

Abstract
Background
Although sperm is transcriptionally and translationally quiescent, complex populations of RNAs, including mRNAs and non-coding RNAs, exist in sperm. Previous microarray analysis of germ cell mutants identified hundreds of sperm genes in Caenorhabditis elegans. To take a more comprehensive view on C. elegans sperm genes, here, we isolate highly pure sperm cells and employ high-throughput technologies to obtain sperm transcriptome and proteome.
Results
First, sperm transcriptome consists of considerable amounts of non-coding RNAs, many of which have not been annotated and may play functional roles during spermatogenesis. Second, apart from kinases/phosphatases as previously reported, ion binding proteins are also enriched in sperm, underlying the crucial roles of intracellular ions in post-translational regulation in sperm. Third, while the majority of sperm genes/proteins have low abundance, a small number of sperm genes/proteins are hugely enriched in sperm, implying that sperm only rely on a small set of proteins for post-translational regulation. Lastly, by extensive RNAi screening of sperm enriched genes, we identified a few genes that control fertility. Our further analysis reveals a tight correlation between sperm transcriptome and sperm small RNAome, suggesting that the endogenous siRNAs strongly repress sperm genes. This leads to an idea that the inefficient RNAi screening of sperm genes, a phenomenon currently with unknown causes, might result from the competition between the endogenous RNAi pathway and the exogenous RNAi pathway.
Conclusions
Together, the obtained sperm transcriptome and proteome serve as valuable resources to systematically study spermatogenesis in C. elegans.
Keywords: C. elegans, Sperm, Transcriptome, Proteome
Background
Spermatogenesis is a process during which undifferentiated germ cells develop into mature sperm cells. A sperm cell has a highly condensed nucleus and lacks many organelles, such as ribosomes and Golgi apparatus; thus, sperm is transcriptionally and translationally quiescent [1-7]. Early observations that RNAs exist in sperm were originally dismissed [8]; however, over the past decade, the view that sperm contains complex RNA populations, including mRNAs and non-coding RNAs, has become established [1-7]. These RNA populations contain not only non-functional remnant spermatogenesis-expressed transcripts, but also RNAs with potential functions during early embryogenesis [9-11]. Recently, the rapid development of microarray and high-throughput sequencing technologies has assisted the profiling of sperm RNAs in a broad range of species, including human [12,13], mouse [13], invertebrates [14-17] and plants [18-22].
The germline of Caenorhabditis elegans is transparent, and is well suited for cellular characterization of spermatogenesis [23]. The transcription of sperm genes is initiated during the pachytene stage; when spermatocytes enter the karyosome stage, global transcription ceases [24-26]. In C. elegans, approximately 60 genes that are essential for normal spermatogenesis have been characterized [23,27]. By microarray analyses, Reinke et al. identified sets of genes that are spermatogenesis-enriched, oogenesis-enriched and sex-regulated [28,29]. Notably, their analyses showed that the sperm-enriched genes encode considerable numbers of kinases and phosphatases, and are depleted from the X-chromosome, echoing the findings of Reuben et al. that the X-chromosome in males exhibits striking H3K9 methylation [30]. These microarray analyses identified sperm genes on a large-scale; however, the analyses were based on comparisons between germ cell mutants, and purified sperm cells were not used. In addition, their microarray analyses only identified sperm-enriched genes, while omitted those sperm genes that are also abundant in oocyte.
Compared with sperm genes, the C. elegans sperm small RNAs have been investigated more extensively. A few studies reported deep sequencing of the small interfering RNAs (siRNAs) of spermatogenic cells [14-17]. It was demonstrated that the ERI-1/RRF-3/ALG-3 pathway is essential for production of the spermatogenesis-specific 26G endogenous siRNAs (endo-siRNAs) [17]. Han et al. also showed that 26G endo-siRNAs regulate spermatogenic gene expressions [15].
In the present study, we purified highly pure C. elegans sperm cells and applied high-throughput approaches to obtain sperm transcriptome and proteome. A large number of long or intermediate-sized non-coding RNAs (lncRNAs hereafter) are found in sperm transcriptome, implying the important roles of lncRNAs during spermatogenesis. We showed that sperm proteome/transcriptome is enriched in not only kinase and phosphatase proteins/genes as previously reported [28,29], but also ion binding proteins/corresponding genes, underlying the crucial roles of intracellular ions in sperm. Our extensive RNAi screening of sperm genes did not produce many defective phenotypes. Our further analysis revealed a tight correlation between sperm transcriptome and sperm small RNAome, which leads to a view that the phenomenon of inefficient RNAi screening might result from the endogenous RNAi pahway that acts strongly during spermatogenesis and is to compete over the exogenous RNAi pathway.
Results
Sequencing C. elegans sperm transcriptome and proteome
We performed large-scale culturing of C. elegans strain him-5, followed by isolating males and purifying sperm, and obtained highly pure sperm cells. These cells contain nearly no residue bodies or primary spermatocytes, as examined by microscope (Figure 1a). To further examine the purity of acquired sperm, these concentrated sperm as well as the whole worms were subjected to Western blot analysis using antibodies against tubulin and H3K4 mono-methylation mark. Trace H3K4 methylation mark has been detected in mammalian sperm [1,5,7]; here, we show C. elegans sperm retain histone methylation mark (Figure 1b). In contrast, tubulin which is not transmitted to mature sperm is not detected in our prepared sperm (Figure 1b). This suggests the purified sperm cells were of high purity.
Figure 1.
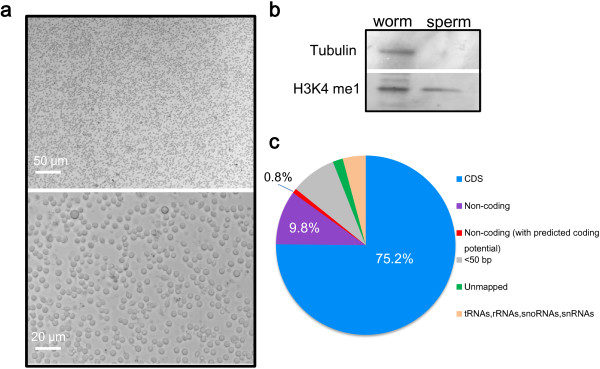
Sequencing of C. elegans sperm transcriptome. (a) Microscopic image of purified sperm cells. (b) Whole worms and concentrated sperm were analyzed by immunoblotting using antibodies against mono-methylated H3K4 and tubulin. No tubulin was detected in the sperm suggesting high purity of the prepared sample. (c) Classification of the sperm transcriptome sequencing reads. Most of the reads are coding sequences (CDS); about ~10% of the reads are non-coding sequences including those with predicted coding potential. Reads shorter than 50 bp, unmapped to the C. elegans genome or coding tRNAs, rRNAs, snoRNAs and snRNAs were filtered out before assembly.
