

Abstract
A fundamental goal of medical genetics is the accurate prediction of genotype–phenotype correlations. As an approach to develop more accurate in silico tools for prediction of disease-causing mutations of structural proteins, we present a gene- and disease-specific prediction tool based on a large systematic analysis of missense mutations from hemophilia A (HA) patients. Our HA-specific prediction tool, HApredictor, showed disease prediction accuracy comparable to other publicly available prediction software. In contrast to those methods, its performance is not limited to non-synonymous mutations. Given the role of synonymous mutations in disease and drug codon optimization, we propose that utilizing a gene- and disease-specific method can be highly useful to make functional predictions possible even for synonymous mutations. Incorporating computational metrics at both nucleotide and amino acid levels along with multiple protein sequence/structure alignment significantly improved the predictive performance of our tool. HApredictor is freely available for download at http://www.ncbi.nlm.nih.gov/CBBresearch/Przytycka/HA_Predict/index.htm.
Keywords: hemophilia A/B, coagulation factor VIII, coagulation factor IX, gene/disease-specific prediction tool, synonymous mutation
Introduction
In recent years, personalized approaches in medical research and drug development have become important research focal points [1]. Such personalized approaches demand development of accurate tools that predict the possibility and severity of damage to protein function caused by a given (point) mutation. Such tools are not only useful when genetic information of newly diagnosed patients is revealed but also indispensable when a newly optimized/altered recombinant protein is developed for pharmaceutical purposes.
Recent progress in genetic screening and analysis of patients with hereditary diseases has unveiled that a variety of diseases are caused not only by missense (non-synonymous) mutations but also by synonymous mutations that do not change amino acid sequence [2]. Additionally, many novel recombinant proteins are being developed and released to the pharmaceutical market as therapeutic drugs. These recombinant therapeutics contain multiple modifications such as missense mutations, synonymous mutations, and deletions [3,4].
Several methods such as PolyPhen-2 [5] (Polymorphism Phenotyping v2), SIFT (Sorting Intolerant From Tolerant) [6], Condel [7] (Consensus Deleteriousness score of missense single-nucleotide variants), and PROVEAN (Protein Variation Effect Analyzer) [8] have been developed that give the possibility of assessing the impact of amino acid substitutions on protein function. Common software tools such as SIFT and PolyPhen-2 are based on multiple sequence alignment and homology modeling. These tools allow relatively accurate prediction of the impact of underlying mutations on protein function, yet the multiple sequence alignment methodology can be limiting when analyzing sequences that are relatively less conserved across species and/or those carrying synonymous mutations that do not alter protein composition. Alternative functional prediction approaches have been developed based on individual amino acid properties such as PASE, which analyzes amino acid substitutions based on the physicochemical changes of the replaced amino acid residue [9].
It should be noted that studies utilizing commonly used prediction software to predict protein deleteriousness have yielded mixed results [10–13]. Gray et al. report an assessment of several algorithms (Condel, PolyPhen-2, and SIFT) used for the analysis of the effects of a large number of mutations reported in the UniProt Knowledgebase (UniProtKB) database. Comparison of experimental profiling results with those from computation predictions showed numerous obstacles in the difficulty of predicting both deleterious and neutral mutations [14].
Better predictive tools would allow not only for more accurate disease forecasting but also for improved screening of suitable DNA constructs for protein therapeutics. Ultimately, this would result in more cost-effective and efficient drug design due to more accurate forecasting of an introduced point mutation’s impact on its protein function. As an alternative to generic methods that attempt to predict the damaging impact of mutations on protein function in a disease- and protein-independent manner, we focus on developing disease-specific approaches. A similar gene- and disease-specific approach has been previously investigated by Crockett et al. using the RET proto-oncogene as a model. However, this approach did not utilize nucleic-acid-based measurements and focused only on predicting phenotypic severity of uncertain gene variants [12].
Previously, we have analyzed correlations between various biochemical parameters of the point mutations in the f9 gene and disease [hemophilia B (HB)] severity. We used the largest possible collection of point mutations found in HB patients [15]. Despite the modest size of the analyzed cohort (131 unique mutations), this study demonstrated positive correlations between the disease severity and certain parameters at the nucleotide level, such as change in free energy of messenger RNA (mRNA) and the change in codon usage frequency as a result of mutation. These findings implied that incorporation of certain information at the nucleotide level into prediction software can improve their predictive power and accuracy. Furthermore, this study indicated that a gene-specific approach or a protein-specific approach to build a mutation prediction tool may be very useful in examining occurrence and severity of a specific disease where gene–disease linkage is clearly shown. Our current study investigates the application of these amino-acid-based and nucleotide-based measures to establish a gene- and disease-specific prediction tool capable of interpreting synonymous mutations, using hemophilia A (HA) as a disease model.
HA is an X-linked disease caused by mutations in blood coagulation factor VIII (FVIII), encoded by the f8 gene. FVIII is one of the key proteins in the blood coagulation cascade that serves as a cofactor of factor IX (encoded by f9 gene) and is stabilized by von Willebrand factor. The FVIII protein is regulated by multiple proteins such as thrombin and protein C. It is synthesized as a 2351-amino-acid pre-propeptide. After secretion, it is cleaved into a leader sequence, heavy chain, and light chain, forming into the mature (heterotrimer) active form FVIIIa. HA is one of the most well studied genetic diseases, as evidenced by the largest available database of f8 mutations that contains more than 2500 patient entries with various types of mutations including more than 1200 missense mutations [CDC Hemophilia A Mutation Project (CHAMP)] [16]. Because of the clear linkage between genetic mutation and disease (monogenic disease linked to the X chromosome), the internationally uniform description of disease severity and the statistically significant number of the known mutations, f8 and its mutations, were used as a model in the present study.
