

Abstract
Spleen Tyrosine Kinase (SYK) was recently identified as a new target in acute myeloid leukemia (AML); however, its mechanistic role in this disease is poorly understood. Based on the known interaction between SYK and mTOR signaling in lymphoma, we hypothesized that SYK may regulate mTOR signaling in AML. Both small-molecule inhibition of SYK and SYK-directed shRNA suppressed mTOR and its downstream signaling effectors, as well as its upstream activator, AKT. Moreover, the inhibition of multiple nodes of the PI3K signaling pathway enhanced the effects of SYK suppression on AML cell viability and differentiation. Evaluation of the collateral MAPK pathway revealed a heterogeneous response to SYK inhibition in AML with down-regulation of MEK and ERK phosphorylation in some AML cell lines but a paradoxical increase in MEK/ERK phosphorylation in RAS-mutated AML. These studies reveal SYK as a regulator of mTOR and MAPK signaling in AML and demonstrate that inhibition of PI3K pathway activity enhances the effects of SYK inhibition on AML cell viability and differentiation.
Keywords: AML, SYK, mTOR, differentiation therapy, RPS6, 4E-BP1, MAPK
Introduction
Acute myeloid leukemia (AML), the most common form of acute leukemia in adults, continues to have poor survival rates. The overall five-year survival for adults under 50 years is approximately 50% and for those over 65 years is less than 20%.1 New and less toxic approaches to treat AML are needed. With the success of all-trans retinoic acid (ATRA) as a differentiating agent for acute promyelocytic leukemia (APL), there has been interest in identifying new pro-differentiating compounds effective in the non-APL AML subtypes.2 Because the targets of differentiation in AML remain largely unknown, or pharmacologically intractable, and because the differentiation phenotype is complex, our laboratory developed a new gene expression-based approach to compound discovery called Gene Expression-based High-throughput Screening (GE-HTS). This chemical genomics approach, integrated with proteomics and shRNA screening, previously identified spleen tyrosine kinase (SYK) as a candidate pro-differentiating target in AML.3–5
SYK is a non-receptor tyrosine kinase that is broadly expressed in hematopoietic cells and is an important mediator of signal transduction and differentiation, particularly in B-cell development.6 The importance of SYK in hematological malignancies has been recognized in lymphoma, leukemia, and myelodysplastic syndrome (MDS). In peripheral T-cell lymphoma, recurrent ITK-SYK translocations have been reported,7 and conditional expression of ITK-SYK in mice induces highly malignant peripheral T-cell lymphoma with 100% penetrance.8 In diffuse large B-cell lymphoma (DLBCL), SYK-dependent tonic B-cell receptor signaling has been recognized as an important survival pathway,9 and in pre-B cells, deregulated SYK has been reported to inhibit differentiation and induce growth factor independence.10 Moreover, in a clinical trial, SYK inhibition had activity in patients with non-Hodgkin's lymphoma and chronic lymphocytic leukemia.11 A role for SYK in myeloid malignancies was first suggested with the report of a fusion of TEL to SYK in a patient with MDS with t(9;12)(q22;p12).12 Importantly, this TEL-SYK fusion transforms the interleukin-3 (IL-3) dependent murine hematopoietic cell line Ba/F3 to growth factor independence.12 We identified AML as another hematologic malignancy in which SYK plays an important role.3
While we have established that targeting SYK reduces viability and promotes differentiation in AML, little is known about the downstream signaling effectors of SYK in AML. There is a significant body of literature documenting the role of SYK in non-Hodgkin's lymphoma, which has served as a useful framework for investigating SYK in AML.8, 9, 11, 13–16 In B-cell lymphoma, SYK has been demonstrated to be a critical regulator of mTOR activity.14, 17 mTOR positively regulates protein synthesis by activating two primary signaling branches: p70S6K/RPS6 and 4E-BP1/eIF4E.18 This regulation, in turn, controls cap-dependent translation of mRNAs with highly structured 5' UTRs, a feature characteristic of transcripts for many oncogenic proteins. There has been much interest in mTOR as a target in AML. mTOR has been found to be constitutively activated in the majority of primary AML blasts, and it has been shown to be important for AML cell survival after etoposide treatment.19 Furthermore, the inhibition of mTOR in AML has been associated with both anti-proliferative and pro-differentiating effects 19–25 and mTOR inhibitors are now being tested in patients with AML.23, 26–29
In light of the evidence that SYK has been shown to activate mTOR in lymphoma and that mTOR plays an important role in AML, we hypothesized that SYK may also regulate mTOR signaling in AML. Here, we test this hypothesis using both chemical and genetic inhibition of SYK to assess the effects on mTOR and its upstream activators and downstream signaling effectors. We demonstrate that inhibition and constitutive activation of SYK lead to corresponding inhibition and activation of mTOR signaling and that concurrent inhibition of SYK and the PI3K pathway can promote differentiation and inhibit viability in AML cells. Moreover, we reveal a heterogeneous response in the collateral MAPK pathway to SYK inhibition in AML, with down-regulation of MEK and ERK phosphorylation in some AML cell lines but a paradoxical increase in phosphorylation in RAS-mutated AML.
Materials and Methods
Cell Culture
HL-60, U937, and KG-1 were purchased from the American Type Culture Collection (ATCC) (Manassas, VA, USA). THP-1 and MOLM-14 were kindly provided by Dr. Scott Armstrong and NOMO-1 by Dr. Ross Levine. Some of the well characterized genetic features of these cell lines include: HL-60 (NRAS mutation30), U937 (CALM-AF10 rearrangement31) KG-1 (FGFR1OP2-FGFR1 rearrangement32), THP-1 (MLL-AF9 rearrangement,33NRAS mutation34), MOLM-14 (MLL-AF9 rearrangement,35FLT-3 ITD36), and NOMO-1 (KRAS mutation37). All cell lines were maintained in RPMI 1640 (Cellgro, Manassas, VA, USA) supplemented with 1% penicillin-streptomycin and 10% fetal bovine serum (Sigma-Aldrich, St. Louis, MO, USA) at 37°C with 5% CO2. Primary patient AML blasts were collected from peripheral blood or bone marrow aspirate after obtaining patient informed consent under Dana-Farber Cancer Institute Internal Review Board-approved protocols. Mononuclear cells were isolated using Ficoll-Paque Plus (Amersham Biosciences) and red blood cells lysed before staining for flow cytometry analysis.
Chemicals
R940406 (R406, the active metabolite of fostamatinib) (supplied by Rigel Pharmaceuticals, Inc., South San Francisco, CA, and AstraZeneca Pharmaceuticals, Wilmington, DE, USA), rapamycin (Santa Cruz Biotechnology, Inc., Dallas, Texas, USA) and PD0325901 (Selleck, Houston, Texas, USA) were resuspended in dimethyl sulfoxide (DMSO) (Sigma-Aldrich) and stored at −80°C. 4EGI-1 (kindly provided by Dr. Gerhard Wagner), ATRA (Sigma-Aldrich), Torin 1 (Tocris, Bristol, UK), GDC-0941 (Selleck) and Syk Inhibitor IV, BAY 61-3606 (EMD Biosciences, Darmstadt, Germany) were dissolved in DMSO and stored at −20°C.
