

Abstract
Background
The reverse rate dependence (RRD) of actions of IKr-blocking drugs to increase action potential duration (APD) and beat-to-beat variability (BVR) of APD is proarrhythmic. Therefore we determined if inhibition of endogenous, physiological late Na+ current (late INa) attenuates the RRD and proarrhythmic effect of IKr inhibition.
Methods and Results
Duration of the monophasic action potential (MAPD) was measured from female rabbit hearts paced at cycle lengths from 400 to 2000 ms and BVR was calculated. In the absence of drug, MAPD90 and BVR increased as the cycle length was increased from 400 to 2000 ms (n=36 and 26, p<0.01). Both E-4031 (20 nmol/L) and d-sotalol (10 μmol/L) increased MAPD90 and BVR at all stimulation rates and the increase was greater at slower than at faster pacing rates (n=19 and 11, 12 and 7, respectively, p<0.01). TTX (1 μmol/L) significantly attenuated the RRD of MAPD90, reduced BVR, (p<0.01), and abolished torsade de pointes (TdP) in 5 of 6 hearts treated with either 20 nmol/L E-4031 or 10 μmol/L d-sotalol. Endogenous late INa in cardiomyocytes stimulated at cycle lengths from 500 to 4000 ms was greater at slower than at faster stimulation rates, and rapidly decreased during the first several beats at faster but not at slower rates (p<0.01, n=8). In a computational model, simulated RRD of APD caused by E-4031 and d-sotalol was attenuated when late INa was inhibited.
Conclusions
Endogenous late INa contributes to the RRD of IKr inhibitor-induced increases in APD and BVR and to bradycardia-related ventricular arrhythmias.
Keywords: Late sodium current, reverse rate dependence, action potential duration, beat-to-beat variability, rabbit heart
Introduction
Reverse rate (or frequency or use)-dependence (RRD) of action potential duration (APD) is an adaptive property of the normal heart wherein the APD shortens as heart rate increases.1 Drug effects may also have a reverse rate-dependency, i.e., the effect of drug to prolong APD may be greater at slow vs. fast heart rates. Drug RRD is associated with ventricular proarrhythmic activity.1, 2 Inhibitions of rapidly-activating delayed rectifier K+ current (IKr) or inward rectifier K+ current (IK1),3 and enhancement of L-type Ca2+-inward current (ICaL)4 have been reported to prolong APD in a RRD manner,5, 6 whereas inhibition of IKs by chromanol 293B prolonged APD in a frequency-independent manner.3, 7. RRD of APD caused by drugs that inhibit IKr is considered to be an undesired response because it results in a reduction of drug efficacy during tachycardia and may lead to proarrhythmic activity when the tachycardia is terminated. 1,8
The mechanisms underlying the RRD of physiological and drug-induced enhanced RRD have not been clearly identified.5 In mammalian hearts in physiological conditions, IKr and IKs are major determinants of APD and IKs is increased at fast heart rates.9 An increase in IKs at faster heart rates due either to incomplete deactivation of current or to accumulation of extracellular K+ is considered a possible mechanism of physiological RRD.10-12 Also, the time- and voltage-dependencies of both drug binding and disassociation from channels are presumed to be causes of drug-induced enhancement of RRD.13, 14 However, none of these mechanisms is fully supported by experimental and/or clinical evidence.5 Furthermore, an increased beat-to-beat variability of repolarization (BVR) is also associated with drug-induced ventricular tachycardia15-19 and the mechanism responsible for the increased BVR and its rate dependency have not been identified.
The endogenous (or physiological) slowly-inactivating component of the inward tetrodotoxin (TTX)-sensitive Na+ current (late INa) in the heart is normally small.20 This current is proarrhythmic when enhanced or when cardiac repolarization reserve is reduced by IKr blockers.16-20 To our knowledge, the role of late INa in RRD of APD has not been previously investigated. We hypothesized that physiological late INa enhances the RRD and BVR of APD prolongation caused by IKr inhibitors in the heart, perhaps because the magnitude of late INa itself may be use-dependent.20 In this study, the contribution of late INa to RRD of the IKr inhibitors sotalol and E-4031 was determined in rabbit isolated hearts and myocytes. TTX (1 μmol/L) and ranolazine (10 μmol/L) were used to provide selective inhibition of INa relative to other ion currents, and selective inhibition of late INa relative to peak INa, respectively.19 The contribution of late INa to RRD of APD was also simulated in a computational model of a rabbit ventricular myocyte AP.
Methods
Female rabbit isolated heart model
The use of animals in this investigation conformed to “Guide for the Care and Use of Laboratory Animals” (NIH publication No. 85-23). Animal use was approved by the Institutional Animal Care and Use Committee of Gilead Sciences (Palo Alto, CA, USA) and by the Administration of Regulation of Laboratory Animals (Hubei Province, China). Hearts from New Zealand White female rabbits weighing 2.5-3.5 kg were isolated, perfused by the method of Langendorff, and instrumented as previously described by Wu et al.17 Monophasic action potentials (MAPs) from the left ventricular epicardium and pseudo 12-lead electrocardiograms (ECGs) were recorded. The AV nodal area was thermally ablated to produce complete atrioventricular block, and hearts were paced at increasing cycle lengths (CLs) of 400, 500, 667, 1000 and 2000 ms for 3-4 minutes at each rate. After control (no drug) measurements were recorded, hearts were exposed to either 20 nmol/L E-4031 or 10 μmol/L d-sotalol in the absence and presence of 1 μmol/L TTX, allowing 20 min between increases in drug concentration to record a steady-state effect. The stimulation protocol at CL from 400 to 2000 ms was repeated before (control) and after each drug treatment.
BVR was determined by analysis of 30 consecutive stimulated beats using the following equation: Σ|MAPDn+1-MAPDn|/[30×√2], as described by Wu et al.16-18 The slope of the restitution curves was calculated based on the fast slope portion (at stimulation CL of 400, 500 and 667 ms) of the relationship between the MAPD90 and diastolic interval.
