

Abstract
Alzheimer’s disease (AD) is a chronic, progressive and irreversible neurodegenerative disease with clinical characteristics of memory loss, dementia and cognitive impairment. Although the pathophysiologic mechanism is not fully understood, inflammation has been shown to play a critical role in the pathogenesis of AD. Inflammation in the central nervous system (CNS) is characterized by the activation of glial cells and release of proinflammatory cytokines and chemokines. Accumulating evidence demonstrates that inflammasomes, which cleaves precursors of interleukin-1β (IL-1β) and IL-18 to generate their active forms, play an important role in the inflammatory response in the CNS and in AD pathogenesis. Therefore, modulating inflammasome complex assembly and activation could be a potential strategy for suppressing inflammation in the CNS. This review aims to provide insight into the role of inflammasomes in the CNS, with respect to the pathogenesis of AD, and may provide possible clues for devising novel therapeutic strategies.
Keywords: Alzheimer’s disease, Inflammasome, Inflammation, Astrocytes, Micrioglia, Interleukins
1. Introduction
Alzheimer’s disease (AD) is an irreversible neurodegenerative disease, characterized by extracellular deposition of Aβ beta (Aβ) plaques and intracellular accumulation of hyper-phosphorylated tau protein and NFTs (neurofibrillary tangles) (Huang and Mucke, 2012). Studies in AD patients demonstrate a significant reduction in the size of specific brain regions through the loss of neurons and synapses as compared with healthy subjects (Desikan et al., 2009; Wenk, 2003). The early stages of AD manifest as mild cognitive decline, with progressive memory loss and impairment of other intellectual abilities as the disease progresses, and at the late stages patients suffering from AD alter their personality and lose their bodily functions(Huang and Mucke, 2012). Currently, AD affects more than 38 million people worldwide and every four seconds a new patient is diagnosed with AD (Alzheimer’s, 2012). The number of AD patients is expected to double every 20 years and is expected to reach 115 million in 2050 (Martin Prince et al., 2012). As the baby boom generation ages, it is anticipated that the number of AD cases will rise dramatically. However, effective treatment of AD remains a challenge, although numerous hypotheses of possible mechanisms have been put forth.
To date, there are several competing hypotheses that attempt to explain the cause of AD, such as the cholinergic hypothesis, Aβ hypothesis, tau hypothesis and inflammation hypothesis. Cholinergic deficiency is the oldest hypothesis, but has not been widely accepted due to unsuccessful clinical and experimental results (Comim et al., 2012; Nelson et al., 2009). The Aβ and tau hypotheses postulate that Aβ deposits or tau protein abnormalities are the fundamental causes of the disease. Nevertheless, Aβ plaques and neurofibrillary tangles are not sufficient in explaining all the features of AD, since abnormal levels of Aβ plaques have been found among normal healthy elderly individuals (Aizenstein et al., 2008). In addition, highly expressed Aβ or tau protein in animal models of AD do not show significant neurodegenerative changes (Johnston et al., 2011). Furthermore, clinical trials with immune-therapeutics to reduce Aβ level in the brain did not improve cognitive function in AD patients, although evidence suggests clearance of the Aβ plaques (Holmes et al., 2008). These findings suggest that other factors might also be involved in the pathogenesis of AD. Inflammation has been proposed as a key effector of AD, and several features of AD, such as microglial activation, reactive astrocytes and elevated cytokine expression, have all been observed in AD patients, leading to an inflammatory hypothesis. Additionally, epidemiological evidence suggests that non-steroidal anti-inflammatory drugs (NSAIDs) could delay the onset and reduce severity of AD symptoms, although several studies have failed to confirm this (Szekely and Zandi, 2010). More recently, analysis of human brain AD samples has revealed highly expressed inflammatory cytokines during the early stages of AD (Sudduth et al., 2013), and genome-wide studies show an upregulation of inflammatory genes, indicating a potential role of inflammation in the progression of AD (Hollingworth et al., 2011; Sudduth et al., 2013).
Considering the importance of inflammation in the pathogenesis of AD, several recent review articles have discussed how inflammatory molecules, signaling pathways and processes could be involved in the pathogenesis of AD (Blasko et al., 2000; Johnston et al., 2011; Lau and Yu, 2001; Li et al., 2011; Liu et al., 2013a). In this review, we focus on specific inflammasomes identified in the central nervous system (CNS), in particular the brain, their potential as therapeutics, and the cytokines released by these inflammasomes, with emphasis on environmental factors, i.e. fatty acids, that trigger the generation of cytokines through inflammasomes.