Immunoblotting
Cells were lysed using Cell Signaling Lysis Buffer (Cell Signaling, Danvers, MA, USA) containing Complete, EDTA-free Protease Inhibitor Cocktail Tablet (Roche, Indianapolis, IN, USA) and PhosSTOP Phosphatase Inhibitor Tablet (Roche) for protein extraction, as per the manufacturer's instructions. Protein was quantified, resolved by gel electrophoresis, and transferred to nitrocellulose membranes (BioRad Laboratories, Hercules, CA, USA). Blots were incubated with primary antibodies to p-SYK (Tyr525/526) (Cell Signaling 2710), p-mTOR (Ser2448) (Cell Signaling 2971), p-RPS6 (Ser240/244) (Cell Signaling 2215), p-4E-BP1 (Thr37/46) (Cell Signaling 2855), p-AKT (Ser473) (Cell Signaling 4060), p-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (Cell Signaling 4370), p-MEK1/2 (Ser217/221) (Cell Signaling 9154), p-c-RAF (Ser338) (Cell signaling 9427), total SYK (Santa Cruz Biotechnology, SC-1240) (Santa Cruz, CA, USA), total mTOR (Cell Signaling 2972), total RPS6 (Cell Signaling 2217), total 4E-BP1 (SC-81149), total 4E-BP1 (Cell Signaling 9644), total AKT (Cell Signaling 2920), total p44/42 MAPK (Erk1/2) (Cell Signaling 4696), total MEK1/2 (Cell Signaling 4694), GAPDH (Cell Signaling 2118), total c-RAF (Cell Signaling 9422), and Vinculin (Abcam, ab18058) (Cambridge, MA, USA). After incubation in primary antibody for two hours at room temperature, blots were washed and incubated in secondary anti-rabbit-HRP (Amersham #NA9340V) (Piscataway, NJ, USA) or anti-mouse-HRP antibodies (Amersham #NA9310V) for one hour at room temperature. Bound antibody was detected by chemiluminescence using either Western Lightning Plus ECL (PerkinElmer, Inc., Waltham, MA, USA) or SuperSignal West Dura Extended Duration Substrate (ThermoScientific, Rockford, IL, USA).
Phospho-specific Flow Cytometry
All flow cytometry studies using fluorophore-conjugated primary antibodies were conducted with reagents and protocols from BD Biosciences (San Jose, CA, USA). The intracellular phospho-specific flow protocol used was Protocol III (Mild or Harsh Alcohol Method) for human peripheral blood mononuclear cells per the manufacturer's instructions. Flow cytometric analysis was performed using the BD FACS Canto II flow cytometer. Cells were gated by forward and side scatter to select viable cells for analysis. Data were analyzed with the FlowJo Tree Star software package (Ashland, OR, USA). Antibodies used were purchased from BD Biosciences and included p-RPS6 (pS240) (560430), p-4E-BP1 (pT36/pT45) (560285), and total-SYK (552476). Antibody anti-p-SYK (Y525/526) was purchased from Cell Signaling Technology (6485). For the primary patient AML/MDS studies, ficoll separated mononuclear cells were incubated for 45 minutes with a combination of anti-human PE-Cy7 CD13 (eBioscience # 25-0138-41) and anti-human PE-Cy7 CD33 (eBioscience # 25-0338-41) (used at 1/50) or PE-Cy7 isotype control antibodies before staining with anti-phospho-antibodies. Effects of R406 on p-RPS6, p-4E-BP1 and pSYK, in the CD13 and/or CD33 positive population of cells, were determined.
Morphological Evaluation
Cytospin preparations were performed with a Thermo Scientific Shandon Cytospin 4 instrument (Rockford, IL, USA). May Grunwald Giemsa staining (Sigma-Aldrich) was used to assess changes in cellular morphology. Stained cells were examined by light microscopy at 400× magnification with an Olympus BX41 microscope (Center Valley, PA, USA) and Q-capture software (Surrey, BC, Canada).
Gene Expression-based High-throughput Screening (GE-HTS)
GE-HTS was conducted using methods previously described to assess for an expression signature composed of genes that distinguish AML from either the neutrophil or monocyte differentiated state.5, 38 The 32 marker genes for myeloid differentiation were chosen using previously published Affymetrix AML-related data sets (Supplementary Table S1).5 These genes have been shown to distinguish AML from either neutrophil or monocyte with p < 0.05 by t-test and to distinguish undifferentiated versus HL-60 differentiated with ATRA (Sigma-Aldrich), phorbol 12-myristate 13-acetate, or 1,25-dihydroxyvitamin D3 with p < 0.05 by t-test. The GE-HTS assay was performed as detailed in the Supplemental Experimental Procedures in Hahn et al., 2009.3 This assay uses ligation-mediated amplification with a fluorescent bead-based detection system to quantify the expression of up to 500 genes in a single well. Two primary scoring methods were used to quantify induction of the 32-gene myeloid differentiation signature.3, 5 The Summed Score combines expression ratios (marker gene/control gene) by summing them with a sign determined by the expected direction of regulation from ATRA-treated positive controls. The Weighted Summed Score combines expression ratios by summing them with a weight and sign determined by the signal-to-noise ratio of each expression ratio for the positive control (ATRA-treated) and negative control (DMSO-treated) samples. To assess the statistical significance of the differences between these differentiation scores a one-way ANOVA with a Bonferroni correction was employed.
Flow Cytometric Analysis of Activated Caspase 3, BrdU Incorporation and CD14/CD11b Expression
Cells were washed with ice-cold PBS and then stained with a cocktail of CD11b-FITC / CD14-PE (Beckman Coulter) antibodies diluted in PBS 2 mM EDTA 0.2% BSA. After fixation and permeabilization with Cytofix/Cytoperm solution (BD Bioscience) and two washes with Perm/Wash solution (BD Bioscience), cells were incubated for 30 min with anti-active-caspase-3-APC monoclonal antibody (BD Pharmingen). Finally, cells were washed and resuspended in Perm/Wash solution before being analyzed using the BD FACS Canto II flow cytometer.
BrdU staining was performed using FITC BrdU Flow Kit (BD Pharmingen) per the manufacturer's instructions.
Viability Assay
Viability experiments were performed using the Promega Cell-Titer Glo ATP-based assay (Madison, WI, USA) per the manufacturer's instructions. Luminescence was measured using a Fluostar Omega by BMG-labtech (Cary, NC, USA). Results were assessed using an excess above Bliss additivism model. This model predicts the combined response, C, of two individual agents with the effects A and B using the formula C = A + B − A × B.39, 40
Lentiviral Vectors and Infection
Oligonucleotides encoding shRNAs were cloned into pLKO.1 as described previously.41 The SYK-directed shRNAs chosen for study included SYK_1, CCGGGCAGGCCATCATCAGTCAGAACTCGAGTTCTGACTGATGATGCCTGCTTTTT and SYK_10, CCGGGCAGCAGAACAGACATGTCAACTCGAGTTGACATGTCTGTTCTGCTGCTTTTTTG, the shRNAs previously confirmed to yield efficient knockdown of SYK,3 and a control shRNA, CCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGGTTTTT
293T cells were used to produce lentivirus containing the desired SYK-directed shRNA plasmid. Specifically, 500,000 293T cells were plated in 6 cm plates and transfected 24 hr later with 1 μg of DNA from the lentiviral backbone vector and packaging plasmids (pCMVdeltaR8.91 and pCMV-VSV-G) according to the FuGENE 6 (Roche) protocol. Medium was changed to RPMI 1640 (Cellgro) 24 hours post-transfection, and viral supernatant was harvested and filtered 48 hours post-transfection. Cells were infected for 2 hr at 37°C with 2 ml of lentivirus and 8 μg/ml polybrene (Sigma-Aldrich). Cells were selected 48 hr later with 1 μg/ml puromycin (Sigma-Aldrich).
To generate the SYK-TEL construct, RT-PCR was used to isolate the sequence encoding amino acids 1–336 of human ETV6 (TEL) and the full length sequence of human SYK. The assembled cDNA was subcloned from the full-length sequence of SYK into a pWZL-Neo Retroviral vector (Cell Biolabs, Inc. San Diego, CA). The SYK-TEL kinase dead (KD) construct was generated by point mutation (K402R) in the kinase domain of SYK, using a QuikChange XL Site-directed mutagenesis kit (Stratagene). The primer sequences used to generate these mutants were 5'-GTG AAA ACC GTG GCT GTG CGA ATA CTG AAA AAC GAG GC and 5'-GCC TCG TTT TTC AGT ATT CGC ACA GCC ACG GTT TTC AC. The pLenti 6.2 vector encoding V5-tagged full length SYK was generated by performing an LR recombination reaction between the pENTR/SD/D-TOPO entry vector containing the wildtype, full length, sequence of SYK and the pLenti 6.2/V5-DEST Gateway destination vector.