Whole cell patch clamp study
Rabbit left ventricular cardiomyocytes were isolated enzymatically as prescribed previously.21 Only cardiomyocytes with smooth and glossy edges and surfaces, clear transverse striations, and that could be stably voltage-clamped for 8 min without significant contraction were used. Seal resistance was above 1GΩ with less than 10 % variability. Currents were obtained with a patch-clamp amplifier (EPC-10, Heka Electronic, Lambrecht, Pfalz, Germany), filtered at 1 kHz and digitized at 10 kHz. During recording of late INa, the bath solution contained (in mmol/L):135 NaCl, 5.4 CsCl2, 1.8 CaCl2, 1 MgCl2, 0.3 BaCl2, 0.33 NaH2PO4, 10 glucose, 10 HEPES, 0.001 nicardipine, pH 7.3. The patch pipette solution contained (in mmol/L):120 CsCl2, 1.0 CaCl2, 5 MgCl2, 5 Na2ATP, 10 TEACl, 11 EGTA, 10 HEPES (pH 7.3, adjusted with CsOH). All experiments were carried out at a temperature of 23±1 °C.
To elicit a late INa in a single cell, twenty 300-ms depolarizing pulses to -20 mV from a holding potential of -90 mV were applied at CLs of 500, 667, 1000, 2000 and 4000 ms using a serial repeated-measurement protocol, with 3 min between each change of stimulation CL, during which membrane potential was held at -90 mV. In cardiomyocytes isolated from 8 rabbit hearts, the amplitude of late INa was measured at 200 ms after initiation of the depolarization step before (control, no drug) and 3 min after exposure of a myocyte to 1 and 4 μmol/L TTX, respectively.
Rabbit ventricular AP simulations
The rabbit ventricular AP was simulated using the Shannon-Bers model.22,23 Simulation of the late INa was added to the original model using a Hodgkin-Huxley formalism as done by Hund et al.24
Late INa activation (gating variable m) mimics the activation of the fast INa, whereas inactivation (gating variable h) was formulated as follow:
Late INa maximal conductance (GNa,L) was set to 0.012 mS/μF (i.e., 0.075% of the fast INa conductance) to simulate a small endogenous late INa in rabbit ventricular myocytes. Model differential equations were implemented in Matlab (Mathworks Inc., Natick, MA, U.S.A.) and solved numerically using a variable order solver (ode15s). The digital cell was stimulated with a current pulse (9.5 A/F, 5 ms) at various CLs from 400 to 2000 ms, and APD was measured as the interval between AP upstroke and 90% repolarization level (APD90).
Based on the results of measurements of late INa in rabbit cardiomyocytes, applications of 1 and 4 μmol/L TTX were simulated by reducing GNaL by 47 and 100 %, respectively. GKr was reduced by 55% to simulate the effect of 20 nmol/L E-4031 and by 45% to simulate the effect of 10 μmol/L d-sotalol application.
Statistical analyses
Data were plotted and analyzed using Prism version 5 (Graph Pad Software, San Diego, CA) and expressed as mean ± SEM. The significance of differences of measures before and after interventions in the same hearts was determined by repeated measure one-way analysis of variance (ANOVA) followed by Student-Newman-Keuls test. When treatment values were obtained at different rates from different groups of hearts, two-way ANOVA of repeated measures was used. A paired or un-paired student t test was used to determine the statistical difference between values of two means obtained from the same or different experiments, respectively.
Results
Relationships between pacing heart rate and MAPD or BVR in control hearts
In the absence of drug (control), MAPD90 and BVR were significantly increased in a reverse rate-dependent manner from 148.7±1.67 and 0.20±0.01 to 192.9±3.1 and 0.31±0.01 ms (n=36 and 26, p<0.01, Figs. 1-3, open symbols) as the pacing CL was prolonged from 400 ms to 2000 ms.
Figure 1.
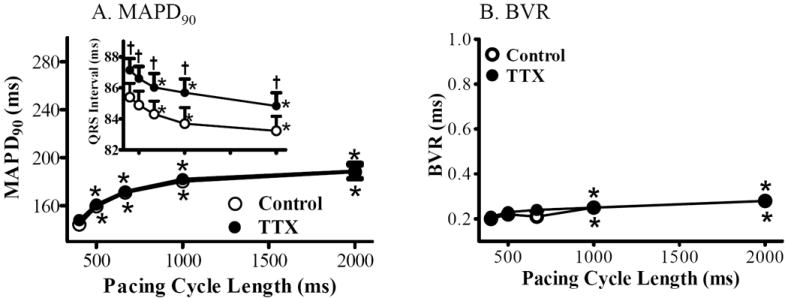
Rate-dependent changes of MAPD and BVR in absence (control, ○) and presence of 1 μmol/L TTX (●). MAPD90 (A), BVR (B) and QRS interval (A, inset) were measured from rabbit isolated hearts paced at CLs of 400, 500, 667, 1000 and 2000 ms, respectively. *, p<0.05 compared with values at pacing CL of 400 ms, p<0.05. †, compared with control at same pacing CL, p<0.05.
Figure 3.
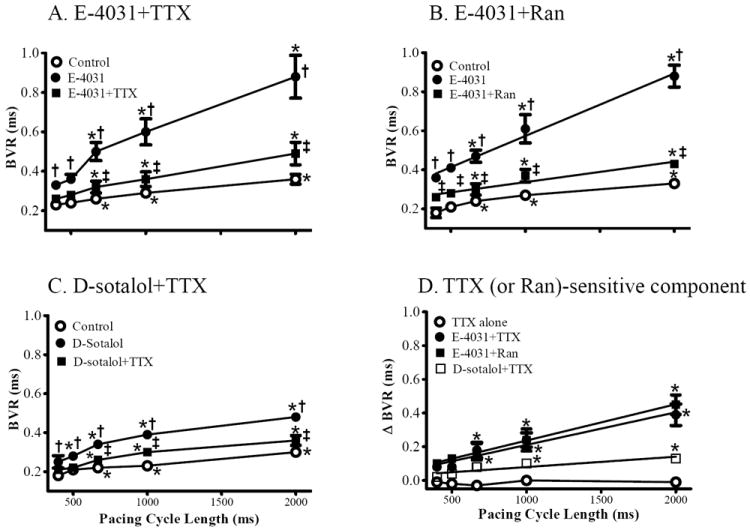
Effects of TTX and ranolazine (Ran) on the reverse rate-dependent changes of BVR of left ventricular MAPD induced by IKr inhibitors. BVR was calculated in absence (control, ○) and presence of either E-4031 (20 nmol/L, ●), E-4031 plus TTX (1 μmol/L, ■, A), E-4031 plus Ran (10 μmol/L, B), or d-sotalol (10 μmol/L) plus TTX (C). Each point indicates a mean of 4, 7 and 11 hearts, respectively. The TTX (or Ran) sensitive components of BVR are plotted in panel D. *, compared to CL of 400 ms, p<0.05-0.001, p<0.05-0.001; †, Compared to control at the same CL; p<0.05-0.001; ‡, compared to either E-4031 (A and B) or d-sotalol (C) at same CL, p<0.05-0.001.