First, we established a training set composed of non-synonymous severe HA-causing f8 point mutations in addition to synonymous and non-synonymous f8 neutral point mutations. A variety of parameters measuring levels of conservation, amino acid properties, genomic DNA, and mRNA properties were examined for associations to disease occurrence using this database of f8 mutations. Next, selected biochemical parameters shown to have strong association with HA were used to build a decision tree classifier for disease-causing mutation prediction. Three test sets were established to test the decision tree: one containing non-synonymous moderate and mild HA-causing f8 point mutations, another one containing non-synonymous and synonymous HA-causing f8 point mutations, and a third test set containing neutral and non-synonymous HB-causing f9 point mutations.
As a consequence of introduction of the information on the nucleotide level, the inferred decision tree is capable of predicting the disease-causing possibility of the synonymous mutations. This is a new important feature that no other prediction tool possesses. Conceivably, the strategy of building the decision tree introduced here can be adapted to any monogenic disease with publically available tools to obtain the same measured parameters and establish alternative gene-specific prediction tools.
Results
Statistical analysis of f8 mutations and HA occurrence
For the correlation analysis between mutation parameters and disease occurrence, neutral and severe disease-causing f8 gene mutations from F8 Training Set were compared. Discrete-valued features are compared by Chi-squared test, and continuous-valued features are compared by unpaired Wilcoxon test. Among the parameters (over 30) tested (Supplemental Table 1), several parameters were found to be statistically significant and different between severe HA-causing and neutral f8 point mutations. Parameters with high association to HA occurrence (p-value < 0.05) are summarized in Table 1. This analysis demonstrates that nucleotide-level parameters have as much association to disease occurrence as protein-level parameters.
Table 1.
Parameters with significant differences among severe disease-causing and neutral f8 mutations (F8 Training Set)
| Feature | Mean (range)
|
p-Values
|
||
|---|---|---|---|---|
| Severe | Neutral | Wilcoxon | Chi-square | |
| Secondary structure | 8.21e-06 | |||
| Domain F5/8 type A | 4.11e-17 | |||
| Domain plastocyanin like | 1.06e-16 | |||
| Sequence conservation score | −0.84 (−1.32 to 1.45) | 0.36 (−1.29 to 2.79) | 8.3e-27 | |
| Structure conservation score | −0.88 (−1.39 to 1.65) | 0.52 (−1.37 to 2.17) | 1.7e-32 | |
| ΔHydrophobicity scale | −0.62 (−9.0 to 8.3) | 0.30 (−5.40 to 7.70) | 0.014 | |
| ΔPhosphorylation potential | 0.05 (−0.997 to 0.995) | −0.06 (−0.99 to 0.99) | 1.06e-3 | |
| ΔChange type | 4.18e-4 | |||
| Cysteine involved? | 7.53e-3 | |||
| Position in codon | 3.54e-13 | |||
| Type of mutation | 0.02 | |||
| Relative entropy, w = 150 nt | 2.77 (0.126–10.42) | 2.14 (0.125–7.72) | 4.93e-05 | |
| MFE (wild type), w = 150 nt | −29.98 (−37.83 to 19.37) | −25.93(−36.27 to 16.40) | 1.26e-14 | |
| GC ratio, w = 150 nt | 0.44 (0.33–0.52) | 0.42 (0.33–0.51) | 6.44e-09 | |
Parameters appeared in feature selection analysis are indicated in boldface.
A Best First feature selection method was used to identify more important features in our training set for the decision tree, Tree A (Fig. 1). The algorithm returned the following list as the result: conservation score without three-dimensional information, conservation score with 2R7E.pdb, Δphosphorylation potential (mut-wild type) position in codon, MFE (minimum free energy) (wild type) and GC ratio around the mutation site, type of nucleotide change, codon usage, hydrophobicity scale (mut), and location in domain F5/8 type A (Table 1). The most informative predictive features were related to both structure-and sequence-based conservation levels of the mutated amino acid. Stacked bar histograms of all analyzed features are shown in Supplemental Fig. 1.
Fig. 1.
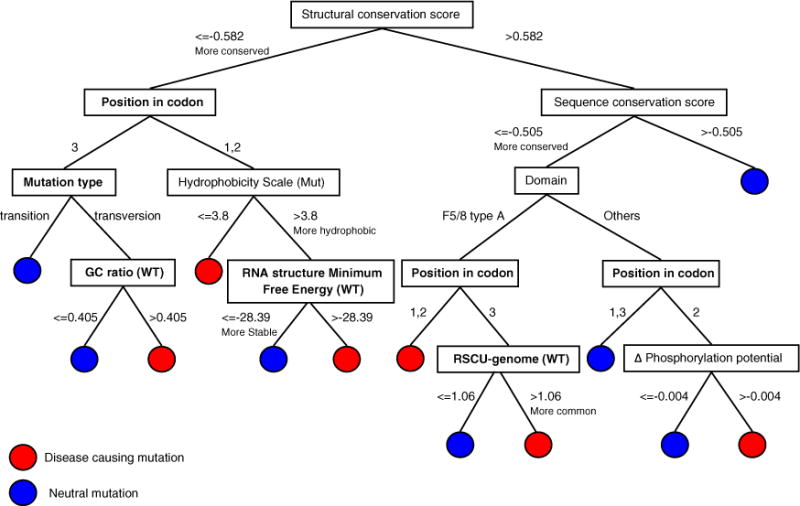
Optimal decision Tree A for HA-causing prediction trained by severe HA-causing mutations and neutral f8 variants. Nucleotide features are shown in boldface. Threshold values were determined using the Best First feature selection method from both disease-causing and neutral mutations.