Results
Overexpression of SYK leads to increased activation of mTOR and MAPK signaling in AML
Because SYK has been reported to regulate mTOR in lymphoma, we investigated whether this relationship was also present in AML where little is known about SYK's downstream effectors. We also tested the effects of SYK signaling on the collateral MAPK pathway, known to interact with PI3K/AKT/mTOR signaling. First, we overexpressed wildtype SYK, SYK-TEL (a constitutively activated form of SYK), and kinase dead SYK-TEL in the AML cell line MOLM-14. Both the mTOR pathway, including the upstream activator AKT and downstream effectors, and the MAPK pathway were activated with expression of either wildtype SYK or SYK-TEL but not kinase-dead SYK-TEL (Figure 1), suggesting that enhanced activation of these two pathways is dependent on the kinase activity of SYK.
Figure 1.
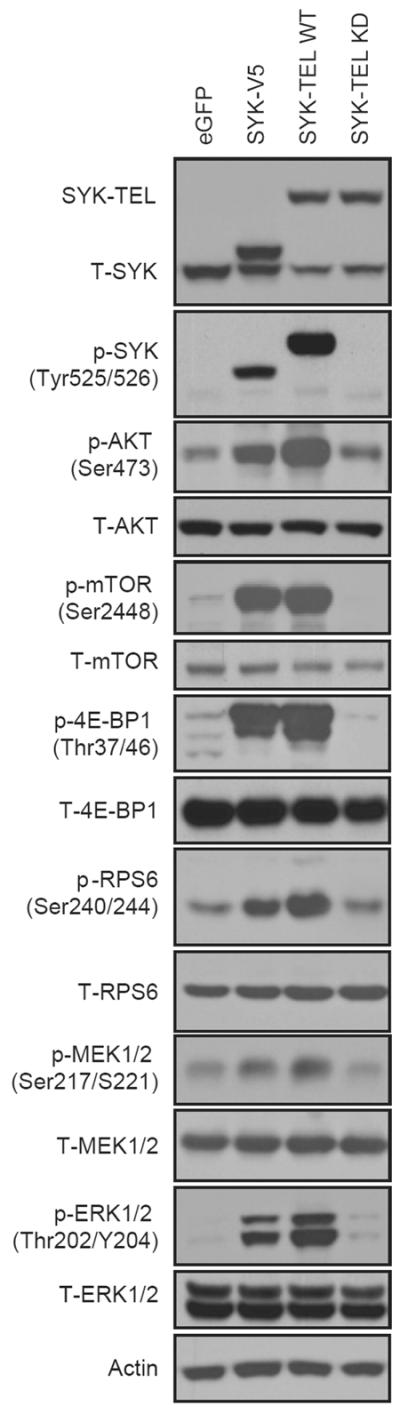
A constitutively activated form of SYK and overexpressed wildtype SYK stimulate AKT/mTOR and MAPK pathways. Western-blot of AKT/mTOR and MAPK downstream effectors in MOLM-14 transduced with V5-tagged SYK, SYK-TEL or SYK-TEL KD (KD = kinase dead).
Chemical or genetic suppression of SYK inhibits AKT/mTOR in AML cell lines
We first identified AML cell lines with high levels of AKT activation (Supplementary Figure S1) and treated two of these (MOLM-14 and U937) with R940406 (R406), an ATP-competitive inhibitor of phosphorylation at the Y525/526 activation site of SYK (Supplementary Figure S2a). R406 is the active metabolite of fostamatinib, a soluble orally dosed prodrug converted to R406.42 Treatment with R406 led to a dose-response in the inhibition of p-AKT and p-mTOR in both cell lines, indicating that SYK may be signaling upstream of p-AKT in AML (Figure 2a and b). In MOLM-14 cells, total AKT was seen to decrease along with p-AKT after a 24 hour treatment. In light of this observation, we evaluated a shorter compound exposure of 6 hours and observed a decrease in phosphorylated, but not total, AKT (Supplementary Figure S2b). This inhibition was seen as early as 30 minutes post-treatment for p-AKT and p-mTOR. mTOR activation is commonly evaluated by assessing the phosphorylation status of its two major downstream targets: p70S6K/RPS6 and 4E-BP1/eIF4E. While inhibition of RPS6 phosphorylation was seen at 30 minutes, phosphorylation of the downstream effector 4E-BP1 was only minimally inhibited until the 24 hour time point (Supplementary Figure S2c).
Figure 2.
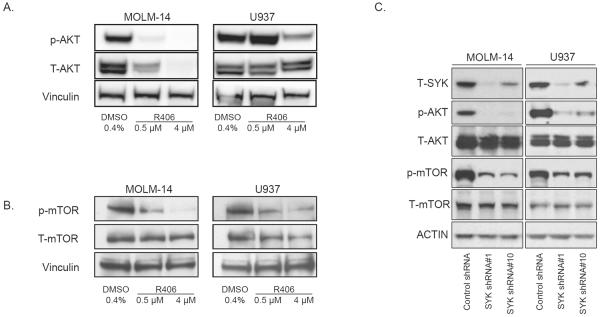
Chemical or genetic manipulation of SYK leads to altered AKT and mTOR activation. Western blot of (a) p-AKT (Ser473) or (b) p-mTOR (Ser2448) in AML cell lines treated with R406 for 24 hours. (c) AML cell lines MOLM-14 and U937 were infected with shRNA targeting SYK or an shRNA control and assessed for levels of p-AKT and p-mTOR 5 days post-infection.
We next investigated the effects of SYK knockdown on AKT/mTOR activation using two previously validated SYK-directed shRNAs.3 Both AKT and mTOR activation were downregulated in parallel with diminished levels of SYK, indicating that this effect is likely on-target for SYK (Figure 2c). In contrast, inhibition of the PI3K/mTOR pathway did not have a positive feedback effect on SYK with no increase in SYK phosphorylation observed with the PI3K inhibitor GDC-0941 nor the mTORC1/2 inhibitor Torin1 at concentrations altering phosphorylation of target effector proteins (Supplementary Figure S3a and b).43
mTOR signaling effectors are differentially regulated by SYK in AML
Effects of SYK inhibition on p70S6K/RPS6 and 4E-BP1/eIF4E were next assessed across a larger panel of AML cell lines by immunoblotting. RPS6 phosphorylation was downregulated by R406 treatment (Figure 3a), while the effects of R406 on the phosphorylation status of 4E-BP1 were more variable across the cell lines (Figure 3b). In order to quantify the phosphorylation status of RPS6 and 4E-BP1, single cell intracellular phospho-specific flow cytometry was used. Again, there was consistent inhibition of RPS6 across all cell lines while the inhibition of 4E-BP1 was more subtle in some cell lines and absent in others (Supplementary Figure S4). In order to ensure that these effects were reflective of primary human AML rather than simply a feature of cultured cell lines, we next extended testing to primary patient AML and MDS. We used intracellular phospho-specific flow cytometry to best quantify the effect of SYK inhibition given the limited number of total cells in each primary sample. As was seen in the AML cell lines, a strikingly similar pattern of consistently altered RPS6 phosphorylation, with more variable inhibition of 4E-BP1 phosphorylation, was observed in five primary patient AML/MDS samples treated with R406 (Figure 3c; Supplementary Table S2).
Figure 3.

