

Abstract
Fabry disease is a rare, X-linked, lysosomal storage disease caused by mutations in the gene encoding the enzyme alpha-galactosidase A. Complete or partial deficiency in this enzyme leads to intracellular accumulation of globotriaosylceramide (Gb3) and related glycosphingolipids in many cell types throughout the body, including the kidney. Progressive accumulation of Gb3 in podocytes, epithelial cells and the tubular cells of the distal tubule and loop of Henle contribute to the renal symptoms of Fabry disease, which manifest as proteinuria and reduced glomerular filtration rate leading to chronic kidney disease and progression to end-stage renal disease. Early diagnosis and timely initiation of treatment of Fabry renal disease is an important facet of disease management. Initiating treatment with enzyme replacement therapy (ERT; agalsidase alfa, Replagal®, Shire; agalsidase beta, Fabrazyme®, Genzyme) as part of a comprehensive strategy to prevent complications of the disease, may be beneficial in stabilizing renal function or slowing its decline. Early initiation of ERT may also be more effective than initiating therapy in patients with more advanced disease. Several strategies are required to complement the use of ERT and treat the myriad of associated symptoms and organ involvements. In particular, patients with renal Fabry disease are at risk of cardiovascular events, such as high blood pressure, cardiac arrhythmias and stroke. This review discusses the management of renal involvement in Fabry disease, including diagnosis, treatments, and follow-up, and explores recent advances in the use of biomarkers to assist with diagnosis, monitoring disease progression and response to treatment.
Keywords: Fabry disease, Diagnosis, Management, Nephropathy, Enzyme replacement therapy
Review
Fabry disease is a rare, X-linked, lysosomal storage disease (OMIM #301500) caused by mutations in the gene encoding the acid hydrolase enzyme alpha-galactosidase A (EC 3.2.1.22), which catalyses removal of a galactose moiety from neutral sphyngolipids, predominantly globotriaosylceramide (Gb3). Deficiency in this enzyme causes intracellular accumulation of Gb3 and related glycosphingolipids in a wide range of cell types throughout the body [1]. The major clinical manifestations include pain in the hands and feet (acroparaesthesia), angiokeratoma, as well as renal, cardiac, and cerebrovascular disease [2].
The Fabry Outcome Survey (FOS; sponsored by Shire) [2], and Fabry Registry (sponsored by Genzyme) [3-5] are databases established to collect longitudinal data on Fabry disease. These databases have shown a long delay between the onset of initial symptoms and a diagnosis. For example, within the Fabry Registry, median ages for initial onset of symptoms and a diagnosis were 9 and 23 years in males, and 13 and 32 years in females, respectively [3]. Many paediatric patients (aged <19 years) with Fabry disease report early symptoms. At enrolment in the Fabry Registry (median age, 12 years; n = 352), 77% of males and 51% of females had symptoms or signs of Fabry disease, including some patients who had already developed stage-2 or -3 chronic kidney disease (CKD; n = 3) [4]. Assessment of the renal morphological changes in Fabry disease using light microscopy and electron microscopy in 9 symptomatic paediatric patients (7 males, 2 females; 7–18 years) revealed glomerular and vascular changes despite normal renal function [6]. The combination of acroparasthesia, mild albuminuria, glomerular endothelial cell deposits, and arteriopathy may indicate a more severe phenotype [6].
Classically affected males with Fabry disease develop overt proteinuria and progressive renal impairment by the second-to-fifth decades of life [5]. Females with Fabry disease also have a significant risk of major organ involvement. Among adult females with estimated glomerular filtration rate (eGFR) data (n = 638), 62.5% had an eGFR <90 ml/min/1.73 m2 and 19.0% had an eGFR <60 ml/min/1.73 m2[5]. Approximately 30–35% of females with Fabry disease have proteinuria [7,8], 13% have stage 3 CKD [9] and 1–4% have end-stage renal disease (ESRD) [5,10].
A wide range of renal histopathology is found in Fabry disease [11]. All cell types within the kidney are affected, even in patients with normal GFR and minimal proteinuria [12]. Vacuolization of podocytes and epithelial cells is characteristic of Fabry disease, with mesangial expansion and progressive segmental and global glomerulosclerosis also observed [12-15]. It is becoming increasingly clear that podocytes have an important role in proteinuria. In a study of renal histology conducted prior to enzyme replacement therapy (ERT) in 14 young patients (age range: 4–19 years), podocyte Gb3 inclusion volume and density was shown to increase with age [16]. Podocyte foot process width also increased with age and was directly correlated with proteinuria. In the same study, endothelial fenestrations were reduced, indicating a central role for podocytes in the development and progression of Fabry nephropathy [16]. Additionally, accumulation of Gb3 in tubular cells in the distal tubule and loop of Henle may also contribute to some of the early renal symptoms of Fabry renal disease [17,18].
The pathogenesis of Fabry renal disease is not fully understood and is likely to be multifactorial (Figure 1). Progressive intracellular accumulation of Gb3 leads to cellular changes and histological damage. Deposition of Gb3 can promote vascular smooth muscle cell proliferation and the release of mediators involved in the pathogenesis of other nephropathies [19]. Other factors considered to be involved in the pathophysiology are the rupture of the lysosome [20] and damage due to local inflammation [21]. Furthermore, addition of Gb3 to cultured endothelial cells has been found to increase oxidative stress as demonstrated by an increase in reactive oxygen species and up-regulation of the expression of cell adhesion molecules [22]. Vasculopathy has also been observed in patients with Fabry disease that may be due to the local up-regulation of the renin–angiotensin system (RAS) [23].
Figure 1.

Pathogenesis of Fabry nephropathy. Gb3, globotriaosylceramide; lyso-Gb3, globotriaosylsphingosine.
Glomerular sclerosis and tubulo-interstitial fibrosis are the histological features that best correlate with the progression of renal disease in humans with Fabry disease [24]. As already mentioned, accumulation of Gb3 in podocytes plays an important role in the pathogenesis of glomerular damage. A human podocyte model of Fabry disease was developed using RNA interference to reduce alpha-galactosidase A expression. Accumulation of Gb3 was accompanied by an increase in autophagosomes, suggesting that deregulated autophagy pathways have some involvement in the pathogenesis of glomerular damage in Fabry disease [25].
Progression to ESRD results in the need for renal replacement therapy (RRT) [26]. The extra-renal manifestations of Fabry disease persist in patients receiving RRT [27]. Renal failure increases the risk of cardiovascular events and, consequently, those who develop ESRD are at a greater risk of experiencing cardiovascular events and strokes than patients with Fabry disease overall [8]. Indeed, cardiovascular disease is the most common cause of death in patients with Fabry disease [28].
Diagnosis and assessment of Fabry nephropathy
The American College of Medical Genetics and the National Society of Genetic Counselors have issued guidelines on the diagnostic confirmation of lysosomal storage disorders, including Fabry disease [29]. Enzymatic analyses of dried blood spots allow population screening and an initial diagnosis in males [30], whilst in heterozygous females, in whom alpha-galactosidase A activity is highly variable, genotyping is essential for a diagnosis. Genotyping can also be useful in male patients to assist with tracing family history of the disease. Urine microscopy may be clinically useful in diagnosing Fabry disease: vacuolated epithelial cells, filled with glycosphingolipids, giving the appearance of a ‘Maltese cross’, can be seen using polarized light microscopy [31]. Assessment of renal Fabry disease involves several assessments and tools (Table 1), with renal biopsy being particularly important to assess and monitor disease progression long term [32]. It has been reported that globotriaosylsphingosine (lyso-Gb3), a product of Gb3 metabolism, is elevated in male patients with Fabry disease and that it can be a useful marker in secondary screening to differentiate mutations of the alpha-galactosidase A gene causing Fabry disease from those not causing disease [33].
Table 1.
Recommendations for the management of Fabry renal disease in adults
| Recommendations | |
|---|---|
|
Diagnosis and assessments |
|
| Confirm diagnosis |
• Confirm presence of Fabry disease (by enzyme analysis in males and by gene mutation studies in females) |
| |
• GFR <90 ml/min/1.73 m2 (CKD stage 1–5) |
| |
• Proteinuria: >30 mg/day or >30 mg/g creatinine (albuminuria); >300 mg/day or >300 mg/g creatinine (proteinuria) |
| |
• Other renal conditions excluded rigorously (even if a renal biopsy is needed to make that exclusion) |
| Kidney biopsy |
• Histological injury can precede clinical signs, and provides a compelling indication for institution of ERT, especially in children and young adults |
| |
• Excludes other conditions (especially in patients with atypical presentations) |
| |
• Confirms the diagnosis and stage and can be used to assess response to therapy |
| Initial assessment and follow-up |
• Measure serum creatinine and use CKD-EPI equation to estimate the GFR |
| |
• Use iohexol plasma clearance or isotopic methods (depending on local availability) for precise measurement of the GFR if the eGFR >60 ml/min/1.73 m2 |
| |
• Standard CKD assessment schedule |
| |
• Quantify urinary albumin and protein levels |
| |
• Calculate eGFR slope |
|
Treatment |
|
| ERT |
• Agalsidase alfa or beta at approved dose |
| |
• Start ERT as soon as the definitive diagnosis has been made in patients with little or no residual enzyme activity |
| |
• Start ERT as soon as the definitive diagnosis has been made in patients with residual enzyme activity if there is evidence of kidney involvement |
| |
• ERT will not reduce proteinuria (in adults) |
| Control of proteinuria |
• Use ACE inhibitors and/or ARBs in addition to ERT |
| |
• Titrate doses to achieve urine protein <500 mg/day, even if blood pressure <130/180 mmHg |
| |
• Effects on progression are likely to occur only in the setting of optimal ERT dosing |
| Other therapy | • All other aspects of standard CKD care apply to the management of Fabry renal disease |
ACE: angiotensin-converting enzyme; ARB: angiotensin-receptor blocker; CKD: chronic kidney disease; CKD-EPI: Chronic Kidney Disease Epidemiology Collaboration; eGFR: estimated glomerular filtration rate; ERT: enzyme replacement therapy; GFR: glomerular filtration rate.
