

Abstract
Chlorite dismutases (Clds) are heme b-containing prokaryotic oxidoreductases that catalyze the reduction of chlorite to chloride with the concomitant release of molecular oxygen. Over time, they are irreversibly inactivated. To elucidate the mechanism of inactivation and investigate the role of the postulated intermediate hypochlorite, the pentameric chlorite dismutase of “Candidatus Nitrospira defluvii” (NdCld) and two variants (having the conserved distal arginine 173 exchanged with alanine and lysine) were recombinantly produced in Escherichia coli. Exchange of the distal arginine boosts the extent of irreversible inactivation. In the presence of the hypochlorite traps methionine, monochlorodimedone, and 2-[6-(4-aminophenoxy)-3-oxo-3H-xanthen-9-yl]benzoic acid, the extent of chlorite degradation and release of molecular oxygen is significantly increased, whereas heme bleaching and oxidative modifications of the protein are suppressed. Among other modifications, hypochlorite-mediated formation of chlorinated tyrosines is demonstrated by mass spectrometry. The data obtained were analyzed with respect to the proposed reaction mechanism for chlorite degradation and its dependence on pH. We discuss the role of distal Arg173 by keeping hypochlorite in the reaction sphere for O–O bond formation.
Chlorite dismutases (Clds, EC 1.13.11.49) are heme b-dependent oxidoreductases that can convert chlorite into chloride and dioxygen. First, biochemical characterizations of Clds demonstrated that Cld does not catalyze a dismutation or disproportionation.1 Thus, the name chlorite “dismutase” is misleading. The reduction of chlorite to chloride has been proposed to occur in three steps, starting with the addition of ClO2– to the heme iron [PorFe(III)]+ to form a [PorFe(III)]+–ClO2– adduct (reaction 1), immediately followed by the oxidation of the ferric enzyme by chlorite to form Compound I {oxoiron(IV) porphyryl radical, i.e., [Por•Fe(IV)=O]+} and HOCl/OCl– (reaction 2). Finally, hypochlorite rebinds to the ferryl oxygen of Compound I, and chloride and dioxygen are released (reaction 3).2 However, a classical Compound I spectrum typical for a heme b enzyme that shows hypochromicity at the Soret maximum could not be trapped upon mixing ferric Cld with chlorite.
| 1 |
| 2 |
| 3 |
Besides this mechanism that involves heterolytic cleavage of chlorite and the formation of Compound I (reactions 1 and 2), density functional theory (DFT) calculations of water-soluble model iron porphyrins3,4 suggest the formation of Compound II [oxoiron(IV), i.e., PorFe(IV)=O] and chlorine monoxide (O–Cl•) through homolytic cleavage of chlorite (reaction 4). Chlorine monoxide then recombines with Compound II forming Cl– and O2 to complete the cycle (reaction 5).
| 4 |
| 5 |
Crystal structures of functional (i.e., chlorite-degrading) Clds5−8 demonstrated that a fully conserved arginine [i.e., Arg173 in chlorite dismutase from “Candidatus Nitrospira defluvii” (NdCld)] is the only charged amino acid at the distal heme side. Extensive characterization of Arg mutants demonstrated that the basic amino acid is catalytically important but not essential for chlorite degradation.7,9,10 Crystal structures suggest that the distal arginine is flexible and may adopt two main conformations pointing either to the entry of the main access channel into the heme cavity or directly to the heme iron. Principally, the guanidinium group could participate in all five reactions depicted above and could support substrate binding as well as potentially keep the postulated reaction intermediate, hypochlorite (or O–Cl•), in the vicinity of the ferryl oxygen for the recombination step and O2 formation. Recent mutational analysis indicated that Arg173 might be more important in stabilizing the Compound I–hypochlorite complex (reaction 2) [or the Compound II–chlorine monoxide complex (reaction 5)] rather than supporting the binding of chlorite to the heme center.10
Additionally, kinetic studies of Clds from different organisms1,2,5−8,11−15 also demonstrated that these oxidoreductases are irreversibly inhibited with time at higher chlorite concentrations. Their ability to convert chlorite to chloride and dioxygen is limited, and an off pathway was postulated on the basis of the formation of tryptophanyl radicals on the proximal heme side of Clds.6 Later, mutational studies of chlorite dismutases from Dechloromonas aromatica (DaCld)16 and “Candidatus Nitrospira defluvii”10 showed that the exchange of those conserved tryptophan residues on the proximal side did not prevent deactivation of Clds. In the corresponding DaCld mutants, the heme binding properties and the oligomerization state were impaired, whereas in the corresponding NdCld mutants, the reduction potential of the Fe(III)/Fe(II) couple was altered.10
In this work, we aim to elucidate the mechanism of irreversible inhibition of chlorite-degrading Clds. We have analyzed the role of the conserved distal arginine and demonstrate the significant impact of traps of hypochlorite like methionine, monochlorodimedon (MCD), and aminophenylfluorescein (APF) on catalysis. We compare the pH dependence of the enzymatic activity and the inhibitory effect as well as analyze heme bleaching and modifications of the protein by time-resolved UV–vis and electron paramagnetic resonance (EPR) spectroscopy as well as mass spectrometry. The data obtained are discussed with respect to the available biochemical and physical properties of Cld and its known high-resolution structure.
Materials and Methods
Expression and Purification
The expression and purification of StrepII-tagged TEV-cleavable wild-type NdCld and its variants were reported recently.10,17
Polarographic Oxygen Measurement
Chlorite dismutase-mediated degradation of chlorite was monitored by measuring the release of O2 using a Clark-type oxygen electrode (Oxygraph Plus, Hansatech Instruments, Norfolk, U.K.) inserted into a stirred water bath kept at 30 °C. We equilibrated the electrode to 100% O2 saturation by bubbling O2 through the reaction mixture and to 0% saturation by bubbling with N2 until plateaus were reached to derive an offset and calibration factor. Reactions for testing the influence of methionine were conducted in O2-free 50 mM phosphate buffer solutions at pH 5.5 and 7.0, with 25–800 μM NaClO2 added from a stock made in the same buffer and eventually with 5.0 mM methionine. Reactions were started by addition of 25 nM wild-type NdCld, 200 nM NdCld R173A, and 200 nM NdCld R173K. It was important to only use the initial linear phase (v0) for the calculation of rates and Michaelis–Menten parameters, because with increasing chlorite concentrations, irreversible inactivation of the enzyme occurred. This was immediately evident as determined by inspection of the respective time traces. Molecular oxygen production rates (micromolar O2 per second) were obtained from initial linear time traces (<10% substrate consumed) and plotted versus chlorite concentration for the determination of catalytic parameters. Reactions were monitored for approximately 2 min to determine the final amount of produced oxygen. The influence of enzyme concentration was tested by starting the reaction with 5–500 nM wild-type NdCld and a chlorite concentration of 340 μM, and eventually adding 5.0 mM methionine. Reactions for the pH dependence of NdCld were conducted in O2-free 50 mM buffer solutions from pH 4.3 (citrate phosphate) to pH 8.3 (phosphate), with 10–1000 μM NaClO2 added from a stock made in the same buffer. Reactions were started via the addition of 50 nM wild-type NdCld.
Spectrophotometric Monitoring of Chlorite Degradation
The conversion of chlorite (ClO2–) into chloride and dioxygen was monitored photometrically by following the decrease in absorbance at 260 nm (ε260 = 155 M–1 s–1)18 on a Hitachi U-3900 spectrophotometer. Reactions were conducted in 50 mM buffer solutions (pH 5.5 and 7.0). The chlorite concentration was 340 μM, and reactions were started by the addition of the enzyme.
