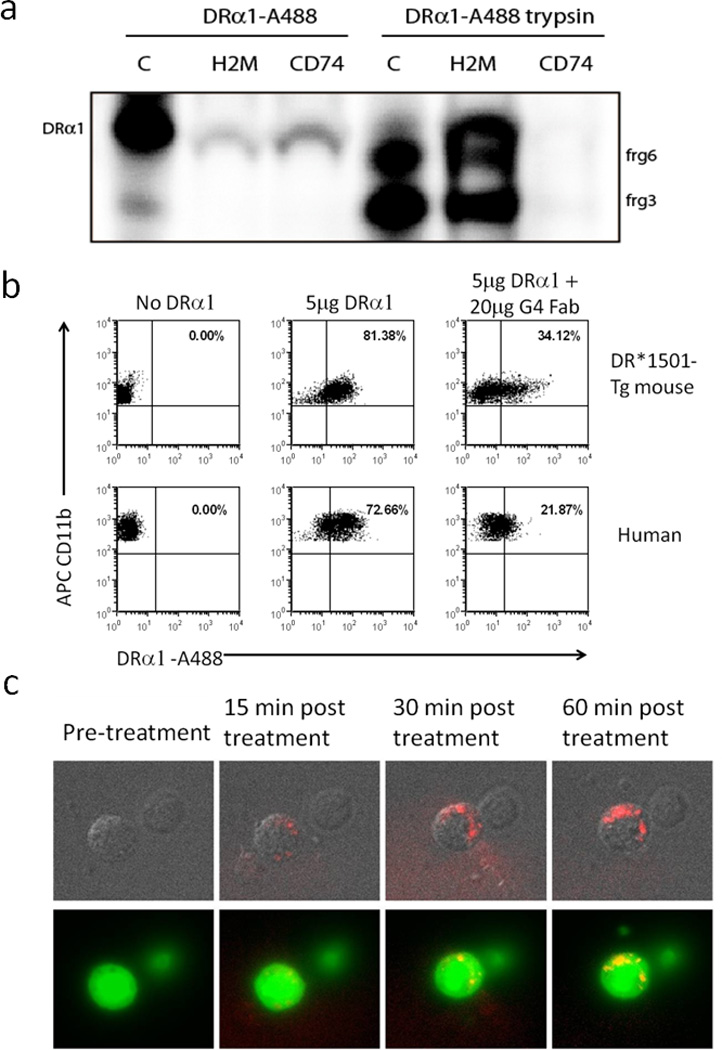
(a) DRα1 domain binds to H2-M (mouse homolog of human HLA-DM) and CD74 but trypsin digested DRα1 fragments bind solely to H2-M. DRα1 domain was labeled with AlexaFluor488 for 2h at RT and digested with trypsin for 2h at room temperature in 20mM Tris pH 8.5. Undigested and digested labeled DRα1were then bound to immunoprecipitated H2M and CD74 for 16–18h at 4°C in 1%CHAPS in TEN buffer and the material bound to immunoprecipitated complexes was visualized by electrophoresis in a 10–20% Tris-Tricine PAGE with 2% SDS. Intact DRα1 binds with higher efficiency to CD74 compared to H2m, but peptides generated by trypsin-digestion of DRα1 bind exclusively to H2-M and not to CD74. Lane C contains labeled undigested and digested proteins that were run as a control. (b) Human PBMC from HC and blood cells from DR*1501-Tg mice were incubated with 5 µg DRα1–Alexa488 for 1 h at 37°C. To neutralize DRα1 binding, 5 µg DRα1–Alexa488 was incubated at a 1:1 molar ratio (20 µg) of FabG4 for 2 h at room temperature prior to incubation with cells. CD11b+ monocytes were then analyzed for DRα1 binding by FACS. (c) Isolated GFP+CD11b+ cells from DR*1501/GFP-Tg mice were treated with 10 µg DRα1–Alexa546 for 60 min and evaluated by fluorescence microscopy.
DRα1 construct binds mouse and human monocytes
To further study the interaction of DRα1 with cell surface CD74 and to determine if DRα1 could bind to mouse and human cells, blood cells from DR*1501-Tg mouse or PBMC from an HLA-DR2+ HC donor were incubated for 1 h with DRα1-A488 with or without an Fab construct that is specific for binding the DRα1 construct (FabG4), as shown in Supplementary Figure 1. Both mouse and human CD11b+ monocytes bound the labeled DRα1 construct (81% and 73% positive, respectively) and this binding was strongly but not completely inhibited (reduced to 34% and 22% positive, respectively) when DRα1 was incubated with FabG4 at a 1:1 molar ratio for 2 h (Fig. 1b) prior to cell binding. In addition, as demonstrated in Supplementary Figure 2, CD3+ T-cells from DR*1501-Tg mice expressed very low levels of cell surface CD74 (about 4.5%) and had low binding levels of DRα1-A488 (about 6%) as compared with CD11b+ cells (97% expression of CD74 and 80% binding of DRα1-A488), demonstrating an association between CD74 expression and degree of DRα1-A488 cell binding.
Direct binding of labeled DRα1 construct to GFP+CD11b+ monocytes, isolated from DR*1501/GFP-Tg mice and visualized by fluorescence microscopy (Fig. 1c), demonstrated cell surface as well as likely-internalized complexes of DRα1, strongly suggesting binding to DRα1 receptors which occurs within minutes after in-vitro treatment.
DRα1 modulates expression of CD74 and directly inhibits binding of MIF and its downstream effects
To evaluate the relationship between binding of DRα1 and expression of CD74 on the cell surface, human PBMC were incubated with 10 µg DRα1, trypsin digested DRα1, DR2β1 or DRα1 incubated with FabG4 at a 1:1 molar ratio for 1 h at 37°C and then evaluated by FACS for cell surface expression of CD74. Results from five HC subjects demonstrated decreased cell surface expression of CD74 when cells were treated with an intact DRα1 construct. Tryptic digestion destroyed the ability of DRα1 to down-regulate cell surface CD74 expression whereas DR2β1 had no effect on CD74 cell surface expression. DRα1-induced down-regulation of CD74 cell surface expression was inhibited by FabG4, suggesting that FabG4 binding to DRα1 blocks the interaction between DRα1 and CD74. Moreover, incubation of PBMC with FabG4 alone significantly increased cell surface expression of CD74 (Fig. 2a), indicating no interaction between the FabG4 and CD74 or other cell surface molecules.
Figure 2. DRα1 modulates the cell surface expression of CD74 and directly inhibits MIF binding.
