

Abstract
Helicobacter pylori establishes a chronic lifelong infection in the human gastric mucosa, which may lead to peptic ulcer disease or gastric adenocarcinoma. The human beta-defensins (hβDs) are antimicrobial peptides, hβD1 being constitutively expressed in the human stomach. We hypothesized that H. pylori may persist, in part, by downregulating gastric hβD1 expression. We measured hβD1 and hβD2 expression in vivo in relation to the presence, density and severity of H. pylori infection, investigated differential effects of H. pylori virulence factors, and studied underlying signalling mechanisms in vitro. Significantly lower hβD1 and higher hβD2 mRNA and protein concentrations were present in gastric biopsies from infected patients. Those patients with higher-level bacterial colonization and inflammation had significantly lower hβD1 expression, but there were no differences in hβD2. H. pylori infection of human gastric epithelial cell lines also downregulated hβD1. Using wild-type strains and isogenic mutants, we showed that a functionalcag pathogenicity island-encoded type IV secretion system induced this downregulation. Treatment with chemical inhibitors or siRNA revealed that H. pylori usurped NF-κB signalling to modulate hβD1 expression. These data indicate that H. pylori downregulates hβD1 expression via NF-κB signalling, and suggest that this may promote bacterial survival and persistence in the gastric niche.
Introduction
Helicobacter pylori persistently infects the stomachs of almost half the world's population. Although the majority of infected people remain asymptomatic, approximately 10–15% go on to develop peptic ulcer disease or gastric cancers. The disease outcome of an infection is determined by a combination of bacterial, host and environmental factors (Blaser and Atherton, 2004; Robinson et al., 2007; Atherton and Blaser, 2009). H. pylori expresses numerous virulence determinants that have been linked to disease, including the polymorphic vacuolating cytotoxin gene A (vacA) and the cag pathogenicity island (cagPAI) (Backert et al., 2010). H. pylori strains possessing toxic alleles of vacA manipulate epithelial and immune cell functions that contribute to disease. The cagPAI encodes a type IV secretion system (T4SS) that binds α5β1 integrin on host cells, penetrates and delivers the bacterial effector protein CagA (Odenbreit et al., 2000; Kwok et al., 2007). Once translocated into the cytosol, CagA activates specific signalling pathways, including MAP kinase and NF-κB-induced signalling. Both NF-κB p50/p65 heterodimers and p65 or p50 homodimers undergo nuclear translocation (Keates et al., 1997; Wada et al., 2001; Saha et al., 2008). This leads to the expression of a variety of pro-inflammatory and immune defence genes. The cagPAI also allows translocation of soluble bacterial cell wall components into the epithelial cytosol. These short-chain peptidoglycan derivatives (disaccharide tripeptides) are generated via activity of the lytic transglycosylase encoded by slt (HP0645), an enzyme normally involved in peptidoglycan remodelling. The disaccharide tripeptides are recognized by nucleotide-binding oligomerization domain 1 (NOD1), an intracellular sensor of Gram-negative bacteria, leading also to NF-κB-induced pro-inflammatory signalling (Viala et al., 2004; Brandt et al., 2005; Boughan et al., 2006). A third cagPAI-mediated pathway has recently been described, where interaction of CagL with the α5β1 integrin on epithelial cells also triggers MAP kinase and NF-κB activation (Gorrell et al., 2013). Bacterial factors therefore manipulate the gastric inflammatory response, which underlies the development of PUD and gastric cancer.
Antimicrobial peptides (AMPs) are important in the host response to infection. These small, cationic peptides are expressed by a number of cell types including epithelial cells. They can be subdivided into several categories, all of which are potent and cytotoxic against bacteria but not against normal mammalian cells (Guani-Guerra et al., 2010). One group, the human β-defensins (hβDs), is a crucial component of the host defence at mucosal epithelia (Zasloff, 2002; O'Neil, 2003). Expression of hβD2 and hβD4 is upregulated during H. pylori infection in a cagPAI-dependent and NF-κB-mediated manner, and these AMPs are known to have antimicrobial activity against the bacterium (George et al., 2003; Boughan et al., 2006; Hornsby et al., 2008; Otte et al., 2009). hβD3 also has bactericidal activity against H. pylori and its expression is initially upregulated by H. pylori infection in vitro (Boughan et al., 2006), but subsequently downregulated in a CagA-dependent manner during prolonged infection (Bauer et al., 2012).
hβD1 (encoded by DEFB1) is constitutively expressed in uninflamed normal tissue (Liu et al., 1997; O'Neil et al., 2000), which highlights its importance in protection against microbial infection. Expression in the GI tract (including the gastric mucosa) is predominantly by epithelial cells rather than inflammatory cells (Frye et al., 2000). One study found increased hβD1 expression in the H. pylori infected human gastric mucosa (Bajaj-Elliott et al., 2002), but a second found decreased expression (Taha et al., 2005). In a more recent study, a non-significant trend towards reduced levels of hβD1 mRNA was found in gastric biopsies from infected patients (Vordenbaumen et al., 2010). These studies, although somewhat contradictory, suggest that H. pylori may modulate hβD1 expression. Consistent with this idea are the observed binding motifs for multiple transcription factors, including NF-κB, in the promoter sequence of the DEFB1 gene (Liu et al., 1997; Zhu et al., 2003; Prado-Montes de Oca et al., 2009).
Many AMPs also have chemotactic activity, working together to direct immune effector cells to the site of infection. Importantly, hβD1, hβD2 and hβD3 are associated with recruiting immature dendritic cells and memory T cells via CC-chemokine receptor 6 (CCR6), hence representing a bridge between the innate and adaptive immune responses (Yang et al., 2002). Cathelicidins have been found to be involved in the recruitment of neutrophils, in addition to circulating and tissue-derived monocytes (De et al., 2000). AMPs therefore act to induce pro-inflammatory immune responses, in some cases inducing immune mediators that further induce the expression of these AMPs, effectively creating a positive feedback loop (Zasloff, 2007). Therefore, downregulation of hβD1 could also mediate persistence of H. pylori infection by modulating the immune response.
The role of hβD1 during H. pylori infection is unclear and modulation of hβD1 expression by both host and bacterial factors may be possible. In this study, we therefore aimed to assess hβD1 expression levels in the H. pylori infected gastric mucosa in comparison with hβD2, to characterize the influence of H. pylori virulence determinants on hβD1 expression, and to determine the signalling pathways involved in regulating expression of this defensin during infection.
Results
H. pylori infection is associated with reduced hβD1 expression in the human stomach in vivo
First, we assessed hβD1 (DEFB1) expression in the human stomach in H. pylori infected and uninfected patients, in comparison with hβD2 (DEFB4A) expression. DEFB1 mRNA expression levels were threefold lower in gastric biopsies from 31 H. pylori infected compared with 23 uninfected patients (P = 0.005; Fig. 1A). In agreement with previous studies (Wada et al., 1999; Hamanaka et al., 2001; Uehara et al., 2003; Boughan et al., 2006; Bauer et al., 2013), DEFB4A expression levels were elevated in H. pylori infected gastric biopsies (P = 0.001; Fig. 1A). Median DEFB1 expression was twofold lower with cagA+ strain infections compared with cagA− infections, while DEFB4A expression was significantly higher (P = 0.028 and P = 0.006 respectively; Fig. 1A). In a manner similar to other studies on gastric mucosal defensins, to determine differences in protein expression, gastric biopsies were lysed and the concentrations of hβD1 and hβD2 were quantified by ELISA (Bauer et al., 2013). As found by RT-qPCR, hβD1 concentrations were significantly lower in biopsies from 10 infected patients compared with five uninfected patients (P = 0.001; Fig. 1B), while hβD2 protein concentrations were higher (P = 0.001). Lower hβD1 and higher hβD2 concentrations were also detected in the presence of a cagA+ infection (P = 0.016 and P = 0.004 respectively; Fig. 1B).
Figure 1.