TTX (1 μmol/L) alone did not change MAPD90 or BVR at any tested pacing rate (n=7 and 6, compared to control, p>0.05, Fig. 1). The QRS interval was prolonged by 1 μmol/L TTX by 2.4±0.3 and 1.7±0.5 ms at CL of 400 and 2,000 ms (n=7, compared to control at same rate, p<0.05, inset in Fig. 1A).
RRD effects of IKr inhibitors to increase MAPD and BVR
E-4031 (20 nmol/L) and d-sotalol (10 μmol/L) significantly prolonged MAPD90 (n=19 and 11, Fig. 2) and increased BVR (n=12 and 7, Fig. 3) at all stimulation rates (p<0.05-0.01) and the increase was greater at longer (2000 ms, 26.2±2.2 and 0.04±0.02 ms for E-4031, 17.3±66.9 and 0.07±0.02 for d-sotalol) than at shorter CLs (400 ms, 82.6±9.2 and 0.56±0.08 ms for E-4031, 66.9±9.2 and 0.19±0.02 for d-sotalol, p<0.01); i.e., the actions of E-4031 and d-sotalol on MAPD and BVR were reverse rate-dependent.
Figure 2.
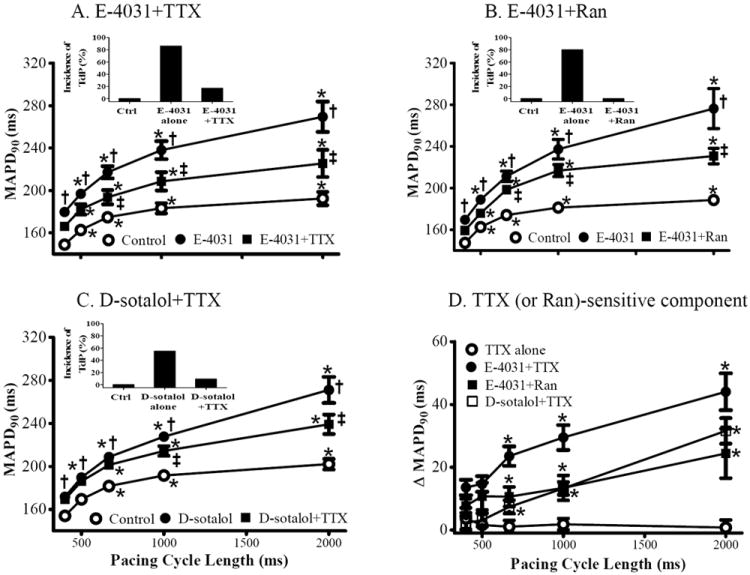
Effect of TTX and ranolazine (Ran) on the reverse rate-dependent changes of left ventricular epicardial MAPD90 caused by IKr inhibitors. Values of MAPD90 were measured in the absence (control, ○) and presence of either E-4031 (20 nmol/L, ●), E-4031 plus TTX (1 μmol/L, ■, A), E-4031 plus Ran (10 μmol/L, B), or d-sotalol (10 μmol/L) plus TTX (C). Each point indicates a mean of 7, 9 and 11 hearts, respectively. The TTX (or Ran) sensitive component of MAPD90 prolongation by each intervention is plotted in panel D. Insets represent the incidence of torsade de pointes in control, E-4031 or d-sotalol in absence and presence of TTX or Ran. *, compared to CL of 400 ms, p<0.05-0.001; †, Compared to control at the same CL; p<0.05-0.001; ‡, compared to either E-4031 (A and B) or d-sotalol (C) alone at the same CL, p<0.05-0.001.
Reversal by TTX and ranolazine of the RRD effects of IK inhibitors
TTX (1 μmol/L) significantly (p<0.05) attenuated the rate-dependent increases in MAPD90 and BVR in the presence of either E-4031(n=9 and 7, Fig. 2A and 3A) or d-sotalol (n=11 and 7, Fig. 2C and 3C) by 36±10 and 68±17%, and 24±8 and 46±17 % at CL of 667 ms (150 bpm), and 63±5 and 75±7 %, and 49±5 and 51±12 %, at CL of 2000 ms (30 bpm), respectively.
The TTX-sensitive component of MAPD and BVR was minimal in control hearts (Figs. 2D and 3D) but was significantly increased in the presence of either E-4031 or d-sotalol, especially at the slower pacing rate of 30 bpm (Figs. 2D and 3D).
In the presence of 20 nmol/L E-4031, the late INa inhibitor ranolazine (Ran, 10 μmol/L) also attenuated the RRD of MAPD and BVR, especially at the longer pacing CLs (n=10 and 6, p<0.05, Figs. 1B and 3B), in spite of the fact that ranolazine is also reported to block IKr as well as late INa.25
Values of QT interval, Tpeak-Tend, and the index of Tpeak-Tend/QT interval had similar RRD as did MAPD and BVR (supplemental figure), and the effects of TTX and ranolazine on these parameters were also similar to the effects of TTX and ranolazine on the rate dependence of MAPD (supplemental figure).
When CL was increased from 500 to 1000 ms (i.e., when stimulation rate was decreased for 120 to 60 beats per minute), MAPD and BVR were increased by 20.6±1.2 (n=36) and 0.04±0.01 ms (n=26, p<0.01) in normal hearts (no drug). In the presence of E-4031 and d-sotalol, MAPD and BVR were further increased to 45.0±4.7 and 0.22±0.04 ms (n=19 and 12, respectively, p<0.01), and 38.1±2.4 and 0.11±0.02 ms (n=11 and 7, p<0.05), respectively. TTX (1 μmol/L) reduced the increases in MAPD and BVR in the continued presence of either E-4031 or d-sotalol to 35.7±4.7 and 0.22±0.08 ms (p<0.05), and 28.0±2.2 and 0.08±0.02 ms (p<0.05), respectively.