Stopped-Flow UV–Visible Spectroscopy
The experiments were conducted with a stopped-flow apparatus (model SX-18MV, Applied Photophysics) equipped for both conventional and sequential measurements. The optical quartz cell with a path length of 10 mm had a volume of 20 μL. The fastest time for mixing two solutions and recording the first data point was 1 or 3 ms. All measurements were performed at 25 °C. To study the impact of hypochlorite traps like methionine19,20 or monochlorodimedone (MCD)21 on the degradation of chlorite by Cld, the conventional stopped-flow mode was used following the decrease in the absorbance of MCD at 290 nm (ε290 = 19000 M–1 s–1).22 Simultaneously, the spectral changes in the Soret region of NdCld were monitored. In a typical experiment, one syringe contained 2 μM enzyme in 50 mM buffer and the second syringe contained 0–1 mM chlorite and eventually a hypochlorite trap (100 μM MCD or 5 mM methionine) in the same buffer. A minimum of three measurements were performed for each substrate concentration, and spectra were recorded for 20 s. To determine the amount of chlorinated MCD by transiently produced HOCl, the difference between the starting concentration and the end concentration of MCD of each measurement was plotted versus chlorite concentration. The effect of HOCl traps on chlorite degradation was also monitored by using the diode array detector (Applied Photophysics), which allowed the synthesis of artificial sets of time-dependent spectra as well as spectral analysis of enzyme intermediates.
Trapping of Hypochlorous Acid with Aminophenylfluorescein
As a further method for detecting hypochlorous acid as an intermediate in Cld catalysis, we used APF, which specifically detects hypohalous acids but does not react with chlorite.23,24 Here the HOCl-derived oxidation of APF was followed at 25 °C by measuring the fluorescence intensity at 522 nm (excitation at 488 nm). The fluorescence spectrophotometer (Hitachi F-7000) was equipped with a thermostatic cell holder for quartz cuvettes with a path length of 10 mm. Instrumental parameters were set as follows. The excitation wavelength was set to 488 nm; excitation and emission bandwidths were set to 5 nm, and the PMT voltage was set to 700 V. Wavelength scans were recorded from 495 to 600 nm (scan speed of 60 nm min–1). In detail, 100 nM NdCld was incubated with 0–750 μM chlorite in 50 mM phosphate buffer (pH 7.0), eventually in the presence of 5 mM methionine. APF (10 μM) was added to the reaction mixture and the mixture incubated in the dark at room temperature for 30 min before measurement.
Mass Spectrometry
Mass spectrometry (MS) was used to detect modifications on NdCld after treatment with either chlorite (in absence and presence of methionine) or hypochlorite. In typical experiments, 100 μM wild-type NdCld in 50 mM phosphate buffer (pH 7.0) was mixed with 500 mM chlorite (in the absence or presence of 25 mM methionine). For protein analysis, 3 μg of treated or untreated wild-type NdCld was directly injected into the LC–MS system, and for peptide analysis, 5 μg of trypsin-digested treated or untreated NdCld was directly injected into the LC–MS system (LC, Dionex Ultimate 3000 LC; MS, Bruker, Maxis 4G, equipped with the standard ESI source). The protein was eluted by developing a linear gradient from 15 to 70% acetonitrile over 42 min (Supelco Discovery Bio Wide Pore C5 column, 50 mm × 0.32 mm, 3 μm packing). Data were processed using Data Analysis 4.0 (Bruker), and spectra were deconvoluted with MaxEnt.
Electron Spin Resonance Spectroscopy
Electron paramagnetic resonance (EPR) spectroscopy was used to determine the effect of chlorite (in the presence or absence of methionine) as well as of HOCl on the electronic structure of the heme iron. Typically, 100 μL samples were prepared in 125 mM MES buffer (final concentration, pH 5.5) containing 30 μM wild-type NdCld, 0–400 mM chlorite (eventually in the presence of 17.5 mM methionine), or 0–23 mM HOCl. After at least 3 min had been allowed to pass (until the reaction was complete), the samples were transferred into Wilmad quartz tubes (3 mm inner diameter) and flash-frozen in liquid N2. Frozen samples were kept frozen on dry ice, while the headspace above the sample was flushed with argon. Oxygen-free samples were frozen at 77 K and transferred into the resonator for 10 K measurements. Spectra were recorded on a Bruker EMX continuous wave (cw) EPR spectrometer, operating at X-band (9 GHz) frequencies, equipped with a high-sensitivity resonator and an Oxford Instruments ESR900 cryostat. EPR spectra were recorded under nonsaturating conditions using a microwave power of 2 mW, a modulation frequency of 100 kHz, a modulation amplitude of 1 mT, a conversion time of 41 ms, a time constant of 41 ms, and 2048 points. Simulations of high-spin and low-spin Fe(III) forms were conducted using EasySpin25 and consist of a weighted sum of simulations of the individual high-spin and low-spin compounds. The rhombicity was obtained from gxeff and gyeff,26 and the relative intensities were calculated on the basis of the simulations.
Molecular Dynamics Simulations
Molecular dynamics simulations were performed with the chlorite dismutase crystal structure from “Candidatus Nitrospira defluvii” (PDB entry 3NN1).7 The GROMOS molecular dynamics simulation package27 was used in conjunction with the GROMOS 54A7 force field.28 Detailed simulation settings and force field parameters for Compound I and hypochlorite were taken from Sündermann et al. (paper submitted to Biochemistry).
Results
pH Dependence of Chlorite Degradation and the Effect of Methionine
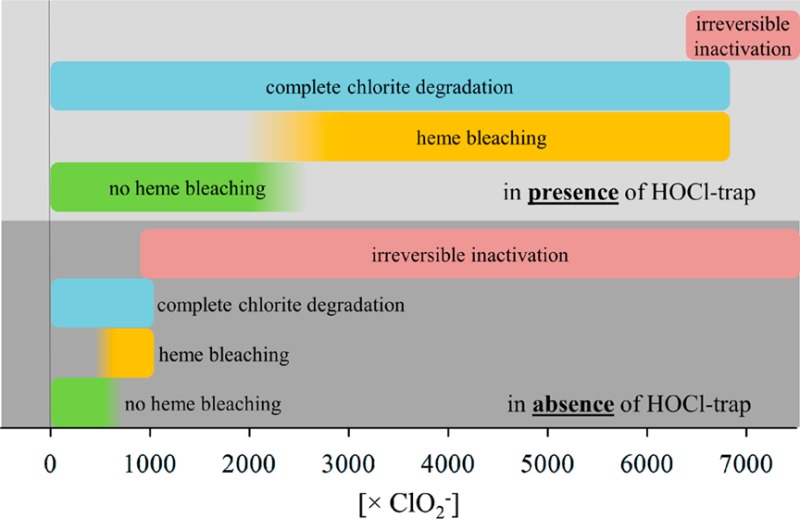
Chlorite dismutases efficiently degrade chlorite to chloride and molecular dioxygen with reported KM values at pH 7.0 varying from 69 to 260 μM, kcat values varying from 43 to 7500 s–1, and kcat/KM values varying from 6.2 × 105 to 3.5 × 107 M–1 s–1.29 Figure 1 shows the pH dependence of these enzymatic parameters for NdCld obtained from polarographic measurements of the release of O2. In contrast to KM (138 ± 20 μM at pH 7.0), kcat (83 ± 4 s–1 at pH 7.0) and the catalytic efficiency (5.9 × 105 M–1 s–1 at pH 7.0) showed a clear pH dependence with an optimum at pH 5.5. Figure 1D clearly depicts that at the pH optimum both the initial rate of O2 release and the total yield of produced dioxygen are highest. With an increase in pH, both the initial reaction velocity (v0) and the yield of O2 (which corresponds to the amount of degraded chlorite) were significantly decreased. The more chlorite was added to 50 nM NdCld, the more pronounced was this effect (Figure 1D). During the reaction, NdCld was irreversibly inhibited. Desalting and buffer exchange did not result in any recovery of enzymatic activity (not shown). The pH optima of NdCld variants R173A and R173K are also in the acidic region around pH 4.5 (Figure 1 of the Supporting Information). As postulated in reactions 2 and 3, HOCl/–OCl is formed during turnover. To test whether hypochlorite is involved in the irreversible inactivation of NdCld, the polarographic measurements were also performed in the presence of 5 mM methionine. The latter is known to react with HOCl very fast (8.7 × 108 M–1 s–1) and efficiently,19 whereas chlorite does not react with methionine.24 Panels A and B of Figure 2 compare the effect of methionine on the degradation of chlorite by wild-type NdCld (20 nM) measured as the amount of O2 released after 120 s at the pH optimum (i.e., pH 5.5) and pH 7.0. At low chlorite concentrations (<50 μM), the effect of methionine was weak. However, at higher chlorite concentrations in the presence of methionine, the yield of O2 increased and this effect was more pronounced at pH 7.0. These data clearly demonstrate that (i) methionine is able to partially protect NdCld from irreversible inhibition and (ii) the inhibition reaction is boosted apart from the pH optimum.