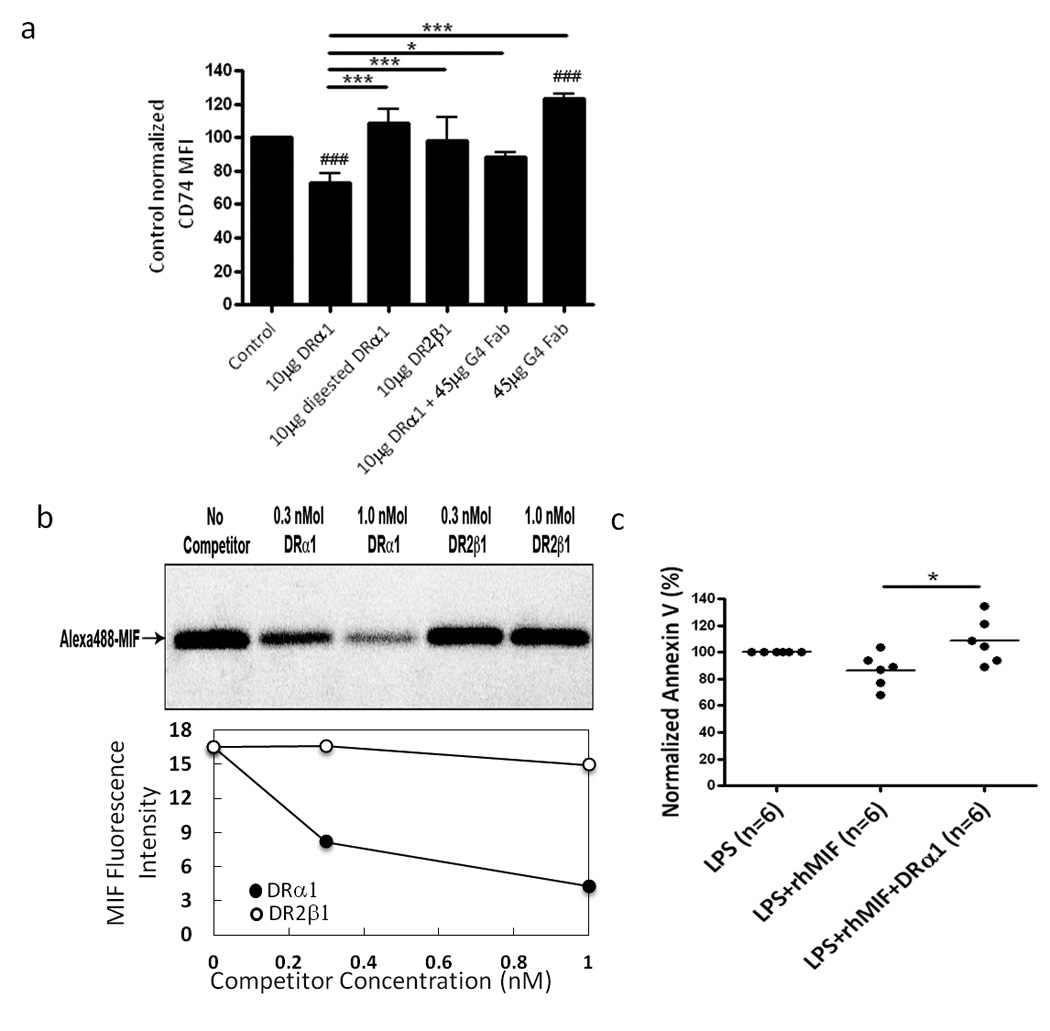
(a) PBMC from HC (n=5) were incubated with 10 µg DRα1, 10 µg Trypsin digested DRα1, 10 µg DR2β1, 10 µg DRα1 that was incubated at a 1:1 molar ratio (20 µg) of FabG4 for 2 h at room temperature prior to incubation with cells and 20 µg of FabG4 alone, for 1 h at 37°C. CD11b+ monocytes analyzed for CD74 expression (CD74 MFI were normalized to the untreated sample (100% of each subject). ###p<0.001 DRα1 or FabG4 vs. Control. *p<0.05, ***p<0.001 One-way Kruskal-Wallis ANOVA with Dunn’s multiple comparisons post test. (b) Unlabeled DRα1 (0.3 nmol or 1 nmol), unlabeled DR2β1 (0.3 nmol or 1 nmol), A488-hMIF (10 pmol) or a mixture of 10 pmol of A488-hMIF and unlabeled constructs were bound for 6 h at 4°C to immune complexes containing mouse CD74 bound to Protein L-adsorbed ln-1 mAb in the presence of 0.01% CHAPS/TEN buffer, pH 7.4. Complexes were then washed extensively with the same buffer and once with TEN buffer only before proteins were eluted with 2% SDS/electrophoresis sample buffer at 95°C for 7 min. Eluates were separated in 10–20% SDS-PAGE and the gel was scanned for the chromophore Alexa488 to visualize rhMIF-A488. The binding of rhMIF-A488 to mCD74 was quantified by relative fluorescence intensive. (c) Human PBMC from 6 HC subjects were stimulated with 10ng/ml LPS with or without 100ng/ml rhMIF and treated with 10 µg/ml DRα1. The cells were incubated at 37°C for 24 h and analyzed for Annexin V staining on CD11b+ monocytes. Results were normalized to the LPS alone treatment of every subject separately. *p<0.05, One-way Kruskal-Wallis ANOVA with Dunn’s multiple comparisons post test.
Binding and down-regulation of CD74 surface expression by DRα1 might have an important regulatory effect on MIF-mediated monocyte activation. In order to determine if DRα1 directly inhibits MIF binding to CD74, we assessed the influence of DRα1 on MIF binding to immunopurified CD74. The immunoprecipitated CD74 complex was incubated for 16 h at 4°C with A488-rhMIF with or without DRα1 or DR2β1. As shown in Fig. 2b, unlabeled DRα1 reduced rhMIF binding to CD74 in a dose dependent manner whereas there was negligible inhibition of MIF binding to immunoprecipitated CD74 by 1 nM DR2β1. The PVDF membrane also was probed with anti-CD74 mAb (ln-1) to confirm the presence of CD74 (data not shown).
We sought to determine if this inhibition of binding also suppressed MIF effects on monocytes, such as inhibition of apoptosis after activation (18). As shown in Figure 2c, human PBMC that were stimulated with LPS and rhMIF and DRα1 for 24 h had significantly higher expression levels of Annexin V on CD11b+ monocytes compared with stimulated cells that were not treated with DRα1, indicating that the DRα1 is blocking the MIF anti-apoptosis effect. The frequency of 7-ADD positive CD11b+ monocytes corresponded with the expression levels of Annexin V staining (Supplementary Figure 3). These results strongly suggest that HLA-DRα1 has a bi-functional regulatory role in reducing both the accessibility and availability of CD74 for MIF binding, thus reversing its consequent downstream signaling, including resistance to apoptosis as assessed by expression of both Annexin V and 7-ADD.
DRα1 treats clinical EAE by inhibiting monocyte infiltration
We have previously shown enhanced CD74 cell surface expression on monocytes in mice undergoing EAE (both in the periphery and the CNS) and in subjects with MS, demonstrating that CD74 expression levels are correlated with induction of inflammation (6, 9, 14). To evaluate the ability of DRα1 to treat EAE, which was recently shown to be influenced by MIF, DR*1501-Tg mice were immunized with mMOG-35–55/CFA/Ptx. Treatment with different concentrations of DRα1 (100 µg, 300 µg, 500 µg or 1 mg daily × 5) after disease onset at a clinical score of 2 significantly reduced clinical EAE scores in a dose-dependent manner, although treatment with 100 µg appeared to have only a transient effect that lasted for 24 h after the last treatment. In contrast, treatment with DR2β1 did not reverse clinical EAE signs (Fig. 3a). In addition, treatment with Trypsin digested DRα1 did not have a therapeutic effect on the disease (Fig. 3b).
Figure 3. DRα1 treats clinical EAE.