Construction of decision tree for prediction of HA-causing mutations
Ten parameters were used in this decision tree to predict disease occurrence in missense mutations. These include five at the protein level (conservation score with and without structural information, hydrophobicity scale, Δphosphorylation potential, and domain F5/8 type A), three at the DNA level [position of the mutation in codon, nucleotide mutation type, and RSCU (relative synonymous codon usage) based on genomic codon usage], and two at the mRNA level [MFE of mRNA (150 nt) and GC ratio of mRNA (150 nt)]. To validate the effectiveness of our tree, we trained the two alternative decision trees with F8 Training Set excluding the 40 neutral synonymous mutations (Alternate Tree B) and with additional mutations from F8 Test Set 1 (Alternate Tree C). These alternate trees showed weaker predictive performance in comparison to Tree A. A user-friendly prediction software “HApredictor” based on optimal construction, Tree A, has been developed and is available for download at http://www.ncbi.nlm.nih.gov/CBBresearch/Przytycka/HA_Predict/index.htm.
Figure 1 describes a detailed scheme of our decision tree. The optimal tree trained with the F8 Training Set resulted in TP (true positive) counts of 205, TN (true negative) counts of 83, FP (false positive) counts of 19, and FN (false negative) counts of 28, resulting in a sensitivity of 88%, a specificity of 81%, and an accuracy of 86% (balanced accuracy 83%) (Table 2).
Table 2.
Comparison of prediction of disease-causing possibility using f8 decision tree developed in this study and other software
|
F8 Training Set
| |||||||
|---|---|---|---|---|---|---|---|
| PolyPhen-2 | SIFT-DNA | PROVEAN | Mutation Assessor | Condel | Decision Tree Aa | Decision Tree Ba | |
| Sensitivity (%) | 94.42 | 89.70 | 90.99 | 87.12 | 87.67 | 87.98 | 88.84 |
| Specificity (%) | 64.52 | 41.94 | 82.26 | 74.19 | 77.42 | 81.37 | 79.03 |
| Accuracy (%) | 88.14 | 79.66 | 89.15 | 84.41 | 85.47 | 85.97 | 86.78 |
| Balanced accuracy (%) | 79.47 | 65.82 | 86.62 | 80.66 | 82.54 | 84.68 | 83.94 |
| Not available | 0 | 0 | 0 | 0 | 6 | 0 | 0 |
| Healthy synonymous mutations predicted | 0/40 | 0/40 | 0/40 | 0/40 | 0/40 | 33/40 | 31/40 |
|
F8 Test Set 1
| |||||||
|---|---|---|---|---|---|---|---|
| PolyPhen-2 | SIFT-DNA | PROVEAN | Mutation Assessor | Condel | Decision Tree A | Decision Tree B | |
| Sensitivity (%) | 92.27 | 82.87 | 83.15 | 71.55 | 72.75 | 80.11 | 77.90 |
| Uninterpretable non-synonymous mutations | 0 | 0 | 0 | 5 | 10 | 0 | 0 |
|
F8 Test Set 2
| |||||||
|---|---|---|---|---|---|---|---|
| PolyPhen-2 | SIFT-DNA | PROVEAN | Mutation Assessor | Condel | Decision Tree A | Decision Tree B | |
| Sensitivity (%) | 89.93 | 79.16 | 80.56 | 69.91 | 71.46 | 74.14 | 74.83 |
| Uninterpretable non-synonymous mutations | 0 | 0 | 0 | 5 | 10 | 0 | 0 |
| Disease-causing synonymous mutations predicted | 0/10 | 0/10 | 0/10 | 0/10 | 0/10 | 3/10 | 6/10 |
Note that performance results for decision trees on F8 Training Set is based on the 10-fold cross-validation and that synonymous mutations were not considered in statistical calculations for other software. Decision tree B was trained without synonymous mutations.
Results of 10-fold cross-validation.
Evaluation of the decision tree and comparison with other software
The optimally trained decision tree was further tested on two training sets of known HA-causing mutations in f8: F8 Test Set 1, which contained moderate and mild non-synonymous mutations, and F8 Test Set 2, which contained severe, moderate, and mild non-synonymous and synonymous mutations. The test resulted in 80% accuracy (TP = 290 and FN = 72) for F8 Test Set 1 and in 74% accuracy (TP = 324 and FN = 113) for F8 Test Set 2 (Table 2). All three f8 mutation datasets can be found in Appendix.
Five commonly used prediction software, PolyPhen-2, SIFT-DNA, PROVEAN, Mutation Assessor, and Condel were examined for disease prediction of hemophilia-causing mutations to compare with our established decision tree method. Default threshold settings were used to determine predictions shown in Table 2. For each method, the corresponding threshold value was altered to generate the ROC (Receiver Operating Characteristics) plot shown in Fig. 2. At default threshold levels, predictions for the F8 Training Set generated by PolyPhen-2 contained a highest calculated sensitivity with 94.42%, but specificity was the second lowest at 64.52% (Table 2). As indicated in Fig. 2, all existing tools showed similar AUC (area under curve) values, further demonstrating similar performance. Our decision tree classifier performed comparably to the best of these tools.
Fig. 2.
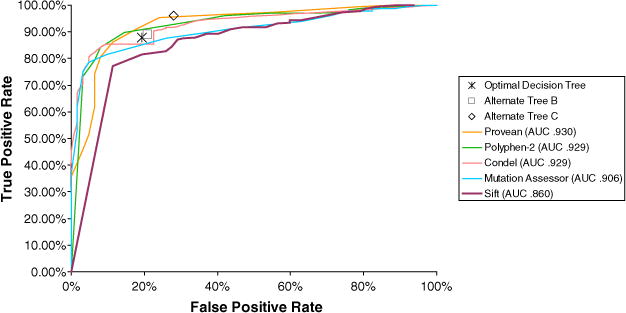
Performance comparison of five point mutation prediction tools. Equations used to calculate TP and FP values are shown below. The mutations in the F8 Training Set were used to establish this ROC curve. Synonymous mutations and unavailable calculations are not taken into account.