Adapted and republished with permission of American Society of Nephrology, from [Enzyme replacement therapy and Fabry renal disease. Warnock DG et al. Clin J Am Soc Nephrol 5: 2010]; permission conveyed through Copyright Clearance Centre, Inc. [34].
A new classification for Fabry disease has recently been proposed based on the functional characterization of GLA mutations. Lucas et al. correlated enzyme activity levels with specific mutational effects on the enzyme’s three-dimensional structure and the resulting alteration in function. It has been proposed that this classification will facilitate the diagnosis of Fabry disease, especially when combined with biomarker data such as lyso-Gb3 levels [35]. The proposed system of classification may help with the so-called late onset single organ phenotypes, some of which may not be clinically insignificant.
Progression of renal disease
In Fabry disease, the decline in renal function over time is related to the degree of proteinuria and, in untreated patients, is more rapid when the eGFR is below 60 ml/min/1.73 m2. Male sex and hypertension are also significant risk factors for development of renal failure [36]. As with all nephropathies, protein overload may cause an increase in the levels of inflammatory mediators, and interstitial accumulation of these mediators may lead to renal scarring. In patients with undiagnosed Fabry renal disease, a significant number of glomeruli may already be sclerotic [37]. Reduced nephron mass thus increases the risk of further renal damage from hyperfiltration, proteinuria, and activation of angiotensin II [37].
Consequently, regular, reproducible estimates of renal function are essential in the management of Fabry disease. Most centres now use the eGFR calculated using the MDRD (modified diet in renal disease) equation to assess renal function during follow-up [38]. There are limitations to current GFR estimates [39] and the CKD-EPI (Chronic Kidney Disease Epidemiology Collaboration) equation is recommended in adults. In children, a CKD-EPI equation developed by Schwartz et al. has been used successfully [40]. GFR can be measured more accurately using the iohexol plasma disappearance curve [40]. Rombach and colleagues suggest that the Stevens’ equation (a creatinine- and cystatin C-based formula) is also helpful, although it is recognized that cystatin C is not uniformly available [39].
Proteinuria
Proteinuria is an early sign of Fabry renal disease in both sexes and is often the most frequent clinical manifestation [5,6]. Importantly, proteinuria is an independent risk factor affecting the extent of renal decline in treated and untreated patients, as well as determining the success of ERT [41-44].
Among patients in the FOS, proteinuria was present in 44–54% of males and 33–41% of females for whom renal data were available [45]. Similarly, data from 1,262 adult patients (585 males, 677 females) in the Fabry Registry demonstrated overt proteinuria (>300 mg/day) in 43% and 26% of males and females with CKD stage 1, respectively, with higher proportions in patients with more advanced kidney involvement [8]. Consequently, proteinuria should be monitored regularly and treated appropriately.
Renal biopsy
Renal biopsy is useful in all patients with any degree of proteinuria/albuminuria and/or renal dysfunction to assess the degree of glomerulosclerosis and interstitial damage because of their prognostic significance [2]. In patients with minimal proteinuria and normal renal function, biopsy can also determine if there is significant Gb3 deposition (especially in podocytes and endothelial cells), or early damage to indicate ERT use. In the classical male patient with signs of renal involvement a renal biopsy can be helpful in providing evidence of prognosis depending on the degree of glomerular sclerosis [41]. In females with even the slightest evidence of Fabry nephropathy, renal biopsy is considered much more important (and by some mandatory) as the presence of significant renal Gb3 deposits is an indication for initiating ERT. Renal biopsy should also be undertaken if there is the possibility of double pathology (e.g. diabetes or other glomerular diseases such as immunoglobulin A [IgA] nephropathy or thin membrane disease if haematuria is present) and if there is a sudden, unexplained decline in renal function [46,47].
Fabry disease can be difficult to diagnose solely from standard light microscopy, so semi-thin toluene blue sections or electron microscopy should be performed particularly if there is doubt about the histological diagnosis [13]. The role of re-biopsy is still undetermined, especially for evaluating the results of ERT. However, a study by Tondel et al. found comparing deposition of Gb3 in glomerular cells from renal biopsies taken at baseline and after 5 years of ERT to be useful in assessing the success of ERT [32]. It is better to compare Gb3 deposition using semi-thin toluene-blue stained sections and/or electron microscopy, rather than with haematoxylin and eosin or periodic acid Schiff stained slides, where the Gb3 deposition is dissolved by solvents during processing of the samples. Figure 2A illustrates how foam cells observed with haematoxylin and eosin staining could be confused with other causes of glomerular nephritis. Typical Fabry lesions observed with toluene-blue staining or electron microscopy are shown in Figures 2B–E.
Figure 2.

Histological diagnosis of Fabry disease. (A) Haematoxylin and eosin stain of tissue from a patient with Fabry disease showing lesions that could be confused with other conditions in which foam cells are present; (B) Semi-thin section stained with toluene blue (the preferred method for diagnosing Fabry disease on renal histology); (C) deposits in the tubules that can give rise to mild renal tubular disorders; (D) a small artery with large degrees of narrowing due to Gb3 deposits; (E) ultrastructural image (electron microscopy) of the glomerulus in Fabry disease shows lamellated lipid inclusions (zebra bodies) within the podocyte cytoplasm (×6,000; stain: UrPb). Reproduced with the permission of Dr. Kostas Giannakakis (Laboratory of Ultrastructural Pathology, Rome La Sapienza University, Rome, Italy). E, eosin; H, haematoxylin; UrPb, uranyl lead.
Renal ultrasound and haematuria
Multiple renal cysts, mostly parapelvic, can be detected using ultrasonography in up to 50% of cases of patients [48]. Although uncommon, haematuria may also occur, but other causes must be excluded [46,47]. The aetiology of cysts is unknown, but the cause of haematuria is similar to that found in other glomerular diseases.
Hypertension
In the FOS Registry, a high prevalence (57% of male and 47% of female patients) of uncontrolled hypertension was reported in patients with Fabry disease which generally increased with worsening CKD stage [49]. Similarly, in the Fabry Registry, hypertension (systemic blood pressure [BP] ≥130/80 mmHg) was observed more often in those with poor renal function (67% vs 48% of patients with eGFR ≥60 ml/min/1.73 m2) [8].
In general, the BP of patients with Fabry disease is lower than that of the general population, predominantly due to the effects of autonomic dysfunction on cardiac and vascular function [50], although the aetiology of this effect is complex and other factors may be involved. Consequently, the current target for controlling hypertension in the general population (systolic BP <130 mmHg or diastolic BP <80 mmHg) may be too high. Maintenance of a BP <125/75 mmHg in patients with proteinuria levels >1 g/day, and <130/80 mmHg in patients with proteinuria of 0.25–1 g/day has been advocated in CKD [37]. Caution is recommended in reducing BP <100/70 mmHg because this has been shown to increase the risk of cardiovascular and/or renal events [31,51,52].
Management of Fabry renal disease
ERT in Fabry disease
Two ERT products are available for treating Fabry disease: agalsidase alfa (Replagal®, Shire), produced in a human cell line by gene activation technology [53], and agalsidase beta (Fabrazyme®, Genzyme), produced in Chinese hamster ovary (CHO) cells [54]. The recommended licensed dose for agalsidase alfa is 0.2 mg/kg/2 weeks and is 1 mg/kg/2 weeks for agalsidase beta.
Current UK guidelines for the management of Fabry disease state that, “No trial has yet addressed the appropriate starting time of treatment or the group of patients most likely to benefit from therapy. However this is a chronic, progressive disorder. The aim of treatment is to prevent progression and where disease is already manifest to try and reverse or stabilise the disease. It is anticipated that treatment will be most successful when started early in the course of the disease. Conversely treatment late in the course of the disease may have limited efficacy.”[38]. Therefore ERT should be started as soon as symptoms/signs occur in an effort to stabilize (or even reverse) pathology. In the UK, ERT treatment is indicated in patients with Fabry disease who have evidence of renal disease (defined as an eGFR <80 ml/min, proteinuria >300 mg/day) and/or microalbuminuria if a renal biopsy shows endothelial deposits [38,51].
ERT has been shown to improve the clinical outcome of patients with Fabry disease, including stabilization of kidney function and reduction in neuropathic pain [41,55-57]. However, because of the heterogeneous disease presentation, no evidence is available to support the optimal timing of ERT or to identify the patient groups in whom disease is likely to be most rapidly progressive or which patients are most likely to gain significant benefit from therapy [58]. Data from the FOS have been used to develop an age- and sex-adjustment of the global Mainz Severity Score Index for Fabry disease. This scoring system corrects for the impact of age and sex on severity. By allowing meaningful comparisons across age and sex [59] it may be possible to determine clinical features that predict future disease severity.
Data from the FOS have also been used to develop organ-specific (cardiac, renal, and neurological) and composite prognostic severity scores [58]. Using the analysis it was possible to differentiate groups of patients with different outcome probabilities. The overall composite score, the Fabry International Prognostic Index (FIPI), distinguished two distinct groups in whom the 50% event-free survival differed by 10 years. The FIPI should prove to be a valuable tool in comparative analyses of outcomes using different therapies [58]. Additionally, a validated disease severity scoring system for Fabry disease has been developed (DS3) to quantify disease burden and monitor disease progression and response to treatment [60].
Impact of ERT on renal function
Data on the renal effects of agalsidase alfa and beta mostly originate from open-label and observational studies, including the FOS and the Fabry Registry [41,45,51,55,61-64], with randomized, long-term, controlled data being limited to one placebo-controlled trial [44] and two studies that utilized historical controls [65,66]. Although registries can provide long-term data on large patient populations, they are limited by their voluntary nature, which can result in incomplete follow-up data [67].
Short-term studies
The renal benefits of ERT at tissue and clinical levels were demonstrated in a double-blind, randomized, placebo-controlled study of 26 patients given agalsidase alfa at 0.2 mg/kg/2 weeks for 6 months [55]. Significant improvements were seen in renal structure (increase in the percentage of mesangium) in patients receiving agalsidase alfa compared with placebo [55].