Figure 1.

pH dependence of enzymatic parameters of wild-type NdCld. Influence of pH on (A) KM values, (B) turnover number (kcat), and (C) catalytic efficiency (kcat/KM). (D) Plot of the initial rate (v0) of release of molecular oxygen as a function of chlorite concentration at pH 4.3–8.3.
Figure 2.

Influence of methionine on the generation of O2 during chlorite degradation mediated by wild-type (A and B) and mutant (C–F) NdCld at pH 5.5 (A, C, and E) and pH 7.0 (B, D, and F). Insets depict typical time traces of oxygen generation.
Next, we wanted to analyze the role of arginine 173 in the inhibition reaction at pH 5.5 and 7.0. Compared to wild-type NdCld (KM = 69 μM, kcat = 43 s–1, kcat/KM = 6.2 × 105 M–1 s–1), variants R173A and R173K exhibited KM values of 90 and 898 μM, kcat values of 2.8 and 14.0 s–1, and kcat/KM values of 3.1 × 104 and 1.5 × 104 M–1 s–1 (pH 7.0), respectively.29 In panels C and D of Figure 2 (200 nM NdCld R173A) and panels E and F of Figure 2 (200 nM NdCld R173K), the amount of produced O2 after 2 min at various chlorite concentrations in the absence and presence (5 mM) of methionine at pH 5.5 and 7.0 is plotted. The data clearly demonstrate that in the absence of the distal arginine 173 NdCld is much more prone to inactivation (despite a 10-fold higher enzyme concentration in the assays) at pH 5.5 and 7.0 ([O2]/[chlorite] ratio decreases in the order wild-type NdCld ≫ R173K > R173A) and that methionine can rescue also the variants at least to some extent.
Besides the amount of O2 produced, methionine also increased the turnover number of wild-type NdCld, R173K, and R173A (Figure 3A), simply because it kept the number of active enzyme molecules higher. This is evident upon degrading a defined amount of chlorite (340 μM) with increasing enzyme concentrations. Figure 3B depicts two representative time traces following O2 release during degradation of 340 μM chlorite by 25 nM NdCld ([chlorite]/[NdCld] ratio of 13600) and 100 nM NdCld ([chlorite]/[NdCld] ratio of 3400) in the absence and presence of methionine (5 mM). In the presence of methionine with 100 nM enzyme, all chlorite was degraded and the theoretical yield of O2 (340 μM) was achieved, whereas at lower enzyme concentrations or in the absence of methionine, the yield of dioxygen was significantly lower. This is summarized in Figure 3C. To produce 340 μM O2 from 340 μM chlorite in the absence of methionine, an enzyme concentration of ∼400 nM was necessary ([chlorite]/[NdCl] ratio of 850), whereas in the presence of 5 mM methionine, complete degradation of chlorite was already achieved by ∼100 nM enzyme ([chlorite]/[NdCl] ratio of 3400).
Figure 3.

Effect of methionine on the kinetics (A) and extent of O2 release (B and C) and chlorite degradation (D and E), varying the enzyme concentration (B–E) and the pH (E).
Very similar results were obtained when the degradation of chlorite was monitored photometrically at 260 nm under identical conditions. Figure 3D depicts two representative time traces from photometric experiments following the decrease in absorbance at 260 nm during degradation of 340 μM chlorite by 25 and 100 nM NdCld in the absence and presence of methionine (5 mM) at pH 7.0. Figure 3E compares the extent of degradation of 340 μM chlorite by 25, 100, and 400 nM NdCld at pH 5.5 and 7.0 in the absence and presence of 5 mM methionine. It demonstrates that 25 nM NdCld ([chlorite]/[NdCld] ratio of 13600) can convert ∼21% of the initial amount of chlorite in the absence and ∼45% of the chlorite in the presence (5 mM) of methionine at pH 5.5. At pH 7.0, those numbers decrease to ∼16 and ∼36%, respectively. When 100 nM NdCld ([chlorite]/[NdCl] ratio of 3400) was used, all chlorite independent of the addition of methionine could be converted at pH 5.5, in contrast to pH 7.0 where only in the presence of 5 mM methionine was the entire initial amount of chlorite converted and only ∼54% in the absence of methionine. At an enzyme concentration of 400 nM NdCld ([chlorite]/[NdCl] ratio of 850), the entire amount of chlorite was converted regardless of pH or the presence of methionine.
These data clearly demonstrate that (i) Cld is irreversibly inhibited during chlorite degradation, (ii) inhibition is weakest at the pH optimum of enzymatic activity, (iii) methionine can rescue the enzyme from inhibition to some extent (depending on the [chlorite]/[NdCl] ratio), and (iv) exchange of the distal arginine amplifies the inhibitory effect.
Quantification of Hypochlorous Acid Released during Catalysis
Next, we aimed to trap hypochlorite with two molecules, namely, MCD and 2-[6-(4-aminophenoxy)-3-oxo-3H-xanthen-9-yl]benzoic acid (APF), that allow the rough calculation of the amount of released hypochlorite by UV–vis21 and fluorescence30 spectroscopy.
In Figure 4A, the kinetics of MCD (55 μM) chlorination during NdCld (100 nM)-mediated chlorite degradation is depicted at various substrate concentrations at pH 5.5 (Figure 4A) and pH 7.0 (Figure 4B). During chlorination of MCD to DCD (dichlorodimedone), the absorbance at 290 nm is lost. Chlorite does not react with MCD.24 Upon plotting the decrease in the absorbance of MCD versus chlorite concentration, we observed a linear dependence at chlorite concentrations of >50 μM at pH 5.5 and >0 μM at pH 7.0 (Figure 4C). On the basis of the assumption that all released hypochlorite reacted with MCD, it can be calculated from the slope of the linear curves that at least ∼7.5 μM hypochlorite/mM chlorite escaped from the reaction sphere at pH 5.5 and 7.0.
Figure 4.

Trapping hypochlorite by monochlorodimedone in wild-type NdCld (A–C) at different pH values and mutant NdClds (D–F).
Similar reactions were performed with 200 nM NdCld R173A (Figure 4D) and 200 nM NdCld R173K (Figure 4E) at pH 5.5. In contrast to the behavior of the wild-type enzyme, a linear dependence of formed DCD on chlorite concentration was observed only over a small concentration range [0–10 μM for R173A and 0–100 μM for R173K (Figure 4F)]. On the basis of the assumption that all released hypochlorite reacted with MCD, it can be estimated that at least ∼68 and ∼31 μM hypochlorite/mM chlorite escaped from the reaction sphere in R173A and R173K at pH 5.5, respectively. These findings clearly suggest that in the absence of R173 the amount of hypochlorous acid released from the reaction sphere is increased significantly.
A further hypochlorite trapping molecule is APF. Upon reaction of –OCl with APF, the fluorescence intensity at 522 nm (excitation at 488 nm) is increased. Figure 2 of the Supporting Information depicts the reaction of 100 nM NdCld at various chlorite concentrations at pH 7.0 followed by fluorescence spectroscopy (10 μM APF). On the basis of a calibration curve (Figure 2A,B of the Supporting Information), the amount of hypochlorite trapped by APF was estimated from its linear dependence of the concentration of chlorite concentration (<500 μM) at pH 7.0 (Figure 2D of the Supporting Information). On the basis of the assumption that all released hypochlorite reacted with APF, it can be calculated that at least ∼4 μM hypochlorite/mM chlorite escaped from the reaction sphere at pH 7.0.
Heme Bleaching during Chlorite Degradation Monitored by EPR and UV–Vis Spectroscopy
Both time-resolved UV–vis spectroscopy and EPR spectroscopy revealed irreversible heme bleaching in NdCld during degradation of chlorite or upon addition of hypochlorite. EPR spectroscopy gives valuable information about the electronic architecture of the paramagnetic ferric heme b center of NdCld. Wild-type NdCld has spectral features that are composed of two high-spin species at pH 5.5 (Figure 5A and Table 1 of the Supporting Information) and two high-spin and two low-spin species at pH 7.0 (Figure 5B and Table 1 of the Supporting Information). Simulation and spin quantification parameters of the experimental spectra are listed in Table 1 of the Supporting Information. The overall high-spin spectrum resembles previously published NdCld spectra.1 It is worth pointing out that differences in buffer conditions and cryo conditions affect the rhombicity in NdCld samples.15
Figure 5.