Analysis of hβD1 and hβD2 expression during H. pylori infection in vivo. Levels of DEFB1 and DEFB4A mRNA were measured in the gastric mucosa of 23 uninfected and 31 H. pylori infected donors (A: *P = 0.005 and P = 0.001 respectively), and compared according to cagA genotype status of the colonizing strain (A: *P = 0.028 and P = 0.006 respectively). hβD1 and hβD2 protein concentrations in gastric biopsies from five uninfected and 10 H. pylori infected donors were measured (B: *P = 0.001 and P = 0.001), and concentrations in five cagA+ and five cagA− biopsies were also compared (B: *P = 0.016 and P = 0.004). Expression levels were also stratified based on histological inflammation scores graded from gastric biopsy tissue sections as mild (score of 1, n = 6), moderate (score of 2, n = 20) or substantial (score of 3, n = 5) (C: *P = 0.045). Data were also stratified according to bacterial density scores: mild (score of 1, n = 13), moderate (score of 2, n = 5) or substantial colonization (score of 3, n = 13) (D: *P = 0.001). RT-qPCR data were normalized against GAPDH and expressed relative to measurements from an uninfected tissue comparator. Protein concentrations were calculated per mg of total protein. Boxes represent the first and third quartiles with median values shown as a horizontal line within the box. Whiskers represent the lowest and highest observations within 1.5 times the first and third quartile.
Next, we examined associations of DEFB1 and DEFB4A expression with the intensity of inflammation as assessed by histopathology, scoring gastric antral tissue sections from the H. pylori infected patients. Sixfold lower DEFB1 mRNA levels were observed in samples with grade 3 inflammation compared with those with grade 1 (P = 0.045; Fig. 1C). There was an opposing trend but no significant differences in DEFB4A expression. Finally, we investigated the relationship between hβD1 and H. pylori colonization density in vivo, also by histopathology. A twofold lower DEFB1 mRNA level was observed in samples with grade 3 density compared with those with grade 1 (P = 0.009; Fig. 1D), suggesting a link between its expression and control of bacterial density. Again, no significant differences were observed for DEFB4A expression.
hβD1 is downregulated in epithelial cells by pathogenic strains of H. pylori in vitro
To assess hβD1 expression by epithelial cells in response to H. pylori infection in vitro, we co-cultured the MKN7 human gastric epithelial cell line [reported to have the most similar characteristics to normal human gastric mucosal cells (Linden et al., 2007)] for 24 h with the cagPAI+ vacA s1/m1 H. pylori strains 60190, 26695, 11637 and P12, and the cagPAI− vacA s2/m2 strains Tx30a, J63 and J68 at a multiplicity of infection (moi) of 100 bacteria per cell. ELISA assays showed that mean hβD1 protein concentrations in culture supernatants were consistently > 73% lower following infection with the cagPAI+ strains compared with uninfected cells (P < 0.001 for each; Fig. 2A), but no effects were induced by any of the cagPAI− strains. This result was confirmed for 60190 and Tx30a strains by RT-qPCR (Fig. 2B). Conversely, in the same experiment the cagPAI+ strains induced marked increases in hβD2 release (P < 0.01 for all; Fig. 2C) as previously reported (Wada et al., 1999; O'Neil et al., 2000; Uehara et al., 2003). To demonstrate that the findings were not a cell line-specific anomaly, we also conducted experiments with AGS cells in parallel and obtained similar results, although lower concentrations of defensins were detected (Fig. 2D–F). These data show that pathogenic H. pylori strains potently downregulate hβD1 expression by different gastric epithelial cell lines.
Figure 2.

Analysis of hβD1 expression during H. pylori infection in vitro. MKN7 (A–C) and AGS (D–F) cell lines were infected with H. pylori strains 60190, 11637, 26695, P12 (all cagA positive and expressing the s1/m1 form of vacA), and Tx30a, J63, J68 (cagPAI−, vacA s2/m2) for 24 h. hβD1 and hβD2 protein concentrations in culture supernatants were measured by ELISA (A, C, D and F). hβD1 mRNA expression was measured by RT-qPCR (B and E), and data presented as fold differences relative to that measured in uninfected cells. Bars depict mean expression levels from three independent experiments and error bars show standard deviations. The asterisk (*) indicates a significant difference in expression compared with uninfected cells (P < 0.01).
The H. pylori cagPAI induces hβD1 downregulation
As we observed hβD1 downregulation in vitro only when cells were infected with cagPAI+ vacA s1/m1 H. pylori strains, we next aimed to determine which bacterial genes influenced the expression of hβD1. To achieve this, hβD1 protein and mRNA expression levels were assessed when MKN7 or AGS cells were co-cultured with the wild-type strain 60190 (60190WT), or its isogenic mutants 60190ΔcagE (which does not express the cagPAI-encoded T4SS), 60190ΔcagA (which expresses the T4SS but does not translocate CagA into host cells) and a vacA null mutant (60190ΔvacA). The reduction in hβD1 in MKN7 and AGS cells was less marked for the 60190ΔcagE mutant than 60190WT (significant difference in AGS cells only, P = 0.01) indicating that the cagPAI contributed to hβD1 downregulation. However, the 60190ΔcagA strain downregulated hβD1 by a similar extent to the wild-type strain, for both mRNA and protein levels, showing that the injected T4SS effector protein CagA was not involved in this process (Fig. 3A, C and D). We also found no difference in hβD1 expression from co-culture of epithelial cell lines with the 60190ΔvacA mutant (Fig. 3A, C and D). As a control for the performance of the mutants in the assays, IL-8 concentrations were also measured. Effects of all mutants were in line with previous reports (Viala et al., 2004; Argent et al., 2008; Gorrell et al., 2013) (Fig. 3B).
Figure 3.
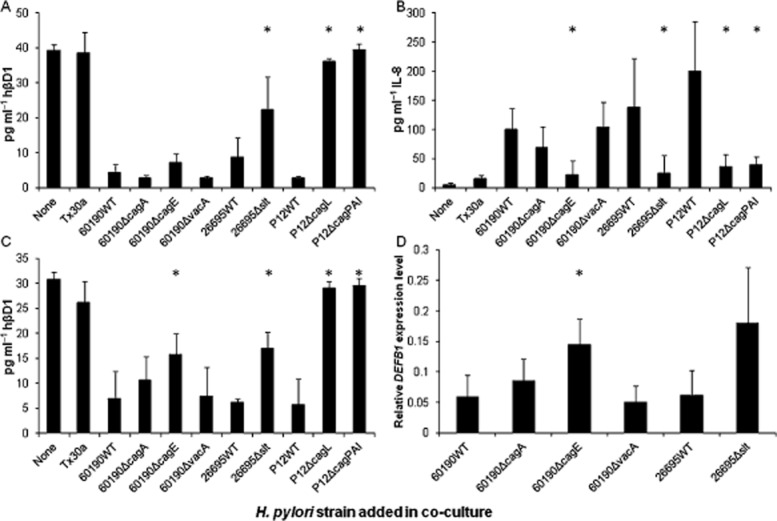
The effect of bacterial virulence factors on hβD1 expression in vitro. MKN7 (A and B) and AGS cells (C and D) were infected with H. pylori strains Tx30a, 60190WT, 60190ΔcagA, 60190ΔcagE, 60190ΔvacA, 26695WT, 26695Δslt, P12WT, P12ΔcagL and P12ΔcagPAI (moi = 100). hβD1 (A and C) and IL-8 (B) concentrations in culture supernatants were measured by ELISA after 24 h. The asterisk (*) indicates a significant difference in concentration when comparing effects of mutant strains to their parental strain 60190WT, 26695WT or P12WT (P < 0.01). Fold differences in hβD1 mRNA expression relative to uninfected cells were quantified by RT-qPCR (D). The asterisk (*) indicates a significant difference in expression level compared with 60190WT-infected cells (P < 0.05). Bars represent the mean of three independent experiments and error bars show standard deviations.