Similarly, agalsidase beta 1 mg/kg/2 weeks resulted in stabilization of the GFR with rapid, marked, reductions in plasma and tissue Gb3 observed by biochemical and histological means [61]. A randomized, double-blind, placebo controlled Phase IV trial investigating the effects of agalsidase beta (1 mg/kg/2 weeks for up to 35 months [mean, 18.4 months]) on clinical outcomes in 82 adult patients with advanced Fabry disease and mild-to-moderate renal dysfunction found an increased time to first clinical event and stabilized renal function compared with placebo when adjusted for the degree of proteinuria [44].
Analyses of data from the FOS showed improvements in renal function based on 1,040 serum creatinine measurements in 201 patients [63]. Time on agalsidase alfa therapy was an independent predictor of serum creatinine levels (inverse association, p = 0.04; multivariate analyses). In patients with CKD stage 2 and 3, ERT halted the progressive decline in renal function within the first year of treatment (longitudinal analyses) [63].
Long-term studies
The benefits of ERT with agalsidase beta over 54 months have also been reported: 41 patients with <50% glomerulosclerosis and ≤1 g proteinuria per 24 hours at baseline showed stable renal function [41].
Long-term positive results with ERT were confirmed in an analysis of renal function in a cohort of males and females (n = 150) in FOS receiving agalsidase alfa and adjunctive therapies for up to 5 years [68]. Annualized rates of decline by CKD stages are shown in Figure 3. Responder analysis showed normalization of the eGFR in 68.8–89.1% of those in CKD stages 1–3. The role of agalsidase alfa in preserving renal function is also supported by pooled data from three prospective, randomized, placebo-controlled trials and their open-label extension studies involving 108 adult male patients treated for 1–4 years [56]. The rate of eGFR loss in patients with a baseline eGFR of 30–135 ml/min per 1.73 m2 was less than that seen during the placebo period. Multivariate analyses revealed that the eGFR and proteinuria (<1 or ≥1 g/day) at baseline significantly predicted the rate of decline of the eGFR during treatment (Figure 4).
Figure 3.
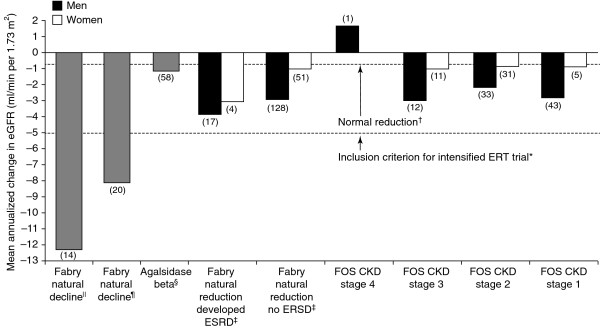
Change in eGFR in patients with Fabry disease and CKD during treatment with agalsidase alfa. Republished with permission of Elsevier, from [Enzyme replacement therapy with agalsidase alfa in patients with Fabry’s disease: an analysis of registry data. Mehta A et al. Lancet 374: 2009]; permission conveyed through Copyright Clearance Centre, Inc. [68]. Data are plotted according to baseline stage of CKD. Patient numbers are shown in parentheses. Data from previous studies for the expected natural fall in renal function in patients with Fabry disease and the effects of agalsidase beta are plotted for reference and comparison. *[69]; †[70]; ‡[18]; §[41]; ¶[63]; ||[26]. CKD, chronic kidney disease; GFR, glomerular filtration rate.
Figure 4.
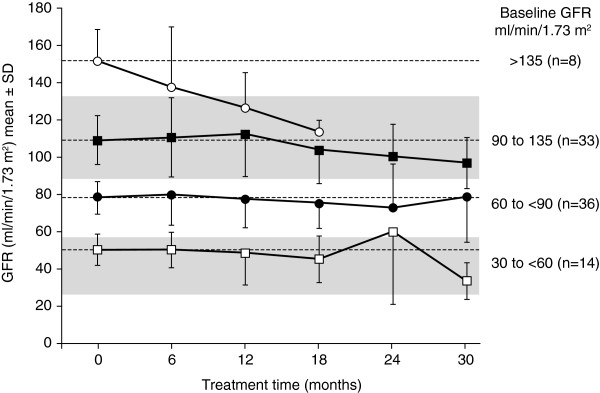
GFR during agalsidase alfa treatment in male patients with Fabry disease (stratified by baseline GFR). Republished with permission of American Society of Nephrology, from [Agalsidase alfa and kidney dysfunction in Fabry disease. West M et al. J Am Soc Nephrol 2009; 20: 2009]; permission conveyed through Copyright Clearance Centre, Inc. [56]. GFR, glomerular filtration rate.
Renal function was evaluated in 151 men and 62 women from the Fabry Registry receiving agalsidase beta at 1 mg/kg/2 weeks for at least 2 years [51]. Multivariate logistic regression analyses were used to identify factors associated with the progression of renal disease. The risk factor most strongly associated with such progression was an averaged urinary protein:creatinine ratio ≥1 g/g and delays in treatment initiation compared with symptom onset.
In the latest long-term study on ERT effectiveness, renal function was assessed in 208 patients (mean time on ERT, 7.4 years). The mean yearly change in eGFR was -2.2 ml/min/1.73 m2 in men and -0.7 ml/min/1.73 m2 in women [62]. In another study by Tondel et al., which monitored the effects of ERT in 12 patients over 5 years using serial renal biopsies, ERT was associated with the clearance of Gb3 deposition from glomerular cells, especially in those receiving high doses of ERT. However, this study was carried out in a small number of patients and these results need to be confirmed in a larger study [32]. It also has been suggested that clearance of Gb3 from podocytes may be affected by heterogeneity between patients in the expression levels of receptors (mannose-6-phosphate receptor, sortilin and megalin) that are responsible for the uptake of alpha-galactosidase A into the lysosomes of podocytes [71].
Early intervention with ERT may preserve renal function
Patients with impaired renal function have a less favourable outcome than those without renal impairment. A 2-year, open-label study of 26 patients with Fabry disease treated with agalsidase beta (1 mg/kg/2 weeks) reported that 9 patients experienced 12 endpoints, including 2 deaths [72]. All clinical endpoints (cerebrovascular and cardiac events, renal failure, and death) occurred in patients with impaired renal function. Despite ERT, renal function deteriorated and left ventricular posterior wall thickness (PWT) was unaltered. In contrast, patients without renal impairment at baseline had a more favourable outcome post-treatment (no clinical events, renal function remained unchanged, and PWT decreased significantly).
Similarly, in a multivariate logistic regression analysis of renal function from 151 men and 62 women from the Fabry Registry who received agalsidase beta (1 mg/kg/2 weeks) for at least 2 years, adults with Fabry disease were at risk of progressive reduction in the eGFR despite ERT if the urinary protein:creatinine ratio was ≥1 g/g [51]. Men with little urinary protein excretion and those who received ERT sooner after symptom onset had stable renal function.
The findings from these studies suggest that there is a strong case for early intervention because ERT seems less effective in more advanced disease [51,72]. Two studies evaluating the occurrence of significant negative clinical events (renal, cardiac or neural) provide further support for starting ERT early. In a prospective study of 75 patients undergoing ERT, prolonged ERT delayed the occurrence of complications [65]. The difference between treated and non-treated patients only became obvious after at least 4 years of ERT, which suggests that the risk of complications decreases with increasing treatment duration. These data also indicated that in patients affected by Fabry disease-related complications, ERT is unlikely to modify outcome. In another prospective study of 40 patients undergoing ERT and 40 matched untreated patients, no differences in the occurrence of renal, cardiac, or neural complications were observed between the cohorts. The authors suggested that a long period of ERT is required to evaluate the results but that in advanced stages of the disease ERT can improve symptoms but not slow down the progression. However, from a nephrological point of view, it is worth noting that the incidence of renal failure was lower in the ERT cohort than in the comparator cohort (10% versus 28%, respectively) [66].
The renal effects of ERT in different disease stages have also been analysed in a meta-analysis of six studies with a mean follow-up of 5 years [67]. This analysis demonstrated a smaller mean decline in renal function in males treated with ERT than in untreated males when the GFR was <60 ml/min/1.73 m2. However, in males with a baseline GFR of >60 ml/min/1.73 m2 and in females the decline in renal function was comparable between the ERT and untreated groups.
Although there is limited long-term, controlled evidence of a clinical benefit of ERT on renal function, we feel that there is adequate, less rigorous evidence to indicate that ERT is of benefit, especially when started early in the course of the disease. However, to optimize the results from ERT, it should be used, when appropriate, with other adjuvant therapies.
Long-term safety of ERT and antibody formation
Long-term ERT can be associated, mostly in male patients, with the occurrence of antibodies against the infused enzyme [73]. While there are problems in interpreting antibody levels and comparing them between the two products due to differences in measurement methodologies and “cut off” points [73], some potentially useful data are available. IgG antibodies occur with both products, but seem to be generally higher with agalsidase beta [73]. In an open-label extension study of agalsidase beta, infusion-related reactions (rigors, temperature-change sensation, fever, nausea, headache and nasal congestion) were the most common enzyme-related adverse events [74]. With the exception of one patient, all the reactions were mild and decreased over time, similar to observations reported from the FOS registry 5-year follow up [68]. The initial increase in frequency of infusion-related reactions seems to parallel the seroconvertion rate [41]. Although high antibody titres to either product could have an inhibitory effect on agalsidase A activity in vitro, the relationship to clinical response is much more difficult to assess [73]. Most studies reported so far have looked at the relationship between antibody titres and surrogate markers such as Gb3 or lyso-Gb3 in urine or plasma [73,75]. Therefore, although the long-term effects of antibodies on ERT have yet to be determined, the prescribing nephrologist should be aware of the issues and potential problems.