Application of decision tree for prediction of HB-causing mutations
As an additional method of testing our decision tree, the tree trained with F8 Training Set was applied to a set of point mutations in f9 (F9 Test Set). The comparison of our decision tree results and prediction by other tools are shown in Table 3. The decision tree trained by F8 Training Set was less accurate in predicting f9 mutations but still gave performance similar to existing tools. This is not a surprise since all parameters except for the “domain” attribute in the 10 parameters are not completely unique to individual protein. The three mild HB-causing synonymous mutations included in this dataset were predicted incorrectly as neutral variations due to extremely low conservation levels; however, all 13 neutral f9 synonymous mutations in this test set were predicted correctly.
Table 3.
Comparison of prediction of HB-causing possibility using decision tree developed in this study and other software
|
F9 Test Set
| ||||||
|---|---|---|---|---|---|---|
| PolyPhen-2 | SIFT-DNA | PROVEAN | Mutation Assessor | Condel | Decision Tree A | |
| Sensitivity (%) | 89.31 | 89.31 | 85.50 | 75.57 | 86.05 | 70.99 |
| Specificity (%) | 75.00 | 66.67 | 83.33 | 83.33 | 83.33 | 88.00 |
| Accuracy (%) | 88.11 | 87.41 | 85.31 | 76.22 | 85.82 | 73.72 |
| Balanced accuracy (%) | 82.16 | 77.99 | 84.41 | 79.45 | 84.69 | 79.50 |
| Healthy synonymous mutations predicted | 0/13 | 0/13 | 0/13 | 0/13 | 0/13 | 13/13 |
Note that synonymous mutations were not considered in statistical calculations for the other software.
Application of decision tree for prediction of disease-causing synonymous mutations
Our decision tree model gave us the opportunity to study the damaging effect of synonymous mutations. Although the very low number of known disease-causing synonymous mutations in f8 (3 of 10) did not allow a strong statement about predicting the disease-causing potential of synonymous mutations, a high percentage of healthy synonymous variations were predicted correctly. Specifically, of the 40 synonymous variations in the F8 Training Set, 33 were correctly predicted by the optimal decision tree. When trained with only non-synonymous mutations, only 31 of 40 neutral synonymous variations were predicted correctly by Alternate Tree B. Furthermore, all 13 neutral synonymous variations in the F9 Test Set were correctly predicted as well. Finally, noting that alternate tree B is over-performed by Tree A, which had synonymous mutations in its training set, suggests that there is valuable information hidden in synonymous mutations that helps to improve our understanding of disease-causing factors.
Discussion
We propose a new gene- and disease-specific strategy of constructing a prediction tool to evaluate the impact of single point mutations on protein function. While many online predictive tools have been available for years, the advantage of this particular predictor is its ability to predict the functional impact of mutations on a disease-specific basis. Most previously available tools utilized forms of multiple sequence alignment methods and protein homology modeling (in the case of PolyPhen-2) to establish predictions calculable for a wide range of proteins. While this allows for universally applicable calculations, two limitations arise: (1) Predictions are of limited value when there is limited homology modeling and sequence alignment available, and (2) synonymous mutations cannot be examined with this method because of amino-acid-based inputs and computations. In contrast, our proposed approach is disease specific and utilizes other parameters not limited to multiple sequence alignment.
We used f8–HA linkage as a proof of concept for the decision-tree-based classifier we developed. HA is historically one of the best-studied genetic diseases, and because it is an X-linked disease, damage on the FVIII protein directly affects the individual as a symptom. Although multiple sequence alignment is also utilized in our decision tree, the majority of the attributes contained in the decision tree assess gene- and protein-specific variations. In the context of our study, this strategy proves beneficial when evaluating mutations located in the B-domain of FVIII, which does not have a structural homolog [17]. As shown in Supplemental Table 2, our decision tree yields the highest combination of sensitivity (36%) and specificity (100%) compared to other tools when examining these B-domain mutations. All neutral B-domain mutations are in the F8 Training Set, and this could have contributed to the high specificity of our decision tree. An important advantage of the construction strategy of the prediction tool is its capability to analyze synonymous mutations due to inclusion of mutational attributes on the nucleotide level. The ability of the proposed prediction tool to interpret and evaluate the disease-causing synonymous mutations is likely to become even more relevant as more examples of disease-causing synonymous mutations are being revealed [18–20]. Understanding the disease-causing properties of synonymous mutations is also important in the context of drug design. Among the modifications practiced to recombinant proteins as therapeutic drugs, synonymous mutations are introduced with the least concern of its impact to the quality and potency of the products. However, understanding the significance of synonymous mutations has been changing in the last decade; it has been shown that synonymous mutations can influence protein folding that is critical for the structure of the whole protein and can be crucial for the protein function [21,2].