RNA was extracted from these cells, followed by cDNA synthesis and amplification, and the purified cDNA was deep sequenced on a 454 GS FLX Titanium platform. High-throughput sequencing produced 367,638 high quality (HQ) reads with an average length of 315 base-pairs (bp) (Additional file 1: Figure S1 and Table S1). These HQ sequences were filtered to remove repeat, tRNAs, rRNA, snoRNA and snRNA sequences (see Methods), and then assembled by Newbler (version 2.3) using the C. elegans coding sequence dataset (WS228) as a reference, and 9,287 coding transcripts were identified (Additional file 2). The sequences unmapped to the C. elegans coding transcriptome were further assembled using the C. elegans genome as a reference to identify possible non-coding transcripts (Additional file 3). In summary, the majority of the sequences (75.2%) are coding transcripts, and 10.6% of the sequences were identified as non-coding transcripts (Figure 1c). A small part of these non-coding sequences (0.8%) are predicted to have coding potential by CPAT (version 1.2.1) (Additional file 3). In the non-coding portion, many novel transcripts supported by overlapping reads were identified (Figure 2a-d, also see next section). Meanwhile, although the sequencing depth is limited, we did find a few new splicing isoforms, e.g., the asterisks in Figure 2e,f point to the introns covered by reads indicating the introns can be transcribed.
Figure 2.

Novel non-coding RNA transcripts and gene splicing isoforms. (a), (b), (c) and (d) illustrate new non-coding RNA transcripts supported by sequencing reads. (e) and (f) show new gene splicing variants. Red asterisks point to the introns covered by reads.
We further assessed the level of possible contaminations for sperm transcriptome by examining 15 somatic marker genes (Additional file 1: Table S2) for their abundances in the trancriptome dataset. Only five (wrt-2, ges-1, hlh-17, myo-2 and myo-3) were detected in sperm transcriptome, and the number of reads for these five genes ranged only from 1 to 3, thus, somatic cell contamination is much low in the prepared sperm cells. The RPKM (Reads Per Kilobase of exon model per Million mapped reads) values for these five genes range from 3.42e-4 to 3.27e-3. To obtain a higher confidence transcriptome dataset, we set the RPKM cut-off to 3.27e-3. The resultant sperm transcriptome dataset contains 3,760 genes (Additional file 4) and our following analyses are based on these 3,760 genes.
In parallel with sperm mRNA sequencing, sperm protein extract was prepared and separated on SDS-polyacrylamide gel prior to removal of the MSP fraction. Subsequently, the protein extract was processed for shotgun LC-MS/MS analysis. LC-MS/MS analysis identified 27,667 peptides, corresponding to 2,994 proteins in the C. elegans protein dataset (WS229), with a probability >95% using percolator scores (data available from http://159.226.118.206/miaolab/C.elegans%20data.htm). None of the above-mentioned somatic cell markers were detected in this proteome dataset. 207 proteins (6.9%) were degenerative (i.e., they cannot be distinguished among homologous proteins).
Next, we compared sperm transcriptome and proteome (MSPs and the predicted proteins were excluded). The result showed that 30.1% of the transcriptome overlaps with 50.2% of the proteome (Figure 3a and Additional file 5). The limited overlap between the two–omes might suggest the different compositions of sperm transcriptome and proteome. Alternatively, it may results from the insufficient sequencing depth. When using RPKM and emPAI (exponentially modified protein abundance index) values to evaluate mRNA and protein abundances, we found that in the overlapping portion, mRNA and protein abundances are significantly correlated (r = 0.58, P < 2.2e-16, Pearson test) (Figure 3b). The overlapping portion contains the most abundant non-MSP sperm genes/proteins.
Figure 3.
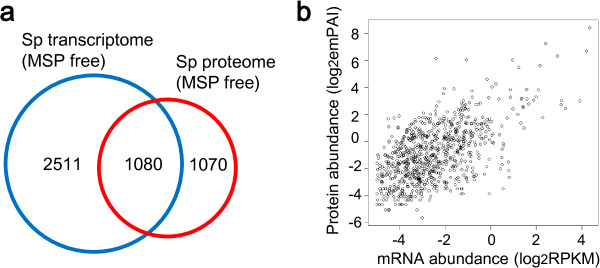
Comparison of sperm transcriptome and proteome. (a) Venn diagram showing the overlap between the sperm transcriptome and the sperm proteome (MSPs and predicted proteins were excluded). The number of genes/proteins in each part is indicated. (b) Plot showing sperm mRNA and protein abundances are well correlated.
Validation of novel lncRNAs
As mentioned above, sperm transcriptome comprises ~10% of non-coding RNAs. It has been proposed that over 26% and 13% of the full-length cDNAs in human and mice, respectively, are mRNA-like ncRNAs [31-33]. In Drosophila, some lncRNAs are thought to be important for male-specific processes [34]. Therefore, we selected 30 lncRNA candidates that have poly(A/T) tail from sperm transcriptome and verified them by 3′RACE. Eight novel lncRNAs (Spnc1-8) were successfully validated by 3′RACE (Figure 4a and Additional file 6), and Spnc7 was expressed specifically in males (Figure 4b, note that at 25°C, the mutant fem-3 overproduces sperm while fem-1 only produces oocytes; fog-2 generates separate male and female offspring).
Figure 4.

Identification of transcripts of novel lncRNAs. (a) Schematic representation of the 3′ structures of eight novel lncRNAs, namely Spnc1-8 (in red), and their poly(A) tails (in purple), which were confirmed by 3′RACE. Known gene models are shown in dark blue. (b) Identification of the male-specific lncRNA Spnc7 and long non-coding RNA linc-40 by RT-PCR. The Act-1 gene was used as control.
Recently, Nam et al. identified ~170 polyadenylated lncRNAs in C. elegans, and many of these lncRNAs are predicted to be associated with male identity and sperm functions [35]. Thus, we compared these lncRNAs with the non-coding portion of our sperm transcriptome dataset by BLAST (E-value = 9e-13, this E-value cutoff allows alignment to have ~60 bp contiguous perfect match). 32 lncRNAs were found in sperm transcriptome, and linc-40 was shown to be male-specific (Figure 4b). Together, these results suggest that apart from coding genes, substantial numbers of lncRNAs are expressed and may play functional roles during spermatogenesis.