Dosage of ERT
The optimal dosage and regimen for ERT in Fabry disease are unclear. The licensed doses for agalsidase alfa and agalsidase beta are 0.2 mg/kg/2 weeks and 1 mg/kg/2 weeks, respectively. Clearance of Gb3 from podocytes has been shown to be dose-dependent, with higher doses associated with greater clearance [32]. Regarding frequency of dosing, in patients demonstrating a continuing decline in renal function, switching from bi-weekly dosing to weekly dosing of agalsidase alfa (0.2 mg/kg) has been found to improve eGFR, with the mean rate of change in eGFR slowing from -8.0 ml/min per 1.73 m2/year to -3.3 ml/min per 1.73 m2/year [69]. A recent short-term study assessing three doses of agalsidase alfa (0.2 mg every 2 weeks; 0.1 mg and 0.2 mg every week) showed a trend for the higher dose to be most effective suggesting a longer-term study was needed [76]. A study of agalsidase beta involving 21 male patients receiving the standard dose of 1 mg/kg body weight for 6 months followed by a reduced dose of 0.3 mg/kg body weight for a further 18 months showed that 100% of patients cleared Gb3 by 6 months while only 70% were still clear at 18 months [77].
From June 2009 to 2012, production and quality issues led to a worldwide shortage of agalsidase beta resulting in adult patients receiving reduced doses of agalsidase beta (approved dose 1 mg/kg/2 weeks, reduced dose 0.3-0.5 mg/kg/2 weeks), or switching to agalsidase alfa (administered dose 0.2 mg/kg/2 weeks) [78]. In 2010, the European Medicines Agency (EMA) focused their attention on patients treated with lower doses of agalsidase beta noting an increase in adverse events in these patients; specifically “a pattern of adverse events resembles the natural, but accelerated, course of Fabry disease”[78]. The effectiveness of specific ERT doses has also been evaluated in groups of patients switched from agalsidase beta to agalsidase alfa during this period. In 2011, Smid et al. reported that there were no relevant clinical differences in patients treated with variable doses of beta and/or alfa agalsidase, although both patients switching to agalsidase alfa (following a dose reduction of agalsidase beta) and those who continued on a reduced agalsidase beta dose had increased levels of lyso-Gb3 suggesting increased disease activity [79]. This study also showed a minimal but significant decrease in two of the quality of life subscales in the SF-36 questionnaire. End-organ damage and clinical symptoms during dose reduction of agalsidase beta or switch to agalsidase alfa has also been assessed in an observational study (N = 105) [80]. During a median follow-up of 12 months, predefined measures of organ function were stable in those continuing on agalsidase beta 1 mg/kg/2 weeks. In contrast, there was a significant decrease in renal function as assessed by eGFR in those receiving a reduced dose of agalsidase beta, as well as an increase in the number of patients experiencing pain attacks or pain crises. There was also an increase in microabuminuria and Fabry disease-related symptoms (pain aggravation and gastrointestinal symptoms) among patients switching to agalsidase alfa. Considering all data published on the clinical effects of agalsidase alfa, it is surprising how in this report the patients treated with agalsidase alfa had such a significant worsening of their symptoms in such a short time.
In other studies from Japan [81] and Italy [82] no clinical and/or laboratory differences (Gb3 levels) were observed after the switch from agalsidase beta to agalsidase alfa. Similar results were reported for a cohort of 40 patients in Australia although lower energy levels were observed in the male patients [83]. It is important to emphasize that all the studies on switching from agalsidase beta to agalsidase alfa were carried out in a small number of patients and for short periods of time (<2 years).
Adjunctive therapy for proteinuria and cardiovascular disease
Reduction of proteinuria using drugs inhibiting the RAS is an essential and potent tool for improving renal outcome in patients with Fabry disease with CKD. Angiotensin-converting enzyme (ACE) inhibitors and angiotensin-II receptor antagonists (ARBs), have been demonstrated to be renoprotective in chronic nephropathy [37]. Protein levels can be reduced with ACE inhibitors/ARBs to <0.5 g/day, and are associated with significant stabilization of renal glomerular function [36,84]. Care should be taken to titrate doses to avoid hypotension [34,51,52]. The additional cardioprotective action of RAS inhibitors provides a further rationale for their use in patients with CKD [37]. These drugs will help control BP and are important for preserving renal function. Management should also include lipid-lowering and anti-platelet agents as indicated, together with appropriate prophylaxis against stroke [38]. Additionally, since cardiac arrhythmias do not respond to ERT, care should be taken to detect arrhythmias, as such events can lead to sudden cardiac death, particularly in women [9,85].
Dialysis and transplantation
Survival of patients on dialysis with Fabry disease and ESRD is poor. Three-year survival has been shown to be 60% in Europe [86] compared with 63% in the USA [87]. Although this was better than 3-year survival in a diabetic control population (53%), it was significantly lower than in the non-diabetic control group (74%) [87]. This is almost certainly due to the added disease burden from the cardiovascular complications of Fabry disease [88]. Thus, ERT should be considered on an individual patient basis in an attempt to reduce the risk of cardiovascular events.
Renal transplantation was initially considered as a potential treatment for alleviating some of the symptoms of Fabry disease by providing a source of alpha-galactosidase A enzyme. However, this was found not to be the case, with circulating alpha-galactosidase A levels remaining low after transplantation [89]. Nevertheless, the results of renal transplantation in patients with Fabry disease are excellent: 5-year graft survival has been reported as 74%, which was superior to that of patients with ESRD due to other causes (69%), but comparable with a matched cohort [90]. Patient survival at 5 years was 81%, which was slightly lower than that of the matched cohort (90%) [90]. The results of this study were comparable with those of an earlier study, in which 5-year graft survival and 5-year patient survival were 75% and 83%, respectively [27]. In fact, the results of renal transplantation are so encouraging that it should be considered as the first-line option to correct renal dysfunction in patients with Fabry disease and ESRD [91]. However, despite the positive data supporting renal transplantation, Fabry disease has been shown to confer a higher risk of death versus diabetic control patients (odds ratio 2.15) [88].
There are few data on ERT in patients with ESRD, either on dialysis or with a functioning renal transplant. However, ERT administered to patients with ESRD on dialysis or with a history of renal transplantation has demonstrated a favourable safety profile and the same dosing regimen can be used as for patients with Fabry disease without ESRD [92]. Furthermore, there is evidence that ERT can be administered during haemodialysis, without loss of the enzyme in the dialysate [93].
Management of Fabry disease in women
Organ involvement occurs later in life and with a lower prevalence in women than in men, but the signs and symptoms of Fabry disease should be treated in women because organ involvement is progressive and causes significant disease burden [5]. Few published studies have been dedicated specifically to women with Fabry disease [94]. However, a 4-year study of agalsidase alfa 0.2 mg/kg/2 weeks in 36 symptomatic women (mean age, 47 years) highlighted its long-term effectiveness [95].
Areas for further research
Chaperon molecules
An investigational therapeutic strategy, which is currently being explored, is the use of chaperon molecules to stabilize the chemical structure of the residual agalsidase alfa or to prolong the half-life of the infused enzyme [96,97]. At present, clinical studies in Fabry patients are very limited and as yet no licensed chaperone drugs are available [98].
Biomarkers
Biomarkers have an important role in the assessment of disease activity and response to treatment in other lysosomal storage diseases. However, despite encouraging research, a biomarker has yet to be identified that is relevant to clinical outcomes in Fabry disease [67,75,99]. Nevertheless, biomarkers may play an important part as indicators of diagnosis and when to commence therapy in Fabry disease patients. They may also be useful in determining the progression of Fabry disease. Urinary protein and albumin excretion are the most important biomarkers of Fabry disease nephropathy [36,44]. Urinary Gb3 levels are consistently elevated in most patients with Fabry disease (although marked inter-patient variability has been observed). Urinary Gb3 levels decrease during ERT in men, women, and children with Fabry disease. However, despite this response, urinary Gb3 cannot be considered to be a suitable biomarker of clinical efficacy because baseline levels do not correlate with disease severity, and do not predict clinical efficacy [100].
Recently, plasma [101] and urinary [102] lyso-Gb3 have been identified as new potential biomarkers. In a study of 10 male and 8 female patients with Fabry disease, plasma lyso-Gb3 was increased in males, and was higher in cases of the classic form of Fabry disease than in variant Fabry hemizygotes [101]. In females, plasma lyso-Gb3 was moderately increased in symptomatic and asymptomatic cases, with a correlation between the increase in plasma lyso-Gb3 and decrease in alpha-galactosidase A activity. Lyso-Gb3 analogues were also isolated in the urine of 63 patients with Fabry disease [102]. Increased urinary excretion of lyso-Gb3 in patients with Fabry disease correlated well with several indicators of disease severity, and may be a reliable independent biomarker for the clinically important characteristics of Fabry disease. However, there was no correlation between urinary lyso-Gb3 and eGFR. Therefore, one cannot assume that lyso-Gb3 is a reliable biomarker of renal involvement [101].
In 2012, Rombach et al. confirmed elevated levels of lyso-Gb3 in the plasma and/or urine of patients with Fabry disease and described a reduction of plasma levels of lyso-Gb3 after 1 year of ERT [75]. This decline in lyso-Gb3 was associated with a reduction in left ventricular mass in females and a lower risk of developing white matter lesions in the nervous system for both sexes [75]. Recently, new potential biomarkers, Gb3 analogues, have been identified in the plasma of untreated male Fabry disease patients, but their clinical meaning still has to be validated [103].
The utility of Cystatin C as a biomarker in Fabry disease has also been evaluated [104]. In an observational, multicentre study of 89 patients (n = 42 females; n = 47 males) with Fabry disease and varying degrees of disease severity, serum Cystatin C levels were measured. The authors concluded that Cystatin C is a good and cost-effective prognostic indicator of early renal dysfunction and/or heart failure in Fabry disease [104].
Urinary uromodulin excretion has been examined in 15 male patients with Fabry disease [105]. In untreated patients, changes ranged from a normal to marked decrease or absence of urinary uromodulin frequently accompanied by aberrantly processed uromodulin. These patterns normalized in all patients on ERT and some on substrate reduction therapy. This suggests that uromodulin may also be a potential biochemical marker of renal therapeutic response to ERT.