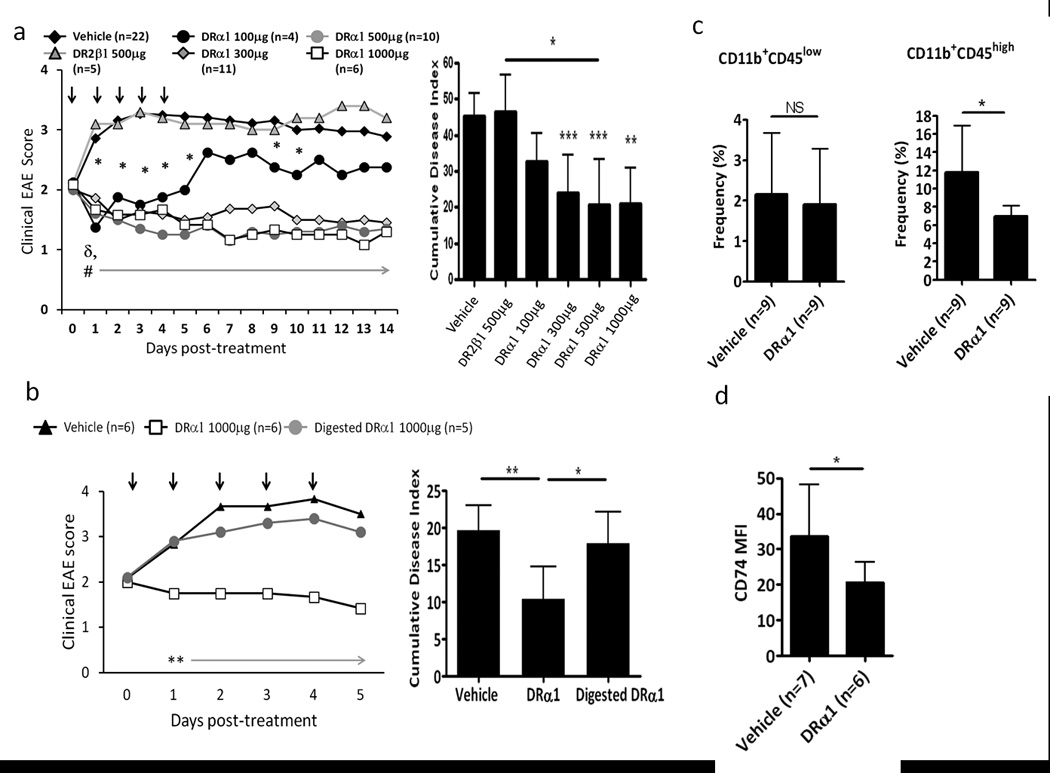
(a) DR*1501-Tg mice with mMOG-35–55 induced EAE were treated after disease onset at a clinical score of 2 with vehicle, DR2β1 (100 µg) or DRα1 (100 µg, 300 µg, 500 µg and 1mg daily × 5, black arrows). Mean clinical EAE daily disease scores (left) and cumulative disease index (right) are shown. (b) DR*1501-Tg mice with mMOG-35–55 induced EAE were treated after disease onset at a clinical score of 2 with vehicle, DRα1 or Trypsin digested DRα1 (1mg daily × 5). Mean clinical EAE daily disease scores (left) and cumulative disease index (right) are shown. *p<0.05 (DR-α1 100 µg vs. Vehicle); #p<0.0003 &δp<0.04 (DR-α1 300–1000 µg vs. Vehicle and DR2-β1, respectively)*p<0.05, **p<0.01, ***p<0.001.Daily mean scores were analyzed by Mann Whitney U and mean CDI by one way ANOVA with Tukey post-test, and nonparametric one way Kruskal Wallis ANOVA with Dunn’s multiple comparisons post-test. (c) Frequency of CD11b+CD45+ cells in spinal cord was analyzed 24 h after last Vehicle or DRα1 treated DR*1501-Tg mice. (d) Frequency of cell surface CD74 on CD11b+CD45+ cells in spinal cord was analyzed 24 h after last Vehicle or DRα1 treated DR*1501-Tg mice. *p<0.05, Student’s t-test (Figure 3c –d).
Analysis of spinal cords from DRα1 (1 mg) and vehicletreated mice 24 h after the last treatment revealed a decreased frequency of CD11b+CD45+cells (infiltrating monocytes and activated microglia) (Fig. 3c). In addition, the expression of cell surface CD74 on CD11b+CD45+ spinal cord cells was significantly lower in DRα1 treated mice compared with vehicle treated mice (p<0.05) (Fig. 3d). Taken together, these results demonstrate the DRα1 can not only down-regulate CD74 expression, but also inhibit cell migration to the CNS which contributes to the reversal of clinical signs of EAE.
DRα1-MOG-35–55 acquires secondary structure and has increased potency for treating EAE
In order to increase the therapeutic efficacy of the DRα1 construct, we produced DRα1 constructs bearing a covalently linked N-terminal mMOG-35–55 (DRα1-MOG-35–55) peptide antigen. A comparison of DRα1-MOG-35–55 to DRα1 in vitro revealed that a concentration of 1 µg DRα1-MOG-35–55 was more effective than DRα1 in downregulating CD74 cell surface expression on human CD11b+ monocytes, with the constructs having equivalent activity at higher concentrations (5 µg and 10 µg, Fig. 4a).
Figure 4. DRα1–MOG-35–55 has increased potency for treating EAE.
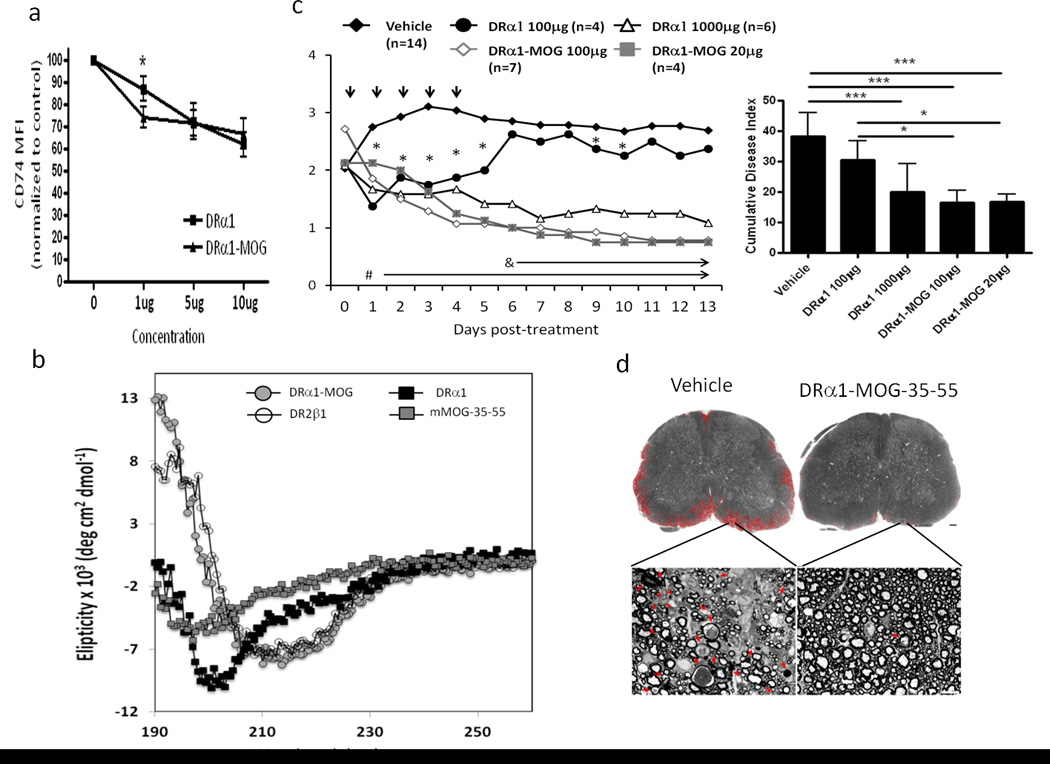
(a) PBMC from HC (n=3) were incubated with DRα1 or DRα1–MOG-35–55 at different concentrations (1 µg, 5 µg, 10 µg) and CD11b+ monocytes analyzed for CD74 expression (CD74 MFI were normalized to the untreated sample- 100 % of each subject). *p<0.05, Student’s t-test. (b) Far UV spectra of DRα1, DRα1–MOG-35–55, DR2β1 constructs and mouse MOG-35–55 peptide in 20mM Tris, pH 8.5 at a concentration of 1mg/ml. Spectra were measured on an AVIV spectropolarimeter at 0.5 nm bandwidth using a 0.1 cm light path. Blank CD spectra (20mM Tris, pH 8.5) were determined and were subtracted from the sample spectra. (c) DR*1501 mice with EAE were treated after disease onset at a clinical score of 2 with Vehicle (5 daily treatments), DRα1 (100 µg × 5), DRα1 (1000 µg × 5), DRα1-MOG-35–55 (100 µg × 5) and DRα1-MOG-35–55 (20 µg × 5). left: Daily mean clinical EAE disease scores. *p<0.05, DRα1(100 µg) vs. Vehicle); #p<0.001 DRα1 (1000 µg), DRα1-MOG-35–55(100 µg) and DRα1-MOG-35–55 (20 µg) vs. Vehicle. δp<0.03, DRα1-MOG-35–55(100 µg) and DRα1-MOG-35–55 (20 µg) vs. DRα1 (100 µg). right: Statistical comparisons of cumulative disease indices (CDI): *p<0.05; ***p<0.0001. Daily mean scores were analyzed by Mann Whitney U and mean CDI by one way ANOVA with Tukey post-test. (d) Spinal cord sections from EAE DR*1501-Tg mice, as visualized after toluidine blue staining, either from Vehicle or DRα1-MOG-35–55 treated mice. The areas of tissue damage in white matter are circled with red (The scale bar is 100 µm). High power view of small areas of lumbar spinal cord (white rectangles) show axons that are undergoing demyelination (red arrow) and cellular infiltration (red arrowhead) (The scale bar is 10 µm).