As H. pylori peptidoglycan processed by the lytic transgycosylase Slt has also been reported to be translocated into epithelial cells via the cagPAI-encoded T4SS, inducing activation of NOD1, NF-κB signalling and secretion of the pro-inflammatory cytokine IL-8 (Viala et al., 2004), we investigated whether this process contributed to hβD1 downregulation. Cells were cultured with an slt (HP0645) null mutant derived from H. pylori strain 26695 (26695Δslt) (Viala et al., 2004; Chaput et al., 2007). This mutant generates up to 40% less cell wall disaccharide tripeptide than the wild-type (26695WT) but has comparable growth rates with the wild-type strain and has no defects in the formation of the T4SS. We showed that the 26695Δslt strain induced significantly less hβD1 downregulation compared with 26695WT in MKN7 and AGS cells (P = 0.01) but this did not completely reverse the effect (Fig. 3A and C).
Finally we co-cultured cells with a complete cagPAI null mutant derived from the P12 strain (P12ΔcagPAI), and confirmed that levels of hβD1 expression were similar to that observed in uninfected cells. Similarly a cagL null mutant (P12ΔcagL), in which the T4SS is incapable of interacting with epithelial cells via α5β1 integrin, did not downregulate hβD1 expression. These results show that the cagPAI induces hβD1 downregulation, possibly through CagL-α5β1 integrin interactions and delivery of cell wall disaccharide tripeptides, rather than via delivery of CagA.
H. pylori usurps NF-κB signalling to downregulate hβD1
We next aimed to determine the intracellular signalling pathways through which H. pylori regulates hβD1 expression. Sequence analysis of the DEFB1 gene identified binding sites in the promoter for NF-κB1 (p50 subunit of NF-κB) and Activator Protein (AP)-1 (Prado-Montes de Oca, 2010), which implies regulation of hβD1 transcription through NF-κB and/or MAP kinase signalling. Given the observed association between the cagPAI and hβD1 expression, we investigated the role of NF-κB and the individual ERK, p38 and JNK MAP kinase signalling pathways in hβD1 downregulation during infection. AGS and MKN7 cells were cultured with H. pylori strain 60190WT in the presence of specific drug inhibitors of each pathway respectively. Effects on DEFB1 mRNA, and hβD1 and hβD2 protein were examined (Fig. 4). Uninfected cells were treated with recombinant TNFα as a positive control for activation of NF-κB, and this reduced DEFB1 expression, reduced hβD1 secretion (P = 0.001), and increased hβD2 release compared with untreated cells (P = 0.01). As previously, the 60190WT strain reduced hβD1 and increased hβD2 expression. The ERK, p38 and JNK kinase inhibitors had a slight but no significant impact on H. pylori-induced hβD1 downregulation. In contrast, the NF-κB inhibitor blocked these effects significantly (two- to fourfold difference in hβD1 concentrations between cultures infected with 60190WT in the presence and absence of NF-κB inhibitor; P = 0.05 and P = 0.01 in AGS and MKN7 cells respectively; Fig. 4B and C). These results confirm the importance of the NF-κB signalling pathway in H. pylori-modulated expression of hβD1 expression.
Figure 4.
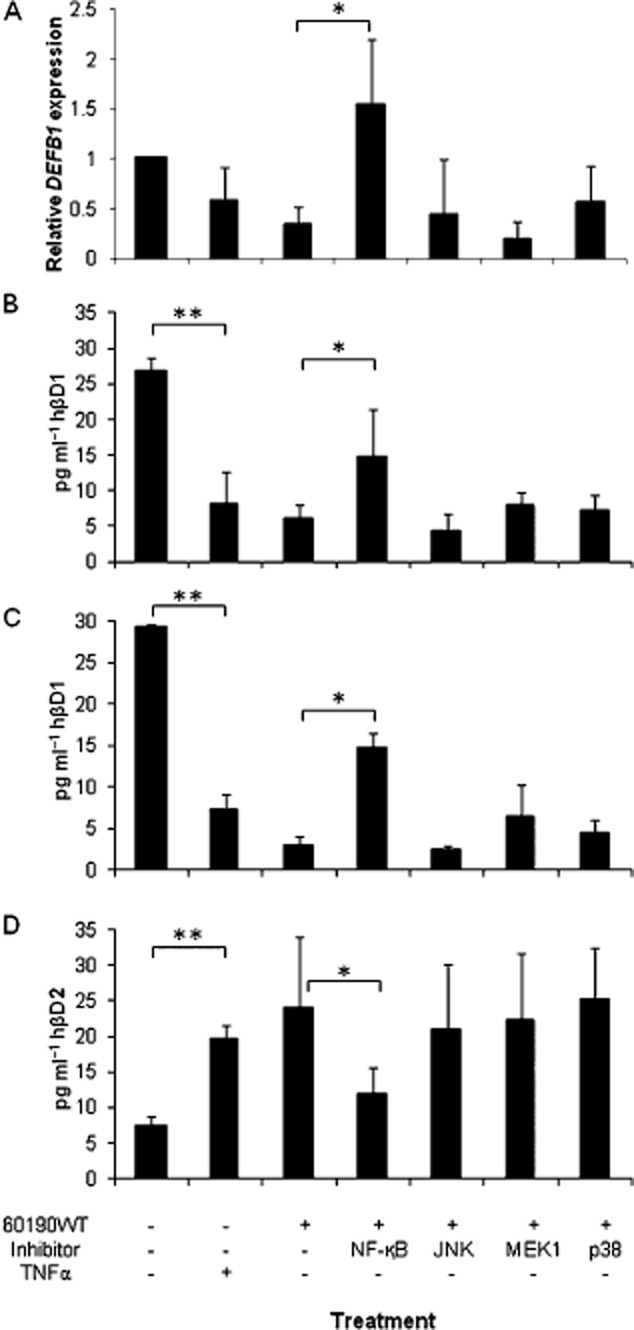
Assessing the signalling pathways involved in modulation of hβD1 expression in vitro, using inhibitor drugs. Expression of hβD1 mRNA (A) and protein (B and C), and also hβD2 protein (D) was assessed after treating 60190WT H. pylori infected AGS (A and B) and MKN7 (C and D) cells with 6-amino-4-(4-phenoxyphenylethylamino)quinazoline (NF-κB activation inhibitor), SP600125 (JNK inhibitor), U0126 (MEK1 inhibitor), or SB203586 (p38 inhibitor), prior to and during incubation. Treatment with the drug diluent alone was included as a negative control. TNFα treatment was included as a positive control inducer of NF-κB activation. mRNA expression levels are given as a fold difference relative to uninfected and untreated cells. *hβD1 significantly higher and hβD2 lower in NF-κB inhibitor-treated cells, compared with controls (P < 0.05). **hβD1 significantly lower and hβD2 higher in TNFα-treated, compared with untreated cells (P < 0.05). Bars represent the mean from three independent experiments and error bars show standard deviations.
To confirm the data and investigate the mechanisms further, small interference RNA (siRNA) experiments were performed to silence expression of NFKB1 (which encodes the NF-κBp50 subunit), and RELA (NF-κBp65 subunit). MAPK1 siRNA duplexes were also tested since the MAP kinase pathway is known to be stimulated by cagA-independent cagPAI signalling. Western blots confirmed the gene knock-downs (Fig. S1). 60190WT-infected cells previously treated with NFKB1 or RELA siRNA expressed two- to fivefold higher concentrations of hβD1 compared with those treated with negative control duplexes (P < 0.05 for both siRNAs in MKN7 and AGS cells; Fig. 5A and D). hβD2 expression in H. pylori-infected MKN45 cells is reportedly controlled by the p65 homodimeric form of NF-κB (Wada et al., 2001). Threefold lower concentrations of hβD2 were detected following RELA silencing in both cell lines (P < 0.05); effects of NFKB1 siRNA were less marked (Fig. 5B and E). RELA silencing also had a dramatic effect on IL-8 responses, but NFKB1 siRNA had no effect (Fig. 5C and F). MAPK1 siRNA treatment also had an effect on H. pylori-induced hβD1 expression, with significantly increased concentrations in AGS cell supernatants (P = 0.05). These data confirm the importance of NF-κB in the H. pylori-mediated downregulation of hβD1 expression and upregulation of hβD2 expression. They also indicate some involvement of the ERK pathway.
Figure 5.