The plot of the dependence of MAPD on the duration of the diastolic interval is known as the restitution curve,26 and is similar to the dependence of MAPD on CL that is shown in Fig. 2. Inhibition of IKr by either E-4031 or d-sotalol increased the steepness of the slope of the restitution curve from 0.103±0.021 (control) to 0.158±0.017 and from 0.112±0.024 to 0.157±0.023, respectively (p<0.05, Fig. 4). TTX (1 μmol/L) flattened the restitution curves in the continuous presence of either E-4031 (Fig. 4A) or d-sotalol (Fig. 4B) and decreased the slope of the curves to 0.109±0.029 and 0.131±0.028, respectively (p<0.05).
Figure 4.
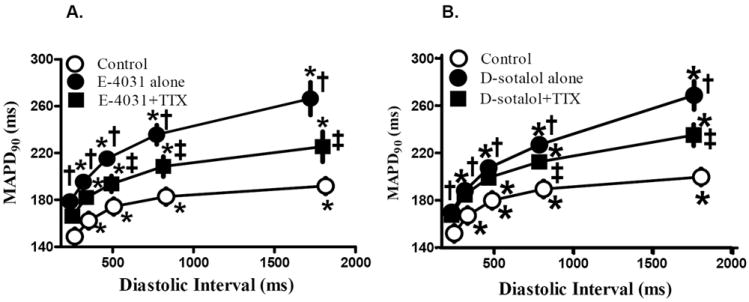
MAPD restitution curve in control (no drug) and in presence of either drug alone (E-4031 in A or d-sotalol in B), or drug plus 1 μmol/L TTX. *, Compared with CL of 400 ms, p<0.05; †, Compared to control at the same CL; p<0.05; ‡, compared to either E-4031 (A and B) or d-sotalol (C) at same CL, p<0.05.
Simulated effect of late INa inhibition on RRD of APD90
We used the Shannon-Bers rabbit ventricular AP model to recapitulate our MAPD90 results from rabbit hearts. Representative AP traces are shown in Figure 5D. The control APD was 252 ms at a CL of 2000 ms, and 185 ms at a CL of 400 ms (Fig 5A, open circles). The rate-dependent properties of late lNa contributed to APD adaptation, as the simulated current amplitude was larger at slow vs. fast pacing rates (not shown). When the effect of TTX was simulated by GNa,L reductions of 47 % (1 μmol/L TTX) and 100 % (4 μmol/L TTX), APD90 was decreased (Fig 5). APD adaptation to change of stimulation rate in the absence and presence of 1 μmol/L TTX was not markedly different (APD90 shortened by ~23 and ~27% with and without TTX respectively, when decreasing the CL from 2000 to 400 ms). TTX shortened APD90 more prominently at longer CLs (1000 and 2000 ms), especially when simulating complete late INa block by 4 μmol/L TTX (Fig. 5A).
Figure 5.
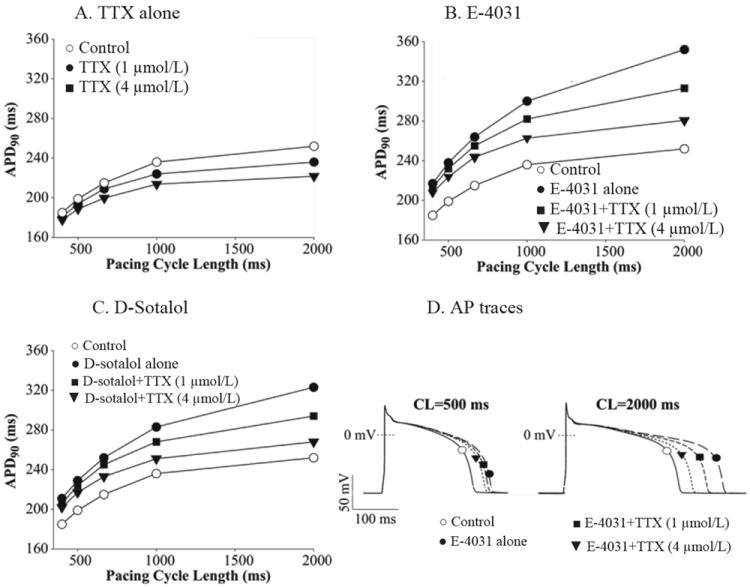
Simulated effect of TTX on the reverse rate-dependent changes of rabbit action potential duration (APD90). A, APD90 prolongation as pacing CL was increased from 400 to 2000 ms in absence (open circles) and presence of TTX (1 and 4 μmol/L). B. RRD of APD90, predicted when reducing IKr conductance to mimic E-4031 administration (solid circles), was greatly attenuated when late INa was inhibited by TTX (1 and 4 μmol/L, solid symbols). C. RRD of APD90 predicted when reducing IKr conductance to mimic d-sotalol administration (solid circles) was reduced or nearly abolished when late INa was inhibited by 1 and 4 μmol/L TTX (solid symbols). D. Representative AP traces obtained when a digital cell was paced at CLs of 500 ms (left) and 2000 ms (right) in control, in presence of IKr block E-4031 alone, and in presence of simultaneous inhibitions of lKr and late INa.
Simulation of IKr block by E-4031 predicted a marked prolongation of the rabbit ventricular AP showing clear RRD (Fig. 5B, squares), being larger at 2000 than at 400 ms CL (100 vs. 32 ms). When late INa inhibition was simultaneously simulated, the model predicted a reduction of RRD in the presence of E-4031 (Fig. 5B, closed circles). Similar results were found when IKr block by d-sotalol was simulated (Fig. 5C), i.e., APD prolongation induced by GKr reduction was more pronounced at slower than at faster heart rates, and this effect was markedly reduced when 47 and 100 % block of late INa was simulated.