Sperm are enriched in ion binding proteins
By microarray analysis, Reinke et al. identified hundreds of hermaphrodite and male germline genes; the hermaphrodite germline genes were further classified as sperm-enriched, oocyte-enriched and germline-intrinsic groups [28]. These gene sets were compared with our sperm transcriptome data. The comparison showed that sperm transcriptome overlaps with 77.8% and 77.2% of the hermaphrodite and male spermatogenesis gene sets, respectively; in contrast, this sperm transcriptome covers only 18.2% and 28% of the oogenesis-enriched and germline-intrinsic gene sets, respectively (Figure 5a,b). This result shows a consistency of our sperm transcriptome data with previous microarray analysis. We were particularly interested in the hermaphrodite and male sperm genes identified in both studies (grey-shaded parts in Figure 5a,b). Gene Ontology (GO) analysis of the two sperm gene sets showed that sperm significantly enrich in kinase/phosphatase (KP) activities and ion binding activity (hypergeometric probability test, P < 0.01) (Figure 5c). KP has been linked to sperm function in C. elegans[28,29,36]; however, enrichment of the genes encoding ion binding proteins was not reported previously. Because Cl-, Na+, K+ and Ca2+ channels were implicated to modulate sperm function [37-41], ion binding proteins may play crucial roles in post-translational regulation in sperm. We also performed GO analysis of the whole sperm transcriptome, and the result resembles the above GO analysis: sperm are significantly enriched in phosphatase and ion binding activities (P < 0.01) (Figure 5d). Additionally, GO analysis of the sperm proteome confirms that KPs and ion binding proteins are three most enriched classes in sperm (Figure 5e). In contrast to KPs and ion binding activities, transcription factor activity and signal transducer activity are significantly low in sperm (P < 0.01) (Figure 5d), verifying the transcriptionally and translationally quiescent nature of sperm.
Figure 5.

Comparison of the sperm transcriptome with the available microarray datasets. (a) Venn diagram showing the overlap between the sperm transcriptome (in red) and the hermaphrodite germline gene datasets obtained by microarray analysis (in blue). The total number of genes in each dataset and the number of genes in the overlapping portion are indicated. (b) Venn diagram showing the overlap between the sperm transcriptome (in red) and the male germline gene datasets (in blue). (c) GO analysis of the male and hermaphrodite spermatogenesis-enriched genes (gray-shaded sets in (a) and (b)) shows that sperm are enriched in ion binding and kinase/phosphatase activities. (d) GO analysis of the sperm transcriptome shows that sperm are enriched in ion binding and phosphatase activities. (e) GO analysis of the sperm proteome shows that sperm are enriched in ion binding and kinase/phosphatase activities. **indicates P < 0.01 (hypergeometric probability test).
We also reasoned the transcriptome dataset has deeper depth than previous microarray data, thus we employed RT-PCR and used the germ cell mutants fem-1, fem-3 and fog-2 to screen for novel sperm specific/enriched genes. The expression of the genes with RPKM above 2.5 (reads counts >100) in sperm transcriptome was surveyed in these mutants (MSP and some well-known sperm-specific genes were excluded in this analysis), and 53 genes that represent 56% of the genes investigated were shown to be sperm-specific/enriched (Figure 6). Of these 53 genes, 12 were not defined to be spermatogenesis-enriched in previous microarray analysis (indicated by asterisks in Figure 6). Expression of the 53 genes is mostly detectable in both males (fem-3 and fog-2♂) and hermaphrodites (strain N2)(Additional file 1: Figure S2), suggesting that they play common roles in the sperm of both sexes. In this screen, we identified sperm-specific/enriched genes in so high efficiency, demonstrating the sperm transcriptome data serves as a valuable resource for studying spermatogenesis.
Figure 6.

Identification of novel sperm-specific/enriched transcripts. RT-PCR analysis identified 53 genes that are highly expressed in males (fog-2♂ and fem-3) compared with in females (fog-2♀ and fem-1). Most of these genes were identified previously as spermatogenesis-enriched [28]; however, 12 genes (indicated by the red asterisks) are newly identified as being sperm-specific/enriched. The Cdc-42 and Act-1 genes were used as controls.
Sperm gene/protein abundance exhibits a skewed distribution
When examining sperm mRNA/protein abundance (MSPs were excluded), we found that the majority of sperm mRNAs/proteins have very low abundance, whereas a small set of mRNAs/proteins are hugely enriched in sperm (Figure 7). We should note that biased expression for male-enriched genes has been observed in zebrafish [42], fly [43], and recently in C. elegans[44]. These results indicate that sperm may only rely on a small number of proteins to complete their post-translational regulations, e.g., signalling cascades regulating sperm activation, motility and interaction with oocyte.
Figure 7.
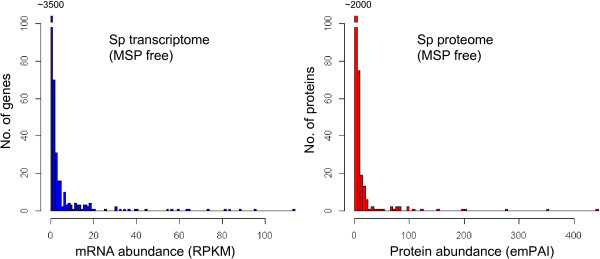
Sperm transcriptome and proteome show skewed abundance. Histograms showing the distributions of sperm gene (left panel) and protein (right panel) abundances.
RNAi analysis of sperm enriched genes
To explore the functions of sperm genes, we firstly tested the RNAi of 85 genes that have read counts above 100 using the rrf-3;him-5 strain. Most of the RNAi-treated hermaphrodites had normal fertility. To examine the fertility of the RNAi males, we crossed them with spe-8;dyp-4 hermaphrodites. The crossing progenies were also normal (without significant numbers of dumpy worms being observed, as compared with the control), suggesting that the sperm of these males had no defect. We identified only one gene, act-4, whose knockdown leads to a significantly reduced brood size (t-test, P < 0.001), as shown in Figure 8g. Act-4 was previously shown to be expressed in spermatheca [45]. This actin is possibly involved in the contraction of spermatheca.
Figure 8.
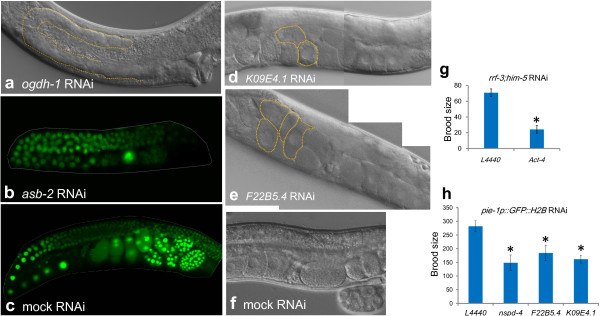
RNAi analysis of sperm genes. (a) Germline of the ogdh-1 RNAi worm. Yellow dashed line marks its immature germline. (b)asb-2 RNAi worm showed retarded growth of the germline (~60 h after egg stage). (c) Mock RNAi control (the same stage as (b)). (d) and (e) show the germlines of F22B5.4 and K09E4.1 RNAi worms. Yellow dashed lines mark the unfertilized oocytes. (f) Mock RNAi control. (g) and (h) show the brood sizes of the act-4, nspd-4, F22B5.4, and K09E4.1 RNAi worms are significantly reduced as compared with the control in which empty vector L4440 was used. *indicates P < 0.05 (Student’s t-test).