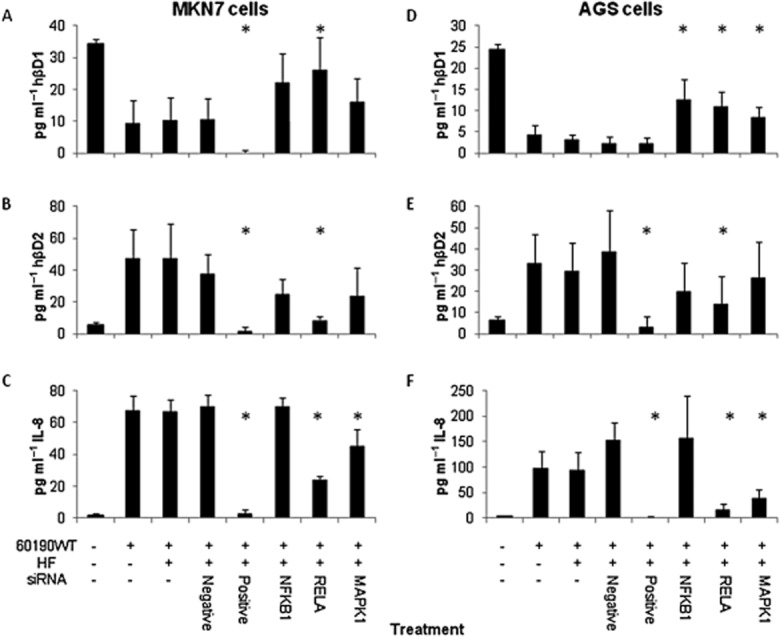
Assessing the signalling pathways involved in modulation of hβD1 expression in vitro, using gene silencing. hβD1 (A and D), hβD2 (B and E) and IL-8 (C and F) concentrations 24 h after infecting MKN7 (A–C) and AGS (D–F) cells with 60190WT H. pylori. Cells were pre-treated 48 h previously with siRNA duplexes in HiPerfect transfection reagent (HF). siRNA treatments targeted the NFKB1 (NFκBp50), RELA (NFκBp65) and MAPK1 genes. Negative control duplexes were non-silencing, whereas positive control duplexes targeted genes necessary for cell survival. *Significantly different concentration compared with cells treated with negative control siRNA (P < 0.05). Bars represent the mean from three independent experiments and error bars show standard deviations.
Discussion
Antimicrobial peptides play a vital role during infection, acting as a key line of defence against invading microbes and also as essential components in modulating the immune response to infections. While expression of hβD2 and hβD4 is inducible and upregulated in response to H. pylori infection, hβD1 is normally constitutively expressed by epithelial cells in the absence of H. pylori. Mice with a deletion in the homologous mBD1 gene have an impaired capacity to combat bacterial infections (Morrison et al., 2002; Moser et al., 2002), reflecting the importance of this AMP as a component of the innate anti-bacterial immune response. However, there is conflict in the literature concerning how hβD1 is differentially expressed during H. pylori infection (Bajaj-Elliott et al., 2002; Taha et al., 2005; Kocsis et al., 2009; Vordenbaumen et al., 2010). In agreement with the study by Taha et al., we found that mRNA expression of hβD1 was downregulated in the H. pylori-infected human gastric mucosa and also in infected gastric epithelial cells in vitro. Two studies reporting upregulated hβD1 expression in infected epithelial cell lines in vitro used the same primer sequences (Bajaj-Elliott et al., 2002; Kocsis et al., 2009). When we performed additional tests using these however, the trends in our data remained the same, i.e. hβD1 expression was downregulated by infection by functional T4SS cagPAI+ H. pylori (data not shown). We were also able to confirm our findings using ELISA to quantify hβD1 protein both in gastric biopsy tissue and in culture supernatants, which validates our mRNA data.
Our data show that hβD1 expression is modulated during H. pylori infection. Downregulation of hβD1 expression has previously been observed in the intestinal mucosa of patients infected with Shigella dysenteriae (Islam et al., 2001), or those with Crohn's disease or ulcerative colitis (Wehkamp et al., 2003). There is also a precedent for hβD1 downregulation in epithelial cells in vitro. Culturing intestinal epithelial cells with the enteric pathogens Vibrio cholerae, enterotoxigenic Escherichia coli and S. dysenteriae suppressed hβD1 expression in a manner involving protein kinase A and ERK MAP kinase signalling (Chakraborty et al., 2008). Infections of airway and gingival epithelial cells with influenza virus, Herpes simplex virus 1 and Sendai virus was also recently reported to downregulate hβD1 expression. This process required live virus, but the mechanism remains unknown (Ryan et al., 2011).
We found that cagPAI+ wild-type strains markedly suppressed hβD1 expression, while three cagPAI− strains consistently did not. Analysis of bacterial factors demonstrated that hβD1 downregulation was cagA independent. Although our gastric biopsy data showed lower hβD1 expression in those infected with cagA+ strains, we have merely used this as a marker for presence of the cagPAI. In vitro, hβD1 downregulation was completely abrogated in cells infected with cagPAI- or cagL-deficient mutants, and partially reversed with the slt mutant. This indicated that the suppression was mediated by T4SS engagement of the α5β1 integrin and NOD1 activation in epithelial cells. We then investigated NF-κB- and MAPK-dependent downregulation of hβD1, given the known action of cagPAI-containing strains upon these signalling pathways (Brandt et al., 2005). Interestingly, increased hβD1 expression was observed when NF-κB signalling was inhibited, and was reduced with TNFα-mediated NF-κB activation. NF-κB response elements have been described in the DEFB1 promoter sequence (Prado-Montes de Oca et al., 2009). The role of H. pylori induced NF-κB signalling in the suppression rather than induction of gene expression is somewhat unusual, but not unknown. For example, suppression of H,K-ATPase expression, the enzyme mediating gastric acid secretion, was observed in H. pylori infected AGS cells and found to involve T4SS-dependent, CagA-independent NF-κB activation (Saha et al., 2008; 2010).
The NF-κB family of transcription factors consists of five members, and NF-κB exists as a homo- or heterodimer of these subunits. Of these, p50 and p52 lack the transcription activation domain necessary for transcription. Binding of these homodimers to a promoter can block transcription of the target gene (Hayden and Ghosh, 2008). Saha et al. showed that infection of AGS cells with a cagPAI+ strain of H. pylori induced transfer of both homodimeric p50/p50 and heterodimeric p65/p50 forms to the nucleus. Expression of H,K-ATPase was repressed by the binding of p50/p50 NF-κB to the HKα promoter (Saha et al., 2008). The DEFB1 gene promoter is known to have a p50-binding domain, therefore p50 homodimers or p65/p50 heterodimers could potentially bind (Prado-Montes de Oca et al., 2009). We found that silencing of NFBK1 and RELA equivalently prevented the inhibition of hβD1 expression, therefore each of these genes plays a role and the inhibitory effect of p50 homodimers appears a less likely explanation. Another possibility is that NF-κB activation (p65/p50) stimulates expression of host factors which then block hβD1 gene expression, for example olfactomedin 4, which inhibits NF-κB activation in a feedback mechanism involving NOD1 (Liu et al., 2010a), and various microRNAs (Xiao et al., 2009; Tang et al., 2010; Liu et al., 2010b). Our finding that hβD1 suppression could be induced by TNFα, which is known to stimulate activation and nuclear translocation of NF-κBp65 in AGS cells (Robinson et al., 2008), is novel and adds weight to this theory. TNFα could also be exerting an effect on defensin expression in the stomach, and it would be interesting to test this using animal models. Incubation of other types of epithelial cells with NF-κB inhibitors or TNFα has not been shown to influence hβD1 expression (Zhao et al., 1996; O'Neil et al., 1999; Joly et al., 2005); however, defensin responses are known to be cell line dependent (Grubman et al., 2010).