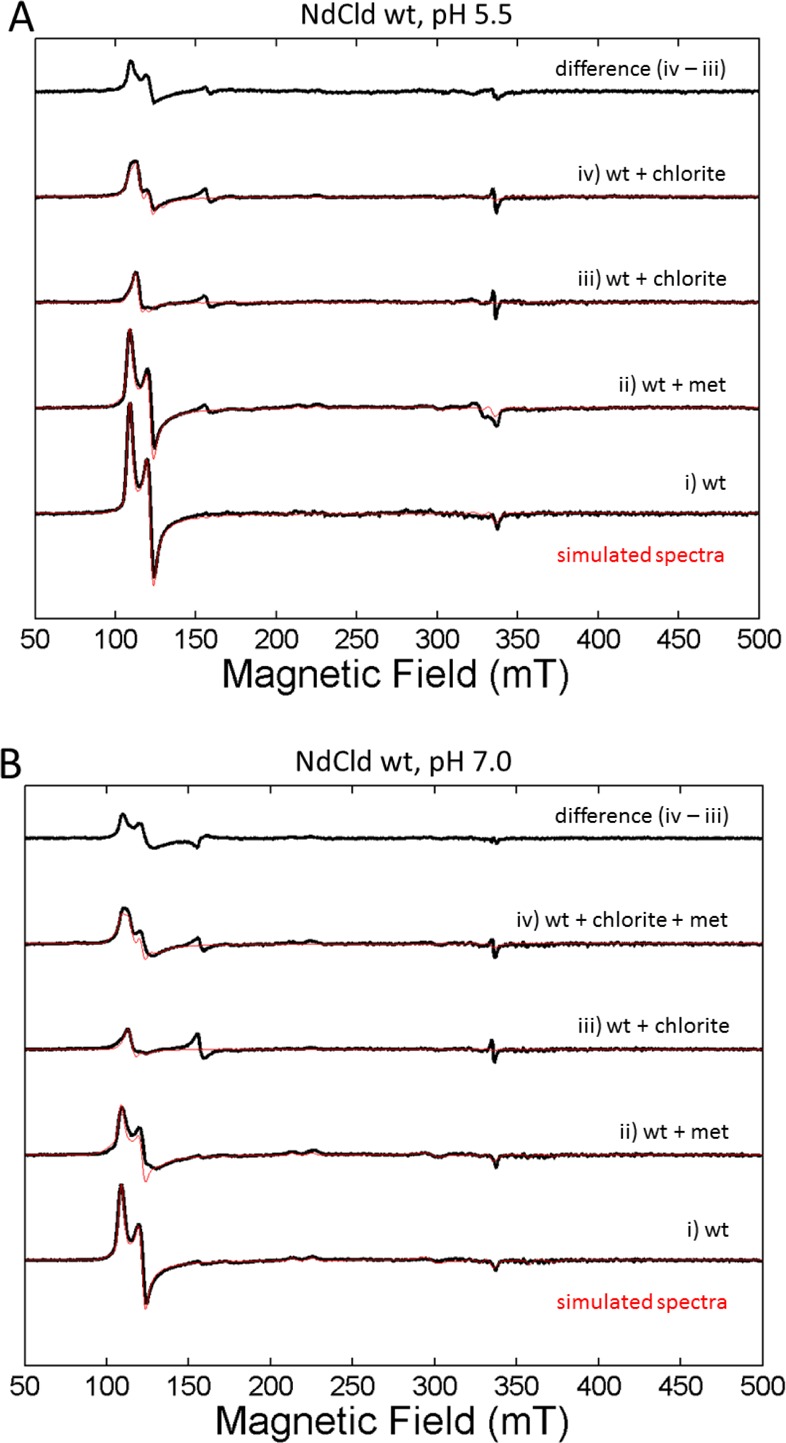
Protein deactivation by chlorite followed by an intensity change of the cw electron paramagnetic resonance high-spin spectra at (A) pH 5.5 and (B) 7.0, detected at 10 K (black, experimental spectrum; red, simulated spectrum).
The intensity of the high-spin signal of wild-type NdCld decreases in the presence of chlorite (Figure 5 and Figure 3 of the Supporting Information). With 100 mM chlorite, the rhombic signal disappears almost completely but part of the axial signal remains (Figure 3A of the Supporting Information). This decrease in the intensity of the high-spin signal with no concomitant formation of a low-spin signal indicates a change in the paramagnetic oxidation state of Fe(III) NdCld and hence an inactivation of the protein. With a large excess of chlorite (∼2500-fold), protein radicals begin to form at a g value of ∼2. As already observed in the steady-state kinetic assays, methionine is able to protect NdCld (Figure 3B of the Supporting Information). It disrupts the heme environment of NdCld [indicated by slight changes in the rhombicity in the spectra and a significant decrease in the intensity of the high-spin ferric signal (see Figure 5A,B and Table 1 of the Supporting Information)], suggesting that it can enter the heme cavity and trap hypochlorite at the site of its production. In the presence of methionine (17.5 mM), the remaining high-spin signal of NdCld (40 μM) incubated with 100 mM chlorite still has a significant amount of rhombic high-spin signal. The protective effect of methionine is more pronounced at pH 7 than at pH 5.5 (see also the difference spectra depicted in Figure 5).
Finally, NdCld was incubated directly with hypochlorous acid (Figure 4 of the Supporting Information). With an increasing amount of hypochlorous acid, the rhombic high-spin compound disappears and the intensity of a non-heme Fe(III) high-spin signal (g = 4.3) representative of degraded protein increases significantly. A small protein radical species is also observed. The effect of HOCl on the deactivation and degradation of NdCld is much more pronounced in this experiment then the effect of the experiment in which HOCl is generated during chlorite turnover because of a much higher HOCl concentration at the heme site.
Besides EPR, we investigated the modification of the spectral signatures of the prosthetic group during chlorite degradation by UV–vis stopped-flow spectroscopy (Figure 6). Figure 6A depicts the spectral changes, when 1 μM wild-type NdCld is mixed with 1 mM chlorite at pH 5.5 in the conventional stopped-flow modus. The ferric resting state with its Soret maximum at 406 nm (green spectrum) is rapidly converted within 1 ms to an intermediate with a Soret maximum of 414 nm and a prominent peak at 535 nm (black spectrum). During chlorite degradation, this species dominated and was converted to the resting state within 10 s. The resulting heme spectrum significantly lost Soret absorbance (maxima at 408 and 545 nm), suggesting heme bleaching (red spectrum in Figure 6A). At pH 7.0, the loss of absorbance at the Soret maximum was more pronounced and the rate and extent of chlorite degradation were smaller (Figure 5 of the Supporting Information). Performing the same reactions in the presence of 5 mM methionine reveals a different outcome. The reaction rate is enhanced, chlorite degradation complete, and heme bleaching less pronounced (Figure 6B and Figure 5D of the Supporting Information).
Figure 6.