We reasoned that the difference in downregulation of CD74 could be the result of conformational differences between the two proteins. As a first approach to test this possibility, we evaluated secondary and tertiary structures of these constructs using circular dichroism (CD). As shown in Fig. 4b, spectrometric characterization in the near UV lightof the DRα1 construct and the MOG-35–55 peptide using circular dichroism (CD) revealed a quasi-unfolded, extended conformation with no content of alpha helix and very low beta sheet secondary structure, given by the absorbances at 193, 208 and 220nm for alpha helix, and 198 and 218nm for beta sheets. In contrast, the DRα1-MOG-35–55 construct showed a high content of alpha helix and beta sheet structures similar to that expected for the MHC class II α1 domain from X-ray crystallographic studies (19–24).
Based on these results, we sought to determine whether DRα1-MOG-35–55 had more potency in treating EAE than DRα1. In initial experiments, DR*1501-Tg mice immunized with mMOG-35–55/CFA/Ptx were treated with different concentrations of DRα1-MOG-35–55 (20 µg, 100 µg, 500 µg daily × 5) after disease onset at a clinical score of 2. All doses of DRα1-MOG-35–55, including 20 µg, significantly reduced clinical EAE scores (p<0.001) (Supplementary Fig. 4). Evaluation of DRα1 and DRα1-MOG-35–55 treatments demonstrated comparable reduction of EAE scores using 1mg DRα1 vs. 20 µg DRα1-MOG-35–55 (a 50X difference) (Fig. 4c), indicating that the activity of DRα1 was indeed strongly potentiated by the amino-terminal MOG-35–55 peptide extension. The DRα1-MOG-35–55 peptide treatment also strongly decreased histological damage of EAE in spinal cord sections (Fig. 4d).
DRα1-MOG-35–55 transiently attenuates MBP-85–99 induced EAE in MBP-TCR/DR2-Tg mice
We previously reported that partial MHC class II constructs containing the MOG-35–55 peptide differed from other constructs in their remarkable ability to treat EAE induced by non-cognate encephalitogenic peptides(9, 14). In order to assess if the DRα1 or DRα1-MOG-35–55 constructs might induce this bystander suppression effect, negatively isolated CD11b+ cells were co-incubated with CFSE labeled CD4+ T-cells obtained from MBP-85–99 peptide-immunized MBP-TCR/DR2-Tg mice. Isolated CD11b+ cells were co-cultured with CFSE labeled transgenic T cells in the presence of free MBP-85–99 peptide, DRα1 or DRα1-MOG-35–55 in 96 well round-bottom plates for 3 days. As expected, CD11b+ cells induced proliferation of CFSE labeled T cells (22% positive) in the presence of added MBP-85–99 peptide (Fig. 5A). However, incubation of CD11b+ APC + CD4+ T-cells with DRα1-MOG-35–55 did not induce a significant proliferation response greater than medium alone. Of critical importance, a significantly reduced proliferation response of activated T-cells was observed when free MBP-85–99 peptide was added to the DRα1 or DRα1-MOG-35–55 armed CD11b+ cells (about 7% positive, Fig. 5A), clearly demonstrating a substantial degree of bystander suppression.
Figure 5. DRα1-MOG-35–55 inhibits MBP-85–99 peptide induced proliferation of activated T-cells and transiently attenuates MBP-85–99 induced EAE in MBP-TCR/DR2-Tg mice.
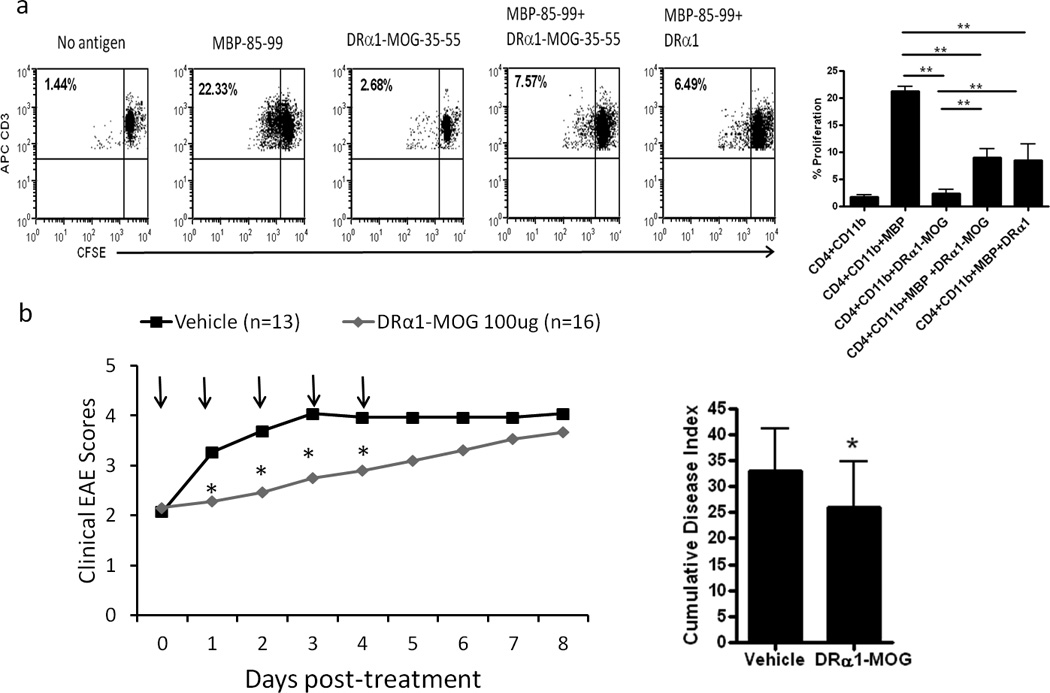
(a) CD4+ and CD11b+ cells were isolated from the spleens of MBP-85–99/CFA-immunized MBP-TCR/DR2 mice. Sorted CD11b+ cells were cultured with CSFE-labeled primed T-cells from the same mice at a 1:2 ratio (CD4+:CD11b+) in the presence or absence of free MBP-85–99 peptide, DRα1-MOG-35–55 or a combination of MBP-85–99 peptide and DRα1-MOG-35–55 or DRα1. After 3 days of culture, cells were washed and evaluated by FACS Calibur for CD3 expression and CFSE dilution. (b) MBP-TCR/DR2-Tg mice with MBP-85–99 induced EAE were treated after disease onset at a clinical score of 2 withVehicle or DRα1-MOG-35–55 (500 µg daily × 5, black arrows). Mean clinical EAE daily disease scores *p<0.05, (left) and cumulative disease index (right) are shown.