As a further control for our experiments, we measured expression of the more widely studied defensin hβD2. In accordance with others, we found this to be increased in response to H. pylori both in vivo and in vitro (Wada et al., 1999; Boughan et al., 2006; Bauer et al., 2013), and increased further with cagPAI+ strains (Hornsby et al., 2008; Grubman et al., 2010). Bauer et al. found that although DEFB4 mRNA was elevated in the infected gastric mucosa, this trend could not be shown with protein concentrations (Bauer et al., 2013). The defensin concentrations detected in our study were lower, possibly because we used a buffer with a lower detergent content when preparing the lysates (Staples et al., 2013). This possible explanation for the discrepant results between the studies warrants further investigation. Our mechanistic data on hβD2 agreed with that of Grubman et al., who found that NOD1 activation induced by cagPAI+ strains induced DEFB4 mRNA expression in AGS cells. Interestingly they showed that DEFB4 expression could also be induced in HEK293 cells by stimulation with TNFα (Grubman et al., 2010). We found similar trends to our in vivo data using two different cell lines, and also confirmed the findings of others. This is very encouraging, but further studies are needed with a wider range of cell types, and using other methods, e.g. luciferase reporter assays of DEFB1 and DEFB4 gene promoter activity, and immunohistochemistry analysis of biopsy tissues. Using a defensin ELISA on whole biopsy lysates does not take account of the possibility that increased inflammatory cells in infected tissue influenced the findings, which were normalized for total protein content. The range of biopsy protein concentrations among the groups, however, were similar.
We have shown that epithelial cell hβD1 expression is downregulated during H. pylori infection, but the importance of such modulation is still not completely clear. It has recently come to light that hβD3 expression is also suppressed during prolonged H. pylori infection of AGS cells via a CagA-dependent mechanism, and that its expression in vivo is also reduced in gastric biopsies from infected patients (Bauer et al., 2012; 2013). The fact that high colonization densities in vivo correspond with lower hβD1 expression indicates that reducing the level of hβD1 may contribute to the persistence of the bacterium in the gastric mucosa, but a role for hβD3 suppression is also likely to be important. Additionally, hβD1 bactericidal activity has been reported to be synergistic with hβD2 and the cathelicidin LL-37 (George et al., 2003; Hase et al., 2003), both of which have bactericidal activity against the bacterium. Therefore, downregulation of hβD1 may also limit the consequences of hβD2 and LL-37 activity, providing an additional benefit over merely reducing hβD1 expression.
In conclusion, we have demonstrated an NF-κB-dependent downregulation of hβD1 expression during H. pylori infection, which was dependent on CagA-independent cagPAI signalling. In agreement with the in vitro experiments, lower-level expression of hβD1 in the infected human gastric mucosa was significantly associated with cagPAI+ strains, more severe inflammation and higher colonization densities. We suggest that H. pylori-induced modulation of hβD1 expression may contribute to the persistence of H. pylori in the gastric mucosa.
Experimental procedures
Tissue samples
Antral gastric biopsies were donated by 31 H. pylori-infected and 23 uninfected patients attending the University Hospital, Nottingham, for routine upper gastrointestinal endoscopy, with informed written consent and approval from the Nottingham Research Ethics Committee. H. pylori status was determined by rapid urease test, bacterial culture and histology. Samples were not collected from patients taking proton pump inhibitors, non-steroidal anti-inflammatory drugs, or antibiotics in the 2 weeks preceding endoscopy. Bacterial isolates were PCR-genotyped for cagA status as previously described (Hussein et al., 2008). Biopsy specimens for histology were formalin-fixed, paraffin-embedded, cut to 4 μm thickness, and stained with haematoxylin and eosin or toluidine blue for assessment of inflammation and H. pylori colonization density respectively. Grading was carried out using the modified Sydney Scoring System (0 = not present, 1 = mild, 2 = moderate and 3 = substantial) by an experienced histopathologist (AMZ) who was blinded to other data (Genta and Dixon, 1995). Biopsies for RNA analysis were immediately preserved in RNAlater (Sigma-Aldrich, UK).
Gastric biopsy lysates
Gastric biopsies from five uninfected and 10 infected patients (five with cagA+ strains) were homogenized according to a previously described method (Staples et al., 2013). Single biopsies were suspended in 300 μl PBS containing 2 mM Mg2+ (Sigma), 25 U ml−1 Benzonase® nuclease (Novagen, Germany), and protease inhibitors (complete mini [EDTA-free], Roche, Germany), processed on ice using disposable pestles and filter tips. Samples were clarified by centrifugation at 10 000 g for 10 min at 4°C. Supernatants were aliquoted into LoBind tubes (Eppendorf), tested for total protein concentration using a bicinchoninic acid (BCA) assay kit (Pierce, IL, USA), and stored at −80°C. Supernatants from infected and uninfected donors contained similar protein concentrations (medians 1.77 and 1.54 mg ml−1 respectively).
Cell lines and bacterial strains
The human gastric epithelial MKN7 cell line (kind gift from Dr Sara Linden, University of Gothenburg, Sweden) was maintained in RPMI1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Sigma-Aldrich). AGS cells (ATCC CRL-1739™) were grown in nutrient mixture F12 Ham supplemented with 10% FBS and 2 mM l-glutamine (Sigma-Aldrich). All cell lines were incubated at 37°C in a 5% CO2 humidified atmosphere. cagPAI+ H. pylori strains 60190, 11637, 26695, P12 and cagPAI− isolates Tx30a, J63 and J68 (Boughan et al., 2006; Corcoran et al., 2007; Keates et al., 2007) were cultured on Blood agar base 2 containing 5% (v/v) horse blood (Oxoid, Cambridge, UK) at 37°C under microaerobic conditions (Argent et al., 2004). Isogenic mutants deficient in vacA (60190ΔvacA), cagA (60190ΔcagA) and cagE (60190ΔcagE) derived from the 60190 strain (Argent et al., 2008), cagPAI- and cagL-deficient mutants (P12ΔcagPAI and P12ΔcagL) derived from the P12 strain (Kwok et al., 2007), and an slt deletion mutant (26695Δslt) derived from the 26695 strain [kindly donated by Dr Richard Ferrero, Monash University, Victoria, Australia (Viala et al., 2004)], were also used.
In vitro culture experiments
Using methods based on those of Bajaj-Elliott et al. (2002), 5 × 104 MKN7 or AGS cells per well were seeded in 24-well culture plates with the appropriate medium and allowed to adhere at 37°C in a 5% CO2 air-humidified atmosphere for 24 h. The medium was replaced with a suspension of H. pylori at a multiplicity of infection of 100 bacteria per epithelial cell, and cultures were incubated for a further 24 h. Multiplicities of infection were confirmed by viable counting. For quantification of defensins and IL-8 concentrations in supernatants, 1 × 105 epithelial cells per well were seeded, and co-cultures were carried out using serum-free F12 medium.
Defensin and IL-8 ELISA assays
After co-culture of epithelial cells with H. pylori, supernatants were aliquoted, frozen at −80°C and thawed once only. Biopsy lysates were thawed and tested immediately for defensins. hβD1 and hβD2 assays were performed using Human BD-1 and BD-2 ELISA Development Kits (PeproTech, UK) and IL-8 concentrations were determined with a Human IL-8 CytoSet™ ELISA (Invitrogen), according to manufacturers' instructions and with a standard curve on each plate. Typical sensitivity limits (mean plus 3 standard deviations of six replicate 0 pg ml−1 control wells) were 0.5 pg ml−1 hβD1, 4.5 pg ml−1 hβD2 and 5.1 pg ml−1 IL-8.
Reverse transcriptase PCR (RT-qPCR)
RNA was extracted from antral gastric biopsies and cell lines using an RNeasy Mini kit (QIAGEN, Crawley, UK) according to the manufacturer's instructions. cDNA was generated from 100 ng RNA using Superscript reverse transcriptase II, with oligo (dT) primers (Invitrogen). Real-time PCR was performed using the Rotor-Gene 3000 real-time PCR system (QIAGEN). First stage RT-PCR samples, produced in the absence of reverse transcriptase from each RNA sample, were tested in parallel to detect genomic DNA contamination. Samples were run in duplicate and the results were analysed using the Pfaffl method (Pfaffl, 2001). Relative gene expression levels were determined by normalizing against human glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mRNA levels, and data were presented as a fold difference in comparison with an uninfected reference sample. For assessing expression in vivo, the uninfected comparator consisted of cDNA synthesized from pooled purified RNA extracted from biopsies of 10 randomly selected H. pylori-negative patients. For in vitro analysis, RNA was purified from epithelial cells cultured under different conditions for 24 h. The uninfected negative controls in each experiment were taken as the negative comparator. A commercial human cDNA standard (BD Biosciences; Oxford, UK) was included as a positive control in all assays.