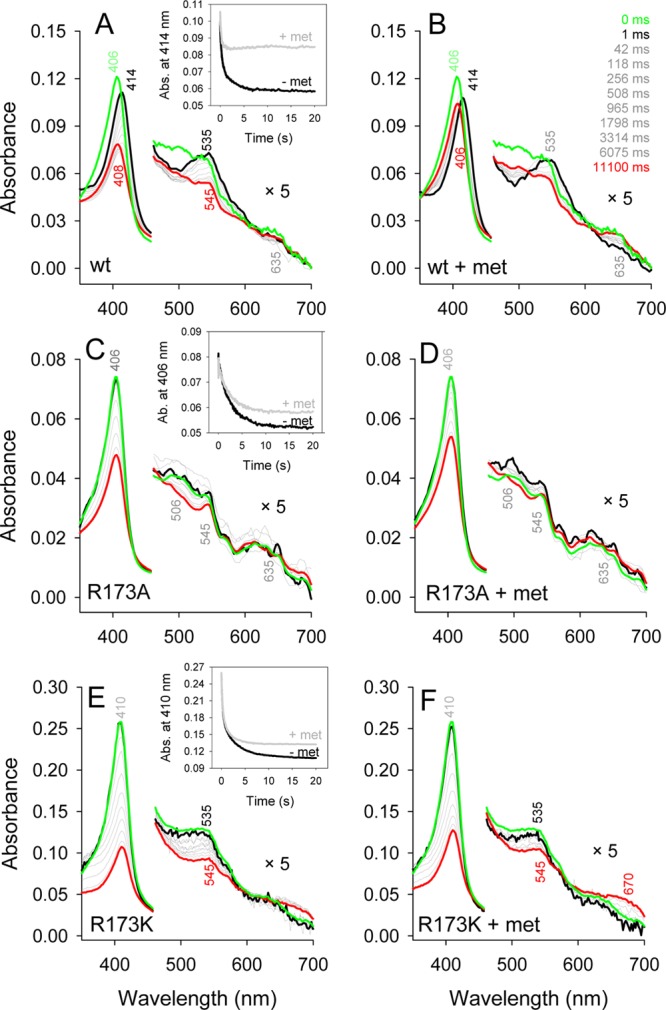
Effect of methionine on the interconversion of redox intermediates and heme bleaching in wild-type NdCld (A and B) and NdCld variants (C–F). Insets depict representative time traces at the Soret maximum.
Mixing 1 μM NdCld R173A with 1 mM chlorite (same condition as the wild-type protein) led to a complete loss of heme absorbance within 2 s. Thus, the variant was mixed with only 50 μM chlorite in the absence (Figure 6C) and presence (Figure 6D) of 5 mM methionine. In contrast to that of the wild-type protein, the Soret maximum remained at 406 nm during chlorite degradation and the effect of methionine on heme bleaching was weak. Mutant R173K was more robust and could be mixed with 1 mM chlorite (Figure 6E). The Soret maximum of ferric R173K is at 410 nm and was recently described as a low-spin species.10 When R173K was mixed with chlorite, no spectral shift occurs and more than 60% of the heme absorbance disappears (Figure 6F).
Mass Spectrometric Analyses of Heme and Protein Modification
Furthermore, we analyzed the modifications of the protein moiety of NdCld incubated with chlorite in the presence and absence of methionine by mass spectrometry. StrepII-tagged NdCld has a theoretical mass of 29751.8 Da without the heme b cofactor (which is lost during sample preparation for MS). Analysis of recombinant NdCld revealed the presence of some heterogeneity at the N-terminus in the region of the StrepII tag and the TEV cleavage site. Six different variants were detected, with the full length protein having the highest intensity (peak maximum at 29751.3 Da and two minor peaks at higher m/z values, reflecting possibly methylation and acetylation) (Figure 7A). Truncated versions start at Met–22, Phe–14, Phe–5, Gly–3, Met–1, and Ala1 (data not shown). Upon treatment with a 5000-fold stoichiometric excess of chlorite, the resulting spectrum (Figure 7B) shows two dominating species representing NdCld oxidized two and three times in addition to other forms having been oxidized up to approximately 10 times. A low-intensity peak at the original mass is still detected, suggesting the presence of some unmodified NdCld after the reaction with chlorite. In the presence of methionine, the mass peak of the unmodified enzyme is still the most prominent one (Figure 7C). Modified (oxidized one to five times) NdCld was also detected in this sample (Figure 7C) but to a much lesser extent compared to the sample from which methionine was absent.
Figure 7.

Modification of the protein during NdCld-mediated degradation of chlorite. Wild-type NdCld (A) was treated with chlorite (B) and with chlorite in the presence of methionine (C). MS2 spectrum of peptide 175–194 of wild-type NdCld upon treatment with chlorite. The inset shows the amino acid sequence of NdCld, including the N-terminal StrepII tag and TEV cleavage site (highlighted in gray).
Finally, to identify modified (oxidized) residues, peptide analysis of NdCld incubated with chlorite was performed. All methionines in the protein are oxidized to a large degree. By contrast, tryptophan and tyrosine (Y189) residues are marginally oxidized (Table 1). Interestingly, two of seven tyrosines (Tyr56 and Tyr176) were found to be chlorinated, as was proven by MS/MS analysis (Figure 7D). The corresponding peptides were present as two species with a mass unit difference of 2 and a 3:1 ratio of intensities, reflecting the occurrence of the two chlorine isotopes, 35Cl (75.8%) and 37Cl (24.2%). 3-Chlorotyrosines are specific fingerprints for the action of hypochlorite.31−33 Both modified tyrosine residues are in the core of the protein on the distal side of the heme, where the hypochlorite is produced during turnover (Figure 8). They are located at rather remote sites with respect to the cofactor but are connected with the heme center by a tunnel as calculated with CAVER34 (Figure 8A).
Table 1. Digestion of 100 μM NdCld, Treated with 500 mM Chlorite in the Absence and Presence of 25 mM Methionine, by GluC/Trypsin and Analysis by LC–MS in 50 mM Phosphate Buffer (pH 7.0).
| NdCld
with chlorite |
NdCld
with chlorite and methionine |
|||||
|---|---|---|---|---|---|---|
| amino acids | peptide sequence | % Tyr-Cl | % total modification | % Tyr-Cl | % total modification | Δ% |
| 6–33 | LLTESGVYGTFATFQMDHDWWDLPGESR | 57.27 | 5.86 | 51.41 | ||
| 34–41 | VISVAEVK | no modification | no modification | |||
| 42–50 | GLVEQWSGK | 22.22 | 2.57 | 19.65 | ||
| 51–59 | ILVESYLLR | 2.53 | 2.53 | 0.04 | 0.04 | 2.50 |
| 60–70 | GLSDHADLMFR | 98.49 | 6.54 | 91.95 | ||
| 75–90 | TLSDTQQFLSAFMGTR | 99.76 | 24.92 | 74.84 | ||
| 94–106 | HLTSGGLLHGVSK | no modification | no modification | |||
| 107–119 | KPTYVAGFPESMK | 96.75 | 18.86 | 77.90 | ||
| 120–140 | TELQVNGESGSRPYAIVIPIK | no modification | no modification | |||
| 142–153 | DAEWWALDQEAR | 20.79 | 1.78 | 19.01 | ||
| 154–169 | TALMQEHTQAALPYLK | 85.43 | 16.74 | 68.69 | ||
| 175–194 | LYHSTGLDDVDFITYFETER | 8.53 | 13.56 | 0.67 | 2.46 | 11.10 |
| 195–203 | LEDFHNLVR | no modification | no modification | |||
| 204–209 | ALQQVK | no modification | no modification | |||
| 217–235 | FGHPTLLGTMSPLDEILEK | 99.88 | 14.34 | 85.54 | ||
Figure 8.
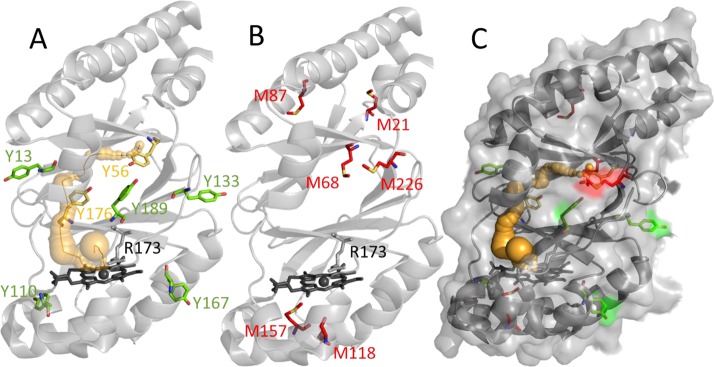
Amino acids modified during NdCld-mediated chlorite degradation. (A) Ribbon representation of a single NdCld subunit. A possible pathway of hypochlorite within the subunit toward modified tyrosine residues is depicted as orange spheres and an orange ribbon. (B) All oxidized methionines are colored red. (C) NdCld with a semitransparent surface. Figures were generated using PyMOL (http://www.pymol.org/).
The addition of free methionine to the reaction mixture significantly reduced the percentage of modifications of methionine, tyrosine, and tryptophan residues of NdCld (Table 1). The fact that methionine both at the surface and in the protein interior is oxidized (Figure 8B,C) demonstrates that hypochlorite escapes from the active site during the degradation of chlorite.