The parameters used in our prediction tool were selected due to their strong associations to HA disease severity and occurrence. Not all parameters with strong associations to HA disease severity and occurrence were used in the decision tree. On the nucleotide level, the mutation position in the codon, type of nucleotide change (transition/transversion), codon usage (RSCU), and Gibbs free energy of mRNA fragments are clearly shown to have highly positive associations. Several of these measurements were also previously indicated to have functional impacts on other proteins [22,15] and have been emphasized further in this study. The significance of codon usage, represented by RSCU, suggests that the balance of supply and demand of specific aminoacyl-tRNA might affect the local translation rate/rhythm. In particular, changes in codon usage could impact local co-translational folding and consequently affect the total folding of the resulting protein. Therefore, it is becoming a common consensus that a change in codon, even a synonymous one, has the possibility to lead to the change in the nature of the protein such as misfolding, aggregation, mislocation, or loss of activity. The local mRNA stability, represented by Gibbs free energy of mRNA fragment, is one of the contributing features in our decision tree components. This parameter suggests the local secondary structure that could affect half-life of the mRNA and/or accessibility of the anticodon to the corresponding aminoacyl-tRNA.
One attribute that was additionally investigated was mutation proximity to splicing sites. Since a splice site disruption would result in the synthesis of an incomplete or dysfunctional protein, this attribute jointly affects the gene and the protein. The distance accountable for splicing disruption was assessed by changing the distance under the range of 20 nt. As shown in Supplemental Fig. 2, variations close to splice sites are more likely to be disease-causing mutations than neutral. The most significant association with HA comparing severe and neutral mutations was observed when distance was set to <8 nt (p-value = 0.09; Supplemental Fig. 2). However, this parameter did not contribute to the decision tree.
Ideally, predicting disease severity is the next goal. However, among the parameters at both the protein level and the nucleotide level used in this study, only conservation score with 2R7E.pdb was significantly differentiable for severe and moderate cases of HA (p-value = 4e-06). In Supplemental Fig. 3, one can observe how the distribution of conservation score changes for different levels of severity. Although the association of the conservation score with 2R7E.pdb information to the HA severity is indeed striking and may prove to be important for disease severity prediction in general, it still did not provide enough information to allow for accurate prediction of disease severity and additional factors still need to be identified.
Interestingly, application of the f8-trained decision tree to the f9 mutation cohort showed reasonable success. Although both are involved in the blood coagulation cascade, factor VIII and factor IX, gene products of f8 and f9, are very different proteins in their nature and function. The good performance of this decision tree on f9 mutations may indicate the possibility of common/similar molecular mechanisms behind the associations for different types of disease-causing proteins. In general, our HA-specific predictor is not expected to provide accurate predictions for other diseases. However, the principle behind our approach is general.
Currently, many medical studies are focused on particular diseases. As more disease-causing mutations are discovered by such studies, disease-specific prediction tools will provide more accurate alternative to generic approaches. This paper presents a proof-of-concept of utilizing gene- and disease-specific parameters to successfully predict HA occurrence. We expect that, in the future, similar analyses would be applied to other diseases.
Materials and methods
Figure 3 illustrates the flow of the study including the materials and the process. The training set contained neutral and disease-causing f8 point mutations. After training, we tested the decision tree using three disjoint sets.
Fig. 3.
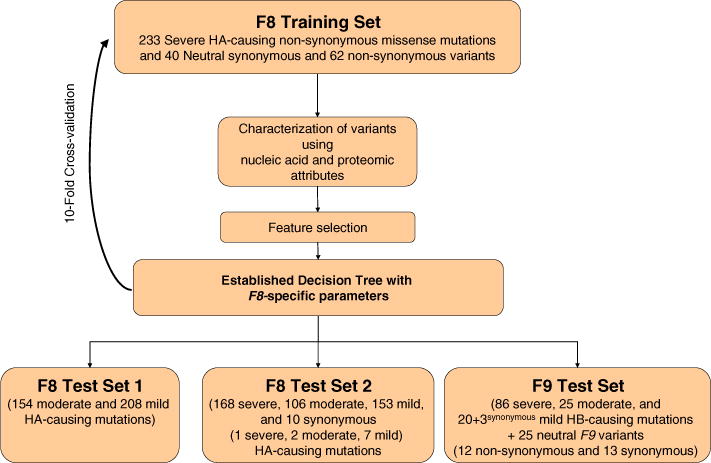
Flow chart of construction of decision tree for prediction of disease-causing mutations.
Mutation/variation datasets
The f8 mutation HA-causing mutations were annotated using the wild-type f8 open reading frame sequence derived from the National Center for Biotechnology Information (NCBI) RefSeq NM_000132.3. The first nucleotide of the start codon was denoted as nucleotide number one, and the first amino acid of the open reading frame, methionine, was denoted as amino acid number one.
The disease-causing f8 mutations were retrieved mainly from CHAMP (released in November 20122) [16]. Several additional mutations were added from individual reports [23,24]. Only point mutations (synonymous and non-synonymous missense mutations) in the coding region were chosen to be able to investigate the relationship between the mutations and their association to HA. Therefore, deletions, insertions, and mutations with undefined nucleotide sequence or severity information were excluded. Patients carrying multiple mutations and carrier females were also excluded from the analysis. The final set of unique HA-causing mutations included 1022 non-synonymous and 10 synonymous mutations.
The neutral (nondisease-causing) f8 mutations were selected from the cSNP list for human f8 from NCBI dbSNP: Short Genetic Variations3. The final set of neutral f8 variations included 62 non-synonymous and 40 synonymous point mutations.
Compiled mutations are grouped into exclusive datasets as summarized in Table 4. Severe disease-causing f8 mutations, which were reported before 2007, along with neutral mutations, have been used to train our classifier model. The remaining mutations are all used to test the classifier. To further evaluate our model, we applied it to the set of HB-causing f9 mutations previously collected, in addition to all neutral f9 variants chosen from the dbSNP database [15]. Composition details of this dataset, which contains both non-synonymous and synonymous point mutations, are also reported in Table 4.
Table 4.