Quantification of hβD1 mRNA was carried out using a QuantiTECT™ SYBR Green PCR kit with commercial primers (QIAGEN). Amplification of hβD2 was carried out over 45 cycles of 15 s at 95°C, 30 s at 61°C and 30 s at 72°C (Primer sequences: hβD2 forward: 5′-CTGATGCCTCTTCCAGGTGTTT-3′; hβD2 reverse: 5′-GAGACCACAGGTGCCAATTTG-3′; GAPDH forward: 5′-CCACATCGCTCAGACACCAT-3′; GAPDH reverse: 5′-GGCAACAATATCCACTTTACCAGAGT-3′). No-template controls were included in each run.
Inhibitor studies
Epithelial cells were pre-treated with specific chemical inhibitors (Merck, Nottingham, UK) for 60 min prior to and during bacterial stimulation. The drugs used were U0126 (10 μM; MEK 1 inhibitor), SP600125 (10 μM; JNK inhibitor), SB203586 (10 μM; p38 inhibitor) and 6-amino-4-(4-phenoxyphenylethylamino)quinazoline (1 μM; NF-κB activation inhibitor). Cultures were incubated as described above and defensin expression levels were assessed. As a positive control inducer of NF-κB activation (Robinson et al., 2008), cells were treated with 50 ng ml−1 recombinant TNFα (PeproTech).
siRNA transfections
Validated siRNA duplexes targeting NFKB1, RELA and MAPK1 mRNA (QIAGEN) were prepared according to the manufacturer's instructions. Non-silencing AllStars Hs Negative Control siRNA and AllStars Hs Cell Death Control siRNA (positive control) (QIAGEN) were tested in parallel. Epithelial cells were seeded at 1 × 105 per well in 24-well plates and treated with 10 nM siRNA suspended in HiPerfect transfection regent (QIAGEN). Controls were treated with HiPerfect only, or PBS. The cells were incubated for 48 h at 37°C in 5% CO2, when a high degree of cell death was observed in the positive control wells. This siRNA construct targets genes that are indispensable for cell survival, thus cell death confirmed successful transfection. NFKB1, RELA and MAPK1 gene knock-down was confirmed by Western blotting (Fig. S1) using rabbit antibodies against NF-κB p50 (Cell Signaling Technology, MA, USA), NF-κB p65 (Millipore, MA, USA), MAPK1/ERK (Source BioScience UK) and actin (Sigma-Aldrich), with an anti-rabbit IgG-peroxidase conjugate (Sigma-Aldrich) and chemiluminescent ECL substrate (GE Healthcare, UK). Medium was removed from the wells before infecting with H. pylori for a further 24 h.
Statistical analysis
Statistical analyses were carried out using GraphPad Prism 6 software. A P value ≤ 0.05 was taken as indicative of a significant difference. In vivo data were displayed in box-and-whisker plots, and compared using a Mann–Whitney U test or, for multiple parameters, Kruskal–Wallis tests with a post hoc Dunn's multiple comparison. In vitro data were described using means and standard deviations, and comparisons between groups were made using one-way anova with a Dunnett's post hoc test for multiple variates.
Acknowledgments
This article presents independent research supported by the Medical Research Council (Project Grant G0601170), by the National Institute for Health Research (NIHR) through the NIHR Biomedical Research Unit in Gastrointestinal and Liver Diseases at Nottingham University Hospitals NHS Trust, and an award from the University of Nottingham. The work of S.B. is supported through a grant by the German Science Foundation (project B10 of CRC-796). The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Confirmation of gene silencing in AGS cells treated with siRNA duplexes by Western blotting. Cells seeded in 24-well plates were transfected with siRNA to target expression of NFKB1, RELA, and MAPK1. Non-silencing negative control duplexes (Neg) were also used. After 48 h, cells were harvested into SDS-PAGE sample buffer. Western blots were probed, stripped and re-probed using antibodies against NF-κB p50 (NFKB1 gene product), NF-κB p65 (RELA gene product), MAPK1/ERK and beta actin.
References
- Argent RH, Kidd M, Owen RJ, Thomas RJ, Limb MC, Atherton JC. Determinants and consequences of different levels of CagA phosphorylation for clinical isolates of Helicobacter pylori. Gastroenterology. 2004;127:514–523. doi: 10.1053/j.gastro.2004.06.006. [DOI] [PubMed] [Google Scholar]
- Argent RH, Thomas RJ, Letley DP, Rittig MG, Hardie KR, Atherton JC. Functional association between the Helicobacter pylori virulence factors VacA and CagA. J Med Microbiol. 2008;57:145–150. doi: 10.1099/jmm.0.47465-0. [DOI] [PubMed] [Google Scholar]
- Atherton JC, Blaser MJ. Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J Clin Invest. 2009;119:2475–2487. doi: 10.1172/JCI38605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backert S, Mimuro H, Israel DA, Peek RM. Virulence factors of Helicobacter pylori. In: Sutton P, Mitchell HM, editors. Helicobacter pylori. Oxford, UK: CABI international; 2010. pp. 212–247. in the 21st Century. [Google Scholar]
- Bajaj-Elliott M, Fedeli P, Smith GV, Domizio P, Maher L, Ali RS, et al. Modulation of host antimicrobial peptide (beta-defensins 1 and 2) expression during gastritis. Gut. 2002;51:356–361. doi: 10.1136/gut.51.3.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer B, Pang E, Holland C, Kessler M, Bartfeld S, Meyer TF. The Helicobacter pylori virulence effector CagA abrogates human beta-defensin 3 expression via inactivation of EGFR signaling. Cell Host Microbe. 2012;11:576–586. doi: 10.1016/j.chom.2012.04.013. [DOI] [PubMed] [Google Scholar]
- Bauer B, Wex T, Kuester D, Meyer T, Malfertheiner P. Differential expression of human beta defensin 2 and 3 in gastric mucosa of Helicobacter pylori-infected individuals. Helicobacter. 2013;18:6–12. doi: 10.1111/hel.12000. [DOI] [PubMed] [Google Scholar]
- Blaser MJ, Atherton JC. Helicobacter pylori persistence: biology and disease. J Clin Invest. 2004;113:321–333. doi: 10.1172/JCI20925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boughan PK, Argent RH, Body-Malapel M, Park JH, Ewings KE, Bowie AG, et al. Nucleotide-binding oligomerization domain-1 and epidermal growth factor receptor: critical regulators of beta-defensins during Helicobacter pylori infection. J Biol Chem. 2006;281:11637–11648. doi: 10.1074/jbc.M510275200. [DOI] [PubMed] [Google Scholar]
- Brandt S, Kwok T, Hartig R, Konig W, Backert S. NF-kappaB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc Natl Acad Sci USA. 2005;102:9300–9305. doi: 10.1073/pnas.0409873102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty K, Ghosh S, Koley H, Mukhopadhyay AK, Ramamurthy T, Saha DR, et al. Bacterial exotoxins downregulate cathelicidin (hCAP-18/LL-37) and human beta-defensin 1 (HBD-1) expression in the intestinal epithelial cells. Cell Microbiol. 2008;10:2520–2537. doi: 10.1111/j.1462-5822.2008.01227.x. [DOI] [PubMed] [Google Scholar]