Urine proteomic analysis based on capillary electrophoresis coupled to mass spectrometry has recently been used to identify a biomarker profile in patients with Fabry disease [106]. In 35 treatment-naïve adult female patients with Fabry disease an abnormal urine profile was identified, which was almost completely corrected following ERT [106]. A further two protein biomarkers have been identified in the urine of children with Fabry disease and type-1 diabetes using label-free quantitative proteomics. Prosaposin and ganglioside GM2 activator protein were significantly elevated in patients with Fabry disease and ERT was associated with a significant reduction in urinary excretion of these proteins. Therefore, these urinary biomarker models may useful as diagnostic tools for patients with Fabry disease, as well as for monitoring response to ERT [107]. However, there is still work to be done to identify a biomarker that truly reflects disease activity and progression, as well as enabling responses to ERT to be measured.
Conclusions
Fabry disease should be considered in the differential diagnosis of proteinuria of uncertain origin to ensure an early diagnosis. Proteinuria is a risk factor for the progression of renal disease and should be managed appropriately. Early detection of renal involvement should be achieved by regular measurement of GFR and urine protein excretion in all patients, male and female, using renal biopsy where indicated. Biomarkers play an important part in the assessment of disease activity and response to treatment in other lysosomal storage diseases, and could potentially aid the diagnosis and management of Fabry renal disease.
Early intervention with ERT may help stabilize renal function or slow its decline, when used as part of a comprehensive management strategy to prevent complications of Fabry disease. Although there are currently limited long-term, controlled effectiveness data, ERT with agalsidase alfa or beta has been shown to reduce the decline in renal function in short- and long-term studies in male, female, and paediatric patients.
Abbreviations
ACE: Angiotensin-converting enzyme; ARBs: Angiotensin-II receptor antagonists; BP: Blood pressure; CHO: Chinese hamster ovary; CKD: Chronic kidney disease; CKD-EPI: Chronic Kidney Disease Epidemiology Collaboration; ESRD: End-stage renal disease; ERT: Enzyme replacement therapy; eGFR: Estimated glomerular filtration rate; FIPI: Fabry international prognostic index; FOS: Fabry outcome survey; Gb3: Globotriaosylceramide; lyso-Gb3: Globotriaosylsphingosine; PWT: Modified diet in posterior wall thickness; MDRD: Modification of Diet in Renal Disease; RAS: Renin–angiotensin system; RRT: Renal replacement therapy.
Competing interests
Stephen Waldek has received speaker fees, travel grants, and research support from Shire and Genzyme. Sandro Feriozzi has received speaker fees, travel grants, and research support from Shire and Genzyme.
Authors’ contributions
SF and SW contributed to the writing of the manuscript, ensuring the inclusion of up-to-date literature. Both authors commented on all drafts and approved the final version of the manuscript.
Authors’ information
Stephen Waldek retired in November 2011 and is working as an independent medical consultant.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Contributor Information
Stephen Waldek, Email: stephenwaldek@yahoo.co.uk.
Sandro Feriozzi, Email: sandro.feriozzi@asl.vt.it.
Acknowledgements
The authors take full responsibility for the content of this contribution and thank Nikki West of Communigen Ltd, Oxford, UK (funded by Shire) for preparing the manuscript drafts.
References
- Desnick RJ, Ioannou YA, Eng CM. In: The metabolic and molecular bases of inherited disease. 8. Scriver CR, Beaudet AL, Sly WS, Valle D, editor. New York, USA: McGraw Hill; 2001. α-Galactosidase A deficiency: Fabry disease; pp. 3733–3774. [Google Scholar]
- Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30. doi: 10.1186/1750-1172-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng CM, Fletcher J, Wilcox WR, Waldek S, Scott CR, Sillence DO, Breunig F, Charrow J, Germain DP, Nicholls K, Banikazemi M. Fabry disease: baseline medical characteristics of a cohort of 1765 males and females in the Fabry Registry. J Inherit Metab Dis. 2007;30:184–192. doi: 10.1007/s10545-007-0521-2. [DOI] [PubMed] [Google Scholar]
- Hopkin RJ, Bissler J, Banikazemi M, Clarke L, Eng CM, Germain DP, Lemay R, Tylki-Szymanska A, Wilcox WR. Characterization of Fabry disease in 352 pediatric patients in the Fabry Registry. Pediatr Res. 2008;64:550–555. doi: 10.1203/PDR.0b013e318183f132. [DOI] [PubMed] [Google Scholar]
- Wilcox WR, Oliveira JP, Hopkin RJ, Ortiz A, Banikazemi M, Feldt-Rasmussen U, Sims K, Waldek S, Pastores GM, Lee P, Eng CM, Marodi L, Stanford KE, Breunig F, Wanner C, Warnock DG, Lemay RM, Germain DP. Females with Fabry disease frequently have major organ involvement: lessons from the Fabry Registry. Mol Genet Metab. 2008;93:112–128. doi: 10.1016/j.ymgme.2007.09.013. [DOI] [PubMed] [Google Scholar]
- Tondel C, Bostad L, Hirth A, Svarstad E. Renal biopsy findings in children and adolescents with Fabry disease and minimal albuminuria. Am J Kidney Dis. 2008;51:767–776. doi: 10.1053/j.ajkd.2007.12.032. [DOI] [PubMed] [Google Scholar]
- Deegan PB, Baehner AF, Barba Romero MA, Hughes DA, Kampmann C, Beck M. Natural history of Fabry disease in females in the Fabry outcome survey. J Med Genet. 2006;43:347–352. doi: 10.1136/jmg.2005.036327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz A, Oliveira JP, Waldek S, Warnock DG, Cianciaruso B, Wanner C. Nephropathy in males and females with Fabry disease: cross-sectional description of patients before treatment with enzyme replacement therapy. Nephrol Dial Transplant. 2008;23:1600–1607. doi: 10.1093/ndt/gfm848. [DOI] [PubMed] [Google Scholar]
- Weidemann F, Niemann M, Beer M, Breunig F, Wanner C. Interdisciplinary approach towards female patients with Fabry disease. Eur J Clin Invest. 2012;42:455–462. doi: 10.1111/j.1365-2362.2011.02614.x. [DOI] [PubMed] [Google Scholar]
- Ortiz A, Cianciaruso B, Cizmarik M, Germain DP, Mignani R, Oliveira JP, Villalobos J, Vujkovac B, Waldek S, Wanner C, Warnock DG. End-stage renal disease in patients with Fabry disease: natural history data from the Fabry Registry. Nephrol Dial Transplant. 2010;25:769–775. doi: 10.1093/ndt/gfp554. [DOI] [PubMed] [Google Scholar]
- Faraggiana T, Churg J, Grishman E, Strauss L, Prado A, Bishop DF, Schuchman E, Desnick RJ. Light- and electron-microscopic histochemistry of Fabry’s disease. Am J Pathol. 1981;103:247–262. [PMC free article] [PubMed] [Google Scholar]
- Gubler MC, Lenoir G, Grunfeld JP, Ulmann A, Droz D, Habib R. Early renal changes in hemizygous and heterozygous patients with Fabry’s disease. Kidney Int. 1978;13:223–235. doi: 10.1038/ki.1978.32. [DOI] [PubMed] [Google Scholar]
- Alroy J, Sabnis S, Kopp JB. Renal pathology in Fabry disease. J Am Soc Nephrol. 2002;13(Suppl 2):134–138. [PubMed] [Google Scholar]
- Fischer EG, Moore MJ, Lager DJ. Fabry disease: a morphologic study of 11 cases. Mod Pathol. 2006;19:1295–1301. doi: 10.1038/modpathol.3800634. [DOI] [PubMed] [Google Scholar]
- Sessa A, Toson A, Nebuloni M, Pallotti F, Giordano F, Battini G, Maglio A, Meroni M, Calconi G, Bertolone G, Gatti P. Renal ultrastructural findings in Anderson-Fabry disease. J Nephrol. 2002;15:109–112. [PubMed] [Google Scholar]
- Najafian B, Svarstad E, Bostad L, Gubler MC, Tondel C, Whitley C, Mauer M. Progressive podocyte injury and globotriaosylceramide (GL-3) accumulation in young patients with Fabry disease. Kidney Int. 2011;79:663–670. doi: 10.1038/ki.2010.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sessa A, Meroni M, Battini G, Maglio A, Brambilla PL, Bertella M, Nebuloni M, Pallotti F, Giordano F, Bertagnolio B, Tosoni A. Renal pathological changes in Fabry disease. J Inherit Metab Dis. 2001;24(Suppl 2):66–70. doi: 10.1023/a:1012423924648. [DOI] [PubMed] [Google Scholar]
- Finn L. In: Heptinstall’s pathology of the kidney. 6. Jennette JC, Olsen JL, Schwartz MM, Silva FG, editor. Philadelphia, PA: Lippincott Williams & Wilkins; 2007. Renal disease caused by familial metabolic and hematologic diseases; pp. 1199–1256. [Google Scholar]