Molecular Dynamics Simulation of the Movement of Hypochlorite
MD simulations support the finding that transiently produced hypochlorite migrates away from the reaction sphere. In this setup, hypochlorite is placed in silico to the distal side of the heme in its Compound I state with Arg173 in an “in” conformation (pointing to the heme iron and not toward the substrate channel).
In MD simulations, hypochlorite cannot further react to form chloride and dioxygen. It leaves the active site rapidly in all five monomers of NdCld. This is illustrated in Figure 9, where the positions of one hypochlorite molecule over time are shown (every 5 ps) over a 20 ns simulation. Positions early in the simulation are colored green, gradually changing to blue, yellow, orange, and red (final position after simulation for 20 ns).
Figure 9.
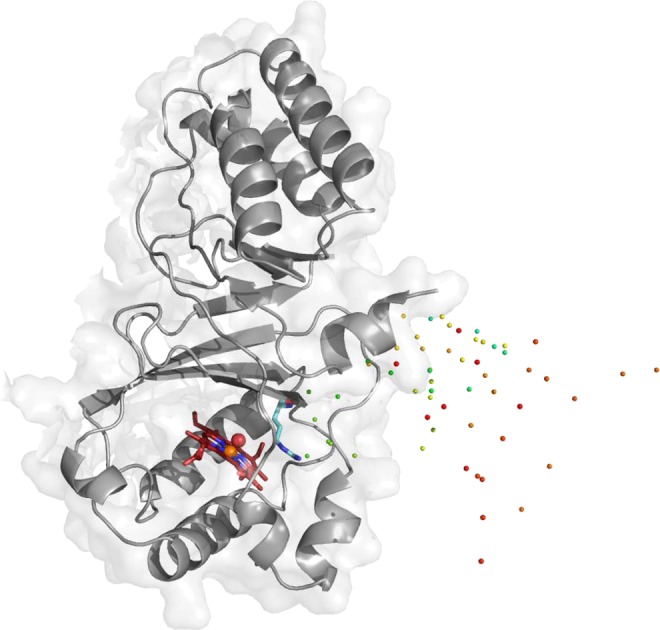
Molecular dynamics simulation of the movement of hypochlorite. A single NdCld monomer is shown, along with the position of one hypochlorite molecule at different time points (0–20 ns; green, blue, yellow, orange, and red spheres). Hypochlorite escapes the active site rapidly. Compound I oxygen is depicted as a red sphere and Compound I heme iron as an orange sphere. Catalytically important R173 is colored cyan and points away from the active site, being unable to keep the transiently produced hypochlorite in the reaction sphere. This figure was generated using PyMOL (http://www.pymol.org/).
Discussion
Understanding the inhibition mechanism of chlorite dismutase (Cld) is fundamental for future enzymatic application of Cld in bioremediation and biotechnology.29 The enzyme from “Candidatus Nitrospira defluvii” (NdCld) was chosen as a model protein for inhibition studies because it has a high thermal and conformational stability17 and thus is the most promising candidate for biotechnological applications.29
Chlorite reacts with a variety of different heme-containing enzymes, including methemoglobin35 or cytochrome P450.36 Among heme peroxidases, chlorite was shown to be utilized by cytochrome c peroxidase, chloroperoxidase, and horseradish peroxidase (HRP).37 With HRP, it was demonstrated that chlorite mediates the two-electron oxidation of ferric HRP to Compound I (similar to postulated reaction 2 for Cld) but also serves as one-electron reductant of both Compound I and Compound II.36 Thereby, HRP is inactivated with time. Importantly, upon being mixed with chlorite, HRP produces a halogenating agent that reacts with MCD and was postulated to be hypochlorite.37 By contrast, chlorite rapidly and irreversibly inactivates human peroxidases, like myeloperoxidase (MPO) and lactoperoxidase,24 which belong to a different heme peroxidase superfamily.38 However, the first heme enzyme that was described to utilize chlorite as a natural substrate and degrade it efficiently was Cld from perchlorate-respiring bacteria that actually produce chlorite as a metabolic intermediate.39
Both the overall fold and the heme cavity architecture of heme peroxidases like HRP or MPO are completely different compared to those of Cld. In heme peroxidases, hydrogen peroxide is the natural oxidant that mediates the two-electron oxidation of the ferric enzyme to Compound I. A basic amino acid, typically histidine, is essential for acting as a proton acceptor forming the anionic peroxide that binds to Fe(III). The following heterolytic cleavage of H2O2 is supported by a conserved arginine. Thus, Compound I formation in a peroxidase typically depends on the pKa of the distal histidine.
In functional Cld, the only charged amino acid in the distal heme cavity is an arginine (Arg173 in NdCld). The natural oxidant [E°′(ClO2/ClO–) = 1175 mV] is chlorite (at least most probably in those Clds found in perchlorate-respiring bacteria), which is deprotonated (pKa = 1.97) in the biological pH range. Thus, a proton acceptor is not necessary for binding to Fe(III) according to reaction 1. This is reflected by the fact that the calculated KM value is independent of pH (Figure 1A). One might speculate about the role of Arg173 in reaction 1. Exchange of R173 with either alanine or glutamine has only a weak effect on the KM value (i.e., increase by a factor of 1.3 in R173A and 1.9 in R173Q compared to that of the wild-type protein),10 suggesting that its role in substrate binding is negligible.
In contrast to the KM, the kcat of NdCld shows a clear dependence on pH. Above the pH optimum of chlorite degradation (i.e., pH 5.5), the turnover number decreases with an increase in pH (Figure 1B). This behavior could be related to a role of Arg173 in (i) the heterolytic cleavage of chlorite (reaction 2) and/or (ii) the recombination reaction between the (postulated) transiently produced intermediate hypochlorite and Compound I (reaction 3). The turnover number (kcat) for chlorite degradation is ∼6.5% (R173A) and 33% (R173Q) of that of the wild-type enzyme. Thus, arginine is not fully essential for catalysis. In its presence, the turnover number is increased and, as demonstrated in this work, inactivation is significantly retarded (see below).
It has been postulated that the pH dependence of chlorite degradation by Cld reflects the protonation state of the distal arginine, which was reported to have its pKa at ∼pH 6.5 in D. aromatica Cld (DaCld).14 The high degree of similarity of (i) the hydrophobic distal site architecture of NdCld7 and DaCld5 and (ii) the pH dependence of chlorite degradation determined in this work (Figure 1B) suggests a similar pKa for Arg173 in NdCld. At pH 5.5, the positively charged guanidinium group of Arg173 is perfectly suited to keep the transiently produced hypochlorite in place for the postulated rebound mechanism of reaction 3. In the close vicinity to the guanidinium group, the HOCl––OCl equilibrium (pKa = 7.54) will be shifted to the anionic conjugated base (i.e., hypochlorite). With an increase in pH, the interaction between hypochlorite and Arg173 will gradually be weakened. However, as demonstrated in this work, this behavior is emphasized by the fact that Cld is irreversibly inactivated during catalysis. This is the main reason why it is problematic to compare the published catalytic properties, because the various enzymes were probed in different chlorite concentration regimes and are differently susceptible to inactivation.29
This work shows that the extent of degraded chlorite (which corresponds to the yield of produced O2) is significantly smaller at alkaline pH (Figure 1D) and that the presence of millimolar methionine can increase both the extent of chlorite degradation and the reaction rate. This was demonstrated both polarographically and spectrophotometrically. The protecting effect of methionine clearly suggests a role of hypochlorite in the inactivation reaction, because it is able to trap the transiently formed intermediate but not the substrate chlorite. Methionine easily can enter the relatively large substrate channel as well as the active site of Cld.7,15 EPR demonstrated that methionine forms a six-coordinated complex with Cld (Figure 5). It does not inhibit the reaction; quite the contrary, its presence increases the lifetime of active Cld molecules. The favorable impact of methionine on catalysis is more pronounced as conditions become more alkaline.