We further tested the RNAi of 486 genes that have read counts above 20 using the strain pie-1::gfp::H2B at 25°C (25°C is a challenging condition for sperm fertility, e.g., fer-1, spe-9 and rrf-3 all control fertility in a temperature-sensitive manner). RNAi was conducted over three generations [46]. However, our RNAi assay did not produce as many defective phenotypes as those collected from the Wormbase RNAi dataset. We describe five unreported genes that control fertility as below. First, RNAi for the gene ogdh-1 or asb-2 resulted in complete sterility. OGDH-1 is a mitochondrial 2-oxoglutarate dehydrogenase. ogdh-1 RNAi worms had severe germline defects without germ cells being produced (Figure 8a). ASB-2 is the subunit of a mitochondrial ATPase. asb-2 RNAi worms failed to produce living embryos and its germline growth was retarded when compared with the RNAi control (Figure 8b,c show the germlines at ~60 h after egg stage). The dissected germline of some asb-2 RNAi worms evidently lacked sperm (Additional file 1: Figure S4). Moreover, RNAi for the genes nspd-4, F22B5.4 and K09E4.1 led to production of unfertilized oocytes (Figure 8d,e and Additional file 1: Figure S5). The brood sizes of the RNAi worms for these three genes were significantly reduced when compared with that of RNAi control (t-test, P < 0.01) (Figure 8h). The observation of unfertilized oocytes in the uterus of RNAi worms as well as reduced brood sizes hints that these genes are involved in spermatogenesis.
Collectively, our RNAi screen identified a few genes that regulate germline development and sperm fertility. It is worth noting that our study, consistent with prior studies, showed that downregulation of sperm genes by RNAi feeding approach is inefficient to produce defective phenotypes [28,47].
Endo-RNAi pathway represses sperm gene expression
Regarding to the phenomenon that RNAi of sperm genes produces fewer defective phenotypes than the corresponding mutants, we speculated it is due to that the exogenously triggered RNAi pathway could not compete over the endogenous RRF-3/ERI-1/ALG-3 RNAi pathway. The competition of these two RNAi pathways has been proposed [14]. Supporting this hypothesis, mutants lacking RRF-3 and ERI-1 have been successfully used as tools for enhanced RNAi screening. When comparing our sperm transcriptome data with the published sperm primary siRNAs data [16], we did find the sperm 19G-28G primary siRNAs have a strong correlation with sperm transcriptome; i.e., for the sperm-enriched mRNAs, the corresponding antisense siRNAs are also enriched (Figure 9a, details in Additional file 7). This result further supports the previous view that the endo-siRNA pathway represses the expression of sperm genes [17].
Figure 9.

Sperm genes are downregulated by endo-siRNA pathway. (a) Heat-map showing the transcripts in the sperm transcriptome are targeted by antisense 19G-28G primary siRNAs. Each row represents a sperm transcript. Abundance of a sperm transcript is quantified by log2RPKM. The sperm transcript targeted by the corresponding siRNAs is indicated by a black line. (b) RT-PCR identification of 32 sperm genes that are expressed at higher levels in the rrf-3;him-5, as compared with the him-5.
We also examined the expressions of the 53 sperm specific/enriched genes (shown in earlier result) between him-5 and him-5;rrf-3 adult worms (him-5;rrf-3 worms produces offspring at 20°C, whereas are sterile at 25°C) by RT-PCR (25 amplification cycles). Notably, of the 53 genes, 32 genes were expressed at elevated levels in the him-5;rrf-3 as compared with him-5 (Figure 9b). This expression elevation is particularly evident for worms grown at restrictive temperature (25°C). This result demonstrated that RRF-3, as a vital component of the endo-siRNA pathway, negatively regulates the expression of sperm genes. Together, we propose that the inefficient RNAi of sperm genes might result from the competition between exogenous RNAi and endogenous RNAi pathways during spermatogenesis (see Discussion).
Discussion and conclusion
The sperm cell has highly condensed nucleus packed with protamines instead of histones and lacks many essential organelles, such as ribosomes and Golgi apparatus [1]. Therefore, sperm cell is devoid of transcriptional and translational activities. The long assumed view was that, at fertilization, sperm transmits solely its DNA to the oocyte, which has all the components necessary for early embryonic development. However, this notion has been changed by recent advancement of high-throughput technologies that made the unravelling of the sperm transcriptome and epigenome possible. It was discovered that sperm not only retains the histones that may play significant roles during early embryogenesis [1,5,7,48-50], but also contains complex RNA species, including mRNAs and non-coding RNAs, some of which are vital for early embryonic development [4,11]. C. elegans is an easily manipulated model organism, and is ideal to study sperm biology. By microarray analysis, Reinke et al. identified spermatogenesis, oogenesis and sex-regulated genes in C. elegans[28]. To improve study of spermatogenesis, here, we isolated highly pure sperm cells and obtained sperm transcriptome and proteome by high-throughput approaches.
First, sequencing sperm transcriptome showed that considerable numbers of lncRNAs are present in sperm. lncRNAs are a major group of ncRNAs whose functions are poorly understood. In Drosophila, some lncRNAs were thought to be involved in male-specific processes related to sexual dimorphism [34]. Recently, Nam et al. showed that, in C. elegans, many lncRNAs are associated with processes such as dauer formation, male identity, sperm formation, and interaction with sperm-specific mRNAs [35]. In our small-scale screen, eight novel lncRNAs were isolated, one of which was specifically expressed in males.
We showed that sperm gene sets are significantly enriched in not only kinase/phosphatase genes as previously reported [28,29], but also genes encoding ion binding proteins. Sperm development is regulated posttranslationally. The Cl-, Na+, K+ and Ca2+ ion channels have been implicated to respond to extracellular signals and mediate sperm activation [36-41]. Therefore, our identification of the sperm genes encoding ion binding proteins will help uncover the intracellular ion regulated pathways associated with sperm function.
We showed that sperm genes or proteins have skewed abundance, i.e., a small set of genes/proteins are greatly enriched in sperm. This supports previous observations that male-enriched genes have biased expressions than female-enriched genes in zebrafish [42], fly [43] and C. elegans[44]. This result may suggest that compared with somatic cells, sperm only require a small number of proteins to complete their post-translational regulations, e.g., signalling factors for sperm activation, MSP dynamics and interaction with oocyte.
By RNAi of hundreds of sperm-enriched genes, we identified a few genes, whose knockdown leads to production of unfertilized oocytes and significantly reduced brood size. It is worth noting that previous study showed that RNAi of sperm genes is inefficient for producing defective phenotypes [28,43]. To our knowledge, the mechanism behind this phenomenon has been unclear. It could be caused by the low penetrance of RNAi to germline tissue. Because endo-RNAi pathway strongly acts during spermatogenesis [15,17], here, we presume that this phenomenon might result from the competition between endogenous and exogenous RNAi pathways. The endogenous RNAi pathway may compete over the exogenous RNAi pathway, leading to the failure of RNAi of sperm genes.