- Sanchez-Niño MD, Sanz AB, Carrasco S, Saleem MA, Mathieson PW, Valdivielso JM, Ruiz-Ortega M, Egido J, Ortiz A. Globotriaosylsphingosine actions on human glomerular podocytes: implications for Fabry nephropathy. Nephrol Dial Transplant. 2011;26:1797–1802. doi: 10.1093/ndt/gfq306. [DOI] [PubMed] [Google Scholar]
- Valbuena C, Carvalho E, Bustorff M, Ganhao M, Relvas S, Nogueira R, Carneiro F, Oliveira JP. Kidney biopsy findings in heterozygous Fabry disease females with early nephropathy. Virchows Arch. 2008;453:329–338. doi: 10.1007/s00428-008-0653-2. [DOI] [PubMed] [Google Scholar]
- Safyan R, Whybra C, Beck M, Elstein D, Altarescu G. An association study of inflammatory cytokine gene polymorphisms in Fabry disease. Eur Cytokine Netw. 2006;17:271–275. [PubMed] [Google Scholar]
- Shen JS, Meng XL, Moore DF, Quirk JM, Shayman JA, Schiffmann R, Kaneski CR. Globotriaosylceramide induces oxidative stress and up-regulates cell adhesion molecule expression in Fabry disease endothelial cells. Mol Genet Metab. 2008;95:163–168. doi: 10.1016/j.ymgme.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rombach SM, Twickler TB, Aerts JM, Linthorst GE, Wijburg FA, Hollak CE. Vasculopathy in patients with Fabry disease: current controversies and research directions. Mol Genet Metab. 2010;99:99–108. doi: 10.1016/j.ymgme.2009.10.004. [DOI] [PubMed] [Google Scholar]
- Fogo AB, Bostad L, Svarstad E, Cook WJ, Moll S, Barbey F, Geldenhuys L, West M, Ferluga D, Vujkovac B, Howie AJ, Burns A, Reeve R, Waldek S, Noel LH, Grunfeld JP, Valbuena C, Oliveira JP, Muller J, Breunig F, Zhang X, Warnock DG. Scoring system for renal pathology in Fabry disease: report of the International Study Group of Fabry Nephropathy (ISGFN) Nephrol Dial Transplant. 2010;25:2168–2177. doi: 10.1093/ndt/gfp528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebau MC, Braun F, Hopker K, Weitbrecht C, Bartels V, Muller RU, Brodesser S, Saleem MA, Benzing T, Schermer B, Cybulla M, Kurschat CE. Dysregulated autophagy contributes to podocyte damage in Fabry’s disease. PLoS One. 2013;8:e63506. doi: 10.1371/journal.pone.0063506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branton MH, Schiffmann R, Sabnis SG, Murray GJ, Quirk JM, Altarescu G, Goldfarb L, Brady RO, Balow JE, Austin Iii HA, Kopp JB. Natural history of Fabry renal disease: influence of alpha-galactosidase A activity and genetic mutations on clinical course. Medicine (Baltimore) 2002;81:122–138. doi: 10.1097/00005792-200203000-00003. [DOI] [PubMed] [Google Scholar]
- Ojo A, Meier-Kriesche HU, Friedman G, Hanson J, Cibrik D, Leichtman A, Kaplan B. Excellent outcome of renal transplantation in patients with Fabry’s disease. Transplantation. 2000;69:2337–2339. doi: 10.1097/00007890-200006150-00020. [DOI] [PubMed] [Google Scholar]
- Waldek S, Patel MR, Banikazemi M, Lemay R, Lee P. Life expectancy and cause of death in males and females with Fabry disease: findings from the Fabry Registry. Genet Med. 2009;11:790–796. doi: 10.1097/GIM.0b013e3181bb05bb. [DOI] [PubMed] [Google Scholar]
- Laney DA, Bennett RL, Clarke V, Fox A, Hopkin RJ, Johnson J, O’Rourke E, Sims K, Walter G. Fabry disease practice guidelines: recommendations of the National Society of Genetic Counselors. J Genet Couns. 2013;22:555–564. doi: 10.1007/s10897-013-9613-3. [DOI] [PubMed] [Google Scholar]
- Caudron E, Dermain DP, Prognon P. Fabry disease: enzymatic screening using dried blood spots on filter paper. Rev Med Interne. 2010;31:S263–S269. doi: 10.1016/S0248-8663(10)70025-4. [DOI] [PubMed] [Google Scholar]
- Selvarajah M, Nicholls K, Hewitson TD, Becker GJ. Targeted urine microscopy in Anderson-Fabry disease: a cheap, sensitive and specific diagnostic technique. Nephrol Dial Transplant. 2011;26:3195–3202. doi: 10.1093/ndt/gfr084. [DOI] [PubMed] [Google Scholar]
- Tondel C, Bostad L, Larsen KK, Hirth A, Vikse BE, Houge G, Svarstad E. Agalsidase benefits renal histology in young patients with Fabry disease. J Am Soc Nephrol. 2013;24:137–148. doi: 10.1681/ASN.2012030316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama H, Takata T, Tsubata Y, Tazawa R, Goto K, Tohyama J, Narita I, Yoshioka H, Ishii S. Screening of male dialysis patients for fabry disease by plasma globotriaosylsphingosine. Clin J Am Soc Nephrol. 2013;8:629–636. doi: 10.2215/CJN.08780812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnock DG, Daina E, Remuzzi G, West M. Enzyme replacement therapy and Fabry nephropathy. Clin J Am Soc Nephrol. 2010;5:371–378. doi: 10.2215/CJN.06900909. [DOI] [PubMed] [Google Scholar]
- Lukas J, Giese AK, Markoff A, Grittner U, Kolodny E, Mascher H, Lackner KJ, Meyer W, Wree P, Saviouk V, Rolfs A. Functional characterisation of alpha-galactosidase a mutations as a basis for a new classification system in fabry disease. PLoS Genet. 2013;9:e1003632. doi: 10.1371/journal.pgen.1003632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffmann R, Warnock DG, Banikazemi M, Bultas J, Linthorst GE, Packman S, Sorensen SA, Wilcox WR, Desnick RJ. Fabry disease: progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. Nephrol Dial Transplant. 2009;24:2102–2111. doi: 10.1093/ndt/gfp031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieppati A, Remuzzi G. Proteinuria and its consequences in renal disease. Acta Paediatr Suppl. 2003;92:9–13. doi: 10.1111/j.1651-2227.2003.tb00213.x. [DOI] [PubMed] [Google Scholar]
- National Health Services. SOP for Anderson-Fabry disease. http://www.webarchive.org.uk/wayback/archive/20130325153347/http://www.specialisedservices.nhs.uk/library/23/SOP_for_Anderson_Fabry_disease.pdf.
- Rombach SM, Baas MC, ten Berge IJM, Krediet RT, Bemelman FJ, Hollak CEM. The value of estimated GFR in comparison to measured GFR for the assessment of renal function in adult patients with Fabry disease. Nephrol Dial Transplant. 2010;25:2549–2556. doi: 10.1093/ndt/gfq108. [DOI] [PubMed] [Google Scholar]
- Schwartz GJ, Munoz A, Schneider MF, Mak RH, Kaskel F, Warady BA, Furth SL. New equations to estimate GFR in children with CKD. J Am Soc Nephrol. 2009;20:629–637. doi: 10.1681/ASN.2008030287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germain DP, Waldek S, Banikazemi M, Bushinsky DA, Charrow J, Desnick RJ, Lee P, Loew T, Vedder AC, Abichandani R, Wilcox WR, Guffon N. Sustained, long-term renal stabilization after 54 months of agalsidase beta therapy in patients with Fabry disease. J Am Soc Nephrol. 2007;18:1547–1557. doi: 10.1681/ASN.2006080816. [DOI] [PubMed] [Google Scholar]
- Schiffmann R, Ries M, Timmons M, Flaherty JT, Brady RO. Long-term therapy with agalsidase alfa for Fabry disease: safety and effects on renal function in a home infusion setting. Nephrol Dial Transplant. 2006;21:345–354. doi: 10.1093/ndt/gfi152. [DOI] [PubMed] [Google Scholar]
- Wanner C, Oliveira JP, Ortiz A, Mauer M, Germain DP, Linthorst GE, Serra AL, Marodi L, Mignani R, Cianciaruso B, Vujkovac B, Lemay R, Beitner-Johnson D, Waldek S, Warnock DG. Prognostic indicators of renal disease progression in adults with Fabry disease: natural history data from the Fabry Registry. Clin J Am Soc Nephrol. 2010;5:2220–2228. doi: 10.2215/CJN.04340510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banikazemi M, Bultas J, Waldek S, Wilcox WR, Whitley CB, McDonald M, Finkel R, Packman S, Bichet DG, Warnock DG, Desnick RJ. Agalsidase-beta therapy for advanced Fabry disease: a randomized trial. Ann Intern Med. 2007;146:77–86. doi: 10.7326/0003-4819-146-2-200701160-00148. [DOI] [PubMed] [Google Scholar]
- Mehta A, Ricci R, Widmer U, Dehout F, Garcia de Lorenzo A, Kampmann C, Linhart A, Sunder-Plassmann G, Ries M, Beck M. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. Eur J Clin Invest. 2004;34:236–242. doi: 10.1111/j.1365-2362.2004.01309.x. [DOI] [PubMed] [Google Scholar]
- Chen HC, Tsai JH, Lai YH, Guh JY. Renal changes in heterozygous Fabry’s disease – a family study. Am J Kidney Dis. 1990;15:180–183. doi: 10.1016/s0272-6386(12)80518-x. [DOI] [PubMed] [Google Scholar]
- Sheu SS, Chan LP, Liao SC, Hsiao KJ, Shu KH, Lu YS, Cheng CH, Lian JD. Fabry’s disease: clinical, pathologic and biochemical manifestations in two Chinese males. Zhonghua Yi Xue Za Zhi (Taipei) 1994;54:368–372. [PubMed] [Google Scholar]
- Ries M, Bettis KEB, Choyke P, Kopp JB, Austin HA, Brady RO, Schiffmann R. Parapelvic kidney cysts: a distinguishing feature with high prevalence in Fabry disease. Kidney Int. 2004;66:978–982. doi: 10.1111/j.1523-1755.2004.00846.x. [DOI] [PubMed] [Google Scholar]