Inhibition of endogenous late INa abolished slow rate-related TdP in the presence of E-4031 and d-sotalol
Neither early afterdepolarizations (EAD), extra-ventricular beats (EVB) nor TdP was observed in 28 control hearts at any tested pacing rate. In contrast, in 15 of 17 (88 %) hearts treated with 20 nmol/L E-4031, EAD, frequent EVBs and episodes of TdP were observed when CL was increased from 400 to 2000 ms. Similar arrhythmic activities were observed 6 of 11 (55 %) hearts treated with d-sotalol. These results are consistent with a previous report of the proarrhythmic effects of QT prolonging drugs. EADs, EVBs and episodes of TdP caused by 20 nmol/L E-4031 were abolished by 1 μmol/L TTX and 10 μmol/L ranolazine in 5 of 6 (83 % Fig. 2A, inset) and 8 of 8 (100 %, Fig. 2B, inset) hearts, respectively. Similarly, 1 μmol/L TTX abolished TdP caused by 10 μmol/L d-sotalol in 5 of 6 hearts (83 %, Fig. 2C, inset).
Effect of stimulation rate on magnitude of endogenous late INa in rabbit ventricular myocytes
The magnitude of late INa in rabbit isolated cardiomyocytes decreased with increasing stimulation rate (CL from 4000 to 500 ms) and with use (i.e., from first pulse to 20th pulse) (see representative traces in Fig. 6 and summary data in Fig. 7). The amplitude of late INa recorded during the first pulse of each pulse train was similar: 48.1±3.8, 48.2±3.8, 48.1±4.3, 48.2±4.3 and 47.9±4.1 pA, (n=8, p=NS, Figs.7, 8). TTX (1 and 4 μmol/L) caused a significant decrease in late INa at all stimulation rates, as the rate-late INa response relationship was shifted down in parallel (p<0.01 for each rate, Figs. 6-8). The steady-state amplitudes of late INa recorded from cells paced at 2.0, 1.5, 1.0, 0.5 and 0.25 Hz were -18.8±3.9, -24.3±3.8, -32.0±4.2 and -41.4±4.4 pA, respectively (n=8, p<0.05-0.01). TTX at concentrations of 1 and 4 μmol/L attenuated late INa at all stimulating rates (Figs. 6-8).
Figure 6.
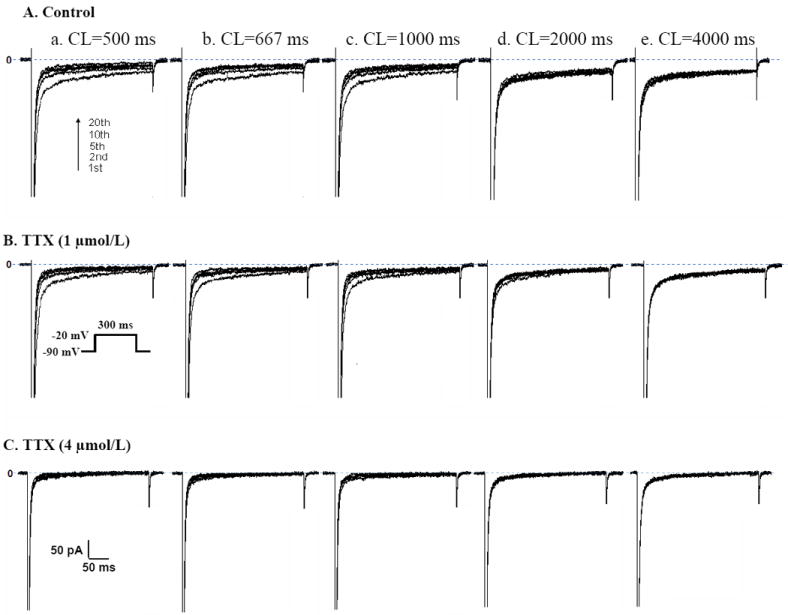
Representative recordings of the rate (2.0, 1.5, 1.0, 0.5 and 0.25 Hz) dependent changes of late Na+ current (late INa) in a single rabbit ventricular myocyte. Traces of INa at different stimulation cycle length (CL in ms) in absence (control, A) and presence of TTX (1 μmol/L in B; 4 μmol/L in C) from impulses # 1, 2, 5, 10 and 20. The depolarization pulse protocol to elicit late INa is shown in the inset.
Figure 7.
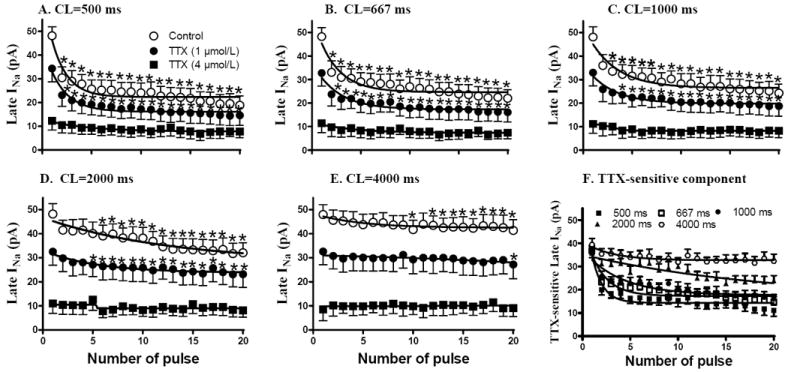
TTX (1 and 4 μmol/L) reduced the beat-to-beat (from pulse 1 to pulse 20) changes of late sodium current (INa) at shorter (from 500 to 2000 ms in CL, A-D) but not at longer (4000 ms in CL, E) stimulation cycle length. The TTX-sensitive component of late INa decreased with a decrease in stimulation CL, as shown in panel F. Each point indicates a mean of measurements from 10 cells. Absolute values of amplitudes of late INa are shown. *, Compared to pulse #1, p<0.05.
Figure 8.
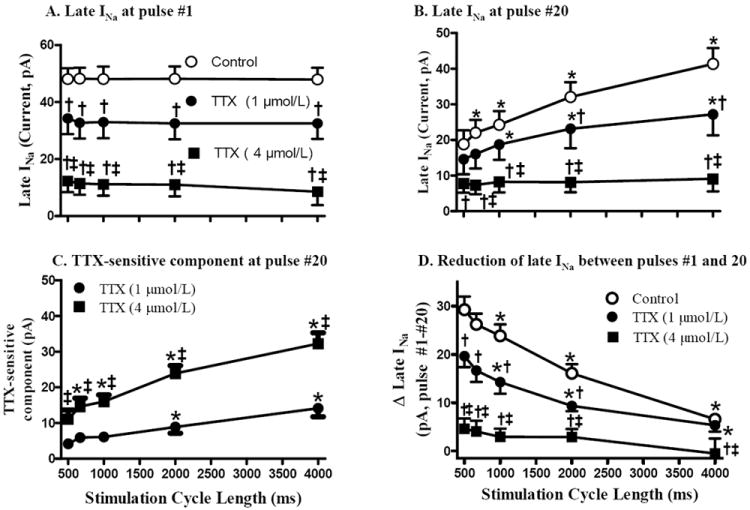
The rate-dependent decrease of late INa (measured as TTX-sensitive current). TTX (1 and 4 μmol/L) was used to inhibit late INa. Late INa at pulse #1 (A) and #20 (B), the TTX sensitive component (control-TTX) at pulse #20 (C), and the reduction of late INa between pulses #1 and 20 (D) are plotted. *, Compared with CL of 500 ms; †, compared with control at same rate; ‡, compared to 1 μmol/L TTX at same rate; p<0.05. Absolute values of amplitudes of late INa are shown.