Methods
C. elegans strains and growth conditions
The following strains were used in this study: wild-type Bristol strain N2, spe-8(hc40)I, rrf-3(pk1426)II, dpy-4(e1166)IV, fem-1(hc17)IV, fem-3(q23)IV, fog-2(q71)V, him-5(e1490)V, pie-1p::gfp::H2B. All the strains were maintained at 16°C or 20°C. The strains fem-1 and fem-3 are temperature-sensitive. At 25°C, fem-1 only produces oocytes, while fem-3 overproduces sperm.
Large-scale culture of C. elegans and purification of mature sperm
Large-scale culture of C. elegans strain him-5 and purification of mature sperm cells were performed using a modified protocol based on published methods [51-53] (detailed protocols in Additional file 1).
cDNA synthesis and 454 pyrosequencing
The total RNA of C. elegans sperm was prepared using a mirVana miRNA Isolation kit (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s instructions. Trace genomic DNA was removed using RNase-free DNase I (Promega, Madison, WI, USA). The DNase I-treated RNA was reverse transcribed to cDNA using a SMARTer PCR cDNA Synthesis kit (Clontech, Mountain View, CA, USA) and then amplified for 20 cycles using an Advantage 2 PCR kit (Clontech). Amplified cDNA was purified using a QIAquick PCR Purification Kit (Qiagen, Hilden, Germany) and 0.5 μg of the cDNA was subjected to 454 pyrosequencing using a GS FLX Titanium General Library Preparation Kit (Roche 454 Company, CT, USA).
Sperm protein preparation and mass spectrometry analysis
C. elegans sperm was suspended in PBS solution, disrupted by sonication for 8 s (repeated three times at intervals of 8 s), mixed with SDS loading buffer and heated for 10 min. After centrifugation, the supernatant was loaded on a 12% SDS-PAGE gel for separation. The resulting gel was silver stained using a Fast Silver Stain kit (Beyotime, Shanghai, China). The major sperm protein (MSP) band was removed prior to tryptic in-gel digestion, according to standard protocols. The tryptic peptides were extracted using 60% acetonitrile in 0.1% formic acid, dried in vacuum and then resuspended in 0.1% formaic acid for mass spectrometry analysis. LC-MS/MS analysis was performed in a LTQ-orbitrap XL (ThermoFinnigan, San Jose, CA) coupled online with an Eksgent Nano 2D LC system. Peptides mixture was first loaded on a trap column (300SB-C18, 5 × 0.3 mm, 5 μm particle) (Agilent Technologies, Santa Clara, CA), and then analysed using a self-packed capillary C18 column (75 μm i.d. × 150 mm, 3 μm particle, C18 resin), and eluted with a gradient of 4-35% of Buffer B (0.5% formic acid in acetonitrile) in Buffer A (0.5% formic acid in water) at a flow rate of ~300 nl/min for 120 min. Data-dependent scanning was incorporated to select the 10 most abundant ions from a full-scan mass spectrum (mass range 200–1800 Da) for fragmentation by collision-induced dissociation.
Mass spectrometry data processing
MS data were analyzed using Mascot (Matrix Science, version 2.3.02) against the C. elegans protein dataset (release WS229) with the following parameters: only tryptic peptides with up to two missed cleavage sites were allowed; 20 ppm mass tolerances for MS; and 0.8 Da for MS/MS fragment ions. Peptides with Mascot Percolator posterior error probability values lower than 0.05 were considered to be potential candidates. Decoy database searches in Mascot revealed a false positive rate of 1.25% at the peptide level. Proteins with ≥2 unique peptides were accepted as matches.
Bioinformatic analyses
The raw 454 sequencing reads were filtered by trimming off the adaptor sequences and removing short reads (<50 bp). Repeat sequences were masked, and tRNA, rRNA, snoRNA and snRNA sequences (WS228) were filtered before reads assembling using Newbler, version 2.3 (Roche). For sequence assembly, the C. elegans coding transcript data (WS228) was used as a reference; the unassembled reads were further assembled using the C. elegans genome as a reference. For the assembly we used the Newbler default parameters (overlap length >40 bp, identities >90%). Sequences in the non-coding portion were evaluated for coding potential using CPAT (http://lilab.research.bcm.edu/cpat/index.php). IGV genome browser (http://www.broadinstitute.org/igv/) was used to view reads assembling information for novel non-coding RNA transcripts. Sperm transcriptome was functionally annotated using the GO Slimmer tool (http://amigo.geneontology.org/cgi-bin/amigo/slimmer?session_id). Sperm primary siRNAs data described previously [16] were downloaded from the NCBI GEO database (http://www.ncbi.nlm.nih.gov/geo/). These siRNAs datasets were compared to our sperm transcriptome data using BLAST, and the siRNAs with a perfect antisense match were retained as the siRNAs targeting sperm genes. C. elegans polyadenylated lncRNA data was previously described [35]. C. elegans genome data, cds transcript dataset and annotation data were all downloaded from Wormbase (ftp://ftp.wormbase.org/pub/wormbase/species/c_elegans/).
3′-rapid amplification of cDNA ends (3′RACE)
DNase I-treated sperm RNA was reverse transcribed into cDNA following the 3′RACE protocol in the FirstChoice RLM-RACE kit (Invitrogen). Nested PCR was conducted using the High Fidelity PCR SuperMix (TransGen, Beijing, China) and the amplified product was cloned into the pEASY-Blunt vector (TransGen) before sequencing to identify its 3′ sequence.
Reverse transcription PCR (RT-PCR)
The N2, fem-1(lf), fem-3(gf) and fog-2(lf) strains were synchronized and the adults were collected and frozen as starting materials. Total RNAs from these strains were isolated using a mirVana miRNA Isolation kit (Invitrogen). Total RNA (2 μg) was reverse transcribed into cDNA using the SuperScript III First-Strand Synthesis System (Invitrogen). The cDNA product was diluted 10-fold and used as the template for RT-PCR. The Act-1 and Cdc-42 genes were used as controls.
Microscopic analysis
The RNAi treated pie-1p::gfp::H2B strain were anesthetized and mounted on agarose pads for visualization using a differential interference contrast (DIC) microscope (Zeiss, Axio Imager M2) as well as a confocal microscope (Zeiss LSM 510 Meta). The gonads of asb-2RNAi worms were dissected, and then fixed by cold methanol method (http://www.wormbook.org/toc_wormmethods.html), followed by DAPI staining and microscopic visualization.
RNA interference screen
The RNAi screen was performed as previously described [54,55]. Briefly, the strain rrf-3;him-5 or pie-1p::gfp::H2B was fed with the RNAi bacteria obtained from the Ahringer RNAi library [56]. Bacteria carrying the empty vector (L4440) were used as the control. The bacteria clones that affect fertility were sequenced to verify the gene sequences.
Brood size assay
The L4 stage hermaphrodites of the RNAi worms were individually picked onto plates, and transferred to new plates daily until no eggs were laid. Brood size was determined by counting the worms on all plates. 50 replications were performed. Worms fed with bacteria carrying L4440 were used as the control.