2. Inflammatory process in AD
Inflammation is a complex cellular and molecular defense mechanism in response to diverse stimuli, including stress, injury and infection. In the brain, an upregulation in inflammatory signaling (i.e. neuroinflammation) is characterized by the activation of astrocytes and microglia, and the release of proinflammatory mediators. Neuroinflammation is involved in neurodegenerative diseases, such as Parkinson’s disease, Huntington’s disease, amyotrophic lateral sclerosis, multiple sclerosis, psychiatric, as well as AD (Craft et al., 2006; Pizza et al., 2011; Sudduth et al., 2013; Varnum and Ikezu, 2012). Mounting evidence indicates an upregulation in the inflammatory molecules and activated glial cells surrounding the senile plaques in brains of AD patients and AD transgenic animal models (Bauer et al., 1991; Cagnin et al., 2001; Fillit et al., 1991). Concomitantly, a downregulation in anti-inflammatory molecules is observed in post mortem brains of AD patients (Walker et al., 2009). These findings implicate an involvement of inflammatory responses in the pathogenesis of AD. In the CNS, activated astrocytes and microglia are major sources of inflammatory molecules, such as cytokines, chemokines, neurotransmitters, reactive oxygen species, and nitric oxide (NO) (Tansey et al., 2007). The released cytokines, in particular IL-1β, IL-6 and TNFα (tumor necrosis factor α), are the major effectors of the neuroinflammatory signals (Allan and Rothwell, 2003; Rothwell, 1999), and can affect neurophysiologic mechanisms regarding cognition and memory (Gemma and Bickford, 2007; Jankowsky and Patterson, 1999). Cytokines could establish a feedback loop, to activate more astrocytes and microglia and lead to further generation of inflammatory molecules. In addition, the secreted inflammatory molecules also recruit other cells such as monocytes and lymphocytes to cross the blood brain barrier to enhance neuroinflammation in the CNS (Das and Basu, 2008).
2.1 Astrocytes and AD
Astrocytes are the most abundant non-neuronal cells in the CNS, constituting about 20–50% of the human brain volume, much higher than microglia (Shimada et al., 2012). Astrocytes have multiple functions, such as regulation of extracellular ionsand energy reserves, clearance and metabolism of neurotransmitters, and facilitating the maintenance of normal neuronal functions in the CNS (Dong and Benveniste, 2001; Rossi and Volterra, 2009). Astrocytes can be activated by numerous factors, including pathogens, lipopolysaccharide, oxidative stress (Querfurth and LaFerla, 2010), free saturated fatty acids (Liu et al., 2013a) as well as Aβ (Jana and Pahan, 2010). The activation of astrocytes is believed to last longer than that of microglia, enabling a prolong engagement of astrocytes in the neuroinflammatory response (Li et al., 2011).
Reactive astrocytes as opposed to quiescent astrocytes, can produce cytokines such as interleukins, TNFα and interferon γ (IFN-γ), etc. (Liu et al., 2013a). They can also generate low amount of Aβ, in addition to neurons, the major producer of Aβ (Blasko et al., 2000; Li et al., 2011). Cytokines, IFN-γ along with TNFα or IL-1β, have been demonstrated to induce the generation of Aβ in primary human astrocytes and astrocytoma cells (Blasko et al., 2000). Proinflammatory molecules secreted by reactive astrocytes can elevate the expression of secretases in neurons to enhance the production of Aβ (Tang, 2009; Yu et al., 2009b), and activate microglia to further produce inflammatory factors (Otth et al., 2002).
2.2 Microglia and AD
Microglia constitute around 20% of the total glial cells in the brain, and 10% of the cells in the CNS (Lawson et al., 1992; Rubio-Perez and Morillas-Ruiz, 2012). They have various functions in the CNS, including phagocytosis and inducing cytotoxicity (Gehrmann et al., 1995). Microglia can be activated by many factors including Aβ. Activated microglia have phagocytic ability by migrating to damaged cells and clearing debris, and like astrocytes, can also generate inflammatory molecules such as cytokines, chemokines, reactive oxygen and nitrogen species.
Activated microglia colocalize in areas of heavy Aβ concentration (Edison et al., 2008), and preferentially associate with certain types of Aβ plaques, such as fibrillar Aβ (D’Andrea et al., 2004). Aβ binds to microglia and induces phagocytosis or expression of proinflammatory molecules (Ho et al., 2005; Pan et al., 2011), and this activation is dependent on the load of Aβ (Permanne et al., 2002). In an earlier study, upon depletion of microglial cells in animal models the Aβ level increased thereby suggesting microglial cells play a role in the clearance of Aβ by phagocytosis (Broussard et al., 2012; Majumdar et al., 2007). However, more recent work confirmed a detrimental role of microglia in AD that depends upon the degree of activation of the microglial cells. They suggest that with moderate activation by light Aβ concentration microglia have strong phagocytic ability to clear Aβ, while heavily activated microglia by high levels of Aβ increase the production of proinflammatory molecules, such as IL-1β and TNFα, and trigger neuronal damage and coincide with a reduced ability to clear Aβ (Hickman et al., 2008; Johnston et al., 2011). The lower clearance is due to a significant decrease in the expression of Aβ binding scavenger receptors and Aβ degrading enzymes in microglia (Hickman et al., 2008; Johnston et al., 2011). Therefore, the exact function of microglia in AD, whether it is beneficial or detrimental, may depend on the activation state of the microglia.