In order to confirm this bystander effect in vivo, EAE was induced with MBP-85–99/CFA/Ptx in MBP-TCR/DR2-Tg mice. Treatment with DRα1-MOG-35–55 (100 µg, daily × 5) after disease onset at a clinical score of 2 significantly reduced clinical EAE scores compared with Vehicle treated mice on days 1–4 post treatment (p<0.05) (Fig. 5B). Thus, DRα1-MOG-35–55 treatment transiently attenuated the MBP induced disease in the MBP-TCR/DR2-Tg mice. Although the treatment effect did not appear to be as potent as in the DR*1501-Tg mice with MOG-35–55 induced EAE, these data demonstrate that DRα1-MOG-35–55 has a significant bystander effect on MBP-TCR transgenic T cells and on MBP-95–99 induced EAE.
Discussion
The major histocompatibility complex (MHC) encodes highly polymorphic proteins that bind peptides for presentation to CD4+ and CD8+ T-cell receptors(25, 26). However, several of the MHC proteins are not polymorphic, including the class II invariant chain (CD74, Ii) and the DRα chain. We demonstrate a here-to-fore unrecognized immunoregulatory interaction between these two highly conserved MHC proteins in which DRα1 constructs, upon binding to CD74, inhibit MIF binding and signaling and treat ongoing clinical and histological signs of experimental autoimmune encephalomyelitis (EAE). We further report that the therapeutic potency of the DRα1 construct could be enhanced by addition of a peptide extension at the N-terminus.
Although the function of the CD74 invariant chain as an MHC class II chaperone has been studied extensively (15, 27), the role of the cell surface form of CD74 in the immune response is not fully understood. It was shown previously that CD74 forms a functional signal transduction complex with CD44, CXCR2, and CXCR4 to transduce MIF signaling(3–5, 28). Recently, we reported that CD74 surface expression on CD11b+ monocytes is up-regulated in mice with EAE and subjects with multiple sclerosis (MS)(6). However, very little is known about the interaction of CD74 and MHC class II on the surface of APC during the steady state or inflammation. Consistent with the prior literature that studied intracellular interactions between CD74, HLA-DM and DRα during peptide loading of nascent MHC class II molecules(15–17, 29–31), we recently demonstrated that DRα1 constructs could bind to immunoprecipitated CD74 (14). Hence, we utilized this construct to study cell surface interactions of DRα1with CD74 and its possible effects on MIF binding and signaling. Here we report that the DRα1 construct can bind to both immunoprecipitated CD74 and H2-M,the mouse homologue of human HLA-DM. The fact that the tryspin digested DRα1 could bind only to H2-M but not to CD74 suggests that the binding of DRα1 to CD74 is either conformationally dependent or that the trypsin digestion destroyed the binding site to CD74. Future experiments will determine if overlapping peptides of the DRα1 domain can bind to CD74. Conformation-dependent binding of DRα1 to CD74 might indicate that binding sites on DRα1 for CD74 and H2-M are distinct. Since H2-M is not expressed on the cell surface and thus would not be available for binding, we focused on the interaction of CD74 with the DRα1 domain. The DRα1 construct not only could bind human and mouse CD11b+ monocytes, but also could down-regulate surface expression of CD74.Interestingly, although DRα1 bound better to human vs. mouse monocytes, there was less efficient down-regulation of CD74 levels on human monocytes, possibly due to a faster recycling of the cell surface CD74 (32). This binding could be inhibited with a specific Fab to DRα1 that apparently could block the binding site on DRα1 for CD74. We further show for the first time that this binding of DRα1 to immunoprecipitated CD74 could inhibit binding of MIF to CD74 and its downstream signaling effects.
It was previously demonstrated that during the assembly of MHC class II in the endoplasmic reticulum, the homotrimeric CD74 invariant chain initially binds single MHC class II α chains and that this complex serves as a scaffold for subsequent binding of the β chains (30). We here demonstrate that the immunoprecipitated cell surface homotrimeric CD74 complex reiterates the initial binding to the DRα1 construct but does not bind to the DR2β1 construct (9, 14). We would thus speculate that the binding of the DRα1 construct to CD74 and subsequent blocking of MIF binding and signaling might represent a natural immunoregulatory role for the DRα chain in terminating MIF-dependent inflammation. This novel concept is supported by the binding of the Staphylococcal toxic shock syndrome toxin 1 (TSST-1) to the DRα chain, wherein the TSST-1 binding site on HLA-DRα1 partially overlaps with the DRα1/CD74 binding site(33). These data, coupled with the report by Calandra et al. showing that MIF is a mediator of the activation of immune cells by TSST-1 (34), suggests the possibility that TSST-1 is blocking the binding of HLA-DRα to CD74, which in turn makes more CD74 available to bind MIF, thus inducing the toxic shock.
We further demonstrate for the first time that the DRα1 construct could be used as a therapeutic reagent in a CNS disease model that was shown to be mediated by MIF (35–37). Treatment with DRα1 reduced the frequency of activated microglia and infiltrating macrophages in the spinal cord and reduced the CD74 level of expression on these cells compared to those from Vehicle-treated mice. We also showed a correlation between the ability of different constructs to bind to and down-regulate CD74 expression on CD11b+ cells in vitro and their ability to treat EAE. Only the DRα1 construct, but not its peptide digest or the DR2β1 construct could bind to and down-regulate CD74 expression and treat EAE. Our data demonstrate that DRα1 constructs have a bi-functional role, down regulating CD74 surface expression and inhibiting MIF binding to CD74. These two functions might have distinct effects on the cell. DRα1 treatment reduces but does not completely down regulate CD74 surface expression (only about 30%), providing a detectable phenotypic change. However, this partially reduced availability of CD74 alone would not seem sufficient to account for observed inhibition of MIF effects. Alternatively, we would speculate that competitive DRα1 inhibition of MIF binding to remaining cell surface CD74 might be the more potent mechanism for therapeutic effects of DRα1 on EAE. Relevant to this issue, only a fraction (~5%) of surface CD74 on monocytes exists free of MHC class II (38), and it is conceivable that the remaining 95% of class II associated CD74 would not be amenable to MIF binding due to occupancy by natural DRα present in cell surface MHC class II. If true, only the remaining class II-unassociated CD74 would mediate MIF effects, thus reducing the scope of regulatory interactions needed for effective DRα1 treatment.