Availability of supporting data
The raw sperm transcriptome sequencing data has been deposited in NCBI SRA database with the accession number of SRA056374. Sperm mRNA-seq assembled data and sperm proteomics data can be downloaded from http://159.226.118.206/miaolab/C.elegans%20data.htm.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
XM and LM designed the study and wrote the manuscript. XM prepared the samples. CL performed 454 pyrosequencing. PX carried out proteomics analysis. XM and YiZ carried out bioinformatic analysis. XM performed RT-PCR analysis. XM and YaZ performed RNAi screening. FY, SC helped to coordinate the experiments. All authors read and approved the final manuscript.
Supplementary Material
Summary of sperm transcriptome sequencing. Table S2. Somatic marker genes used to evaluate contamination levels in the sperm transcriptome. Figure S1. Length distribution of the sequencing reads in the sperm mRNAome. Most of the reads are longer than 400 bp suggesting high quality sequencing. Purple line indicates average length. Figure S2. RT-PCR expression analysis of 51 genes in the N2, fem-3 and fem-1 strains. These genes have biased expressions in the male (fem-3) and hermaphrodite (N2) compared with their expressions in the female (fem-1), suggesting that they are sperm-specific/enriched. The genes Cdc-42 and Act-1 were used as controls. Figure S3. DAPI staining of the gonad of the RNAi control and one asb-2 RNAi worm. White arrows indicate that sperm are present in the mock RNAi control; yellow arrow indicates the absence of sperm in one asb-2 RNAi worm. Figure S4.F22B5.4 RNAi worm produces unfertilized oocytes. Arrows indicate the unfertilized oocytes from the cracked body. Supplementary methods. Large-scale culture of C. elegans and purification of mature sperms.
Assemblies mapped to C. elegans CDS transcriptome. Sequences available from http://www.ibp.cas.cn/MiaolLab/index.html.
Assemblies mapped to C. elegans genome, but unmapped to CDS transcriptome. Sequences available from http://159.226.118.206/miaolab/index.htm.
List of sperm genes with RPKM above 3.27e-3.
Genes/Proteins found in C. elegans sperm transcriptome and proteome.
Sequences of eight novel lncRNAs confirmed by 3′RACE.
C. elegans sperm genes targeted primary siRNAs.
Contributor Information
Xuan Ma, Email: xuanma@genetics.ac.cn.
Yingjie Zhu, Email: yjzhu@implad.ac.cn.
Chunfang Li, Email: cfli@implad.ac.cn.
Peng Xue, Email: xuepeng@moon.ibp.ac.cn.
Yanmei Zhao, Email: zhaoym@moon.ibp.ac.cn.
Shilin Chen, Email: slchen@implad.ac.cn.
Fuquan Yang, Email: fqyang@ibp.ac.cn.
Long Miao, Email: lmiao@moon.ibp.ac.cn.
Acknowledgements
This work was supported by the National Basic Research Program of China (Grants 2012CB945022 and 2010CB912303 to L.M.; grant 2009CB825503 to Y.Z.; grants 2010CB833703, 2012CB966803 and 2011CB915501 to F.Y.) and the National Natural Science Foundation of China (Grants 31171337 and 30971648 to L.M., grant 90919047 to F.Y.). We thank Ping Huang, Zheng Cao, Zhiyu Liu, Wei Zhuang and Chao Sun for assistances with this work.
References
- Krawetz SA. Paternal contribution: new insights and future challenges. Nat Rev Genet. 2005;6(8):633–642. doi: 10.1038/nrg1654. [DOI] [PubMed] [Google Scholar]
- Boerke A, Dieleman SJ, Gadella BM. A possible role for sperm RNA in early embryo development. Theriogenology. 2007;68:S147–S155. doi: 10.1016/j.theriogenology.2007.05.058. [DOI] [PubMed] [Google Scholar]
- Lalancette C, Miller D, Li Y, Krawetz SA. Paternal contributions: new functional insights for spermatozoal RNA. J Cell Biochem. 2008;104(5):1570–1579. doi: 10.1002/jcb.21756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadoune JP. Spermatozoal RNAs: what about their functions? Microsc Res Tech. 2009;72(8):536–551. doi: 10.1002/jemt.20697. [DOI] [PubMed] [Google Scholar]
- Jenkins TG, Carrell DT. The paternal epigenome and embryogenesis: poising mechanisms for development. Asian J Androl. 2011;13(1):76–80. doi: 10.1038/aja.2010.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson GD, Lalancette C, Linnemann AK, Leduc F, Boissonneault G, Krawetz SA. The sperm nucleus: chromatin, RNA, and the nuclear matrix. Reproduction. 2011;141(1):21–36. doi: 10.1530/REP-10-0322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi Y, Shaman JA, Ward WS. Non-genetic contributions of the sperm nucleus to embryonic development. Asian J Androl. 2011;13(1):31–35. doi: 10.1038/aja.2010.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betlach CJ, Erickson RP. A unique RNA species from maturing mouse spermatozoa. Nature. 1973;242(5393):114–115. doi: 10.1038/242114a0. [DOI] [PubMed] [Google Scholar]
- Rassoulzadegan M, Grandjean V, Gounon P, Vincent S, Gillot I, Cuzin F. RNA-mediated non-mendelian inheritance of an epigenetic change in the mouse. Nature. 2006;441(7092):469–474. doi: 10.1038/nature04674. [DOI] [PubMed] [Google Scholar]
- Bayer M, Nawy T, Giglione C, Galli M, Meinnel T, Lukowitz W. Paternal control of embryonic patterning in Arabidopsis thaliana. Science. 2009;323(5920):1485–1488. doi: 10.1126/science.1167784. [DOI] [PubMed] [Google Scholar]
- Liu WM, Pang RTK, Chiu PCN, Wong BPC, Lao KQ, Lee KF, Yeung WSB. Sperm-borne microRNA-34c is required for the first cleavage division in mouse. Proc Natl Acad Sci U S A. 2011;109(2):490–494. doi: 10.1073/pnas.1110368109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostermeier GC, Dix DJ, Miller D, Khatri P, Krawetz SA. Spermatozoal RNA profiles of normal fertile men. Lancet. 2002;360(9335):772–777. doi: 10.1016/S0140-6736(02)09899-9. [DOI] [PubMed] [Google Scholar]