2.3 Neurons and AD
Neurons at around 100 billion, are the core components of the brain (Williams and Herrup, 1988). They are electrically excitable and have diverse functions, including processing and transmitting information through electrical and chemical signals. In AD, a significant loss of neurons in the cerebral cortex and certain subcortical regions have been reported (Annaert and De Strooper, 2002). Both extracellular deposition of amyloid plaques and intracellular aggregation of neurofibrillary tangles are hallmarks of AD. Aβ peptides and hyperphosphorylated tau protein are major components of amyloid plaques and neurofibrillary tangles, respectively (Annaert and De Strooper, 2002). Both are toxic to the cells and are believed to lead to neuronal dysfunction and death (Price and Sisodia, 1994).
Inflammatory processes are thought to play a role in neuronal degeneration (Meng et al., 2013; Morales et al., 2010). In addition to astrocytes and microglia, neurons also contribute to the inflammatory response in the CNS by releasing cytokines, e.g. IL-1β and IL-18. Inflammasomes involve in the maturation of IL-1β and IL-18 are expressed in neurons (de Rivero Vaccari et al., 2008; Yang-Wei Fann et al., 2013; Zou and Crews, 2012). Trauma, a risk factor for AD, increases inflammasome expression in rat neurons (de Rivero Vaccari et al., 2009; de Rivero Vaccari et al., 2008). Furthermore, postmortem brain tissues from stroke patients show increase protein levels of inflammasomes in primary cortical neurons (Yang-Wei Fann et al., 2013). Currently, the expression levels of inflammasomes in neurons of AD patients are not extensively studied, and much needed insight into the pathogenesis of AD could be gained through further studies in this area.
3. Inflammatory cytokine molecules in AD
Elevated levels of inflammatory cytokines, TNFα, IFNγ, and interleukins, are found in the brain, in particular near the Aβ plaques, of Alzheimer’s patients and AD transgenic animals (Johnston et al., 2011). Cytokines are also detected in the peripheral blood and cerebrospinal fluid of AD patients (Bossu et al., 2008; Di Rosa et al., 2006; Rubio-Perez and Morillas-Ruiz, 2012). Proinflammatory cytokines have multiple functions, which include stimulating or inhibiting cell proliferation, differentiation and apoptosis, as well as inducing inflammation (Rubio-Perez and Morillas-Ruiz, 2012). In the CNS, proinflammatory cytokines induce the release of a number of other proinflammatory molecules from the same or different cell types, amplifying the cytokine effects. Although these studies do not demonstrate whether inflammation is an initiator, contributor or side effect in the pathophysiological changes associated with AD (Li et al., 2011; Sudduth et al., 2013), it (i.e. cytokines) nevertheless is involved in regulating Aβ plaque deposition and BACE1 expression in AD transgenic mice (Yamamoto et al., 2007). Interleukins, in particular IL-1β and IL-18, are upregulated in AD brain, and the overexpression of IL-1β or IL-18 is critical for the onset of the inflammatory process (Rubio-Perez and Morillas-Ruiz, 2012), and both mediate the expression of a vast array of inflammatory genes (Weber et al., 2010). IL-1β and IL-18 are synthesized as inactive precursors, proIL-1β and proIL-18, respectively, and require inflammasomes for their maturation.