SYK inhibition with R406 inhibits downstream mTOR signaling targets in AML cell lines and primary AML. Four AML cell lines (a and b) were treated for 24 hours with vehicle versus R406. Western blots of (a) p-RPS6 (Ser240/244) and (b) p-4E-BP1 (Thr37/46) (c) Intracellular phospho-flow cytometry of p-SYK, p-4E-BP1 and p-RPS6 in primary patient AML and MDS cells treated for 6 hours with R406. The x-axis denotes expression (log scale) for each protein of interest, and each is labeled with the median fluorescent intensity (MFI) per condition.
To investigate the specificity of SYK's differential effects on RPS6 and 4E-BP1, the phosphorylation status of these targets was interrogated using two SYK-directed shRNAs. While RPS6 was consistently inhibited with SYK knockdown, 4E-BP1 was more minimally inhibited and in only a subset of the cell lines analyzed (Supplementary Figure S5).
Chemical inhibition of PI3K pathway signaling enhances the effects of SYK suppression on differentiation and viability in AML
Because SYK inhibition appeared to affect the p70S6K/RPS6 mTOR axis to a greater degree than the 4E-BP1/eIF4E axis in some AML cell lines, we hypothesized that more complete inhibition of PI3K/mTOR signaling would synergize with SYK inhibition in AML in both its pro-differentiation and anti-viability effects. We thus evaluated in AML cell lines the combination of SYK suppression with small-molecule modulators of the PI3K/mTOR pathway: PI3K (GDC-0941), mTORC1/2 complex (Torin 1), and 4E-BP1 (4EGI-1). We chose compound concentrations reported and confirmed to inhibit the intended pathways targeted by the molecules (Supplementary Figures S3 and S6).43, 44 While Torin 1 treatment led to a marked decrease in 4E-BP1 phosphorylation in AML, GDC-0941 treatment led to only a modest reduction in phosphorylation of 4E-BP1 despite near complete inhibition of AKT phosphorylation (Supplementary Figures S3 and S6). The compound 4EGI-1 mimics hypophosphorylated 4E-BP1 by binding and inhibiting eIF4E's association with other factors in the initiation complex, thus suppressing cap-dependent translation.45 Previous studies demonstrated that 4EGI-1 displaces eIF4G from eIF4E at approximately 25–50 μM.45 Dose-finding analyses using an ATP-based viability assay revealed the IC50's for 4EGI-1 in the majority of AML cell lines tested to be within this range (data not shown).
As shown in Figure 4a, by day 6 there was a viability disadvantage to those cells with both SYK knockdown and exposure to the small-molecule treatments greater than with any one perturbation alone. We next evaluated the combined effects on differentiation focusing on the small molecule expected to be most specific for inhibition of 4E-BP1, 4EGI-1, and AML cell lines known to differentiate with ATRA and SYK inhibition, HL-60 and U937.3 Morphologic analyses reveal an enhanced level of nuclear condensation and cytoplasmic ruffling in the conditions combining SYK knockdown and chemical inhibition of 4E-BP1 (Figure 4b). To more effectively quantify the induction of differentiation, we utilized an assay measuring a 32-gene expression signature for myeloid differentiation previously developed by our laboratory (Supplementary Table S1).3, 5 The combination of SYK-directed shRNAs and 4EGI-1 resulted in a myeloid differentiation score higher than control shRNA with 4EGI-1 or the SYK-directed shRNA with DMSO in HL-60 and U937 cells consistent with the morphological findings (Figure 4c). In both cell lines the combination score approached that of the positive control, ATRA.
Figure 4.

Pharmacologic inhibition of AKT/mTOR enhances the effect of genetic inhibition of SYK on viability and differentiation in AML. (a) AML cell lines infected with SYK-directed shRNA or control shRNA were exposed to either DMSO, GDC-0941 (1 μM), Torin 1 (250 nM for HL-60 or 50 nM for U937), or 4EGI-1 (25 μM) and viability was assessed at 0, 3 and 6 days post treatment. Error bars denote the mean ±SD of 8 replicates with statistical significance evaluated using a one-way ANOVA with a Bonferroni correction for each compound at day 6. In each subpanel, all three brackets are significant with *** P < 0.001. A summary of P values for Bonferroni multiple pairwise comparisons test is shown in Supplementary Table S3. (b) HL-60 and U937 cells were infected with control shRNA or SYK-directed shRNA and exposed to either DMSO or 4EGI-1 for three days. May Grunwald Giemsa staining of HL-60 and U937 cells for each condition is depicted. Evidence of myeloid maturation included a reduction in the nuclear to cytoplasmic ratio and an increase in cytoplasmic ruffling. Images were acquired with an Olympus BX41 microscope, 400× magnification, and Qcapture software. (c) HL-60 and U937 cells were infected with control shRNA and SYK-directed shRNA and then exposed to either DMSO, 4EGI-1, or ATRA for three days. The Differentiation Score (Weighted Summed Score) was quantified by the GE-HTS approach. Error bars denote the mean ±SD of 8 replicates, and statistical significance of the differences between these differentiation scores was derived using a one-way ANOVA with a Bonferroni correction.
Pharmacologic inhibition of SYK and eIF4E enhances differentiation and has a synergistic effect on viability in AML
We next evaluated the effects of the most direct inhibitor of eIF4E, 4EGI-1 in combination with R406. As in the combination of shRNA-directed against SYK with 4EGI-1 treatment, a greater proportion of cells exposed to both compounds showed morphological evidence of myeloid differentiation and an increased differentiation score compared to cells exposed to R406 alone or 4EGI-1 alone (Figures 5a and b). When evaluated on a gene-by-gene level, the effects of the compounds varied. In some cases, they affected the same genes with an amplified effect when combined, and in other cases, they altered the expression of a unique set of genes in the differentiation signature (Supplementary Figure S7). Combination treatment with R406 and 4EGI-1 in AML cell lines was also performed across a range of concentrations in 384-well format and synergy assessed using excess over Bliss additive synergy analysis.40, 46 The Bliss model calculates an expected effect of the combined response, C, of two individual agents with the effects A and B using the formula C = A + B − A × B. Figure 5c shows, for a range of concentrations of the combination of the two agents, the values for excess over Bliss or the difference between the effect measured and the effect predicted by the Bliss model.39, 40 Interestingly, this synergy did not occur through enhancement of apoptosis but was dependent on an induction of CD11b/CD14-positive myeloid differentiation and a decrease in proliferation as evidenced by diminished BrdU incorporation (Supplementary Figure S8).
Figure 5.

Chemical inhibition of eIF4E by 4EGI-1 enhances the effect of chemical inhibition of SYK on AML differentiation. (a) May Grunwald Giemsa staining of HL-60 and U937 cells treated for 5 days with DMSO, R406, 4EGI-1, and in combination is depicted. (b) HL-60 and U937 cells were treated for three days with DMSO, R406, 4EGI-1, or the combination of both compounds. A 32-gene differentiation signature was quantified by the LMA/bead-based approach and a Weighted Summed Score (Differentiation Score) calculated for all genes was determined for each condition. Error bars denote the mean ±SD of 8 replicates and statistical significance of the differences between these differentiation scores was derived using a one-way ANOVA with a Bonferroni correction. (c) The effects of combining small-molecule inhibitors against SYK and eIF4E were assessed with an ATP-based viability assay in two AML cell lines. Viability effects were assessed three days after treatment with R406, 4EGI-1, both in combination and vehicle (DMSO) in replicates of four for each dose combination for HL-60 while U937 was assessed at five days. The data was analyzed using a Bliss additivism model.