- Kleinert J, Dehout F, Schwarting A, de Lorenzo AG, Ricci R, Kampmann C, Beck M, Ramaswami U, Linhart A, Gal A, Houge G, Widmer U, Mehta A, Sunder-Plassmann G. Prevalence of uncontrolled hypertension in patients with Fabry disease. Am J Hypertens. 2006;19:782–787. doi: 10.1016/j.amjhyper.2006.01.011. [DOI] [PubMed] [Google Scholar]
- Jain G, Warnock DG. Blood pressure, proteinuria and nephropathy in Fabry disease. Nephron Clin Pract. 2011;118:43–48. doi: 10.1159/000320903. [DOI] [PubMed] [Google Scholar]
- Warnock DG, Ortiz A, Mauer M, Linthorst GE, Oliveira JP, Serra AL, Marodi L, Mignani R, Vujkovac B, Beitner-Johnson D, Lemay R, Cole JA, Svarstad E, Waldek S, Germain DP, Wanner C. Renal outcomes of agalsidase beta treatment for Fabry disease: role of proteinuria and timing of treatment initiation. Nephrol Dial Transplant. 2012;27:1042–1049. doi: 10.1093/ndt/gfr420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fervenza FC, Torra R, Lager DJ. Fabry disease: an underrecognized cause of proteinuria. Kidney Int. 2008;73:1193–1199. doi: 10.1038/sj.ki.5002677. [DOI] [PubMed] [Google Scholar]
- Rozenfeld P, Neumann PM. Treatment of Fabry disease: current and emerging strategies. Curr Pharm Biotechnol. 2011;12:916–922. doi: 10.2174/138920111795542705. [DOI] [PubMed] [Google Scholar]
- Lidove O, Joly D, Barbey F, Bekri S, Alexandra JF, Peigne V, Jaussaud R, Papo T. Clinical results of enzyme replacement therapy in Fabry disease: a comprehensive review of literature. Int J Clin Pract. 2007;61:293–302. doi: 10.1111/j.1742-1241.2006.01237.x. [DOI] [PubMed] [Google Scholar]
- Schiffmann R, Kopp JB, Austin HA, Sabnis S, Moore DF, Weibel T, Balow JE, Brady RO. Enzyme replacement therapy in Fabry disease: a randomized controlled trial. JAMA. 2001;285:2743–2749. doi: 10.1001/jama.285.21.2743. [DOI] [PubMed] [Google Scholar]
- West M, Nicholls K, Mehta A, Clarke JTR, Steiner R, Beck M, Barshop BA, Rhead W, Mensah R, Ries M, Schiffmann R. Agalsidase alfa and kidney dysfunction in Fabry disease. J Am Soc Nephrol. 2009;20:1132–1139. doi: 10.1681/ASN.2008080870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta A, Clarke JT, Giugliani R, Elliott P, Linhart A, Beck M, Sunder-Plassmann G. Natural course of Fabry disease: changing pattern of causes of death in FOS – Fabry Outcome Survey. J Med Genet. 2009;46:548–552. doi: 10.1136/jmg.2008.065904. [DOI] [PubMed] [Google Scholar]
- Hughes DA, Malmenas M, Deegan PB, Elliott PM, Ginsberg L, Hajioff D, Ioannidis AS, Orteu CH, Ramaswami U, West M, Pastores GM, Jenkinson C. Fabry International Prognostic Index: a predictive severity score for Anderson-Fabry disease. J Med Genet. 2012;49:212–220. doi: 10.1136/jmedgenet-2011-100407. [DOI] [PubMed] [Google Scholar]
- Hughes DA, Ramaswami U, Barba Romero MÃ, Deegan P. Age adjusting severity scores for Anderson-Fabry disease. Mol Genet Metab. 2010;101:219–227. doi: 10.1016/j.ymgme.2010.06.002. [DOI] [PubMed] [Google Scholar]
- Giannini EH, Mehta AB, Hilz MJ, Beck M, Bichet DG, Brady RO, West M, Germain DP, Wanner C, Waldek S, Clarke JT, Mengel E, Strotmann JM, Warnock DG, Linhart A. A validated disease severity scoring system for Fabry disease. Mol Genet Metab. 2010;99:283–290. doi: 10.1016/j.ymgme.2009.10.178. [DOI] [PubMed] [Google Scholar]
- Eng CM, Guffon N, Wilcox WR, Germain DP, Lee P, Waldek S, Caplan L, Linthorst GE, Desnick RJ. Safety and efficacy of recombinant human alpha-galactosidase A – replacement therapy in Fabry’s disease. N Engl J Med. 2001;345:9–16. doi: 10.1056/NEJM200107053450102. [DOI] [PubMed] [Google Scholar]
- Feriozzi S, Torras J, Cybulla M, Nicholls K, Sunder-Plassmann G, West M. The effectiveness of long-term agalsidase alfa therapy in the treatment of Fabry nephropathy. Clin J Am Soc Nephrol. 2012;7:60–69. doi: 10.2215/CJN.03130411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarting A, Dehout F, Feriozzi S, Beck M, Mehta A, Sunder-Plassmann G. Enzyme replacement therapy and renal function in 201 patients with Fabry disease. Clin Nephrol. 2006;66:77–84. [PubMed] [Google Scholar]
- Pisani A, Visciano B, Roux GD, Sabbatini M, Porto C, Parenti G, Imbriaco M. Enzyme replacement therapy in patients with Fabry disease: state of the art and review of the literature. Mol Genet Metab. 2012;107:267–275. doi: 10.1016/j.ymgme.2012.08.003. [DOI] [PubMed] [Google Scholar]
- Rombach SM, Smid BE, Bouwman MG, Linthorst GE, Dijkgraaf MG, Hollak CE. Long term enzyme replacement therapy for Fabry disease: effectiveness on kidney, heart and brain. Orphanet J Rare Dis. 2013;8:47. doi: 10.1186/1750-1172-8-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidemann F, Niemann M, Störk S, Breunig F, Beer M, Sommer C, Herrmann S, Ertl G, Wanner C. Long-term outcome of enzyme-replacement therapy in advanced Fabry disease: evidence for disease progression towards serious complications. J Intern Med. 2013;274:331–341. doi: 10.1111/joim.12077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rombach SM, Smid BE, Linthorst GE, Dijkgraaf MG, Hollak CE. Natural course of Fabry disease and the effectiveness of enzyme replacement therapy: a systematic review and meta-analysis : effectiveness of ERT in different disease stages. J Inherit Metab Dis. 2014. [Epub ahead of print] [DOI] [PubMed]
- Mehta A, Beck M, Elliott P, Giugliani R, Linhart A, Sunder-Plassmann G, Schiffmann R, Barbey F, Ries M, Clarke JTR. Enzyme replacement therapy with agalsidase alfa in patients with Fabry’s disease: an analysis of registry data. Lancet. 2009;374:1986–1996. doi: 10.1016/S0140-6736(09)61493-8. [DOI] [PubMed] [Google Scholar]
- Schiffmann R, Askari H, Timmons M, Robinson C, Benko W, Brady RO, Ries M. Weekly enzyme replacement therapy may slow decline of renal function in patients with Fabry disease who are on long-term biweekly dosing. J Am Soc Nephrol. 2007;18:1576–1583. doi: 10.1681/ASN.2006111263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeman RD, Tobin J, Shock NW. Longitudinal studies on the rate of decline in renal function with age. J Am Geriatr Soc. 1985;33:278–285. doi: 10.1111/j.1532-5415.1985.tb07117.x. [DOI] [PubMed] [Google Scholar]
- Prabakaran T, Nielsen R, Larsen JV, Sorensen SS, Feldt-Rasmussen U, Saleem MA, Petersen CM, Verroust PJ, Christensen EI. Receptor-mediated endocytosis of alpha-galactosidase A in human podocytes in Fabry disease. PLoS One. 2011;6:e25065. doi: 10.1371/journal.pone.0025065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breunig F, Weidemann F, Strotmann J, Knoll A, Wanner C. Clinical benefit of enzyme replacement therapy in Fabry disease. Kidney Int. 2006;69:1216–1221. doi: 10.1038/sj.ki.5000208. [DOI] [PubMed] [Google Scholar]
- Deegan PB. Fabry disease, enzyme replacement therapy and the significance of antibody responses. J Inherit Metab Dis. 2012;35:227–243. doi: 10.1007/s10545-011-9400-y. [DOI] [PubMed] [Google Scholar]
- Wilcox WR, Banikazemi M, Guffon N, Waldek S, Lee P, Linthorst GE, Desnick RJ, Germain DP. Long-term safety and efficacy of enzyme replacement therapy for Fabry disease. Am J Hum Genet. 2004;75:65–74. doi: 10.1086/422366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rombach SM, Aerts JM, Poorthuis BJ, Groener JE, Donker-Koopman W, Hendriks E, Mirzaian M, Kuiper S, Wijburg FA, Hollak CE, Linthorst GE. Long-term effect of antibodies against infused alpha-galactosidase A in Fabry disease on plasma and urinary (lyso)Gb3 reduction and treatment outcome. PLoS One. 2012;7:e47805. doi: 10.1371/journal.pone.0047805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes DA, Deegan PB, Milligan A, Wright N, Butler LH, Jacobs A, Mehta AB. A randomised, double-blind, placebo-controlled, crossover study to assess the efficacy and safety of three dosing schedules of agalsidase alfa enzyme replacement therapy for Fabry disease. Mol Genet Metab. 2013;109:269–275. doi: 10.1016/j.ymgme.2013.04.015. [DOI] [PubMed] [Google Scholar]
- Lubanda JC, Anijalg E, Bzduch V, Thurberg BL, Benichou B, Tylki-Szymanska A. Evaluation of a low dose, after a standard therapeutic dose, of agalsidase beta during enzyme replacement therapy in patients with Fabry disease. Genet Med. 2009;11:256–264. doi: 10.1097/GIM.0b013e3181981d82. [DOI] [PubMed] [Google Scholar]
- European Medicines Agency. Assessment report on the shortage of Fabrazyme. Overview of shortage period: spontaneous reports from June 2009 through 15 September 2010 and registry data from June 2009 through 05 August 2010. http://www.ema.europa.eu/docs/en_GB/document_library/Other/2010/11/WC500099241.pdf Accessed 16-9-2013.