- Chaput C, Labigne A, Boneca IG. Characterization of Helicobacter pylori lytic transglycosylases Slt and MltD. J Bacteriol. 2007;189:422–429. doi: 10.1128/JB.01270-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corcoran PA, Atherton JC, Kerrigan SW, Wadstrom T, Murray FE, Peek RM, et al. The effect of different strains of Helicobacter pylori on platelet aggregation. Can J Gastroenterol. 2007;21:367–370. doi: 10.1155/2007/490852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Y, Chen Q, Schmidt AP, Anderson GM, Wang JM, Wooters J, et al. LL-37, the neutrophil granule- and epithelial cell-derived cathelicidin, utilizes formyl peptide receptor-like 1 (FPRL1) as a receptor to chemoattract human peripheral blood neutrophils, monocytes, and T cells. J Exp Med. 2000;192:1069–1074. doi: 10.1084/jem.192.7.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye M, Bargon J, Lembcke B, Wagner TO, Gropp R. Differential expression of human alpha- and beta-defensins mRNA in gastrointestinal epithelia. Eur J Clin Invest. 2000;30:695–701. doi: 10.1046/j.1365-2362.2000.00696.x. [DOI] [PubMed] [Google Scholar]
- Genta RM, Dixon MF. The Sydney System revisited: the Houston International Gastritis Workshop. Am J Gastroenterol. 1995;90:1039–1041. [PubMed] [Google Scholar]
- George JT, Boughan PK, Karageorgiou H, Bajaj-Elliott M. Host anti-microbial response to Helicobacter pylori infection. Mol Immunol. 2003;40:451–456. doi: 10.1016/s0161-5890(03)00158-5. [DOI] [PubMed] [Google Scholar]
- Gorrell RJ, Guan J, Xin Y, Tafreshi MA, Hutton ML, McGuckin MA, et al. A novel NOD1- and CagA-independent pathway of interleukin-8 induction mediated by the Helicobacter pylori type IV secretion system. Cell Microbiol. 2013;15:554–570. doi: 10.1111/cmi.12055. [DOI] [PubMed] [Google Scholar]
- Grubman A, Kaparakis M, Viala J, Allison C, Badea L, Karrar A, et al. The innate immune molecule, NOD1, regulates direct killing of Helicobacter pylori by antimicrobial peptides. Cell Microbiol. 2010;12:626–639. doi: 10.1111/j.1462-5822.2009.01421.x. [DOI] [PubMed] [Google Scholar]
- Guani-Guerra E, Santos-Mendoza T, Lugo-Reyes SO, Teran LM. Antimicrobial peptides: general overview and clinical implications in human health and disease. Clin Immunol. 2010;135:1–11. doi: 10.1016/j.clim.2009.12.004. [DOI] [PubMed] [Google Scholar]
- Hamanaka Y, Nakashima M, Wada A, Ito M, Kurazono H, Hojo H, et al. Expression of human beta-defensin 2 (hBD-2) in Helicobacter pylori induced gastritis: antibacterial effect of hBD-2 against Helicobacter pylori. Gut. 2001;49:481–487. doi: 10.1136/gut.49.4.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hase K, Murakami M, Iimura M, Cole SP, Horibe Y, Ohtake T, et al. Expression of LL-37 by human gastric epithelial cells as a potential host defense mechanism against Helicobacter pylori. Gastroenterology. 2003;125:1613–1625. doi: 10.1053/j.gastro.2003.08.028. [DOI] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132:344–362. doi: 10.1016/j.cell.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Hornsby MJ, Huff JL, Kays RJ, Canfield DR, Bevins CL, Solnick JV. Helicobacter pylori induces an antimicrobial response in rhesus macaques in a cag pathogenicity island-dependent manner. Gastroenterology. 2008;134:1049–1057. doi: 10.1053/j.gastro.2008.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussein NR, Mohammadi M, Talebkhan Y, Doraghi M, Letley DP, Muhammad MK, et al. Differences in virulence markers between Helicobacter pylori strains from Iraq and those from Iran: potential importance of regional differences in H. pylori-associated disease. J Clin Microbiol. 2008;46:1774–1779. doi: 10.1128/JCM.01737-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam D, Bandholtz L, Nilsson J, Wigzell H, Christensson B, Agerberth B, Gudmundsson G. Downregulation of bactericidal peptides in enteric infections: a novel immune escape mechanism with bacterial DNA as a potential regulator. Nat Med. 2001;7:180–185. doi: 10.1038/84627. [DOI] [PubMed] [Google Scholar]
- Joly S, Organ CC, Johnson GK, McCray PB, Jr, Guthmiller JM. Correlation between beta-defensin expression and induction profiles in gingival keratinocytes. Mol Immunol. 2005;42:1073–1084. doi: 10.1016/j.molimm.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Keates S, Hitti YS, Upton M, Kelly CP. Helicobacter pylori infection activates NF-kappa B in gastric epithelial cells. Gastroenterology. 1997;113:1099–1109. doi: 10.1053/gast.1997.v113.pm9322504. [DOI] [PubMed] [Google Scholar]
- Keates S, Keates AC, Katchar K, Peek RM, Jr, Kelly CP. Helicobacter pylori induces up-regulation of the epidermal growth factor receptor in AGS gastric epithelial cells. J Infect Dis. 2007;196:95–103. doi: 10.1086/518440. [DOI] [PubMed] [Google Scholar]
- Kocsis AK, Kiss ZF, Tiszlavicz L, Tiszlavicz Z, Mandi Y. Potential role of human beta-defensin 1 in Helicobacter pylori-induced gastritis. Scand J Gastroenterol. 2009;44:289–295. doi: 10.1080/00365520802530879. [DOI] [PubMed] [Google Scholar]
- Kwok T, Zabler D, Urman S, Rohde M, Hartig R, Wessler S, et al. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature. 2007;449:862–866. doi: 10.1038/nature06187. [DOI] [PubMed] [Google Scholar]
- Linden SK, Driessen KM, McGuckin MA. Improved in vitro model systems for gastrointestinal infection by choice of cell line, pH, microaerobic conditions, and optimization of culture conditions. Helicobacter. 2007;12:341–353. doi: 10.1111/j.1523-5378.2007.00509.x. [DOI] [PubMed] [Google Scholar]
- Liu L, Zhao C, Heng HH, Ganz T. The human beta-defensin-1 and alpha-defensins are encoded by adjacent genes: two peptide families with differing disulfide topology share a common ancestry. Genomics. 1997;43:316–320. doi: 10.1006/geno.1997.4801. [DOI] [PubMed] [Google Scholar]
- Liu W, Yan M, Liu Y, Wang R, Li C, Deng C, et al. Olfactomedin 4 down-regulates innate immunity against Helicobacter pylori infection. Proc Natl Acad Sci USA. 2010a;107:11056–11061. doi: 10.1073/pnas.1001269107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Xiao B, Tang B, Li B, Li N, Zhu E, et al. Up-regulated microRNA-146a negatively modulate Helicobacter pylori-induced inflammatory response in human gastric epithelial cells. Microbes Infect. 2010b;12:854–863. doi: 10.1016/j.micinf.2010.06.002. [DOI] [PubMed] [Google Scholar]
- Morrison G, Kilanowski F, Davidson D, Dorin J. Characterization of the mouse beta defensin 1, Defb1, mutant mouse model. Infect Immun. 2002;70:3053–3060. doi: 10.1128/IAI.70.6.3053-3060.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moser C, Weiner DJ, Lysenko E, Bals R, Weiser JN, Wilson JM. beta-Defensin 1 contributes to pulmonary innate immunity in mice. Infect Immun. 2002;70:3068–3072. doi: 10.1128/IAI.70.6.3068-3072.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neil DA. Regulation of expression of beta-defensins: endogenous enteric peptide antibiotics. Mol Immunol. 2003;40:445–450. doi: 10.1016/s0161-5890(03)00161-5. [DOI] [PubMed] [Google Scholar]
- O'Neil DA, Porter EM, Elewaut D, Anderson GM, Eckmann L, Ganz T, Kagnoff MF. Expression and regulation of the human beta-defensins hBD-1 and hBD-2 in intestinal epithelium. J Immunol. 1999;163:6718–6724. [PubMed] [Google Scholar]