Effects of SYK signaling on the MAPK pathway are heterogeneous in AML
The MAPK pathway is known to interact with PI3K/mTOR signaling. Given that constitutive activation of SYK resulted in activation of the MAPK pathway in AML we explored the effects of SYK inhibition on MAPK effectors in a panel of AML cell lines. Treatment of AML cell lines with R406 resulted in a decrease in phosphorylation of MEK and ERK in many of the AML cell lines, while an unexpected increase in phosphorylation was seen in HL-60 (Figure 6a). Because HL-60 is an N-RAS mutated cell line we hypothesized that the increase in output through MAPK was related to the RAS status of this cell line.30 Indeed, in both the N-RAS mutated cell line THP-134 and the K-RAS mutated line NOMO-1,37 we observed a similar increase in signaling through the MAPK pathway with R406 treatment (Figure 6b). This increase in MAPK activation seen in RAS-mutated AML cell lines was observed with a second SYK inhibitor, BAY 61-3606 (data not shown). In order to explore the potential mechanism for this paradoxical increase in ERK signaling in RAS-mutated AML, we next assessed the involvement of c-RAF activation by R406. We observed a decrease in c-RAF phosphorylation in the RAS-wild-type cell line MOLM-14, which was absent in the RAS-mutated AML cell line HL-60 (Figure 6c) suggesting the possibility of a feedback loop through c-RAF. MEK inhibition with PD0325901 blocks this feedback activation of ERK, while the PI3K inhibitor GDC-0941 does not (Supplementary Figure S9).
Figure 6.

SYK inhibition with R406 alters downstream MAPK signaling targets. Six AML cell lines were treated with vehicle versus R406. Western blots of p-MEK1/2 (Ser217/221) and p-ERK1/2 (Thr202/Tyr204) and p-c-RAF (Ser338) are depicted. (a) HL-60, KG-1, MOLM-14 and U937 cells were treated for 6 hours for p-MEK1/2 analysis and 24 hours for p-ERK1/2 analysis. (b) NOMO-1 and THP-1 cells were treated for 24 hours. (c) Effect of SYK inhibition on c-RAF in AML cells. AML cells were grown in the presence of 4 μM R406 for the indicated times and assessed for activation of c-RAF and ERK1/2.
Discussion
Much progress has been made in understanding the biological mechanisms underlying the pathogenesis of AML, yet poor survival rates indicate that new treatment strategies are needed. Our laboratory recently validated SYK as a new target in AML. While little is known about the downstream signaling effectors of SYK in AML, we leveraged knowledge derived from studies of SYK in B-cell signaling and lymphoma to generate hypotheses about its role in AML. One SYK target reported in non-Hodgkin's lymphoma, mTOR, was of particular interest given the growing evidence of the critical role of mTOR in AML. Moreover, previous studies of pathway activation downstream of the constitutively active TEL-SYK fusion (identified in a patient with MDS) implicated a number of AML-relevant pathways, including PI3K, MAPK, and JAK-STAT.47 Here, we provide evidence that SYK regulates mTOR signaling in AML.
mTOR is a serine/threonine kinase that plays an important role in the regulation of a variety of cellular processes required for tumor development, such as growth, proliferation, and translational initiation of key proteins, including numerous, well-known oncogenes.48 mTOR is frequently deregulated in the majority of hematologic malignancies.19, 23, 49 There has been growing interest in the role of mTOR in AML, where mTOR inhibition has been associated with both antiproliferative and pro-differentiating effects. Specifically, mTOR inhibitors have been shown to reduce AML clonogenicity and proliferation in vitro, as well as regulate AML stem cell survival in animal models of engraftment.19, 23–25, 48, 50 Moreover, these molecules are being tested in patients with either refractory/relapsed, de novo, or secondary AML with promising results in early clinical trials.23, 26–29 Furthermore, blockade of mTOR signaling has been shown in multiple studies to potentiate the ability of histone deacetylase inhibitors, 1,25-dihydroxyvitamin D3 and ATRA to induce differentiation in AML.21, 22, 51 The precise mechanism by which mTOR inhibition leads to this enhanced differentiation is not yet established although there have been some recent clues regarding potential mechanisms, such as mTOR's regulation of c-MYC and Programmed Cell Death-4 (PDCD4).52–54
Given the mounting interest in targeting mTOR in AML, as well as previous reports of SYK regulation of mTOR in lymphoma, we chose to test whether SYK regulates mTOR in AML. We demonstrated that chemical inhibition of SYK by R406 leads to the inhibition of p-mTOR in multiple AML cell lines in a dose-dependent manner. Moreover, we showed that genetic inhibition of SYK by shRNA and expression of wildtype SYK or constitutively active SYK-TEL leads to inhibition and up-regulation of p-mTOR, respectively. We further studied the activation of specific mTOR signaling branches, including p70S6K/RPS6 and 4E-BP1/eIF4E. Interestingly, the p70S6K/RPS6 signaling pathway was affected by manipulations in SYK activity to a greater extent than the 4E-BP1/eIF4E pathway in some AML cell lines and in primary patient AML and MDS cells. Within the framework of canonical mTOR signaling diagrams, our data suggest a variable effect of SYK on p70S6K/RPS6 and 4E-BP1 activation. This is not the first observation of downstream targets of mTOR being regulated asymmetrically. For instance, in an mTOR independent manner, phospholipase D-2-derived phosphatidic acid directly binds and activates p70S6K,55 and p90 ribosomal S6 kinases directly phosphorylate RPS6.56 Others have identified the activation of 4E-BP1 by the kinase PIM-2,57, 58 and regulation of eIF4E by MAP kinase-interacting kinase 1 (MNK1).59 It is also apparent that diverse stimuli, such as insulin or amino acid withdrawal, affect these branches in different ways depending on the cellular context.60 It has been reported that in AML, mTORC1 inhibition by either the rapamycin derivative, RAD001, or an siRNA directed against RAPTOR fails to effectively inhibit 4E-BP1 phosphorylation.58 We have confirmed this finding in two AML cell lines using rapamycin (Supplementary Figure S10a). Thus, regulation of mTOR signaling is quite complex, and it is likely that there are different signals working with SYK to affect the activation of p70S6K/RPS6 and 4E-BP1/eIF4E. While it is unclear how SYK regulates p70S6K/RPS6 with greater magnitude and consistency than 4E-BP1 in some AML cell lines and primary patient samples, this observation allowed the assessment of combining a SYK inhibitor and an eIF4E inhibitor on AML viability and differentiation.
Our results revealed that combined chemical inhibition of SYK and eIF4E had a synergistic effect on AML viability across multiple AML cell lines. Furthermore, eIF4E inhibition enhanced the inhibitory effects of SYK knockdown on cell viability, suggesting that this synergistic relationship with small-molecule inhibition of SYK is on-target. eIF4E inhibition also enhanced AML differentiation when combined with SYK inhibition by either chemical or SYK-directed shRNA. There is evidence that the 4E-BPs and eIF4E play pivotal roles in myeloid differentiation,61–63 and our own data suggests that inhibition of 4E-BP1/eIF4E promotes greater differentiation than inhibition of RPS6 alone (Supplementary Figure S10b). The mechanism responsible for this synergistic effect, however, is not clear. More complete inhibition of both arms of the PI3K/mTOR signaling pathway could explain these findings as both PI3K and mTORC1/2 small-molecule inhibitors also enhanced the anti-AML effects of SYK-directed shRNA. It is also possible that there are downstream targets upon which SYK and mTOR converge and that this combined inhibition confers a more potent AML differentiation effect. Indeed, while SYK inhibition leads to a decrement in RPS6 phosphorylation in U937 and HL-60, SYK inhibition promotes greater differentiation than RPS6 inhibition with rapamycin, suggesting that there are likely other RPS6 independent effects contributing to the differentiation induced by SYK inhibition in AML.