In order to enhance the potency of the DRα1 construct and potentially engage neuroantigen peptide-dependent effects, we covalently linked a peptide extension of MOG-35–55 to the N-terminus of DRα1. We have previously reported that partial MHC class II β1α1 constructs containing the MOG-35–55 peptide linked to the N-terminus of DR2β1α1 are more potent therapeutic reagents than other peptide extensions or “empty” constructs (9, 14). Our current report clearly demonstrates that a relative low dose of 1 µg of the DRα1-MOG-35–55 construct in vitro is more potent in down-regulating CD74 surface expression than the same concentration of the DRα1 construct.
The secondary structures of DRα1, DRα1-MOG-35–55, DR2β1 and free MOG-35–55 peptidewere explored by circular dichroism (CD) (Fig. 4b). Crystal structures determined for MHC class II proteins show that they are organized into two well defined regions, each adopting specific folded structures. The α1 and β1 domains, which contain alpha-helices sitting atop a beta-pleated sheet structure, contribute to the peptide antigen binding groove and are located distal to the membrane, whereas the α2 and β2 domain areIg-like structures positioned proximal to the cell membrane (19–24). We here present new data indicating that DRα1 in the absence of covalently-tethered peptide antigen lacks the well defined secondary structure characteristic of the α1 domain within MHC class II, while the DR2β1 domain retains much of the secondary structure of the β1-domain within the MHC class II molecule. When linked to the N-terminus of the DRα1 protein, the MOG peptide induced changes in secondary structure, including increased alpha helix and beta sheet content that are not present in the DRα1 or the MOG-35–55 peptide alone. To this point, other studies have shown that the MOG peptide displays an extended conformation in solution with a not-fully-formed alpha-helix at the N-terminus (39). To the best of our knowledge, ourCD spectroscopy study is the first demonstration of a peptide-triggered conformational change observed in an isolated MHC class II domain.
Taken in the context of an empty MHC class II molecule, these results might suggest that the α1 is present as an extended entity and that the β1 might exist as a partially folded structural domain. If this is actually the case, it might also explain why it has been difficult in practice to crystallize empty MHC class II molecules (since its success depends on properly folded polypeptides) unless they are provided with an antigenic peptide bound to the antigen-binding groove. There is ample evidence suggesting that upon peptide binding, class II molecules undergo dramatic conformational changes induced by the peptide (21, 40–44). Other studies have relied on molecular probes such as monoclonal antibodies and toxins that bind specifically to MHC class II in order to explore structural changes in the involved MHC class II molecules (45, 46). The region that undergoes the most striking changes in conformation is the α1 domain and to a lesser extent, the β1 domain. By a combination of molecular dynamics simulation and biochemical experimentation, Painter et. al. showed that when the peptide is removed, substantial changes in structure occur in the α1 domain between residues 15 and 90, but only local changes occur in the β1 domain, mostly within residues 50–70 at the center of the alpha-helix(42). The results described in these studies are confirmed by our CD spectroscopic data showing a loss of overall structure within the α1 domain and loss of alpha-helix conformation with a high content of anti-parallel beta-sheet region in the β1 domain.
Many cellular proteins are intrinsically disordered and undergo folding, complete or partial, upon binding to their physiological partners and a great majority of these disordered proteins play a relevant role in signaling cascades, acting as hubs of protein interaction networks enabling them to interact promiscuously with a wide variety of targets (47–49). How theMOG-35–55 peptide triggers this global conformational change in the DRα1 domain is the subject of our ongoing research.
Treatment of clinical EAE with the DRα1-MOG-35–55 construct also revealed at least a 50× increase in potency compared to the DRα1 construct (Fig. 4c,20 µg vs. 1000 µg, respectively). In a concurrent study we recently demonstrated that the DRα1-MOG-35–55 construct could treat experimental stroke. Four daily treatments with DRα1-MOG-35–55 reduced infarct size by 40% in the cortex, striatum and hemisphere, inhibited the migration of activated CD11b+CD45high cells from the periphery to the brain and reversed splenic atrophy(50). Hence, this novel construct could treat different CNS diseases that involve migration of immune cells from the periphery to both the brain (stroke) and spinal cord (EAE). In addition, we report here that the DRα1 and DRα1-MOG-35–55 constructs could inhibit MBP-85–99 peptide induced proliferation of activated T- cells when co-cultured with primed CD11b+ cells as well as transiently attenuate MBP-85–99 induced EAE for several days in MBP-TCR/DR2-Tg mice. We have shown recently that the RTL1000 construct containing the MOG-35–55 peptide could treat EAE in this same model, probably through a similar bystander tolerance mechanism(9). The lack of complete treatment of the DRα1-MOG-35–55 construct compared to the RTL constructs might be attributed to the difference in the disease severity between the two experiments (Average peak of disease for the Vehicle treated mice in the RTL experiment was 3.5±0.5 versus 4.4±0.6 in the current experiment).
In summary, we demonstrate that that the DRα1 domain could treat EAE by inhibiting MIF binding to CD74 on monocytes, thus blocking its pro-inflammatory effects. Because the DRα1 amino acid sequence is conserved in humans,the recombinant DRα1-MOG-35–55 construct potentially represents an immunotherapy that would not require HLA screening prior to use. Potency of the DRα1 domain could be destroyed by trypsin digestion, and could be enhanced by addition of a peptide extension (MOG-35–55 peptide) that we hypothesize induced alpha-helix and beta-strand structures not present in DRα1 alone. In addition, this novel therapeutic DRα1-MOG-35–55 construct could represent a new approach for regulating MIF and the immune response in CNS and other inflammatory conditions.
Supplementary Material
Acknowledgments
The authors wish to thank Melissa S. Barber for assistance with manuscript submission.
This work was supported by NIH grants NS47661 (to AAV), AR050498 (to RB), National Multiple Sclerosis Society grant RG3794-B-6 (to AAV), postdoctoral fellowship from the National Multiple Sclerosis Society (to GB) and the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development. The contents do not represent the views of the Department of Veterans Affairs or the United States Government.
Footnotes
No Conflicts of Interestto report.