3.1 Inflammasome
Inflammasome, the platform for proIL-1β and proIL-18 processing, is an intracellular multiprotein complex. To date, the most well-characterized inflammasomes are NLRP1 (nucleotide-binding oligomerization domain (NOD)-like receptor protein 1), NLRP2, (NOD-like receptor protein 2), NLRP3 (NOD-like receptor protein 3), and NLRC4 (CARD domain-containing protein 4, also called IPAF (ICE-protease activating factor)) inflammasome (Martinon et al., 2009; Minkiewicz et al., 2013). The basic components of inflammasomes include a NOD-like receptor (NLR) that recognizes danger signals or ligands, and procaspase-1 which is central to inflammasome activation. In addition to these basic components, other factors could also be involved in the assembly or activity of inflammasomes, depending on the type of cell and stimulus. They include the adaptor protein, ASC (apoptosis-associated speck-like protein containing a caspase recruitment domain), which is an essential component of the NLRP1, NLRP2 and NLRP3 inflammasomes, but not of the more complex NLRC4 inflammasome (Martinon et al., 2009; Minkiewicz et al., 2013; Schroder and Tschopp, 2010). Under certain but not all conditions ASC (Martinon et al., 2009) or Naip5 (NLR family, apoptosis inhibitory protein 5) (Lightfield et al., 2011) is required for the activity of the NLRC4 inflammasome. The receptors of the inflammsomes, NLRs, that sense the pathogen associated and damage associated molecules (Martinon et al., 2009), are predominantly cytosolic proteins. They are structurally characterized by three distinct domains: a domain of leucine rich repeats (LRRs) for ligand sensing, a NACHT domain (also called nucleotide-binding oligomerization domain (NOD)) that facilitates oligomerization, and an effector domain which consists of one of three domains, i.e. a pyrin (PYD), CARD (caspase activation and recruitment domains), or BIR (baculovirus IAP repeat) domain (Martinon et al., 2009). In response to danger signals, inflammasomes assemble by self-oligomerizing the NLRs through interactions with the NACHT domain (van de Veerdonk et al., 2011). As for the NLRP1 and NLRP3 inflammasomes, the oligomerzied NLRs recruit and interact with the adaptor protein ASC, which in turn recruits the effector protein procaspase-1that is central to the activation of inflammasomes (Huang et al., 2013). Once procaspase-1 is recruited to assemble the inflammasome, it self-cleaves to form the mature caspase-1. Caspase-1 is the protease that cleaves the precursor of the proinflammatory molecules to form their mature form, such as IL-1β and IL-18 (Schroder and Tschopp, 2010). The activators of the inflammasomes can be divided into two categories; pathogen associated molecular patterns (PAMPs) activate a host-defense reaction, and damage associated molecular patterns (DAMPs) activate a self-defense mechanism in response to danger signals (Salminen et al., 2008). Activators include bacteria, virus, fungus, protoza, microbial proteins, crystalline urea, RNA, Alum, ATP, potassium efflux, fatty acids, Aβ, and most recently, degraded mitochondrial DNA (Liu et al., 2013a; Mathew et al., 2012; Schmidt and Lenz, 2012). Overall the assembly and activation of inflammasomes are cell type and stimulus specific (Martinon et al., 2009; Schroder and Tschopp, 2010).
3.2 Proinflammatory cytokine: Interleukin-1β
IL-1β is expressed in many cell types including macrophages, monocytes and neurtrophils, and in the CNS IL-1β can be released from astrocytes, microglia and neurons (Dinarello, 2010). IL-1β induces diverse signalings which are cell type specific (Srinivasan et al., 2004), for example, in glial cells IL-1β activates NFκB (nuclear factor kappa B) signaling to upregulate cytokine production. In contrast, in neurons IL-1β activates the MAPK-p38 signaling cascade to increase the secreted APP fragment cleaved by BACE1 to enhance the ability to form Aβ (Salminen et al., 2008; Srinivasan et al., 2004; Sun et al., 2003).
3.2.1 IL-1β relationship to AD
IL-1β plays a key role in the onset and development of diverse diseases, including neurodegenerative diseases such as AD (Dinarello, 2010). The IL-1β gene and eight other interleukin 1 family genes form a cytokine gene cluster on chromosome 2 (Webb et al., 1986). Elevated IL-1β has been observed in the serum, cerebrospinal fluid and brain of patients with AD as well as other dementia (Blum-Degen et al., 1995; Cacabelos et al., 1991; Deniz-Naranjo et al., 2008). Elevated levels also have been reported in the cerebrospinal fluid and brain parenchyma of both humans and rodents shortly after traumatic brain injury, the latter is an independent risk factor for AD (Emmanouilidou et al., 2011; Shaftel et al., 2008; Yamasaki et al., 1995). IL-1β can activate other cell types, in particular astrocytes and microglia, to further induce cytokine release (e.g. IL-1β, IL-6 and IL-18), as well as inducible nitric oxide synthase activity to produce the free radical NO, leading to neurotoxicity (Rossi and Bianchini, 1996; Rubio-Perez and Morillas-Ruiz, 2012). IL-1β secreted from astrocytes has been shown to enhance the production of APP and Aβ from the neurons (Blasko et al., 2000; Bonifati and Kishore, 2007; Li et al., 2011). Additionally, IL-1β has been found to increase the release of S100B from an astroglioma cell line and the plaque-associated activated astrocytes in the primary cortex that promote neuronal APP synthesis (Li et al., 2011; Liu et al., 2005). Injecting IL-1β into the cerebral hemisphere upregulates the levels of APP and amyloidogenesis (Sheng et al., 1996). Furthermore, several studies demonstrate that IL-1β can induce the phosphorylation of the tau protein and mediate the formation of neurofibrillary tangles through the MAPK-p38 pathway (Griffin et al., 2006; Salminen et al., 2008). Blocking IL-1β signaling in the brain of an AD mouse model is able to alter the inflammatory responses of the brain, rescue cognition, attenuate tau pathology, and reduce fibrillar Aβ level (Kitazawa et al., 2011). Conversely, knocking out the IL-1β receptor antagonist in mice increases the neuronal damage induced by Aβ (Craft et al., 2005). These studies implicate a proinflammatory role of IL-1β in the pathogenesisof AD.