Mutation composition of training and test sets
| Data set | Gene | Hemophilia-causing mutations
|
Neutral variants | Mutation source | ||
|---|---|---|---|---|---|---|
| Severe | Moderate | Mild | ||||
| F8 Training Set | f8 | 233 | 0 | 0 | 102 + (40a) | CHAMP, Santacroce et al. [24], dbSNP |
| F8 Test Set 1 | f8 | 0 | 154 | 208 | 0 | CHAMP |
| F8 Test Set 2 | f8 | 169 + (1a) | 108 + (2a) | 160 + (7a) | 0 | CHAMP |
| F9 Test Set | f9 | 86 | 25 | 23 + (3ab) | 25 + (13a) | Katagiri et al. [15], dbSNP |
While the intron 22 inversion is found in 30–45% of severe HA patients, missense mutations are found in about 40% of all the HA patients, and they comprise nearly half of all unique mutations found among all types of mutations in HA [16].
Severity determination
According to International Society of Thrombosis and Haemostasis criteria, severity of HA is categorized into three levels: “severe” (clotting activity level,<1% of normal level), “moderate” (clotting activity level 2, <5%), and “mild” (clotting activity level 6, <40%), where normal level of clotting activity is 1 U/mL. Mutations were divided into these three groups based upon the FVIII activity levels associated with the patients carrying the mutation (Table 4). For the mutations that did not have a corresponding activity level listed in the databases, data from the article in which the mutation was originally described were retrieved. In some cases where patients with different severities were reported with the same mutation, the most common severity was chosen. When the number of patients with different severities was equal, the more severe phenotype was recorded.
Mutation characterization
After the construction of mutation datasets, each mutation was characterized with multiple nucleotide-, amino-acid-, and structure-based features. The characteristics included those at the protein level (nature of individual amino acid, location in secondary structure or domain, conservation in primary sequence and structure, and possible impact in phosphorylation or structure), those at the DNA level (possible impact to splicing, codon usage reflected by RSCU, codon position of the mutation, and type of nucleotide change), and those at the mRNA level (free energy of local RNA secondary structure around the mutation locus and GC content of wild-type mRNA fragment). Some of the f8 mutation data source reported inhibitor development, an issue in HA treatment, but the report rate was low (4.2%). These parameters are summarized in Supplemental Table 1. The amino acid conservation with and without consideration of structural information (2R7E.pdb) was obtained using the ConSurf program4 [25]. Hydrophobicity scales were calculated based on the method of Kyte and Doolittle [26]; phosphorylation potential, using NetPhos5 [27]; N-linked glycosylation potential, using NetNGlyc6 [28]; and N-linked salfation score, using Sulfinator7 [29]. Information about the secondary structure elements and FVIII functional domains were obtained from UniProtKB Web site8 [30]. The charge of the amino acid was determined as follows: positively charged residues lysine, arginine, and histidine, 1; negatively charged residues glutamic acid and aspartic acid, −1; all others, 0.
mRNA characterization
Secondary structure of mRNA and associated Gibbs free energy (AG) predictions were performed using the mfold software [31] based on the nearest neighbor free energy model. The associated MFE value for each nucleotide is the average MFE for all subsequences of size w that includes the mutated nucleotide. The RNA structure with the lowest free energy structure of a given mRNA is the most stable one in the ensemble of all possible structures. The difference in mRNA MFE between mutant and wild type was calculated using the formula ΔMFE = MFEmutant − MFEwild type. Relative entropy between the Boltzmann structural ensembles of the native and mutant RNA was also calculated for each mutation [32]. GC ratio of each size of mRNA fragment was measured. We examined mRNA segments of w= 25, 50, 75, and 150 nt lengths.
The distance from splice site of the neutral and HA-causing mutation sites was examined using a range 0–20 nt from a splice site. All the mutations in our mutation datasets were in exon regions, and mutations within 8 nt from the splice junction were scored as “near splicing sites”.
Codon usage represented by RSCU
RSCU values were calculated as performed previously [2]. RSCU is a measure of codon usage bias and may be indicative of translation rate around a particular codon triplet. ΔRSCU = RSCUmutant − RSCUwild type represents a change in the RSCU values as a consequence of the specific mutation in the gene. The RSCU values were calculated using codon usages of both the entire human genome and the human f8 gene. A negative ΔRSCU value suggests that the mutant codon is less common than the wild-type codon.
Decision tree classifier for disease-causing prediction
To understand the predictive power of the collected features for disease-causing impact, we built a classifier based on the C4.5 decision tree induction algorithm (using package J48 in WEKA) [33]. The decision tree classifier is a graph-shaped model where each node represents a decision. Each path from root to a leaf in the tree structure determines a course of actions that leads to a possible classification.
We constructed our decision tree classifier using a 10-fold cross-validation technique on the F8 Training Set. This optimal tree (Tree A) was trained by only “severe” mutations in order to benefit from high feature scores as well as the balanced volume of data. Note that training the tree using more disease-causing mutations can lead to an overfitting problem due to the unbalanced ratio of known neutral f8 mutations to disease-causing mutations (it has been tested using alternative Tree C).
The decision tree classifier performed as accurately as or more accurately than other machine-learning approaches including Naïve Bayes, logistic regression, and random forests. We chose the decision tree because of its simplicity and intuitiveness properties.
Use of publically available prediction software
Five publically available online tools, PolyPhen-2 (Version 2.1.0)9, SIFT dbSNP (build 132)10, PROVEAN (Version 1.1)11, Condel (Version 1.5)12, and Mutation Assessor (Version 2)13, used for prediction of amino acid substitutions in proteins were tested with our HA-causing and HB-causing mutation training and test sets. Point mutations were entered as amino acid substitutions, and NCBI reference sequence IDs for FVIII (NP_000123) and FIX (NP_000124) were implemented. Software was used with default settings for the performance test.