References
- 1.Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, Delohery T, Chen Y, Mitchell RA, Bucala R. MIF signal transduction initiated by binding to CD74. J Exp Med. 2003;197:1467–1476. doi: 10.1084/jem.20030286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cresswell P. Invariant chain structure and MHC class II function. Cell. 1996;84:505–507. doi: 10.1016/s0092-8674(00)81025-9. [DOI] [PubMed] [Google Scholar]
- 3.Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, Dewor M, Georgiev I, Schober A, Leng L, Kooistra T, Fingerle-Rowson G, Ghezzi P, Kleemann R, McColl SR, Bucala R, Hickey MJ, Weber C. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. 2007;13:587–596. doi: 10.1038/nm1567. [DOI] [PubMed] [Google Scholar]
- 4.Schwartz V, Lue H, Kraemer S, Korbiel J, Krohn R, Ohl K, Bucala R, Weber C, Bernhagen J. A functional heteromeric MIF receptor formed by CD74 and CXCR4. FEBS Lett. 2009;583:2749–2757. doi: 10.1016/j.febslet.2009.07.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shi X, Leng L, Wang T, Wang W, Du X, Li J, McDonald C, Chen Z, Murphy JW, Lolis E, Noble P, Knudson W, Bucala R. CD44 is the signaling component of the macrophage migration inhibitory factor-CD74 receptor complex. Immunity. 2006;25:595–606. doi: 10.1016/j.immuni.2006.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benedek G, Meza-Romero R, Andrew S, Leng L, Burrows GG, Bourdette D, Offner H, Bucala R, Vandenbark AA. Partial MHC class II constructs inhibit MIF/CD74 binding and downstream effects. Eur J Immunol. 2013;43:1309–1321. doi: 10.1002/eji.201243162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Metodieva G, Nogueira-de-Souza NC, Greenwood C, Al-Janabi K, Leng L, Bucala R, Metodiev MV. CD74-dependent deregulation of the tumor suppressor scribble in human epithelial and breast cancer cells. Neoplasia. 2013;15:660–668. doi: 10.1593/neo.13464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stein R, Mattes MJ, Cardillo TM, Hansen HJ, Chang CH, Burton J, Govindan S, Goldenberg DM. CD74: a new candidate target for the immunotherapy of B-cell neoplasms. Clin Cancer Res. 2007;13:5556s–5563s. doi: 10.1158/1078-0432.CCR-07-1167. [DOI] [PubMed] [Google Scholar]
- 9.Vandenbark AA, Meza-Romero R, Benedek G, Andrew S, Huan J, Chou YK, Buenafe AC, Dahan R, Reiter Y, Mooney JL, Offner H, Burrows GG. A novel regulatory pathway for autoimmune disease: binding of partial MHC class II constructs to monocytes reduces CD74 expression and induces both specific and bystander T-cell tolerance. J Autoimmun. 2013;40:96–110. doi: 10.1016/j.jaut.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vandenbark AA, Rich C, Mooney J, Zamora A, Wang C, Huan J, Fugger L, Offner H, Jones R, Burrows GG. Recombinant TCR ligand induces tolerance to myelin oligodendrocyte glycoprotein 35–55 peptide and reverses clinical and histological signs of chronic experimental autoimmune encephalomyelitis in HLA-DR2 transgenic mice. J Immunol. 2003;171:127–133. doi: 10.4049/jimmunol.171.1.127. [DOI] [PubMed] [Google Scholar]
- 11.Bebo BF, Jr, Vandenbark AA, Offner H. Male SJL mice do not relapse after induction of EAE with PLP 139–151. J Neurosci Res. 1996;45:680–689. doi: 10.1002/(SICI)1097-4547(19960915)45:6<680::AID-JNR4>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 12.Bernhagen J, Mitchell RA, Calandra T, Voelter W, Cerami A, Bucala R. Purification, bioactivity, and secondary structure analysis of mouse and human macrophage migration inhibitory factor (MIF) Biochemistry. 1994;33:14144–14155. doi: 10.1021/bi00251a025. [DOI] [PubMed] [Google Scholar]
- 13.Sinha S, Miller L, Subramanian S, McCarty OJ, Proctor T, Meza-Romero R, Huan J, Burrows GG, Vandenbark AA, Offner H. Binding of recombinant T cell receptor ligands (RTL) to antigen presenting cells prevents upregulation of CD11b and inhibits T cell activation and transfer of experimental autoimmune encephalomyelitis. J Neuroimmunol. 2010;225:52–61. doi: 10.1016/j.jneuroim.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vandenbark AA, Meza-Romero R, Benedek G, Andrew S, Huan J, Chou YK, Buenafe AC, Dahan R, Reiter Y, Mooney JL, Offner H, Burrows GG. A novel regulatory pathway for autoimmune disease: Binding of partial MHC class II constructs to monocytes reduces CD74 expression and induces both specific and bystander T-cell tolerance. J Autoimmun. 2012 doi: 10.1016/j.jaut.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cresswell P. Assembly, transport, and function of MHC class II molecules. Annu Rev Immunol. 1994;12:259–293. doi: 10.1146/annurev.iy.12.040194.001355. [DOI] [PubMed] [Google Scholar]
- 16.Fling SP, Arp B, Pious D. HLA-DMA and -DMB genes are both required for MHC class II/peptide complex formation in antigen-presenting cells. Nature. 1994;368:554–558. doi: 10.1038/368554a0. [DOI] [PubMed] [Google Scholar]
- 17.Morris P, Shaman J, Attaya M, Amaya M, Goodman S, Bergman C, Monaco JJ, Mellins E. An essential role for HLA-DM in antigen presentation by class II major histocompatibility molecules. Nature. 1994;368:551–554. doi: 10.1038/368551a0. [DOI] [PubMed] [Google Scholar]
- 18.Mitchell RA, Liao H, Chesney J, Fingerle-Rowson G, Baugh J, David J, Bucala R. Macrophage migration inhibitory factor (MIF) sustains macrophage proinflammatory function by inhibiting p53: regulatory role in the innate immune response. Proc Natl Acad Sci U S A. 2002;99:345–350. doi: 10.1073/pnas.012511599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gunther S, Schlundt A, Sticht J, Roske Y, Heinemann U, Wiesmuller KH, Jung G, Falk K, Rotzschke O, Freund C. Bidirectional binding of invariant chain peptides to an MHC class II molecule. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:22219–22224. doi: 10.1073/pnas.1014708107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim CY, Quarsten H, Bergseng E, Khosla C, Sollid LM. Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:4175–4179. doi: 10.1073/pnas.0306885101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Murthy VL, Stern LJ. The class II MHC protein HLA-DR1 in complex with an endogenous peptide: implications for the structural basis of the specificity of peptide binding. Structure. 1997;5:1385–1396. doi: 10.1016/s0969-2126(97)00288-8. [DOI] [PubMed] [Google Scholar]
- 22.Painter CA, Negroni MP, Kellersberger KA, Zavala-Ruiz Z, Evans JE, Stern LJ. Conformational lability in the class II MHC 310 helix and adjacent extended strand dictate HLA-DM susceptibility and peptide exchange. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:19329–19334. doi: 10.1073/pnas.1108074108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Scott CA, Peterson PA, Teyton L, Wilson IA. Crystal structures of two I-Ad-peptide complexes reveal that high affinity can be achieved without large anchor residues. Immunity. 1998;8:319–329. doi: 10.1016/s1074-7613(00)80537-3. [DOI] [PubMed] [Google Scholar]