3.2.2 Inflammasomes involved in IL-1β maturation in the CNS
In the CNS, the production of IL-1β by inflammasomes, specifically NLRP1, NLRP2, NLRP3 and NLRC4, is well-characterized as compared to other interleukins (Minkiewicz et al., 2013; Trendelenburg, 2008). The NLRP1 inflammasome is present in the neurons, astrocytes, oligodendrocytes, and microglia (Abulafia et al., 2009; de Rivero Vaccari et al., 2008; Kummer et al., 2007; Silverman et al., 2009). Spinal cord injury can activate the NLRP1 inflammasome to produce IL-1β in rat spinal cord neurons (de Rivero Vaccari et al., 2008). A subsequent study demonstrated that P2X7 purinergic receptor is involved in the activation of NLRP1 inflammasome (Silverman et al., 2009). Given that P2X4 and P2X7 are the major purinergic P2X receptor subtypes, a study of spinal cord injury in P2X4 knock-out mice showed a significant reduction in inflammasome activation and proinflammatory cytokine production as compared to wild type (de Rivero Vaccari et al., 2012), supporting the involvement of purinergic receptor P2X4 in the activation of the NLRP1 inflammasome. Similarly, the P2X7 purinergic receptor has been shown to activate the NLRP1 inflammasome in primary neurons (Silverman et al., 2009).
ATP, a danger-associated molecular pattern that is released from damaged cells after brain injury, activates the NLRP2 inflammasome, which consists of the NLRP2 receptor, ASC and caspase-1, in human astrocytes (Minkiewicz et al., 2013). The ATP-induced activation of the NLRP2 inflammasome interacts with the ATP-release pannexin 1 channel and ATP-gated P2X7 receptor leading to the maturation of IL-1β (Minkiewicz et al., 2013).
In vivo and cell studies demonstrate that fibrillar Aβ activates the NLRP3 inflammasome which is composed of the NLRP3 receptor, ASC and caspase-1, to produce IL-1β in microglia (Halle et al., 2008). Phagocytosis and subsequent lysosomal damage trigger by Aβ initiate the activation of the NLRP3 inflammasome in the microglia (Halle et al., 2008). In support, a recent study in APP/PS1 mice confirms that the NLRP3 inflammasome contributes to the AD pathology (Heneka et al., 2013). Deficiency of the NLRP3 gene reduces Aβ deposition and plays a protective role on memory and behavior (Heneka et al., 2013). Similarly, inhibiting the NLRP3 inflammasome reduces the neuritic plaque burden in an AD transgenic mouse model (Shi et al., 2013).
Palmitate, a fatty acid, activates the NLRC4 inflammasome in primary astrocytes leading to the release of IL-1β (Liu and Chan, 2014). The adaptor protein ASC is important for the activation of NLRC4 inflammasome in astrocytes, while Naip 5 is not (Liu and Chan, 2014). The IL-1β release is upregulated by the astrocytes cultured in palmitate and contributes to a higher production of both BACE1 and Aβ by primary neurons (Liu and Chan, 2014; Liu et al., 2013a; Liu et al., 2013b).
3.3 Proinflammatory cytokine: Interleukin-18
Interleukin-18 (IL-18), belonging to the IL-1 superfamily, is constitutively expressed in several cell types, and the active form of IL-18 is generated by cleavage of the precursor, proIL-18. IL-18 has several similarities in their properties with IL-1β, such as, an inactive precursor, is activated by pathogen and danger associated factors, involves inflammasomes, and induces similar signaling events (Das et al., 2008; Huang et al., 2013). However, there are considerable differences between the two cytokines, such as their expression levels, regulation and action (Das et al., 2008; Huang et al., 2013). In normal brain tissue, IL-18 is constitutively and highly expressed, whereas IL-1β is lowly expressed (Culhane et al., 1998). In peripheral immune cells, IL-1β increases rapidly after ischemia, while IL-18 increases much slower (Jander et al., 2002), suggesting differential regulations of the two cytokines. IL-1β activates NFκB and p38 signaling pathways (Griffin et al., 2006; Salminen et al., 2008; Srinivasan et al., 2004) while IL-18 binds to the receptor IL-18R and activates NFκB, STAT3 (Signal transducer and activator of transcription 3) and NFATc4 (Nuclear factor of activated T-cells, cytoplasmic 4) (Suk et al., 2001; Sutinen et al., 2012). Moreover, it activates both Fas and Fas-L promoter activities, and thus has been suggested to be an apoptosis inducer and initiator of atherosclerosis and cardiovascular diseases (Chandrasekar et al., 2006). IL-18 can modulate neuronal excitability (Kanno et al., 2004), and inhibit long term-potentiation, a form of a neuronal plasticity considered to underlie learning and memory (Curran and O’Connor, 2003).