The fact that transiently formed hypochlorite is able to react with methionine suggests that reaction 3 is slower than reaction 2 and that not all hypochlorite generated in reaction 2 reacts with Compound I. A small fraction escapes from the active site. The species formed within 1 ms by mixing of Cld with chloride in the stopped-flow apparatus with absorbance maxima at 414 and 535 nm most probably represents {[Por•Fe(IV)=O]+···[O–Cl–]}. As long as chlorite is available and Cld is active, its spectral signatures dominate. Note that a Compound I spectrum {i.e., [Por•Fe(IV)=O]+, produced by mixing Cld with peracetic acid} exhibits a blue-shifted and hypochromic Soret band at 395 nm and further bands at 525, 550, 600, and 645 nm.40
To further confirm the production and release of hypochlorite, two additional traps (MCD21 and APF23,30) were used. They allowed the rough calculation of the amount of released hypochlorite by spectroscopic means. At the pH optimum (kcat ∼ 180 s–1) and with a stoichiometric excess of chlorite of 500–1000-fold, ∼7.7 μM hypochlorite/mM chlorite could be trapped by MCD. This is a minimal number because it is based on the assumption that all released HOCl molecules reacted with MCD. In the absence of the distal arginine, the amount of released hypochlorite is significantly larger, i.e., 31 μM/mM chlorite (R173K) and 68 μM/mM chlorite (R173A). With an increase in pH, the rate of turnover of Cld gradually decreased in contrast to the amount of trapped HOCl. These findings strongly indicate that (i) hypochlorite is formed during catalysis, (ii) Arg173 helps to keep it in the reaction sphere for the recombination reaction, and (iii) the decrease in the amount of degraded chlorite (or released O2) is related to the promoted inhibition of Cld by escaped HOCl under more alkaline conditions.
The question about the target(s) and the inhibitory effect of hypochlorite remains. Both UV–vis and EPR spectroscopy demonstrated that the heme of Cld is severely modified during catalysis. The loss of heme absorbance and the intensity of the high-spin signal together with the release of free iron occurs. In the absence of Arg173 and at higher pH values, these reactions are more pronounced, whereas in the presence of HOCl traps, they are partially suppressed. Additionally, the protein moiety is modified. Protein radicals are formed, and amino acids, mostly methionine and a few tyrosines and tryptophans, were oxidatively modified. Most importantly, 3-chlorotyrosine is produced, which is a typical fingerprint for the action of hypochlorite.31−33 Solvent-exposed tyrosines and tyrosines in the interface of pentameric NdCld subunits did not show this modification, whereas two chlorinated tyrosines in the core of the protein were found. Their molecular surface accessibility is 0 Å2 (according to the WHATIF Web server, http://swift.cmbi.ru.nl/servers/html/index.html), but they may be reached by HOCl by a small tunnel. On this path, Tyr176 is the first target (8.5% chlorination) and Tyr56 the second (2.5% chlorination). Importantly, all these modifications could be significantly suppressed in the presence of methionine (Table 1).
In summary, the data presented in this work support the postulation that degradation of chlorite by Cld follows reactions 1–3. Upon reduction of chlorite, hypochlorite is formed and kept in the reaction sphere for recombination with the oxoiron(IV) group of Compound I. The guanidinium group of a fully conserved arginine with a pKa of ∼6.5 supports this reaction. However, approximately one molecule of HOCl per 100 full cycles escapes and reacts with both the prosthetic group and the protein moiety. As a consequence, irreversible inactivation of Cld is observed and this reaction is more pronounced with an increase in pH. Importantly, HOCl traps like methionine can rescue the enzyme from inactivation.
Glossary
Abbreviations
- Cld
chlorite dismutase
- NdCld
chlorite dismutase from “Candidatus Nitrospira defluvii”
- DaCld
chlorite dismutase from D. aromatica
- HRP
horseradish peroxidase
- MPO
myeloperoxidase
- MCD
monochlorodimedon
- APF
2-[6-(4-aminophenoxy)-3-oxo-3H-xanthen-9-yl]benzoic acid
- MD
molecular dynamics
- PDB
Protein Data Bank.
Supporting Information Available
pH dependence of the reaction of NdCld variants with chlorite, hypochlorite trapping with APF, additional EPR spectra of chlorite titrations, effect of methionine on heme bleaching, and chlorite degradation at pH 7.0. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
This project was supported by the Austrian Science Foundation, FWF [Doctoral Program BioToP-Molecular Technology of Proteins (W1224) and Projects P25270 and P22276].
Supplementary Material
References
- Hagedoorn P. L.; de Geus D. C.; Hagen W. R. (2002) Spectroscopic characterization and ligand-binding properties of chlorite dismutase from the chlorate respiring bacterial strain GR-1. Eur. J. Biochem. 269, 4905–4911. [DOI] [PubMed] [Google Scholar]
- Lee A. Q.; Streit B. R.; Zdilla M. J.; Abu-Omar M. M.; Dubois J. L. (2008) Mechanism of and exquisite selectivity for O-O bond formation by the heme dependent chlorite dismutase. Proc. Natl. Acad. Sci. U.S.A. 105, 15654–15659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith J. M.; Abu-Omar M. M.; Hall M. B. (2011) Computational investigation of the concerted dismutation of chlorite ion by water-soluble iron porphyrins. Inorg. Chem. 50, 7928–7930. [DOI] [PubMed] [Google Scholar]
- Sun S.; Li Z. S.; Chen S. L. (2014) A dominant homolytic O-Cl bond cleavage with low-spin triplet-state Fe(IV)=O formed is revealed in the mechanism of heme-dependent chlorite dismutase. Dalton Trans. 43, 973–981. [DOI] [PubMed] [Google Scholar]
- de Geus D. C.; Thomassen E. A. J.; Hagedoorn P.-L.; Pannu N. S.; van Duijn E.; Abrahams J. P. (2009) Crystal structure of chlorite dismutase, a detoxifying enzyme producing molecular oxygen. J. Mol. Biol. 387, 192–206. [DOI] [PubMed] [Google Scholar]
- Goblirsch B. R.; Streit B. R.; DuBois J. L.; Wilmot C. M. (2010) Structural features promoting dioxygen production by Dechloromonas aromatica chlorite dismutase. JBIC, J. Biol. Inorg. Chem. 15, 879–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostan J.; Sjoeblom B.; Maixner F.; Mlynek G.; Furtmüller P. G.; Obinger C.; Wagner M.; Daims H.; Djinovic-Carugo K. (2010) Structural and functional analysis of the chlorite dismutase from the nitrite-oxidizing bacterium Candidatus “Nitrospira defluvii”: Identification of a catalytically important amino acid residue. J. Struct. Biol. 172, 331–342. [DOI] [PubMed] [Google Scholar]
- Mlynek G.; Sjöblom B.; Kostan J.; Füreder S.; Maixner F.; Gysel K.; Furtmüller P. G.; Obinger C.; Wagner M.; Daims H.; Djinović-Carugo K. (2011) Unexpected diversity of chlorite dismutases: A catalytically efficient dimeric enzyme from Nitrobacter winogradskyi. J. Bacteriol. 193, 2408–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc B.; Mayfield J. A.; McDonald C. A.; Lukat-Rodgers G. S.; Rodgers K. R.; Dubois J. L. (2012) Understanding how the distal environment directs reactivity in chlorite dismutase: Spectroscopy and reactivity of Arg183 mutants. Biochemistry 51, 1895–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofbauer S.; Gysel K.; Bellei M.; Hagmüller A.; Schaffner I.; Mlynek G.; Kostan J.; Pirker K. F.; Daims H.; Furtmüller P. G.; Battistuzzi G.; Djinovic-Carugo K.; Obinger C. (2014) Manipulating conserved heme cavity residues of Chlorite dismutase: Effect on structure, redox chemistry and reactivity. Biochemistry 53, 77–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenklo K.; Thorell D.; Bergius H.; Aasa R.; Nilsson T. (2001) Chlorite dismutase from Ideonella dechloratans. JBIC, J. Biol. Inorg. Chem. 6, 601–607. [DOI] [PubMed] [Google Scholar]