- Smid BE, Rombach SM, Aerts JM, Kuiper S, Mirzaian M, Overkleeft HS, Poorthuis BJ, Hollak CE, Groener JE, Linthorst GE. Consequences of a global enzyme shortage of agalsidase beta in adult Dutch Fabry patients. Orphanet J Rare Dis. 2011;6:69. doi: 10.1186/1750-1172-6-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidemann F, Kramer J, Duning T, Lenders M, Canaan-Kuhl S, Krebs A, Gonzalez HG, Sommer C, Uceyler N, Niemann M, Stork S, Schelleckes M, Reiermann S, Stypmann J, Brand SM, Wanner C, Brand E. Patients with Fabry disease after enzyme replacement therapy dose reduction versus treatment switch. J Am Soc Nephrol. 2014;25:837–849. doi: 10.1681/ASN.2013060585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuboi K, Yamamoto H. Clinical observation of patients with Fabry disease after switching from agalsidase beta (Fabrazyme) to agalsidase alfa (Replagal) Genet Med. 2012;14:779–786. doi: 10.1038/gim.2012.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisani A, Spinelli L, Visciano B, Capuano I, Sabbatini M, Riccio E, Messalli G, Imbriaco M. Effects of switching from agalsidase beta to agalsidase alfa in 10 patients with anderson-fabry disease. JIMD Rep. 2013;9:41–48. doi: 10.1007/8904_2012_177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghali J, Nicholls K, Denaro C, Sillence D, Chapman I, Goldblatt J, Thomas M, Fletcher J. Effect of reduced agalsidase beta dosage in fabry patients: the Australian experience. JIMD Rep. 2012;3:33–43. doi: 10.1007/8904_2011_44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahir H, Jackson LL, Warnock DG. Antiproteinuric therapy and fabry nephropathy: sustained reduction of proteinuria in patients receiving enzyme replacement therapy with agalsidase-beta. J Am Soc Nephrol. 2007;18:2609–2617. doi: 10.1681/ASN.2006121400. [DOI] [PubMed] [Google Scholar]
- Niemann M, Herrmann S, Hu K, Breunig F, Strotmann J, Beer M, Machann W, Voelker W, Ertl G, Wanner C, Weidemann F. Differences in Fabry cardiomyopathy between female and male patients: consequences for diagnostic assessment. JACC Cardiovasc Imaging. 2011;4:592–601. doi: 10.1016/j.jcmg.2011.01.020. [DOI] [PubMed] [Google Scholar]
- Tsakiris D, Simpson HK, Jones EH, Briggs JD, Elinder CG, Mendel S, Piccoli G, dos Santos JP, Tognoni G, Vanrenterghem Y, Valderrabano F. Report on management of renale failure in Europe, XXVI, 1995. Rare diseases in renal replacement therapy in the ERA-EDTA Registry. Nephrol Dial Transplant. 1996;11(Suppl 7):4–20. doi: 10.1093/ndt/11.supp7.4. [DOI] [PubMed] [Google Scholar]
- Thadhani R, Wolf M, West ML, Tonelli M, Ruthazer R, Pastores GM, Obrador GT. Patients with Fabry disease on dialysis in the United States. Kidney Int. 2002;61:249–255. doi: 10.1046/j.1523-1755.2002.00097.x. [DOI] [PubMed] [Google Scholar]
- Mignani R, Feriozzi S, Schaefer RM, Breunig F, Oliveira JP, Ruggenenti P, Sunder-Plassmann G. Dialysis and transplantation in Fabry disease: indications for enzyme replacement therapy. Clin J Am Soc Nephrol. 2010;5:379–385. doi: 10.2215/CJN.05570809. [DOI] [PubMed] [Google Scholar]
- Van den Bergh FA, Rietra PJ, Kolk-Vegter AJ, Bosch E, Tager JM. Therapeutic implications of renal transplantation in a patient with Fabry’s disease. Acta Med Scand. 1976;200:249–256. [PubMed] [Google Scholar]
- Shah T, Gill J, Malhotra N, Takemoto SK, Bunnapradist S. Kidney transplant outcomes in patients with Fabry disease. Transplantation. 2009;87:280–285. doi: 10.1097/TP.0b013e318191a842. [DOI] [PubMed] [Google Scholar]
- Cybulla M, Kurschat C, West M, Nicholls K, Torras J, Sunder-Plassmann G, Feriozzi S. Kidney transplantation and enzyme replacement therapy in patients with Fabry disease. J Nephrol. 2013;26:645–651. doi: 10.5301/jn.5000214. [DOI] [PubMed] [Google Scholar]
- Pastores GM, Boyd E, Crandall K, Whelan A, Piersall L, Barnett N. Safety and pharmacokinetics of agalsidase alfa in patients with Fabry disease and end-stage renal disease. Nephrol Dial Transplant. 2007;22:1920–1925. doi: 10.1093/ndt/gfm096. [DOI] [PubMed] [Google Scholar]
- Kosch M, Koch HG, Oliveira JP, Soares C, Bianco F, Breuning F, Rasmussen AK, Schaefer RM. Enzyme replacement therapy administered during hemodialysis in patients with Fabry disease. Kidney Int. 2004;66:1279–1282. doi: 10.1111/j.1523-1755.2004.00883.x. [DOI] [PubMed] [Google Scholar]
- Parini R, Feriozzi S. Females and children with Anderson–Fabry disease: diagnosis, monitoring, benefits of enzyme replacement therapy (ERT) and considerations on timing of starting ERT. Expert Opin Orphan Drugs. 2013;1:315–330. doi: 10.1517/21678707.2013.776957. [DOI] [Google Scholar]
- Whybra C, Miebach E, Mengel E, Gal A, Baron K, Beck M, Kampmann C. A 4-year study of the efficacy and tolerability of enzyme replacement therapy with agalsidase alfa in 36 women with Fabry disease. Genet Med. 2009;11:441–449. doi: 10.1097/GIM.0b013e3181a23bec. [DOI] [PubMed] [Google Scholar]
- Ishii S, Chang HH, Yoshioka H, Shimada T, Mannen K, Higuchi Y, Taguchi A, Fan JQ. Preclinical efficacy and safety of 1-deoxygalactonojirimycin in mice for Fabry disease. J Pharmacol Exp Ther. 2009;328:723–731. doi: 10.1124/jpet.108.149054. [DOI] [PubMed] [Google Scholar]
- Porto C, Pisani A, Rosa M, Acampora E, Avolio V, Tuzzi MR, Visciano B, Gagliardo C, Materazzi S, la Marca G, Andria G, Parenti G. Synergy between the pharmacological chaperone 1-deoxygalactonojirimycin and the human recombinant alpha-galactosidase A in cultured fibroblasts from patients with Fabry disease. J Inherit Metab Dis. 2012;35:513–520. doi: 10.1007/s10545-011-9424-3. [DOI] [PubMed] [Google Scholar]
- Giugliani R, Waldek S, Germain DP, Nicholls K, Bichet DG, Simosky JK, Bragat AC, Castelli JP, Benjamin ER, Boudes PF. A Phase 2 study of migalastat hydrochloride in females with Fabry disease: selection of population, safety and pharmacodynamic effects. Mol Genet Metab. 2013;109:86–92. doi: 10.1016/j.ymgme.2013.01.009. [DOI] [PubMed] [Google Scholar]
- Mehta AB. Anderson-Fabry disease: developments in diagnosis and treatment. Int J Clin Pharmacol Ther. 2009;47(Suppl 1):66–74. doi: 10.5414/cpp47066. [DOI] [PubMed] [Google Scholar]
- Mehta A, West ML, Pintos-Morell G, Reisin R, Nicholls K, Figuera LE, Parini R, Carvalho LR, Kampmann C, Pastores GM, Lidove O. Therapeutic goals in the treatment of Fabry disease. Genet Med. 2010;12:713–720. doi: 10.1097/GIM.0b013e3181f6e676. [DOI] [PubMed] [Google Scholar]
- Togawa T, Kodama T, Suzuki T, Sugawara K, Tsukimura T, Ohashi T, Ishige N, Suzuki K, Kitagawa T, Sakuraba H. Plasma globotriaosylsphingosine as a biomarker of Fabry disease. Mol Genet Metab. 2010;100:257–261. doi: 10.1016/j.ymgme.2010.03.020. [DOI] [PubMed] [Google Scholar]
- Auray-Blais C, Boutin M, Gagnon R, Dupont FO, Lavoie P, Clarke JTR. Urinary globotriaosylsphingosine-related biomarkers for Fabry disease targeted by metabolomics. Anal Chem. 2012;84:2745–2753. doi: 10.1021/ac203433e. [DOI] [PubMed] [Google Scholar]
- Manwaring V, Boutin M, Auray-Blais C. A metabolomic study to identify new globotriaosylceramide-related biomarkers in the plasma of Fabry disease patients. Anal Chem. 2013;85:9039–9048. doi: 10.1021/ac401542k. [DOI] [PubMed] [Google Scholar]
- Torralba-Cabeza MÃ, Olivera S, Hughes DA, Pastores GM, Mateo RN, Perez-Calvo JI. Cystatin C and NT-proBNP as prognostic biomarkers in Fabry disease. Mol Genet Metab. 2011;104:301–307. doi: 10.1016/j.ymgme.2011.06.021. [DOI] [PubMed] [Google Scholar]
- Vylet’al P, Hulkova H, Zivna M, Berna L, Novak P, Elleder M, Kmoch S. Abnormal expression and processing of uromodulin in Fabry disease reflects tubular cell storage alteration and is reversible by enzyme replacement therapy. J Inherit Metab Dis. 2008;31:508–517. doi: 10.1007/s10545-008-0900-3. [DOI] [PubMed] [Google Scholar]
- Kistler AD, Siwy J, Breunig F, Jeevaratnam P, Scherl A, Mullen W, Warnock DG, Wanner C, Hughes DA, Mischak H, Wuthrich RP, Serra AL. A distinct urinary biomarker pattern characteristic of female Fabry patients that mirrors response to enzyme replacement therapy. PLoS One. 2011;6:e20534. doi: 10.1371/journal.pone.0020534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manwaring V, Heywood WE, Clayton R, Lachmann RH, Keutzer J, Hindmarsh P, Winchester B, Heales S, Mills K. The identification of new biomarkers for identifying and monitoring kidney disease and their translation into a rapid mass spectrometry-based test: evidence of presymptomatic kidney disease in pediatric Fabry and type-I diabetic patients. J Proteome Res. 2013;12:2013–2021. doi: 10.1021/pr301200e. [DOI] [PubMed] [Google Scholar]