3.3.1 IL-18 relationship to AD
IL-18 is believed to play an important role in various diseases, in particular AD. The IL-18 gene is located in the 11q22.2–22.3 region close to the dopamine receptor D2 locus, near chromosome 11, a chromosomal region of interested in AD as defined by genome studies and suggested as a linkage area for AD pathology in familial AD (Blacker et al., 2003). Moreover, IL-18 promoter polymorphism has been shown to increase the risk of developing sporadic late onset AD in Han Chinese and Italian populations (Bossu et al., 2007; Yu et al., 2009a). In the CNS, IL-18 is produced by astrocytes, microglia and ependymal cells, and has been also detected in neurons (Conti et al., 1999; Ojala et al., 2008; Sugama et al., 2002). The mRNA and protein levels of IL-18 increase significantly in astrocytes, microglia and neurons that co-localized with Aβ plaques in the brains of AD patients (Ojala et al., 2009). IL-18 is elevated significantly in the plasma of mild cognitively impaired and AD patients (Malaguarnera et al., 2006; Ozturk et al., 2007). Moreover, a significant increase in IL-18 is observed in stimulated mononuclear cells and macrophages of peripheral blood from AD patients (Bossu et al., 2008; Di Rosa et al., 2006), as well as in the blood of patients with ischemic heart disease, type-2 diabetes, and obesity, which are risk factors for AD (Sutinen et al., 2012). These results support that IL-18 is involved in AD disease pathology. Mild AD patients show significant increase in IL-18 level, but slightly lower than moderate AD patients, whereas no significant upregulation is observed in severe AD patients as compared to age-matched control subjects (Motta et al., 2007). This gradual decline in immune response in AD suggests that the immune response, in particular IL-18, could be an initiator of AD pathogenesis. Studies demonstrate that IL-18 triggers an elevation in the protein levels of APP, BACE1, and the N-terminal fragment of presenilin-1 and presenilin enhancer 2 which are components of the γ-secretase complex (Sutinen et al., 2012), suggesting IL-18 accelerates Aβ genesis. Additionally, IL-18 increases the expression of glycogen synthase kinase 3β (GSK-3β) and cyclin dependent kinase 5 (Cdk5), which are involved in hyperphosphorylation of the tau protein (Ojala et al., 2008). These studies support an important role of IL-18 in AD.
3.3.2 Inflammasomes involved in IL-18 maturation in the CNS
Like IL-1β, in most cases, the mature secretable form of IL-18 is generated by caspase-1 through the activation of inflammasome. In contrast, the mature secretable form of IL-18 also can be derived from various extracellular enzymes, including protease 3, serine protease, elastase and cathepsin G (Alboni et al., 2010; Gracie, 2004; Sugawara et al., 2001). However the maturation of IL-18 andIL-1β could be regulated by the same type of inflammasome. For example, down-regulation of NLRP1 in macrophages trigger by Cordyceps sinensis mycelium reduces both IL-18 and IL-1β levels (Huang et al., 2013). The release of IL-18 andIL-1β could also be regulated by different inflammasomes even though they are in the same cell type and expose to the same stimuli. A recent report shows that IL-18 and IL-1β are secreted from primed murine dendritic cells in response to Listeria protein p60, but inhibiting NLRP3 reduces the production of IL-1β, but not IL-18 secretion (Schmidt and Lenz, 2012). This suggests that the maturation of IL-18 and IL-1β is regulated condition-specifically by different signaling mechanisms upon exposure to similar stimulus.
Spinal cord injury causes IL-18 and IL-1β release from neuronal cells through the activation of the NLRP1 inflammasome, composed of receptor NLRP1, adaptor protein ASC, caspase-1, caspase-11 and X-linked inhibitor of apoptosis protein (de Rivero Vaccari et al., 2008). ASC neutralization reduces the upregulation in IL-18 and IL-1β levels (de Rivero Vaccari et al., 2008). Spinal cord injury elevates extracellular ATP levels during neuroinflammation, which may act on purinergic receptors to trigger the activation of inflammasome (de Rivero Vaccari et al., 2012; Minkiewicz et al., 2013). However, upon further study of purinergic receptor P2X4 knockout mice with spinal cord injury, the production of IL-1β but not of IL-18 reduces in the neurons as compared with wild-type mice (de Rivero Vaccari et al., 2012). This further supports the differential regulations of IL-18 and IL-1β expression.
In human astrocytes, ATP released from damaged or dying cells after traumatic brain injury activates the NLRP2 inflammasome, leading to the maturation of both IL-1β and IL-18 (Minkiewicz et al., 2013). Inhibiting pannexin 1 by probenecid or Brilliant Blue G significantly reduces the increase levels of IL-1β and IL-18 triggered by ATP (Minkiewicz et al., 2013), suggesting that the regulation of IL-1β and IL-18 expression in the situation could be similar.