- 24.Stern LJ, Brown JH, Jardetzky TS, Gorga JC, Urban RG, Strominger JL, Wiley DC. Crystal structure of the human class II MHC protein HLA-DR1 complexed with an influenza virus peptide. Nature. 1994;368:215–221. doi: 10.1038/368215a0. [DOI] [PubMed] [Google Scholar]
- 25.Klein J, Sato A. The HLA system. Second of two parts. N Engl J Med. 2000;343:782–786. doi: 10.1056/NEJM200009143431106. [DOI] [PubMed] [Google Scholar]
- 26.Klein J, Sato A. The HLA system. First of two parts. N Engl J Med. 2000;343:702–709. doi: 10.1056/NEJM200009073431006. [DOI] [PubMed] [Google Scholar]
- 27.Sant AJ, Miller J. MHC class II antigen processing: biology of invariant chain. Curr Opin Immunol. 1994;6:57–63. doi: 10.1016/0952-7915(94)90034-5. [DOI] [PubMed] [Google Scholar]
- 28.Calandra T, Roger T. Macrophage migration inhibitory factor: a regulator of innate immunity. Nat Rev Immunol. 2003;3:791–800. doi: 10.1038/nri1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koch N, Zacharias M, Konig A, Temme S, Neumann J, Springer S. Stoichiometry of HLA class II-invariant chain oligomers. PLoS One. 2011;6:e17257. doi: 10.1371/journal.pone.0017257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neumann J, Koch N. Assembly of major histocompatibility complex class II subunits with invariant chain. FEBS Lett. 2005;579:6055–6059. doi: 10.1016/j.febslet.2005.09.070. [DOI] [PubMed] [Google Scholar]
- 31.Pos W, Sethi DK, Call MJ, Schulze MS, Anders AK, Pyrdol J, Wucherpfennig KW. Crystal structure of the HLA-DM-HLA-DR1 complex defines mechanisms for rapid peptide selection. Cell. 2012;151:1557–1568. doi: 10.1016/j.cell.2012.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moldenhauer G, Henne C, Karhausen J, Moller P. Surface-expressed invariant chain (CD74) is required for internalization of human leucocyte antigen-DR molecules to early endosomal compartments. Immunology. 1999;96:473–484. doi: 10.1046/j.1365-2567.1999.00676.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jasanoff A, Wagner G, Wiley DC. Structure of a trimeric domain of the MHC class II-associated chaperonin and targeting protein Ii. Embo J. 1998;17:6812–6818. doi: 10.1093/emboj/17.23.6812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Calandra T, Spiegel LA, Metz CN, Bucala R. Macrophage migration inhibitory factor is a critical mediator of the activation of immune cells by exotoxins of Gram-positive bacteria. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:11383–11388. doi: 10.1073/pnas.95.19.11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cox GM, Kithcart AP, Pitt D, Guan Z, Alexander J, Williams JL, Shawler T, Dagia NM, Popovich PG, Satoskar AR, Whitacre CC. Macrophage migration inhibitory factor potentiates autoimmune-mediated neuroinflammation. J Immunol. 2013;191:1043–1054. doi: 10.4049/jimmunol.1200485. [DOI] [PubMed] [Google Scholar]
- 36.Denkinger CM, Denkinger M, Kort JJ, Metz C, Forsthuber TG. In vivo blockade of macrophage migration inhibitory factor ameliorates acute experimental autoimmune encephalomyelitis by impairing the homing of encephalitogenic T cells to the central nervous system. J Immunol. 2003;170:1274–1282. doi: 10.4049/jimmunol.170.3.1274. [DOI] [PubMed] [Google Scholar]
- 37.Powell ND, Papenfuss TL, McClain MA, Gienapp IE, Shawler TM, Satoskar AR, Whitacre CC. Cutting edge: macrophage migration inhibitory factor is necessary for progression of experimental autoimmune encephalomyelitis. J Immunol. 2005;175:5611–5614. doi: 10.4049/jimmunol.175.9.5611. [DOI] [PubMed] [Google Scholar]
- 38.Wraight CJ, van Endert P, Moller P, Lipp J, Ling NR, MacLennan IC, Koch N, Moldenhauer G. Human major histocompatibility complex class II invariant chain is expressed on the cell surface. J Biol Chem. 1990;265:5787–5792. [PubMed] [Google Scholar]
- 39.Albouz-Abo S, Wilson JC, Bernard CC, von Itzstein M. A conformational study of the human and rat encephalitogenic myelin oligodendrocyte glycoprotein peptides 35–55. Eur J Biochem. 1997;246:59–70. doi: 10.1111/j.1432-1033.1997.t01-2-00059.x. [DOI] [PubMed] [Google Scholar]
- 40.Chervonsky AV, Medzhitov RM, Denzin LK, Barlow AK, Rudensky AY, Janeway CA., Jr Subtle conformational changes induced in major histocompatibility complex class II molecules by binding peptides. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:10094–10099. doi: 10.1073/pnas.95.17.10094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neveu R, Auriault C, Angyalosi G, Georges B. Evidences of conformational changes in class II Major Histocompatibility Complex molecules that affect the immunogenicity. Molecular immunology. 2002;38:661–667. doi: 10.1016/s0161-5890(01)00106-7. [DOI] [PubMed] [Google Scholar]
- 42.Painter CA, Cruz A, Lopez GE, Stern LJ, Zavala-Ruiz Z. Model for the peptide-free conformation of class II MHC proteins. PLoS One. 2008;3:e2403. doi: 10.1371/journal.pone.0002403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sato AK, Zarutskie JA, Rushe MM, Lomakin A, Natarajan SK, Sadegh-Nasseri S, Benedek GB, Stern LJ. Determinants of the peptide-induced conformational change in the human class II major histocompatibility complex protein HLA-DR1. J Biol Chem. 2000;275:2165–2173. doi: 10.1074/jbc.275.3.2165. [DOI] [PubMed] [Google Scholar]
- 44.Zarutskie JA, Sato AK, Rushe MM, Chan IC, Lomakin A, Benedek GB, Stern LJ. A conformational change in the human major histocompatibility complex protein HLA-DR1 induced by peptide binding. Biochemistry. 1999;38:5878–5887. doi: 10.1021/bi983048m. [DOI] [PubMed] [Google Scholar]
- 45.Carven GJ, Chitta S, Hilgert I, Rushe MM, Baggio RF, Palmer M, Arenas JE, Strominger JL, Horejsi V, Santambrogio L, Stern LJ. Monoclonal antibodies specific for the empty conformation of HLA-DR1 reveal aspects of the conformational change associated with peptide binding. J Biol Chem. 2004;279:16561–16570. doi: 10.1074/jbc.M314315200. [DOI] [PubMed] [Google Scholar]
- 46.McCormick JK, Tripp TJ, Llera AS, Sundberg EJ, Dinges MM, Mariuzza RA, Schlievert PM. Functional analysis of the TCR binding domain of toxic shock syndrome toxin-1 predicts further diversity in MHC class II/superantigen/TCR ternary complexes. J Immunol. 2003;171:1385–1392. doi: 10.4049/jimmunol.171.3.1385. [DOI] [PubMed] [Google Scholar]
- 47.Cumberworth A, Lamour G, Babu MM, Gsponer J. Promiscuity as a functional trait: intrinsically disordered regions as central players of interactomes. Biochem J. 2013;454:361–369. doi: 10.1042/BJ20130545. [DOI] [PubMed] [Google Scholar]
- 48.Dunker AK, Cortese MS, Romero P, Iakoucheva LM, Uversky VN. Flexible nets. The roles of intrinsic disorder in protein interaction networks. Febs J. 2005;272:5129–5148. doi: 10.1111/j.1742-4658.2005.04948.x. [DOI] [PubMed] [Google Scholar]
- 49.Wright PE, Dyson HJ. Linking folding and binding. Curr Opin Struct Biol. 2009;19:31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Benedek G, Zhu W, Libal N, Casper A, Yu X, Meza-Romero R, Vandenbark AA, Alkayed NJ, Offner H. A novel HLA-DRalpha1-MOG-35–55 construct treats experimental stroke. Metab Brain Dis. 2013 doi: 10.1007/s11011-013-9440-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.