The amplitude of late INa during a train of 20 pulses decreased from the first to the 20th pulse, and the decrease was greater when the stimulation rate was increased (see Fig. 8D for summary data). Compared to pulse #1, the amplitude of late INa at pulse #20 was significantly decreased to 19.8±4.0 (Fig. 7A), 22.6±4.0 (Fig. 7B), 25.2±4.0 (Fig. 7C) and 32.4±4.4 (Fig. 7D) pA at CLs of 500, 667, 1000, and 2000 Hz, respectively, (n=8, p<0.05) but did not change at CL of 4000 ms (42.5±4.6 pA, Fig. 7E, n=8, p=NS). A higher rate of stimulation was associated with a faster decrease in late INa. The greatest decrease of late INa occurred during the first few pulses at each rate (Fig. 7).
Discussion
The findings in this study suggest that physiological late INa may play an important role in the enhanced RRD of APD and BVR caused by QT-prolonging agents that inhibit IKr. RRD of MAPD and BVR were demonstrated using female rabbit isolated hearts, and RRD of APD was simulated in a computer model. In the continuous presence of IKr inhibitors, TTX (1 μmol/L) and ranolazine (10 μmol/L) reduced the RRD of MAPD and BVR induced by IKr inhibitors, and abolished slow rate-related episodes of TdP in the presence of IKr inhibitors. Indeed, the amplitude of endogenous late INa was larger at slower than faster heart rates. Because TTX and ranolazine are known to block cardiac late INa (NaV1.5) at concentrations of 1 and 10 μmol/L, respectively, 19, 20 these results suggest that inhibition of late INa may diminish the RRD of the APD/QT interval prolongation and BVR, and therefore may antagonize the slow rate- and pause-triggered ventricular arrhythmias that may be caused by disease or drug-induced inhibition of IKr.
RRD is an implicit property of the heart that reflects the underlying rate dependence of individual cardiac ion channel currents 5, 27, and thus their contribution to membrane resistance during the AP plateau. In this study, the magnitude of late INa was also found to be reverse rate-dependent. Thus, reduction of the reverse rate-dependent inward late INa by TTX or ranolazine “offset” reduction of outward K+ current by reverse rate-dependent IKr-blocking drugs. Inhibition of late INa by either 1 μmol/L TTX or 10 μmol/L ranolazine reduced the RRD of MAPD in hearts treated with the IKr blockers E-4031 and d-sotalol (Figs. 2, 3). Simulated data from a computer model of rabbit heart electrical activity (including changes in intracellular Na+, Ca2+ and late INa) recapitulated the RRD of APD in the presence of IKr blockers, and predicted that a reduction of IKr accentuates the RRD of APD. Thus, both experimental data and model predictions provide evidence that the RRD of effects of E-4031 and d-sotalol were reduced when late INa was inhibited, and that the contribution of late INa to prolongation of ventricular repolarization was greater at slower heart rates. The latter result is consistent with a previous study of LQT3 ΔKPQ mutant Na+ channels expressed in HEK293 cells, wherein it was reported that the amplitude of ΔKPQ late INa was strongly rate-dependent.28
Increased BVR is proarrhythmic and has been associated with interventions that either block IK or augment late INa, especially at slower heart rates.15 Late INa blockade with 1 μmol/L TTX did not significantly alter baseline RRD of BVR in rabbit hearts, but attenuated the increased RRD of BVR during administration of E-4031 and d-sotalol. Although inhibition of physiological late INa by TTX and lidocaine is sufficient to shorten APD in normal cardiac tissue.19, 20, 29, our findings suggest that late INa contributes more to BVR when repolarization reserve is reduced and the net transmembrane current is small (i.e., during the long AP plateau in the presence of an IKr inhibitor) and support previous reports that endogenous late INa is proarrhythmic in hearts with reduced repolarization reserve.16, 18, 20 We speculate that reduction of a depolarizing current during the AP plateau shortens the ‘vulnerable phase’ in which stochastic fluctuations in ion channel or transporter currents, or local Ca2+ concentrations, can have significant impact on the duration of ventricular repolarization.15 Indeed, Ca2+-dependent mechanisms are thought to underlie BVR, as exogenous intracellular Ca2+ buffering suppresses it. 30
The increased density of late INa recorded from rabbit cardiomyocytes paced at slow vs. fast rates is consistent with the RRD of MAPD and BVR in isolated hearts and in the computational model.17 The increase in late INa at slower heart rates may be attributed to decreased inactivation of Na+ channels at these slow rates,30 as the greatest decrease in late INa was found during the first several beats when the pacing rate was increased (Fig. 7). In the computer model, APD adaptation (i.e., shortening) to an increase of pacing rate was linked to increased intracellular Na+ accumulation at fast rates, and its consequences to increase outward Na+ pump and NCX currents.27, 31 A potential positive feedback between increased late INa and reduced IKr can explain the effect of an enhanced late INa to cause arrhythmias when repolarization reserve is reduced.19 An increase in late INa may also cause changes in Ca2+ handling that alter APD adaptation by multiple mechanisms.20, 27, 32
The observation that late INa plays an important role in the enhanced RRD of IKr inhibitors may explain previous experimental and clinical findings that RRD is greater in Purkinje fibers and mid-myocardial cells where the late INa is greater than in epicardial cells,33 and that IKr-blocking drugs such as amiodarone and ranolazine that also block late INa have no RRD and no or minimal proarrhythmic risk.16, 17, 34 On the other hand, due to the synergistic effect of increased late INa and inhibition of IKr to increase APD and BVR,17, 35 an increase of late INa, as occurs in the ischemic heart and other structure heart diseases,36 can potentiate the proarrhythmic effect of an IKr blocker.