- Sun YQ, Koo S, White N, Peralta E, Esau C, Dean NM, Perera RJ. Development of a micro-array to detect human and mouse microRNAs and characterization of expression in human organs. Nucleic Acids Res. 2004;32(22):e188. doi: 10.1093/nar/gnh186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gent JI, Schvarzstein M, Villeneuve AM, Gu SG, Jantsch V, Fire AZ, Baudrimont A. A Caenorhabditis elegans RNA-directed RNA polymerase in sperm development and endogenous RNA interference. Genetics. 2009;183(4):1297–1314. doi: 10.1534/genetics.109.109686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han T, Manoharan AP, Harkins TT, Bouffard P, Fitzpatrick C, Chu DS, Thierry-Mieg D, Thierry-Mieg J, Kim JK. 26G endo-siRNAs regulate spermatogenic and zygotic gene expression in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2009;106(44):18674–18679. doi: 10.1073/pnas.0906378106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gent JI, Lamm AT, Pavelec DM, Maniar JM, Parameswaran P, Tao L, Kennedy S, Fire AZ. Distinct phases of siRNA synthesis in an endogenous RNAi pathway in C. elegans Soma. Mol Cell. 2010;37(5):679–689. doi: 10.1016/j.molcel.2010.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conine CC, Batista PJ, Gu WF, Claycomb JM, Chaves DA, Shirayama M, Mello CC. Argonautes ALG-3 and ALG-4 are required for spermatogenesis-specific 26G-RNAs and thermotolerant sperm in Caenorhabditis elegans. Proc Natl Acad Sci U S A. 2009;107(8):3588–3593. doi: 10.1073/pnas.0911685107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei LQ, Yan LF, Wang T. Deep sequencing on genome-wide scale reveals the unique composition and expression patterns of microRNAs in developing pollen of Oryza sativa. Genome Biol. 2011;12(6):R53. doi: 10.1186/gb-2011-12-6-r53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pina C, Pinto F, Feijo JA, Becker JD. Gene family analysis of the Arabidopsis pollen transcriptome reveals biological implications for cell growth, division control, and gene expression regulation. Plant Physiol. 2005;138(2):744–756. doi: 10.1104/pp.104.057935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grennan AK. An analysis of the Arabidopsis pollen transcriptome. Plant Physiol. 2007;145(1):3–4. doi: 10.1104/pp.104.900237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honys D, Twell D. Comparative analysis of the Arabidopsis pollen transcriptome. Plant Physiol. 2003;132(2):640–652. doi: 10.1104/pp.103.020925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafidh S, Breznenova K, Ruzicka P, Fecikova J, Capkova V, Honys D. Comprehensive analysis of tobacco pollen transcriptome unveils common pathways in polar cell expansion and underlying heterochronic shift during spermatogenesis. BMC Plant Biology. 2012;12:24. doi: 10.1186/1471-2229-12-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- L’Hernaults S. Spermatogenesis. Wormbook. 2006. http://wormbook.org/chapters/www_spermatogenesis/spermatogenesis.html. [DOI] [PMC free article] [PubMed]
- Shakes DC, Wu JC, Sadler PL, LaPrade K, Moore LL, Noritake A, Chu DS. Spermatogenesis-specific features of the meiotic program in Caenorhabditis elegans. Plos Genetics. 2009;5(8):1000611. doi: 10.1371/journal.pgen.1000611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Castillo-Olivares A, Kulkarni M, Smith HE. Regulation of sperm gene expression by the GATA factor ELT-1. Dev Biol. 2009;333(2):397–408. doi: 10.1016/j.ydbio.2009.06.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni M, Shakes DC, Guevel K, Smith HE. SPE-44 implements sperm cell fate. Plos Genetics. 2012;8(4):587–600. doi: 10.1371/journal.pgen.1002678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura H, L’Hernault SW. Spermatogenesis-defective (spe) mutants of the nematode Caenorhabditis elegans provide clues to solve the puzzle of male germline functions during reproduction. Dev Dyn. 2010;239(5):1502–1514. doi: 10.1002/dvdy.22271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinke V, Gil IS, Ward S, Kazmer K. Genome-wide germline-enriched and sex-biased expression profiles in Caenorhabditis elegans. Development. 2004;131(2):311–323. doi: 10.1242/dev.00914. [DOI] [PubMed] [Google Scholar]
- Reinke V, Smith HE, Nance J, Wang J, Van Doren C, Begley R, Jones SJM, Davis EB, Scherer S, Kim SK, Ward S. A global profile of germline gene expression in C. elegans. Mol Cell. 2000;6(3):605–616. doi: 10.1016/S1097-2765(00)00059-9. [DOI] [PubMed] [Google Scholar]
- Reuben M, Lin R. Germline X chromosomes exhibit contrasting patterns of histone H3 methylation in Caenorhabditis elegans. Dev Biol. 2002;245(1):71–82. doi: 10.1006/dbio.2002.0634. [DOI] [PubMed] [Google Scholar]
- Numata K, Kanai A, Saito R, Kondo S, Adachi J, Wilming LG, Hume DA, Hayashizaki Y, Tomita M, Grp RG, Members GSL, Hayashizaki Y, Tomita M. RIKEN GER Group. Identification of putative noncoding RNAs among the RIKEN mouse full-length cDNA collection. Genome Res. 2003;13(6B):1301–1306. doi: 10.1101/gr.1011603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki Y, Furuno M, Kasukawa T, Adachi J, Bono H, Kondo S, Nikaido I, Osato N, Saito R, Suzuki H, Yamanaka I, Kiyosawa H, Yagi K, Tomaru Y, Hasegawa Y, Nogami A, Schönbach C, Gojobori T, Baldarelli R, Hill DP, Bult C, Hume DA, Quackenbush J, Schriml LM, Kanapin A, Matsuda H, Batalov S, Beisel KW, Blake JA, Bradt D. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature. 2002;420(6915):563–573. doi: 10.1038/nature01266. [DOI] [PubMed] [Google Scholar]
- Ota T, Suzuki Y, Nishikawa T, Otsuki T, Sugiyama T, Irie R, Wakamatsu A, Hayashi K, Sato H, Nagai K, Kimura K, Makita H, Sekine M, Obayashi M, Nishi T, Shibahara T, Tanaka T, Ishii S, Yamamoto J, Saito K, Kawai Y, Isono Y, Nakamura Y, Nagahari K, Murakami K, Yasuda T, Iwayanagi T, Wagatsuma M, Shiratori A, Sudo H. Complete sequencing and characterization of 21,243 full-length human cDNAs. Nat Genet. 2004;36(1):40–45. doi: 10.1038/ng1285. [DOI] [PubMed] [Google Scholar]
- Jiang ZF, Croshaw DA, Wang Y, Hey J, Machado CA. Enrichment of mRNA-like Noncoding RNAs in the divergence of Drosophila Males. Mol Biol Evol. 2011;28(4):1339–1348. doi: 10.1093/molbev/msq293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam J-W, Bartel D. Long non-coding RNAs in C. elegans. Genome Res. 2012. published online June 15, 2012. [DOI] [PMC free article] [PubMed]
- Wu JC, Go AC, Samson M, Cintra T, Mirsoian S, Wu TF, Jow MM, Routman EJ, Chu DS. Sperm development and motility are regulated by PP1 phosphatases in Caenorhabditis elegans. Genetics. 2012;190(1):143–157. doi: 10.1534/genetics.111.135376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machaca K, DeFelice LJ, Lhernault SW. A novel chloride channel localizes to Caenorhabditis elegans spermatids and chloride channel blockers induce spermatid differentiation. Dev Biol. 1996;176(1):1–16. doi: 10.1006/dbio.1996.9999. [DOI] [PubMed] [Google Scholar]