PolyPhen-2: To assess prediction accuracy, we used the “pph2_class” parameter, which labeled mutations as “deleterious” or “neutral”. Under default thresholds, PolyPhen-2 classifies any mutation with pph_FPR < 0.1 to be deleterious and those with pph_FPR ≥ 0.1 to be neutral.
SIFT: Functional predictions were assessed using the “prediction” parameter using the SIFT batch protein tool available online, which divides mutations into tolerant and damaging mutations. Default threshold values set SIFT score < 0.05 to be damaging.
PROVEAN: Variant predictions are based on a Δalignment score value of −2.5, where predicted deleterious mutations have lower scores and neutral mutations have higher scores.
Condel: The default threshold value for the Condel Web service is set at approximately 0.467, where mutations with a Condel score greater than 0.467 will be assigned as “deleterious” and with others assigned as “neutral”. Mutation Assessor: Functional predictions of Mutation Assessor are given with a quaternary classification system based on a “functional impact score” of increasing likelihood of functional damage, where functional impact scores greater than 1.938 resulted in functional damage and those below 1.938 resulted in non-functional damage.
The PolyPhen-2 Web server utilizes protein sequences from the UniProtKB Release 2011_0 and protein structure from PDB/DSSP 06-Apr-2011. The SIFT dbSNP Protein Tool (using NCBI dbSNP build 132) was used to obtain prediction results. The PROVEAN Protein Tool utilizes NCBI dbSNP build 137 to obtain variants. The Mutation Assessor release 2 that was used for computations included data from Pfam 26, Nov 2011, PDB 13-Jul-2012, UniProtKB 2012_07, RefSeq release 54, and NCBI build 37 version 3. Condel scores were obtained from the Condel Web server, which creates a consensus score derived from SIFT, PolyPhen-2, and Mutation Assessor.
Supplementary Material
Acknowledgments
We would like to thank Ms. Erin Needlman, Ms. Natasha Nelson, Mr. Max Katz, Mr. Jeong Lee, Mr. Stephen Gross, and Mr. Matt Iandoli for helping with construction of datasets. We are grateful to Ms. Sandra C. Tseng, Dr. Ryan Hunt, Dr. Zuben Sauna (Center for Biologics Evaluation and Research, Food and Drug Administration), Dr. Deanna Church, and Mr. John Garner (NCBI, National Institutes of Health) for insightful commentary. We thank Dr. Geoffrey Kemball-Cook (University College London) for maintaining HADB, data from which is now included in CHAMP. The findings and conclusions in this article have not been formally disseminated by the Food and Drug Administration and should not be construed to represent any agency determination or policy. T.M.P. is supported by National Institutes of Health Intramural Research Funding (National Library of Medicine). R.S. was funded partially by Natural Sciences and Engineering Research Council Postdoctoral Fellowships.
Abbreviations used
- HA
hemophilia A
- HB
hemophilia B
- UniProtKB
UniProt Knowledgebase
- NCBI
National Center for Biotechnology Information
- RSCU
relative synonymous codon usage
Footnotes
Conflict of Interest: The authors stated that they had no interests that might be perceived as posing a conflict or bias.
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jmb.2013.07.037.
References
- 1.Rodriguez LL, Brooks DL, Greenberg HJ, Green DE. The complexities of genomic identifiability. Science. 2013;339:275–6. doi: 10.1126/science.1234593. [DOI] [PubMed] [Google Scholar]
- 2.Sauna ZE, Kimchi-Sarfaty C. Understanding the contribution of synonymous mutations to human disease. Nat Rev Genet. 2011;12:683–91. doi: 10.1038/nrg3051. [DOI] [PubMed] [Google Scholar]
- 3.Houde D, Berkowitz SA. Conformational comparability of factor IX-Fc fusion protein, factor IX, and purified Fc fragment in the absence and presence of calcium. J Pharm Sci. 2012;101:1688–700. doi: 10.1002/jps.23064. [DOI] [PubMed] [Google Scholar]
- 4.Turecek PL, Bossard MJ, Graninger M, Gritsch H, Hollriegl W, Kaliwoda M, et al. BAX 855, a PEGylated rFVIII product with prolonged half-life development, functional and structural characterisation. Hamostaseologie. 2012;32:S29–38. [PubMed] [Google Scholar]
- 5.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–82. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 7.Gonzalez-Perez A, Lopez-Bigas N. Improving the assessment of the outcome of nonsynonymous SNVs with a consensus deleteriousness score, Condel. Am J Hum Genet. 2011;88:440–9. doi: 10.1016/j.ajhg.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choi Y, Sims GE, Murphy S, Miller JR, Chan AP. Predicting the functional effect of amino acid substitutions and indels. PLoS One. 2012;7:e46688. doi: 10.1371/journal.pone.0046688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li X, Kierczak M, Shen X, Ahsan M, Carlborg Ö, Marklund S. PASE: a novel method for functional prediction of amino acid substitutions based on physicochemical properties. Front Genet. 2013;4:21–6. doi: 10.3389/fgene.2013.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lannoy N, Lambert C, Vermylen C, Hermans C. Mutations screening in Belgium patients with haemophilia A: identification of 28 new genetic alterations and study of causal effect of 15 unreported missense mutations. Haemophilia. 2011;17:379–379. [Google Scholar]