Conclusion
Endogenous physiological late INa plays an important role in the RRD of APD and BVR caused by IK inhibitors. Inhibition of endogenous late INa may diminish the rate-dependent prolongation of the APD/QT interval by either drugs or pathological conditions that decrease IKr, and may decrease the occurrence of slow rate- or pause-triggered cardiac arrhythmias. If late INa were to be exacerbated by pathological conditions, it is possible that it’s impact to increase the RRD of IKr blocking drugs would also be augmented.
Supplementary Material
Acknowledgments
Funding Sources
Rabbit isolated heart and patch clamp studies were supported by Gilead Sciences.
Footnotes
Disclosures
Drs. Wu, Li, Shryock and Belardinelli are employees of Gilead Sciences; Dr. Ma received a research grant from Gilead; Dr. Grandi and Bers have no disclosures.
References
- 1.Dorian P, Newman D. Rate dependence of the effect of antiarrhythmic drugs delaying cardiac repolarization: an overview. Europace. 2000;2:277–285. doi: 10.1053/eupc.2000.0114. [DOI] [PubMed] [Google Scholar]
- 2.Hondeghem LM. Use and abuse of QT and TRIaD in cardiac safety research: importance of study design and conduct. Eur J Pharmacol. 2008;584:1–9. doi: 10.1016/j.ejphar.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 3.Bosch RF, Gaspo R, Busch AE, Lang HJ, Li GR, Nattel S. Effects of the chromanol 293B, a selective blocker of the slow, component of the delayed rectifier K+ current, on repolarization in human and guinea pig ventricular myocytes. Cardiovasc Res. 1998;38:441–450. doi: 10.1016/s0008-6363(98)00021-2. [DOI] [PubMed] [Google Scholar]
- 4.Bril A, Forest MC, Cheval B, Faivre JF. Combined potassium and calcium channel antagonistic activities as a basis for neutral frequency dependent increase in action potential duration: comparison between BRL-32872 and azimilide. Cardiovasc Res. 1998;37:130–140. doi: 10.1016/s0008-6363(97)00216-2. [DOI] [PubMed] [Google Scholar]
- 5.Banyasz T, Horvath B, Virag L, Barandi L, Szentandrassy N, Harmati G, Magyar J, Marangoni S, Zaza A, Varro A, Nanasi PP. Reverse rate dependency is an intrinsic property of canine cardiac preparations. Cardiovasc Res. 2009 doi: 10.1093/cvr/cvp213. [DOI] [PubMed] [Google Scholar]
- 6.Virag L, Acsai K, Hala O, Zaza A, Bitay M, Bogats G, Papp JG, Varro A. Self-augmentation of the lengthening of repolarization is related to the shape of the cardiac action potential: implications for reverse rate dependency. Br J Pharmacol. 2009;156:1076–1084. doi: 10.1111/j.1476-5381.2009.00116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bauer A, Becker R, Freigang KD, Senges JC, Voss F, Hansen A, Muller M, Lang HJ, Gerlach U, Busch A, Kraft P, Kubler W, Schols W. Rate- and site-dependent effects of propafenone, dofetilide, and the new I(Ks)-blocking agent chromanol 293b on individual muscle layers of the intact canine heart. Circulation. 1999;100:2184–2190. doi: 10.1161/01.cir.100.21.2184. [DOI] [PubMed] [Google Scholar]
- 8.Grom A, Faber TS, Brunner M, Bode C, Zehender M. Delayed adaptation of ventricular repolarization after sudden changes in heart rate due to conversion of atrial fibrillation. A potential risk factor for proarrhythmia? Europace. 2005;7:113–121. doi: 10.1016/j.eupc.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 9.Rocchetti M, Besana A, Gurrola GB, Possani LD, Zaza A. Rate dependency of delayed rectifier currents during the guinea-pig ventricular action potential. The Journal of physiology. 2001;534:721–732. doi: 10.1111/j.1469-7793.2001.00721.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jurkiewicz NK, Sanguinetti MC. Rate-dependent prolongation of cardiac action potentials by a methanesulfonanilide class III antiarrhythmic agent. Specific block of rapidly activating delayed rectifier K+ current by dofetilide. Circ Res. 1993;72:75–83. doi: 10.1161/01.res.72.1.75. [DOI] [PubMed] [Google Scholar]
- 11.Tsujimae K, Suzuki S, Murakami S, Kurachi Y. Frequency-dependent effects of various IKr blockers on cardiac action potential duration in a human atrial model. Am J Physiol Heart Circ Physiol. 2007;293:H660–669. doi: 10.1152/ajpheart.01083.2006. [DOI] [PubMed] [Google Scholar]
- 12.Lu Z, Kamiya K, Opthof T, Yasui K, Kodama I. Density and kinetics of I(Kr) and I(Ks) in guinea pig and rabbit ventricular myocytes explain different efficacy of I(Ks) blockade at high heart rate in guinea pig and rabbit: implications for arrhythmogenesis in humans. Circulation. 2001;104:951–956. doi: 10.1161/hc3401.093151. [DOI] [PubMed] [Google Scholar]
- 13.Hondeghem LM, Katzung BG. Time- and voltage-dependent interactions of antiarrhythmic drugs with cardiac sodium channels. Biochim Biophys Acta. 1977;472:373–398. doi: 10.1016/0304-4157(77)90003-x. [DOI] [PubMed] [Google Scholar]
- 14.Stork D, Timin EN, Berjukow S, Huber C, Hohaus A, Auer M, Hering S. State dependent dissociation of HERG channel inhibitors. Br J Pharmacol. 2007;151:1368–1376. doi: 10.1038/sj.bjp.0707356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Johnson DM, Heijman J, Pollard CE, Valentin JP, Crijns HJ, Abi-Gerges N, Volders PG. I(Ks) restricts excessive beat-to-beat variability of repolarization during beta-adrenergic receptor stimulation. J Mol Cell Cardiol. 48:122–130. doi: 10.1016/j.yjmcc.2009.08.033. [DOI] [PubMed] [Google Scholar]