- O'Neil DA, Cole SP, Martin-Porter E, Housley MP, Liu L, Ganz T, Kagnoff MF. Regulation of human beta-defensins by gastric epithelial cells in response to infection with Helicobacter pylori or stimulation with interleukin-1. Infect Immun. 2000;68:5412–5415. doi: 10.1128/iai.68.9.5412-5415.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odenbreit S, Puls J, Sedlmaier B, Gerland E, Fischer W, Haas R. Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science. 2000;287:1497–1500. doi: 10.1126/science.287.5457.1497. [DOI] [PubMed] [Google Scholar]
- Otte JM, Neumann HM, Brand S, Schrader H, Schmidt WE, Schmitz F. Expression of beta-defensin 4 is increased in human gastritis. Eur J Clin Invest. 2009;39:126–138. doi: 10.1111/j.1365-2362.2008.02071.x. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prado-Montes de Oca E. Human beta-defensin 1: a restless warrior against allergies, infections and cancer. Int J Biochem Cell Biol. 2010;42:800–804. doi: 10.1016/j.biocel.2010.01.021. [DOI] [PubMed] [Google Scholar]
- Prado-Montes de Oca E, Velarde-Felix JS, Rios-Tostado JJ, Picos-Cardenas VJ, Figuera LE. SNP 668C (-44) alters a NF-kappaB1 putative binding site in non-coding strand of human beta-defensin 1 (DEFB1) and is associated with lepromatous leprosy. Infect Genet Evol. 2009;9:617–625. doi: 10.1016/j.meegid.2009.03.006. [DOI] [PubMed] [Google Scholar]
- Robinson K, Argent RH, Atherton JC. The inflammatory and immune response to Helicobacter pylori infection. Best Pract Res Clin Gastroenterol. 2007;21:237–259. doi: 10.1016/j.bpg.2007.01.001. [DOI] [PubMed] [Google Scholar]
- Robinson K, Kenefeck R, Pidgeon EL, Shakib S, Patel S, Polson RJ, et al. Helicobacter pylori-induced peptic ulcer disease is associated with inadequate regulatory T cell responses. Gut. 2008;57:1375–1385. doi: 10.1136/gut.2007.137539. [DOI] [PubMed] [Google Scholar]
- Ryan LK, Dai J, Yin Z, Megjugorac N, Uhlhorn V, Yim S, et al. Modulation of human beta-defensin-1 (hBD-1) in plasmacytoid dendritic cells (PDC), monocytes, and epithelial cells by influenza virus, Herpes simplex virus, and Sendai virus and its possible role in innate immunity. J Leukoc Biol. 2011;90:343–356. doi: 10.1189/jlb.0209079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha A, Hammond CE, Trojanowska M, Smolka AJ. Helicobacter pylori-induced H,K-ATPase alpha-subunit gene repression is mediated by NF-kappaB p50 homodimer promoter binding. Am J Physiol Gastrointest Liver Physiol. 2008;294:G795–G807. doi: 10.1152/ajpgi.00431.2007. [DOI] [PubMed] [Google Scholar]
- Saha A, Backert S, Hammond CE, Gooz M, Smolka AJ. Helicobacter pylori CagL activates ADAM17 to induce repression of the gastric H, K-ATPase alpha subunit. Gastroenterology. 2010;139:239–248. doi: 10.1053/j.gastro.2010.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staples E, Ingram RJM, Atherton JC, Robinson K. Optimising the quantification of cytokines present at low concentrations in small human mucosal tissue samples using Luminex assays. J Immunol Methods. 2013;394:1–9. doi: 10.1016/j.jim.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taha AS, Faccenda E, Angerson WJ, Balsitis M, Kelly RW. Gastric epithelial anti-microbial peptides – histological correlation and influence of anatomical site and peptic ulcer disease. Dig Liver Dis. 2005;37:51–56. doi: 10.1016/j.dld.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Tang B, Xiao B, Liu Z, Li N, Zhu ED, Li BS, et al. Identification of MyD88 as a novel target of miR-155, involved in negative regulation of Helicobacter pylori-induced inflammation. FEBS Lett. 2010;584:1481–1486. doi: 10.1016/j.febslet.2010.02.063. [DOI] [PubMed] [Google Scholar]
- Uehara N, Yagihashi A, Kondoh K, Tsuji N, Fujita T, Hamada H, Watanabe N. Human beta-defensin-2 induction in Helicobacter pylori-infected gastric mucosal tissues: antimicrobial effect of overexpression. J Med Microbiol. 2003;52:41–45. doi: 10.1099/jmm.0.04985-0. [DOI] [PubMed] [Google Scholar]
- Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol. 2004;5:1166–1174. doi: 10.1038/ni1131. [DOI] [PubMed] [Google Scholar]
- Vordenbaumen S, Pilic D, Otte JM, Schmitz F, Schmidt-Choudhury A. Defensin-mRNA expression in the upper gastrointestinal tract is modulated in children with celiac disease and Helicobacter pylori-positive gastritis. J Pediatr Gastroenterol Nutr. 2010;50:596–600. doi: 10.1097/MPG.0b013e3181cd26cd. [DOI] [PubMed] [Google Scholar]
- Wada A, Mori N, Oishi K, Hojo H, Nakahara Y, Hamanaka Y, et al. Induction of human beta-defensin-2 mRNA expression by Helicobacter pylori in human gastric cell line MKN45 cells on cag pathogenicity island. Biochem Biophys Res Commun. 1999;263:770–774. doi: 10.1006/bbrc.1999.1452. [DOI] [PubMed] [Google Scholar]
- Wada A, Ogushi K, Kimura T, Hojo H, Mori N, Suzuki S, et al. Helicobacter pylori-mediated transcriptional regulation of the human beta-defensin 2 gene requires NF-kappaB. Cell Microbiol. 2001;3:115–123. doi: 10.1046/j.1462-5822.2001.00096.x. [DOI] [PubMed] [Google Scholar]
- Wehkamp J, Harder J, Weichenthal M, Mueller O, Herrlinger KR, Fellermann K, et al. Inducible and constitutive beta-defensins are differentially expressed in Crohn's disease and ulcerative colitis. Inflamm Bowel Dis. 2003;9:215–223. doi: 10.1097/00054725-200307000-00001. [DOI] [PubMed] [Google Scholar]
- Xiao B, Liu Z, Li BS, Tang B, Li W, Guo G, et al. Induction of microRNA-155 during Helicobacter pylori infection and its negative regulatory role in the inflammatory response. J Infect Dis. 2009;200:916–925. doi: 10.1086/605443. [DOI] [PubMed] [Google Scholar]
- Yang D, Biragyn A, Kwak LW, Oppenheim JJ. Mammalian defensins in immunity: more than just microbicidal. Trends Immunol. 2002;23:291–296. doi: 10.1016/s1471-4906(02)02246-9. [DOI] [PubMed] [Google Scholar]
- Zasloff M. Antimicrobial peptides of multicellular organisms. Nature. 2002;415:389–395. doi: 10.1038/415389a. [DOI] [PubMed] [Google Scholar]
- Zasloff M. Antimicrobial peptides, innate immunity, and the normally sterile urinary tract. J Am Soc Nephrol. 2007;18:2810–2816. doi: 10.1681/ASN.2007050611. [DOI] [PubMed] [Google Scholar]
- Zhao C, Wang I, Lehrer RI. Widespread expression of beta-defensin hBD-1 in human secretory glands and epithelial cells. FEBS Lett. 1996;396:319–322. doi: 10.1016/0014-5793(96)01123-4. [DOI] [PubMed] [Google Scholar]
- Zhu BD, Feng Y, Huang N, Wu Q, Wang BY. Mycobacterium bovis bacille Calmette-Guerin (BCG) enhances human beta-defensin-1 gene transcription in human pulmonary gland epithelial cells. Acta Pharmacol Sin. 2003;24:907–912. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Confirmation of gene silencing in AGS cells treated with siRNA duplexes by Western blotting. Cells seeded in 24-well plates were transfected with siRNA to target expression of NFKB1, RELA, and MAPK1. Non-silencing negative control duplexes (Neg) were also used. After 48 h, cells were harvested into SDS-PAGE sample buffer. Western blots were probed, stripped and re-probed using antibodies against NF-κB p50 (NFKB1 gene product), NF-κB p65 (RELA gene product), MAPK1/ERK and beta actin.