In evaluating pathways collaborating with PI3K, specifically MAPK, we made the observation that with exogenous expression of activated SYK, MAPK signaling increases. In contrast, with pharmacological inhibition of SYK, there is a decrease in phosphorylation of MEK and ERK in a subset of AML cells, but in RAS-mutated AML cell lines, there is a paradoxical increase in MEK/ERK activation seen with two structurally distinct SYK inhibitors. This activation was inhibited by the MEK inhibitor PD0325901 but not the PI3K inhibitor GDC-0941 (Supplementary Figure S9). Moreover, these RAS-mutated AML cell lines are more resistant to the effects of pharmacologic inhibition of SYK on AML cell viability.3 There have been similar observations of feedback activation of MAPK reported in the context of other malignancies. For example, Carracedo et al. reported that inhibition of mTORC1 leads to MAPK activation through a PI3K dependent feedback loop through S6K-PI3K-RAS pathway in solid tumor samples from patients and in cancer cell lines and mouse models of prostate cancer.64 In the case of SYK inhibition in AML, however, MEK inhibition, but not PI3K inhibition, attenuates the feedback activation of MAPK. It is also possible that this observation is secondary to an off-target effect of both of these small-molecule inhibitors of SYK. It is noteworthy that RAF inhibitors can transactivate RAF dimers and ERK signaling in the context of wildtype RAF expression, particularly in RAS-mutated cell lines.65 While the precise mechanism of this observed phenomenon in AML with SYK inhibition is still under consideration, these results suggest a cautionary use of SYK inhibitors in patients with RAS-mutated AML, but also raise the possibility of combining SYK inhibition with MEK inhibition for these patients, particularly because MEK inhibition appears to attenuate this positive feedback on the MAPK pathway.
Here, we provide the first documentation that SYK regulates mTOR in AML and demonstrate that combined inhibition of SYK and mTOR signaling has a synergistic effect in AML. Moreover, the data suggest that the effects of SYK inhibition on MAPK are variable with downregulation of MAPK signaling in some AML and a paradoxical upregulation in RAS-mutated AML. This data may be of particular relevance for clinical trials of SYK inhibitors in AML and for trials attempting to maximize SYK-inhibitory effects with a combinatorial approach.
Supplementary Material
Acknowledgments
We thank Gerhard Wagner for his generous contribution of the 4EGI-1 molecule and Rigel Pharmaceuticals and AstraZeneca for their kind gift of R406. We thank Margaret Shipp for her scientific advice, John Daley, Susan Lazo-Kallanian, and Giovanni Roti, for guidance regarding flow cytometry studies, Ilene Galinsky for primary patient sample support, and Stacey Frumm for technical support. We thank Aaron Thorner for critical reading of the manuscript. This research was supported with grants from the National Cancer Institute (R01 CA140292), American Cancer Society, the Starr Cancer Consortium, and Project Cupid (KS). JC was a Howard Hughes Medical Institute Medical Student Fellow and AP a Leukemia and Lymphoma Society Fellow.
Footnotes
Conflict of Interest The authors have no relevant conflicts of interest to declare.
References
- 1.Juliusson G, Antunovic P, Derolf A, Lehmann S, Mollgard L, Stockelberg D, et al. Age and acute myeloid leukemia: real world data on decision to treat and outcomes from the Swedish Acute Leukemia Registry. Blood. 2009;113:4179–4187. doi: 10.1182/blood-2008-07-172007. [DOI] [PubMed] [Google Scholar]
- 2.Castaigne S, Chomienne C, Daniel MT, Ballerini P, Berger R, Fenaux P, et al. All-trans retinoic acid as a differentiation therapy for acute promyelocytic leukemia. I. Clinical results. Blood. 1990;76:1704–1709. [PubMed] [Google Scholar]
- 3.Hahn CK, Berchuck JE, Ross KN, Kakoza RM, Clauser K, Schinzel AC, et al. Proteomic and genetic approaches identify Syk as an AML target. Cancer Cell. 2009;16:281–294. doi: 10.1016/j.ccr.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stegmaier K, Corsello SM, Ross KN, Wong JS, Deangelo DJ, Golub TR. Gefitinib induces myeloid differentiation of acute myeloid leukemia. Blood. 2005;106:2841–2848. doi: 10.1182/blood-2005-02-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stegmaier K, Ross KN, Colavito SA, O'Malley S, Stockwell BR, Golub TR. Gene expression-based high-throughput screening(GE-HTS) and application to leukemia differentiation. Nat Genet. 2004;36:257–263. doi: 10.1038/ng1305. [DOI] [PubMed] [Google Scholar]
- 6.Cheng AM, Rowley B, Pao W, Hayday A, Bolen JB, Pawson T. Syk tyrosine kinase required for mouse viability and B-cell development. Nature. 1995;378:303–306. doi: 10.1038/378303a0. [DOI] [PubMed] [Google Scholar]
- 7.Streubel B, Vinatzer U, Willheim M, Raderer M, Chott A. Novel t(5;9)(q33;q22) fuses ITK to SYK in unspecified peripheral T-cell lymphoma. Leukemia. 2006;20:313–318. doi: 10.1038/sj.leu.2404045. [DOI] [PubMed] [Google Scholar]
- 8.Pechloff K, Holch J, Ferch U, Schweneker M, Brunner K, Kremer M, et al. The fusion kinase ITK-SYK mimics a T cell receptor signal and drives oncogenesis in conditional mouse models of peripheral T cell lymphoma. J Exp Med. 207:1031–1044. doi: 10.1084/jem.20092042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen L, Monti S, Juszczynski P, Daley J, Chen W, Witzig TE, et al. SYK-dependent tonic B-cell receptor signaling is a rational treatment target in diffuse large B-cell lymphoma. Blood. 2008;111:2230–2237. doi: 10.1182/blood-2007-07-100115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wossning T, Herzog S, Kohler F, Meixlsperger S, Kulathu Y, Mittler G, et al. Deregulated Syk inhibits differentiation and induces growth factor-independent proliferation of pre-B cells. J Exp Med. 2006;203:2829–2840. doi: 10.1084/jem.20060967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Friedberg JW, Sharman J, Sweetenham J, Johnston PB, Vose JM, Lacasce A, et al. Inhibition of Syk with fostamatinib disodium has significant clinical activity in non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood. 115:2578–2585. doi: 10.1182/blood-2009-08-236471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kuno Y, Abe A, Emi N, Iida M, Yokozawa T, Towatari M, et al. Constitutive kinase activation of the TEL-Syk fusion gene in myelodysplastic syndrome with t(9;12)(q22;p12) Blood. 2001;97:1050–1055. doi: 10.1182/blood.v97.4.1050. [DOI] [PubMed] [Google Scholar]
- 13.Gururajan M, Dasu T, Shahidain S, Jennings CD, Robertson DA, Rangnekar VM, et al. Spleen tyrosine kinase (Syk), a novel target of curcumin, is required for B lymphoma growth. J Immunol. 2007;178:111–121. doi: 10.4049/jimmunol.178.1.111. [DOI] [PubMed] [Google Scholar]