- 16.Wu L, Rajamani S, Shryock JC, Li H, Ruskin J, Antzelevitch C, Belardinelli L. Augmentation of late sodium current unmasks the proarrhythmic effects of amiodarone. Cardiovasc Res. 2008;77:481–488. doi: 10.1093/cvr/cvm069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu L, Shryock JC, Song Y, Belardinelli L. An increase in late sodium current potentiates the proarrhythmic activities of low-risk QT-prolonging drugs in female rabbit hearts. J Pharmacol Exp Ther. 2006;316:718–726. doi: 10.1124/jpet.105.094862. [DOI] [PubMed] [Google Scholar]
- 18.Wu L, Guo D, Li H, Hackett J, Yan GX, Jiao Z, Antzelevitch C, Shryock JC, Belardinelli L. Role of late sodium current in modulating the proarrhythmic and antiarrhythmic effects of quinidine. Heart Rhythm. 2008;5:1726–1734. doi: 10.1016/j.hrthm.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wu L, Rajamani S, Li H, January CT, Shryock JC, Belardinelli L. Reduction of repolarization reserve unmasks the proarrhythmic role of endogenous late Na(+) current in the heart. Am J Physiol Heart Circ Physiol. 2009;297:H1048–1057. doi: 10.1152/ajpheart.00467.2009. [DOI] [PubMed] [Google Scholar]
- 20.Zaza A, Belardinelli L, Shryock JC. Pathophysiology and pharmacology of the cardiac “late sodium current”. Pharmacol Ther. 2008;119:326–339. doi: 10.1016/j.pharmthera.2008.06.001. [DOI] [PubMed] [Google Scholar]
- 21.Zheng J, Ma J, Zhang P, Hu L, Fan X, Tang Q. Milrinone inhibits hypoxia or hydrogen dioxide-induced persistent sodium current in ventricular myocytes. Eur J Pharmacol. 2009;616:206–212. doi: 10.1016/j.ejphar.2009.06.021. [DOI] [PubMed] [Google Scholar]
- 22.Shannon TR, Wang F, Puglisi J, Weber C, Bers DM. A mathematical treatment of integrated Ca dynamics within the ventricular myocyte. Biophys J. 2004;87:3351–3371. doi: 10.1529/biophysj.104.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Grandi E, Puglisi JL, Wagner S, Maier LS, Severi S, Bers DM. Simulation of Ca-calmodulin-dependent protein kinase II on rabbit ventricular myocyte ion currents and action potentials. Biophys J. 2007;93:3835–3847. doi: 10.1529/biophysj.107.114868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hund TJ, Decker KF, Kanter E, Mohler PJ, Boyden PA, Schuessler RB, Yamada KA, Rudy Y. Role of activated CaMKII in abnormal calcium homeostasis and I(Na) remodeling after myocardial infarction: insights from mathematical modeling. J Mol Cell Cardiol. 2008;45:420–428. doi: 10.1016/j.yjmcc.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Antzelevitch C, Belardinelli L, Zygmunt AC, Burashnikov A, Di Diego JM, Fish JM, Cordeiro JM, Thomas G. Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation. 2004;110:904–910. doi: 10.1161/01.CIR.0000139333.83620.5D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Franz MR. The electrical restitution curve revisited: steep or flat slope--which is better? Journal of cardiovascular electrophysiology. 2003;14:S140–147. doi: 10.1046/j.1540.8167.90303.x. [DOI] [PubMed] [Google Scholar]
- 27.Grandi E, Pasqualini FS, Bers DM. A novel computational model of the human ventricular action potential and Ca transient. J Mol Cell Cardiol. 48:112–121. doi: 10.1016/j.yjmcc.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagatomo T, January CT, Ye B, Abe H, Nakashima Y, Makielski JC. Rate-dependent QT shortening mechanism for the LQT3 deltaKPQ mutant. Cardiovasc Res. 2002;54:624–629. doi: 10.1016/s0008-6363(02)00265-1. [DOI] [PubMed] [Google Scholar]
- 29.Elharrar V, Atarashi H, Surawicz B. Cycle length-dependent action potential duration in canine cardiac Purkinje fibers. Am J Physiol. 1984;247:H936–945. doi: 10.1152/ajpheart.1984.247.6.H936. [DOI] [PubMed] [Google Scholar]
- 30.Zaniboni M, Pollard AE, Yang L, Spitzer KW. Beat-to-beat repolarization variability in ventricular myocytes and its suppression by electrical coupling. Am J Physiol Heart Circ Physiol. 2000;278:H677–687. doi: 10.1152/ajpheart.2000.278.3.H677. [DOI] [PubMed] [Google Scholar]
- 31.Carmeliet E. Intracellular Ca(2+) concentration and rate adaptation of the cardiac action potential. Cell Calcium. 2004;35:557–573. doi: 10.1016/j.ceca.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 32.Zaza A. Control of the cardiac action potential: The role of repolarization dynamics. J Mol Cell Cardiol. 2009;48:106–111. doi: 10.1016/j.yjmcc.2009.07.027. [DOI] [PubMed] [Google Scholar]
- 33.Zygmunt AC, Eddlestone GT, Thomas GP, Nesterenko VV, Antzelevitch C. Larger late sodium conductance in M cells contributes to electrical heterogeneity in canine ventricle. Am J Physiol Heart Circ Physiol. 2001;281:H689–697. doi: 10.1152/ajpheart.2001.281.2.H689. [DOI] [PubMed] [Google Scholar]
- 34.Sager PT, Uppal P, Follmer C, Antimisiaris M, Pruitt C, Singh BN. Frequency-dependent electrophysiologic effects of amiodarone in humans. Circulation. 1993;88:1063–1071. doi: 10.1161/01.cir.88.3.1063. [DOI] [PubMed] [Google Scholar]
- 35.Song Y, Shryock JC, Wu L, Belardinelli L. Antagonism by ranolazine of the proarrhythmic effects of increasing late INa in guinea pig ventricular myocytes. J Cardiovasc Pharmacol. 2004;44:192–199. doi: 10.1097/00005344-200408000-00008. [DOI] [PubMed] [Google Scholar]
- 36.Sadanaga T, Ogawa S. Bepridil produces prominent bradycardia-dependent QT prolongation in patients with structural heart disease. J Electrocardiol. 2007;40:426–431. doi: 10.1016/j.jelectrocard.2007.02.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.