- Mehboob F.; Wolterink A. F.; Vermeulen A. J.; Jiang B.; Hagedoorn P. L.; Stams A. J.; Kengen S. W. (2009) Purifcation and characterization of a chlorite dismutase from Pseudomonas chloritidismutans. FEMS Microbiol. Lett. 293, 115–121. [DOI] [PubMed] [Google Scholar]
- Streit B. R.; DuBois J. L. (2008) Chemical and steady-state analysis of a heterologously expressed heme dependent chlorite dismutase. Biochemistry 47, 5271–5280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit B. R.; Blanc B.; Lukat-Rodgers B. S.; Rodgers K. R.; DuBois J. L. (2010) How active-site protonation state influences the reactivity and ligation of the heme in chlorite dismutase. J. Am. Chem. Soc. 132, 5711–5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofbauer S.; Bellei M.; Sündermann A.; Pirker K. F.; Hagmüller A.; Mlynek G.; Kostan J.; Daims H.; Furtmüller P. G.; Djinovic-Carugo K.; Oostenbrink C.; Battistuzzi G.; Obinger C. (2012) Redox thermodynamics of high-spin and low-spin forms of chlorite dismutases with diverse subunit and oligomeric structures. Biochemistry 51, 9501–9512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanc B.; Rodgers K. R.; Lukat-Rodgers G. S.; DuBois J. L. (2013) Understanding the roles of strictly conserved tryptophan residues in O2 producing chlorite dismutases. Dalton Trans. 42, 3156–3131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofbauer S.; Gysel K.; Mlynek G.; Kostan J.; Hagmüller A.; Daims H.; Furtmüller P. G.; Djinovic-Carugo K.; Obinger C. (2012) Impact of subunit and oligomeric structure on the thermal and conformational stability of chlorite dismutases. Biochim. Biophys. Acta 1824, 1031–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippi M.; dos Santos H. S.; Martins A. O.; Azevedo C. M.; Pires M. (2007) Alternative spectrophotometric method for standardization of chlorite aqueous solutions. Anal. Chim. Acta 585, 361–365. [DOI] [PubMed] [Google Scholar]
- Armesto X. L.; Canle M. L.; Fernandez M. I.; Garcia M. V.; Santaballa J. A. (2000) First steps in the oxidation of sulfur-containing amino acids by hypohalogenation: Very fast generation of intermediate sulfenyl halides and halosulfonium cations. Tetrahedron 56, 1103–1109. [Google Scholar]
- Peskin A. V.; Turner R.; Maghzal G. J.; Winterbourn C. C.; Kettle A. J. (2009) Oxidation of methionine to dehydromethionine by reactive halogen species generated by neutrophils. Biochemistry 48, 10175–10182. [DOI] [PubMed] [Google Scholar]
- Tachikawa M.; Sayama C.; Saita K.; Tezuka M.; Sawamura R. (2002) Effects of isocyanuric acid on the monochlorodimedone chlorinating rates with free chlorine and ammonia chloramine in water. Water Res. 10, 2547–2554. [DOI] [PubMed] [Google Scholar]
- Kettle A. J.; Winterbourn C. C. (1988) The mechanism of myeloperoxidase-dependent chlorination of monochlorodimedon. Biochim. Biophys. Acta 957, 185–191. [DOI] [PubMed] [Google Scholar]
- Flemmig J.; Zschaler J.; Remmler J.; Arnhold J. (2012) The fluorescein-derived dye aminophenyl fluorescein is a suitable tool to detect hypobromous acid (HOBr)-producing activity in eosinophils. J. Biol. Chem. 287, 27913–27923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakopitsch C.; Pirker K. F.; Flemmig J.; Hofbauer S.; Schlorke D.; Furtmüller P. G.; Arnhold J.; Obinger C. (2014) Mechanism of reaction of chlorite with mammalian heme peroxidases. J. Inorg. Biochem. 135, 10–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoll S.; Schweiger A. (2006) EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J. Magn. Reson. 178, 42–55. [DOI] [PubMed] [Google Scholar]
- Peisach J.; Blumberg W. E.; Ogawa S.; Rachmilewitz E. A.; Oltzik R. (1971) The effects of protein conformation on the heme symmetry in high spin and ferric heme proteins as studied by electron paramagnetic resonance. J. Biol. Chem. 246, 3342–3355. [PubMed] [Google Scholar]
- Schmid N.; Christ C. D.; Christen M.; Eichenberger A. P.; van Gunsteren W. F. (2012) Architecture, implementation and parallelisation of the GROMOS software for biomolecular simulation. Comput. Phys. Commun. 183, 890–903. [Google Scholar]
- Schmid N.; Eichenberger A. P.; Choutko A.; Riniker S.; Winger M.; Mark A. E.; van Gunsteren W. F. (2011) Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 40, 846–856. [DOI] [PubMed] [Google Scholar]
- Hofbauer S.; Schaffner I.; Furtmüller P. G.; Obinger C. (2014) Chlorite dismutases: A heme enzyme family for use in bioremediation and generation of molecular oxygen. Biotechnol. J. 9, 461–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setsukinai K.; Urano Y.; Kakinuma K.; Majima H. J.; Nagano T. (2003) Development of novel fluorescence probes that can reliably detect reactive oxygen species and distinguish specific species. J. Biol. Chem. 278, 3170–3175. [DOI] [PubMed] [Google Scholar]
- Delporte C.; Zouaoui Boudjeltia K.; Noyon C.; Furtmüller P. G.; Slomianny M.-C.; Dufour D.; Vanhamme L.; Reyé F.; Rousseau A.; Vanhaeverbeek M.; Ducobu J.; Michalski J.-C.; Nève J.; Obinger C.; Malle E.; Van Antwerpen P. (2014) Impact of interaction between low-density lipoproteins and myeloperoxidase on specific enzymatic activity and post-translational oxidative modifications of apoB100. J. Lipid Res. 55, 747–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazen S. L.; Heinecke J. W. (1997) 3-Chlorotyrosine, a specific marker of myeloperoxidase-catalyzed oxidation, is markedly elevated in low density lipoprotein isolated from human artherosclerotic intima. J. Clin. Invest. 99, 2075–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazen S. L.; Crowley J. R.; Mueller M. D.; Heinecke J. W. (1997) Mass spectrometric quantification of 3-chlorotyrosine in human tissues with attomole sensitivity: A sensitive and specific marker for myeloperoxidase-catalyzed chlorination at sites of inflammation. Free Radical Biol. Med. 23, 909–916. [DOI] [PubMed] [Google Scholar]
- Petrek M.; Otyepka M.; Banás P.; Kosinová P.; Koca J.; Damborský J. (2006) CAVER: A new tool to explore routes from protein clefts, pockets and cavities. BMC Bioinf. 7, 316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French C. L.; Yaun S. S.; Baldwin L. A.; Leonhard D. A.; Zhao X. Q.; Calabrese E. J. (1995) Potency ranking of methemoglobin-forming agents. J. Appl. Toxicol. 15, 167–174. [DOI] [PubMed] [Google Scholar]
- Hrycay E. G.; Gustafsson J. A.; Ingelman-Sundberg M.; Ernster L. (1975) Sodium periodate, sodium chloride, and organic hydroperoxides as hydroxylating agents in hepatic microsomal steroid hydroxylation reactions catalyzed by cytochrome P450. FEBS Lett. 56, 161–165. [DOI] [PubMed] [Google Scholar]
- Jakopitsch C.; Spalteholz H.; Furtmüller P. G.; Arnhold J.; Obinger C. (2008) Mechanism of reaction of horseradish peroxidase with chlorite and chlorine dioxide. J. Inorg. Biochem. 102, 293–302. [DOI] [PubMed] [Google Scholar]
- Zamocky M.; Jakopitsch C.; Furtmüller P. G.; Dunand C.; Obinger C. (2008) The peroxidase-cyclooxygenase superfamily. Reconstructed evolution of critical enzymes of the innate immune system. Proteins 71, 589–605. [DOI] [PubMed] [Google Scholar]
- van Ginkel C. G.; Rikken G. B.; Kroon A. G. M.; Kengen S. W. M. (1996) Purification and characterization of chlorite dismutase: A novel oxygen-generating enzyme. Arch. Microbiol. 166, 321–326. [DOI] [PubMed] [Google Scholar]
- Mayfield J. A.; Blanc B.; Rodgers K. R.; Lukat-Rodgers G. S.; DuBois J. L. (2013) Peroxidase-type reactions suggest a heterolytic/nucleophilic O-O joining mechanism in the heme-dependent chlorite dismutase. Biochemistry 52, 6982–6994. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.