- Nelson GA, Ward S. Vesicle fusion, pseudopod extension and ameboid motility are induced in nematode spermatids by the inophore monensin. Cell. 1980;19(2):457–464. doi: 10.1016/0092-8674(80)90520-6. [DOI] [PubMed] [Google Scholar]
- Bandyopadhyay J, Lee J, Il Lee J, Yu JR, Jee C, Cho JH, Jung S, Lee MH, Zannoni S, Singson A, Kim DH, Koo HS, Ahnn J. Calcineurin, a calcium/calmodulin-dependent protein phosphatase, is involved in movement, fertility, egg laying, and growth in Caenorhabditis elegans. Mol Biol Cell. 2002;13(9):3281–3293. doi: 10.1091/mbc.E02-01-0005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park BJ, Lee DG, Yu JR, Jung SK, Choi K, Lee J, Kim YS, Il Lee J, Kwon JY, Singson A, Song WK, Eom SH, Park CS, Kim DH, Bandyopadhyay J, Ahnn J. Calreticulin, a calcium-binding molecular chaperone, is required for stress response and fertility in Caenorhabditis elegans. Mol Biol Cell. 2001;12(9):2835–2845. doi: 10.1091/mbc.12.9.2835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington NL, Ward S. FER-1 regulates Ca2+ -mediated membrane fusion during C. elegans spermatogenesis. J Cell Sci. 2006;119(12):2552–2562. doi: 10.1242/jcs.02980. [DOI] [PubMed] [Google Scholar]
- Small CM, Carney GE, Mo Q, Vannucci M, Jones AG. A microarray analysis of sex- and gonad-biased gene expression in the zebrafish: evidence for masculinization of the transcriptome. BMC Genomics. 2009;10(579):1471–2164. doi: 10.1186/1471-2164-10-579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YSD, Parisi M, Kumar S, Oliver B. Constraint and turnover in sex-biased gene expression in the genus Drosophila. Nature. 2007;450(7167):233–237. doi: 10.1038/nature06323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CGLR, Smith HE, Woodruff GC, Oliver B, Haag ES. Simplification and desexualization of gene expression in self-fertile nematodes. Curr Biol. 2012;22(22):2167–2172. doi: 10.1016/j.cub.2012.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone S, Shaw JE. A Caernorhabditis elegans Act-4::lacZ fusion: use as a transformation marker and analysis of tissue specific expression. Gene. 1993;131(2):167–173. doi: 10.1016/0378-1119(93)90290-J. [DOI] [PubMed] [Google Scholar]
- Ramani AK, Chuluunbaatar T, Verster AJ, Na H, Vu V, Pelte N, Wannissorn N, Jiao A, Fraser AG. The majority of animal genes are required for wild-type fitness. Cell. 2012;148(4):792–802. doi: 10.1016/j.cell.2012.01.019. [DOI] [PubMed] [Google Scholar]
- Zhu GD, Salazar G, Zlatic SA, Fiza B, Doucette MM, Heilman CJ, Levey AI, Faundez V, L’Hernault SW. SPE-39 family proteins interact with the HOPS complex and function in lysosomal delivery. Mol Biol Cell. 2009;20(4):1223–1240. doi: 10.1091/mbc.E08-07-0728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliva R, Ballesca JL. Altered histone retention and epigenetic modifications in the sperm of infertile men. Asian J Androl. 2012;14(2):239–240. doi: 10.1038/aja.2011.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammoud SS, Nix DA, Zhang HY, Purwar J, Carrell DT, Cairns BR. Distinctive chromatin in human sperm packages genes for embryo development. Nature. 2009;460(7254):473–U447. doi: 10.1038/nature08162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrell DT, Hammoud SS. The human sperm epigenome and its potential role in embryonic development. Mol Hum Reprod. 2012;16(1):37–47. doi: 10.1093/molehr/gap090. [DOI] [PubMed] [Google Scholar]
- L’Hernault SW, Roberts TM. Cell biology of nematode sperm. Methods in Cell Biology. 1995. pp. 273–301. [DOI] [PubMed]
- Chu DS, Liu H, Nix P, Wu TF, Ralston EJ, Yates JR III, Meyer BJ. Sperm chromatin proteomics identifies evolutionarily conserved fertility factors. Nature. 2006;443(7107):101–105. doi: 10.1038/nature05050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M. Sperm and oocyte isolation methods for biochemical and proteomic analysis. Methods in Molecular Biology. Methods in Molecular Biology. 2006. pp. 193–201. [DOI] [PubMed]
- Timmons L, Court DL, Fire A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene. 2001;263(1-2):103–112. doi: 10.1016/S0378-1119(00)00579-5. [DOI] [PubMed] [Google Scholar]
- Simmer F, Moorman C, van der Linden AM, Kuijk E, van den Berghe PVE, Kamath RS, Fraser AG, Ahringer J, Plasterk RHA. Genome-wide RNAi of C. elegans using the hypersensitive rrf-3 strain reveals novel gene functions. Plos Biol. 2003;1(1):77–84. doi: 10.1371/journal.pbio.0000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath RS, Fraser AG, Dong Y, Poulin G, Durbin R, Gotta M, Kanapin A, Le Bot N, Moreno S, Sohrmann M, Welchman DP, Zipperlen P, Ahringer J. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature. 2003;421(6920):231–237. doi: 10.1038/nature01278. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of sperm transcriptome sequencing. Table S2. Somatic marker genes used to evaluate contamination levels in the sperm transcriptome. Figure S1. Length distribution of the sequencing reads in the sperm mRNAome. Most of the reads are longer than 400 bp suggesting high quality sequencing. Purple line indicates average length. Figure S2. RT-PCR expression analysis of 51 genes in the N2, fem-3 and fem-1 strains. These genes have biased expressions in the male (fem-3) and hermaphrodite (N2) compared with their expressions in the female (fem-1), suggesting that they are sperm-specific/enriched. The genes Cdc-42 and Act-1 were used as controls. Figure S3. DAPI staining of the gonad of the RNAi control and one asb-2 RNAi worm. White arrows indicate that sperm are present in the mock RNAi control; yellow arrow indicates the absence of sperm in one asb-2 RNAi worm. Figure S4.F22B5.4 RNAi worm produces unfertilized oocytes. Arrows indicate the unfertilized oocytes from the cracked body. Supplementary methods. Large-scale culture of C. elegans and purification of mature sperms.
Assemblies mapped to C. elegans CDS transcriptome. Sequences available from http://www.ibp.cas.cn/MiaolLab/index.html.
Assemblies mapped to C. elegans genome, but unmapped to CDS transcriptome. Sequences available from http://159.226.118.206/miaolab/index.htm.
List of sperm genes with RPKM above 3.27e-3.
Genes/Proteins found in C. elegans sperm transcriptome and proteome.
Sequences of eight novel lncRNAs confirmed by 3′RACE.
C. elegans sperm genes targeted primary siRNAs.