Diverse factors upregulate IL-18 level in the CNS. Kainic acid has been shown to induce IL-18 production from microglia (Jeon et al., 2008). Stress, a risk factor of AD, has been confirmed to promote the synthesis and maturation of IL-18 (Sugama and Conti, 2008). Aging, another risk factor of AD, has been found to activate the NLRP1 inflammasome and upregulateIL-18 and IL-1β levels in the hippocampus of aged mice (Mawhinney et al., 2011). Currently information on the type and composition of the inflammasomes involved in the maturation of IL-18 in the CNS is largely uncharacterized. Further studies are required to reveal the role of IL-18 in AD pathogenesis.
4. Fatty acids involvement in inflammasome activation and pathogenesis of AD
Environmental factors, in particular, saturated fatty acids have been reported to induce the production of proinflammatory cytokines, such as IL-1β, IL-6 and TNFα, the major effectors of neuroinflammatory cascade, in astrocytes and microglia (Gupta et al., 2012; Liu et al., 2013a; Wang et al., 2012). Palmitate, the most common saturated fatty acid in the diet (Guo et al., 2007), triggers the production of cytokines, in particular IL-1β, from many cell types, including astrocytes (Liu et al., 2013a; Luo et al., 2012). Recently we identify that palmitate activates the NLRC4 inflammasomein primary astrocytes to release IL-1β, and ASC participates in the activation of the NLRC4 inflammasome (Liu and Chan, 2014). Reducing NLRC4 or ASC levels in the palmitate (PA)-treated astrocytes significantly reduces IL-1β production (Liu and Chan, 2014). In addition, NLRC4 and ASC levels are upregulated in the brains of AD patients (Liu and Chan, 2014), suggesting a possible role of the NLRC4 inflammasome in AD pathogenesis. In support, palmitate induces IL-1β release from HepG2 (hepatocellular carcinoma cells), and knockdown of NLRC4 inhibits the palmitate induced inflammation and cytokine release (Cho et al., 2013; Luo et al., 2012). Fatty acids can also cause the release of IL-1β from microglia, but the specific inflammasome that regulates this process has not been identified (Wang et al., 2012). These studies highlighted the role of saturated fatty acids in the production of IL-1β by inflammasomes, i.e. NLRC4.
Fatty acids are known to cross the blood-brain barrier through passive diffusion (Dhopeshwarkar and Mead, 1973; Smith and Nagura, 2001), and diets high in saturated fats increase brain uptake of fatty acids from the plasma (Karmi et al., 2010; Wang et al., 1994). It is possible that increase uptake of fatty acids by the brain from the plasma (Karmi et al., 2010) could lead to enhance cytokine release and inflammatory response in the brain. Fatty acid metabolism has been suggested as a risk factor for the development of AD (Di Paolo and Kim, 2011; Morris and Tangney, 2010), and epidemiological studies suggest that consumption of saturated fatty acids increases while consumption of unsaturated fatty acids decreases the risk of AD (Scarmeas et al., 2006; Solfrizzi et al., 2005; Takechi et al., 2010). AD brains show high fatty acid content as compared with normal healthy subjects (Roher et al., 2002). Elevated saturated free fatty acids (FFAs) have been found in the plasma of patients with diabetes mellitus, hypertension and obesity, which are risk factors for AD (Monti et al., 2004; Whitmer et al., 2005). Traumatic brain injury, an independent risk factor for AD (de Rivero Vaccari et al., 2009), induces higher levels of palmitate and stearate in the brain (de Rivero Vaccari et al., 2009; Homayoun et al., 1997; Lipton, 1999). Many in vivo studies report that high fat diet induces Aβ deposition and memory deficits in APP transgenic mice (Julien et al., 2010; Levin-Allerhand et al., 2002; Maesako et al., 2012a; Maesako et al., 2012b; Oksman et al., 2006). Concomitantly, our group has demonstrated that palmitate triggers the upregulation of AD-like characteristics, such as elevated BACE1, Aβ, and hyperphosphorylated tau levels in neurons, mediated by conditioned media from astrocytes cultured in palmitate (Liu et al., 2013a; Patil et al., 2007; Patil et al., 2006). A subsequent study shows the cytokines, in particular IL-1β, secreted by the palmitate-treated astrocytes, contribute to the upregulation of BACE1 and Aβ42 levels in the primary neurons (Liu and Chan, 2014; Liu et al., 2013a). However, palmitate does not directly induce the AD-like changes in neurons (Patil and Chan, 2005; Patil et al., 2006), which could be attributed to the lower ability of neurons to take up and metabolize long chain fatty acids (Blazquez et al., 2000; Qin et al., 2010). In contrast, astrocytes readily metabolize fatty acids, and furthermore peripheral administration of fatty acids is found to accumulate primarily in astrocytes (Bernoud et al., 1998; Morand et al., 1979).