- 11.Doss CGP. In silico profiling of deleterious amino acid substitutions of potential pathological importance in haemophlia A and haemophlia B. J Biomed Sci. 2012;19 doi: 10.1186/1423-0127-19-30. http://dx.doi.org/10.1186/1423-0127-19-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crockett DK, Piccolo SR, Ridge PG, Margraf RL, Lyon E, Williams MS, et al. Predicting phenotypic severity of uncertain gene variants in the RET proto-oncogene. PLoS One. 2011;6:e18380. doi: 10.1371/journal.pone.0018380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bergman JEH, Janssen N, van der Sloot AM, de Walle HEK, Schoots J, Rendtorff ND, et al. A novel classification system to predict the pathogenic effects of CHD7 missense variants in CHARGE syndrome. Hum Mutat. 2012;33:1251–60. doi: 10.1002/humu.22106. [DOI] [PubMed] [Google Scholar]
- 14.Gray VE, Kukurba KR, Kumar S. Performance of computational tools in evaluating the functional impact of laboratory-induced amino acid mutations. Bioinformatics. 2012;28:2093–6. doi: 10.1093/bioinformatics/bts336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hamasaki-Katagiri N, Salari R, Simhadri VL, Tseng SC, Needlman E, Edwards NC, et al. Analysis of F9 point mutations and their correlation to severity of haemophilia B disease. Haemophilia. 2012;18:933–40. doi: 10.1111/j.1365-2516.2012.02848.x. [DOI] [PubMed] [Google Scholar]
- 16.Payne AB, Miller CH, Kelly FM, Soucie JM, Hooper WC. The CDC Hemophilia A Mutation Project (CHAMP) mutation list: a new online resource. Hum Mutat. 2013;34:E2382–91. doi: 10.1002/humu.22247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen BW, Spiegel PC, Chang CH, Huh JW, Lee JS, Kim J, et al. The tertiary structure and domain organization of coagulation factor VIII. Blood. 2008;111:1240–7. doi: 10.1182/blood-2007-08-109918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kimchi-Sarfaty C. A “silent” polymorphism in the MDR1 gene changes substrate specificity. Science. 2007;318:1382–3. doi: 10.1126/science.1135308. [DOI] [PubMed] [Google Scholar]
- 19.Knobe KE, Sjorin E, Ljung RCR. Why does the mutation G17736A/Val107Val (silent) in the F9 gene cause mild haemophilia B in five Swedish families? Haemophilia. 2008;14:723–8. doi: 10.1111/j.1365-2516.2008.01753.x. [DOI] [PubMed] [Google Scholar]
- 20.Bartoszewski RA, Jablonsky M, Bartoszewska S, Stevenson L, Dai Q, Kappes J, et al. A synonymous single nucleotide polymorphism in delta F508 CFTR alters the secondary structure of the mRNA and the expression of the mutant protein. J Biol Chem. 2010;285:28741–8. doi: 10.1074/jbc.M110.154575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Komar AA. A pause for thought along the co-translational folding pathway. Trends Biochem Sci. 2009;34:16–24. doi: 10.1016/j.tibs.2008.10.002. [DOI] [PubMed] [Google Scholar]
- 22.Edwards NC, Hing ZA, Perry A, Blaisdell A, Kopelman DB, Fathke R, et al. Characterization of coding synonymous and non-synonymous variants in ADAMTS13 using ex vivo and in silico approaches. PLoS One. 2012;7:e38864. doi: 10.1371/journal.pone.0038864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guillet B, Lambert T, d’Oiron R, Proulle V, Plantier J-L, Rafowicz A, et al. Detection of 95 novel mutations in coagulation factor VIII gene F8 responsible for hemophilia A: results from a single institution. Hum Mutat. 2006;27:676–85. doi: 10.1002/humu.20345. [DOI] [PubMed] [Google Scholar]
- 24.Santacroce R, Acquila M, Belvini D, Castaldo G, Garagiola I, Giacomelli SH, et al. Identification of 217 unreported mutations in the F8 gene in a group of 1,410 unselected Italian patients with hemophilia A. J Hum Genet. 2008;53:275–84. doi: 10.1007/s10038-007-0238-y. [DOI] [PubMed] [Google Scholar]
- 25.Landau M, Mayrose I, Rosenberg Y, Glaser F, Martz E, Pupko T, et al. ConSurf 2005: the projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005;33:W299–302. doi: 10.1093/nar/gki370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982;157:105–32. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- 27.Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;294:1351–62. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- 28.Blom N, Sicheritz-Ponten T, Gupta R, Gammeltoft S, Brunak S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics. 2004;4:1633–49. doi: 10.1002/pmic.200300771. [DOI] [PubMed] [Google Scholar]
- 29.Monigatti F, Gasteiger E, Bairoch A, Jung E. The Sulfinator: predicting tyrosine sulfation sites in protein sequences. Bioinformatics. 2002;18:769–70. doi: 10.1093/bioinformatics/18.5.769. [DOI] [PubMed] [Google Scholar]
- 30.Apweiler R, Martin MJ, O’Donovan C, Magrane M, Alam-Faruque Y, Antunes R, et al. Ongoing and future developments at the Universal Protein Resource. Nucleic Acids Res. 2011;39:D214–9. doi: 10.1093/nar/gkq1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zuker M. On finding all suboptimal foldings of an RNA molecule. Science. 1989;244:48–52. doi: 10.1126/science.2468181. [DOI] [PubMed] [Google Scholar]
- 32.Salari R, Kimchi-Sarfaty C, Gottesman MM, Przytycka TM. Sensitive measurement of single-nucleotide polymorphism-induced changes of RNA conformation: application to disease studies. Nucleic Acids Res. 2013;41:44–53. doi: 10.1093/nar/gks1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hall M, Frank E, Holmes G, Pfahringer B, Reutemann P, Witten HI. The WEKA Data Mining Software: an update. SIGKDD Explor. 2009;11:10–8. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.