- 14.Leseux L, Hamdi SM, Al Saati T, Capilla F, Recher C, Laurent G, et al. Syk-dependent mTOR activation in follicular lymphoma cells. Blood. 2006;108:4156–4162. doi: 10.1182/blood-2006-05-026203. [DOI] [PubMed] [Google Scholar]
- 15.Rinaldi A, Kwee I, Taborelli M, Largo C, Uccella S, Martin V, et al. Genomic and expression profiling identifies the B-cell associated tyrosine kinase Syk as a possible therapeutic target in mantle cell lymphoma. Br J Haematol. 2006;132:303–316. doi: 10.1111/j.1365-2141.2005.05883.x. [DOI] [PubMed] [Google Scholar]
- 16.Young RM, Hardy IR, Clarke RL, Lundy N, Pine P, Turner BC, et al. Mouse models of non-Hodgkins lymphoma reveal Syk as an important therapeutic target. Blood. 2008;113:2508–2516. doi: 10.1182/blood-2008-05-158618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fruchon S, Kheirallah S, Al Saati T, Ysebaert L, Laurent C, Leseux L, et al. Involvement of the Syk-mTOR pathway in follicular lymphoma cell invasion and angiogenesis. Leukemia. 2012;26:795–805. doi: 10.1038/leu.2011.248. [DOI] [PubMed] [Google Scholar]
- 18.Ma XM, Blenis J. Molecular mechanisms of mTOR-mediated translational control. Nature reviews. 2009;10:307–318. doi: 10.1038/nrm2672. [DOI] [PubMed] [Google Scholar]
- 19.Xu Q, Thompson JE, Carroll M. mTOR regulates cell survival after etoposide treatment in primary AML cells. Blood. 2005;106:4261–4268. doi: 10.1182/blood-2004-11-4468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chapuis N, Tamburini J, Green AS, Vignon C, Bardet V, Neyret A, et al. Dual inhibition of PI3K and mTORC1/2 signaling by NVP-BEZ235 as a new therapeutic strategy for acute myeloid leukemia. Clin Cancer Res. 2010;16:5424–5435. doi: 10.1158/1078-0432.CCR-10-1102. [DOI] [PubMed] [Google Scholar]
- 21.Nishioka C, Ikezoe T, Yang J, Gery S, Koeffler HP, Yokoyama A. Inhibition of mammalian target of rapamycin signaling potentiates the effects of all-trans retinoic acid to induce growth arrest and differentiation of human acute myelogenous leukemia cells. Int J Cancer. 2009;125:1710–1720. doi: 10.1002/ijc.24472. [DOI] [PubMed] [Google Scholar]
- 22.Nishioka C, Ikezoe T, Yang J, Koeffler HP, Yokoyama A. Blockade of mTOR signaling potentiates the ability of histone deacetylase inhibitor to induce growth arrest and differentiation of acute myelogenous leukemia cells. Leukemia. 2008;22:2159–2168. doi: 10.1038/leu.2008.243. [DOI] [PubMed] [Google Scholar]
- 23.Recher C, Beyne-Rauzy O, Demur C, Chicanne G, Dos Santos C, Mas VM, et al. Antileukemic activity of rapamycin in acute myeloid leukemia. Blood. 2005;105:2527–2534. doi: 10.1182/blood-2004-06-2494. [DOI] [PubMed] [Google Scholar]
- 24.Willems L, Chapuis N, Puissant A, Maciel TT, Green AS, Jacque N, et al. The dual mTORC1 and mTORC2 inhibitor AZD8055 has anti-tumor activity in acute myeloid leukemia. Leukemia. 2012;26:1195–1202. doi: 10.1038/leu.2011.339. [DOI] [PubMed] [Google Scholar]
- 25.Zeng Z, Shi YX, Tsao T, Qiu Y, Kornblau SM, Baggerly KA, et al. Targeting of mTORC1/2 by the mTOR kinase inhibitor PP242 induces apoptosis in AML cells under conditions mimicking the bone marrow microenvironment. Blood. 2012;120:2679–2689. doi: 10.1182/blood-2011-11-393934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amadori S, Stasi R, Martelli AM, Venditti A, Meloni G, Pane F, et al. Temsirolimus, an mTOR inhibitor, in combination with lower-dose clofarabine as salvage therapy for older patients with acute myeloid leukaemia: results of a phase II GIMEMA study (AML-1107) Br J Haematol. 2012;156:205–212. doi: 10.1111/j.1365-2141.2011.08940.x. [DOI] [PubMed] [Google Scholar]
- 27.Perl AE, Kasner MT, Shank D, Luger SM, Carroll M. Single-cell pharmacodynamic monitoring of S6 ribosomal protein phosphorylation in AML blasts during a clinical trial combining the mTOR inhibitor sirolimus and intensive chemotherapy. Clin Cancer Res. 2012;18:1716–1725. doi: 10.1158/1078-0432.CCR-11-2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Park S, Chapuis N, Saint Marcoux F, Recher C, Prebet T, Chevallier P, et al. A phase Ib GOELAMS study of the mTOR inhibitor RAD001 in association with chemotherapy for AML patients in first relapse. Leukemia. 2013 doi: 10.1038/leu.2013.17. [DOI] [PubMed] [Google Scholar]
- 29.Perl AE, Kasner MT, Tsai DE, Vogl DT, Loren AW, Schuster SJ, et al. A phase I study of the mammalian target of rapamycin inhibitor sirolimus and MEC chemotherapy in relapsed and refractory acute myelogenous leukemia. Clin Cancer Res. 2009;15:6732–6739. doi: 10.1158/1078-0432.CCR-09-0842. [DOI] [PubMed] [Google Scholar]
- 30.Murray MJ, Cunningham JM, Parada LF, Dautry F, Lebowitz P, Weinberg RA. The HL-60 transforming sequence: a ras oncogene coexisting with altered myc genes in hematopoietic tumors. Cell. 1983;33:749–757. doi: 10.1016/0092-8674(83)90017-x. [DOI] [PubMed] [Google Scholar]
- 31.Dreyling MH, Martinez-Climent JA, Zheng M, Mao J, Rowley JD, Bohlander SK. The t(10;11)(p13;q14) in the U937 cell line results in the fusion of the AF10 gene and CALM, encoding a new member of the AP-3 clathrin assembly protein family. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:4804–4809. doi: 10.1073/pnas.93.10.4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gu TL, Goss VL, Reeves C, Popova L, Nardone J, Macneill J, et al. Phosphotyrosine profiling identifies the KG-1 cell line as a model for the study of FGFR1 fusions in acute myeloid leukemia. Blood. 2006;108:4202–4204. doi: 10.1182/blood-2006-06-026666. [DOI] [PubMed] [Google Scholar]
- 33.Odero MD, Zeleznik-Le NJ, Chinwalla V, Rowley JD. Cytogenetic and molecular analysis of the acute monocytic leukemia cell line THP-1 with an MLL-AF9 translocation. Genes, chromosomes & cancer. 2000;29:333–338. [PubMed] [Google Scholar]
- 34.Lubbert M, Mertelsmann R, Herrmann F. Detection of allele-specific expression of N-RAS oncogenes in human leukaemia cells. Br J Haematol. 1992;81:370–373. doi: 10.1111/j.1365-2141.1992.tb08241.x. [DOI] [PubMed] [Google Scholar]
- 35.Matsuo Y, MacLeod RA, Uphoff CC, Drexler HG, Nishizaki C, Katayama Y, et al. Two acute monocytic leukemia (AML-M5a) cell lines (MOLM-13 and MOLM-14) with interclonal phenotypic heterogeneity showing MLL-AF9 fusion resulting from an occult chromosome insertion, ins(11;9)(q23;p22p23) Leukemia. 1997;11:1469–1477. doi: 10.1038/sj.leu.2400768. [DOI] [PubMed] [Google Scholar]
- 36.Furukawa Y, Vu HA, Akutsu M, Odgerel T, Izumi T, Tsunoda S, et al. Divergent cytotoxic effects of PKC412 in combination with conventional antileukemic agents in FLT3 mutation-positive versus -negative leukemia cell lines. Leukemia. 2007;21:1005–1014. doi: 10.1038/sj.leu.2404593. [DOI] [PubMed] [Google Scholar]
- 37.Scholl C, Frohling S, Dunn IF, Schinzel AC, Barbie DA, Kim SY, et al. Synthetic lethal interaction between oncogenic KRAS dependency and STK33 suppression in human cancer cells. Cell. 2009;137:821–834. doi: 10.1016/j.cell.2009.03.017. [DOI] [PubMed] [Google Scholar]
- 38.Peck D, Crawford ED, Ross KN, Stegmaier K, Golub TR, Lamb J. A method for high-throughput gene expression signature analysis. Genome Biol. 2006;7:R61. doi: 10.1186/gb-2006-7-7-r61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berenbaum MC. Criteria for analyzing interactions between biologically active agents. Advances in cancer research. 1981;35:269–335. doi: 10.1016/s0065-230x(08)60912-4. [DOI] [PubMed] [Google Scholar]
- 40.Borisy AA, Elliott PJ, Hurst NW, Lee MS, Lehar J, Price ER, et al. Systematic discovery of multicomponent therapeutics. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:7977–7982. doi: 10.1073/pnas.1337088100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283–1298. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 42.Braselmann S, Taylor V, Zhao H, Wang S, Sylvain C, Baluom M, et al. R406, an orally available spleen tyrosine kinase inhibitor blocks fc receptor signaling and reduces immune complex-mediated inflammation. J Pharmacol Exp Ther. 2006;319:998–1008. doi: 10.1124/jpet.106.109058. [DOI] [PubMed] [Google Scholar]
- 43.Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Folkes AJ, Ahmadi K, Alderton WK, Alix S, Baker SJ, Box G, et al. The identification of 2-(1H-indazol-4-yl)-6-(4-methanesulfonyl-piperazin-1-ylmethyl)-4-morpholin-4-yl-t hieno[3,2-d]pyrimidine (GDC-0941) as a potent, selective, orally bioavailable inhibitor of class I PI3 kinase for the treatment of cancer. Journal of medicinal chemistry. 2008;51:5522–5532. doi: 10.1021/jm800295d. [DOI] [PubMed] [Google Scholar]
- 45.Moerke NJ, Aktas H, Chen H, Cantel S, Reibarkh MY, Fahmy A, et al. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell. 2007;128:257–267. doi: 10.1016/j.cell.2006.11.046. [DOI] [PubMed] [Google Scholar]
- 46.Bliss C. The toxicity of poisons applied jointly. Ann Appl Biol. 1939;26:585–615. [Google Scholar]
- 47.Kanie T, Abe A, Matsuda T, Kuno Y, Towatari M, Yamamoto T, et al. TEL-Syk fusion constitutively activates PI3-K/Akt, MAPK and JAK2-independent STAT5 signal pathways. Leukemia. 2004;18:548–555. doi: 10.1038/sj.leu.2403266. [DOI] [PubMed] [Google Scholar]
- 48.Chapuis N, Tamburini J, Green AS, Willems L, Bardet V, Park S, et al. Perspectives on inhibiting mTOR as a future treatment strategy for hematological malignancies. Leukemia. 24:1686–1699. doi: 10.1038/leu.2010.170. [DOI] [PubMed] [Google Scholar]
- 49.Dos Santos C, Demur C, Bardet V, Prade-Houdellier N, Payrastre B, Recher C. A critical role for Lyn in acute myeloid leukemia. Blood. 2008;111:2269–2279. doi: 10.1182/blood-2007-04-082099. [DOI] [PubMed] [Google Scholar]
- 50.Altman JK, Sassano A, Kaur S, Glaser H, Kroczynska B, Redig AJ, et al. Dual mTORC2/mTORC1 targeting results in potent suppressive effects on acute myeloid leukemia (AML) progenitors. Clin Cancer Res. 2011;17:4378–4388. doi: 10.1158/1078-0432.CCR-10-2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang J, Ikezoe T, Nishioka C, Ni L, Koeffler HP, Yokoyama A. Inhibition of mTORC1 by RAD001 (everolimus) potentiates the effects of 1,25-dihydroxyvitamin D(3) to induce growth arrest and differentiation of AML cells in vitro and in vivo. Exp Hematol. 2010;38:666–676. doi: 10.1016/j.exphem.2010.03.020. [DOI] [PubMed] [Google Scholar]
- 52.Wall M, Poortinga G, Hannan KM, Pearson RB, Hannan RD, McArthur GA. Translational control of c-MYC by rapamycin promotes terminal myeloid differentiation. Blood. 2008;112:2305–2317. doi: 10.1182/blood-2007-09-111856. [DOI] [PubMed] [Google Scholar]
- 53.Carayol N, Katsoulidis E, Sassano A, Altman JK, Druker BJ, Platanias LC. Suppression of programmed cell death 4 (PDCD4) protein expression by BCR-ABL-regulated engagement of the mTOR/p70 S6 kinase pathway. J Biol Chem. 2008;283:8601–8610. doi: 10.1074/jbc.M707934200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ozpolat B, Akar U, Steiner M, Zorrilla-Calancha I, Tirado-Gomez M, Colburn N, et al. Programmed cell death-4 tumor suppressor protein contributes to retinoic acid-induced terminal granulocytic differentiation of human myeloid leukemia cells. Mol Cancer Res. 2007;5:95–108. doi: 10.1158/1541-7786.MCR-06-0125. [DOI] [PubMed] [Google Scholar]
- 55.Lehman N, Ledford B, Di Fulvio M, Frondorf K, McPhail LC, Gomez-Cambronero J. Phospholipase D2-derived phosphatidic acid binds to and activates ribosomal p70 S6 kinase independently of mTOR. Faseb J. 2007;21:1075–1087. doi: 10.1096/fj.06-6652com. [DOI] [PubMed] [Google Scholar]
- 56.Roux PP, Shahbazian D, Vu H, Holz MK, Cohen MS, Taunton J, et al. RAS/ERK signaling promotes site-specific ribosomal protein S6 phosphorylation via RSK and stimulates cap-dependent translation. J Biol Chem. 2007;282:14056–14064. doi: 10.1074/jbc.M700906200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fox CJ, Hammerman PS, Cinalli RM, Master SR, Chodosh LA, Thompson CB. The serine/threonine kinase Pim-2 is a transcriptionally regulated apoptotic inhibitor. Genes Dev. 2003;17:1841–1854. doi: 10.1101/gad.1105003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tamburini J, Green AS, Bardet V, Chapuis N, Park S, Willems L, et al. Protein synthesis is resistant to rapamycin and constitutes a promising therapeutic target in acute myeloid leukemia. Blood. 2009;114:1618–1627. doi: 10.1182/blood-2008-10-184515. [DOI] [PubMed] [Google Scholar]
- 59.Waskiewicz AJ, Johnson JC, Penn B, Mahalingam M, Kimball SR, Cooper JA. Phosphorylation of the cap-binding protein eukaryotic translation initiation factor 4E by protein kinase Mnk1 in vivo. Mol Cell Biol. 1999;19:1871–1880. doi: 10.1128/mcb.19.3.1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wang X, Beugnet A, Murakami M, Yamanaka S, Proud CG. Distinct signaling events downstream of mTOR cooperate to mediate the effects of amino acids and insulin on initiation factor 4E-binding proteins. Mol Cell Biol. 2005;25:2558–2572. doi: 10.1128/MCB.25.7.2558-2572.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Grolleau A, Sonenberg N, Wietzerbin J, Beretta L. Differential regulation of 4E-BP1 and 4E-BP2, two repressors of translation initiation, during human myeloid cell differentiation. J Immunol. 1999;162:3491–3497. [PubMed] [Google Scholar]
- 62.Olson KE, Booth GC, Poulin F, Sonenberg N, Beretta L. Impaired myelopoiesis in mice lacking the repressors of translation initiation, 4E-BP1 and 4E-BP2. Immunology. 2009;128:e376–384. doi: 10.1111/j.1365-2567.2008.02981.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Topisirovic I, Guzman ML, McConnell MJ, Licht JD, Culjkovic B, Neering SJ, et al. Aberrant eukaryotic translation initiation factor 4E-dependent mRNA transport impedes hematopoietic differentiation and contributes to leukemogenesis. Mol Cell Biol. 2003;23:8992–9002. doi: 10.1128/MCB.23.24.8992-9002.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118:3065–3074. doi: 10.1172/JCI34739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.