Palmitate and its metabolites, such as ceramides, regulate many gene expression and immunological pathways (Ajuwon and Spurlock, 2005; Haughey et al., 2010; He et al., 2010; Jana and Pahan, 2010; Katsel et al., 2007; Little et al., 2012; Puglielli et al., 2003; Saddoughi and Ogretmen, 2013). Serine palmitoyltransferase (SPT), the rate-limiting enzyme that synthesizes ceramide, is upregulated in palmitate-treated astrocytes. Downregulating SPT to decrease ceramide levels mitigates the increase in IL-1β and TNFα that aresecreted by the astrocytes (Liu et al., 2013a; Patil et al., 2007), suggesting that ceramide is involved in the cytokine production. Neutralizing TNFα and IL-1β in the conditioned media significantly reduce the upregulation of BACE1 in the neurons (Liu et al., 2013a). Moreover, our group and others have found the levels of SPT are upregulated in human AD brains as compared to age-matched controls (Geekiyanage and Chan, 2011; He et al., 2010). Our group observe that inhibiting SPT decreases Aβ42 and tau hyperphosphorylation in an AD mouse model (Geekiyanage et al., 2013). Taken together, the aforementioned studies suggest an involvement of saturated fatty acids in the neuroinflammation mediated by glial cells. Due to the quantity, location, and function of the astrocytes, they are likely major players in the induction of inflammation in the CNS generated by saturated fatty acids.
5. Therapeutic for inflammasome-mediated inflammation
Interleukins, in particular, IL-1β and IL-18, have been implicated in neuronal damage in chronic and progressive neuronal diseases, such as AD, Parkinson’s disease, Huntington’s disease, and Amyotrophic lateral sclerosis. Several approaches, described below, have been evaluated for their ability to reduce the deleterious effects of excessive inflammatory cytokine production and signaling. Secreted extracellular domains of soluble IL-1 receptor and IL-1 receptor antagonist (IL-1Ra) have been shown to mitigate IL-1β signaling by binding IL-1β (Chakraborty et al., 2010; Eisenberg et al., 1990). Similarly, the IL-1 receptor II acts as a decoy receptor to inhibit IL-1β signaling (Colotta et al., 1993). Constitutively expressing IL-18 binding protein, which binds IL-18, prevents the binding of IL-18 to its receptor and its downstream signaling (Kim et al., 2003). Consequently, therapeutic approaches have been developed to mimic the actions of these intrinsic factors. To date, blocking IL-1 signaling is the most effective therapy in many auto-inflammatory disorders such as familial cold autoinflammatory syndrome, Muckle-Wells syndrome, chronic infantile neurologic cutaneous and articular syndrome, and TNF-receptor associated periodic syndrome (Moll and Kuemmerle-Deschner, 2013). Cytokine traps such as rIL-Ra, which recognizes IL-1β, are being tested in clinical trials for several autoinflammatory disorders (Hawkins et al., 2003; Hoffman et al., 2004). In support, inhibitors of IL-1 are effective in treating cryopyrin associated periodic syndromes (CAPS) and gout (Lachmann et al., 2011; So et al., 2013). Although inhibitors of IL-1β have been used to systemically treat inflammation (Hoffman et al., 2004), this type of therapy has not been reported for treating CNS inflammation. Non-steroidal anti-inflammatory drugs (NASIDs), used to suppress inflammation (Hayden et al., 2007; Lee et al., 2010), appear to reduce the risk of developing AD after 2–3 years of treatment based on cognitive decline if taken prior to age 65 (Broussard et al., 2012; Hayden et al., 2007). This is controversial, since there are other reports of their increasing the risk of developing AD (Breitner et al., 2009). The diverse outcomes could be the result of different conditions, such as the age of the patients at treatment, dose of the drugs, duration of the treatment, and the severity of the neurodegeneration (Lee et al., 2010). Nevertheless there are promising NSAIDs and combination of NSAIDs currently in clinical trial for AD (Galimberti and Scarpini, 011).
Inflammasome is required for the maturation of IL-1β and IL-18. Probenecid, an inhibitor of pannexin 1, has been shown to significantly inhibit the expression and activation of the NLRP2 inflammasome, and the maturation of bothIL-1β and IL-18 in human astrocytes induced by ATP (Minkiewicz et al., 2013). Probenecid has also been demonstrated to reduce the activation of the NLRP1 inflammasome, and improve the learning performance in age-related cognitive decline (Mawhinney et al., 2011). In addition, Brilliant Blue G (BBG), a P2X7 receptor antagonist, inhibits ATP-induced activation of the NLRP2 inflammasome in human astrocytes (Minkiewicz et al., 2013). P2X4 knock-out mice has been shownto decrease the level of IL-1β and to have impair inflammasome signaling (de Rivero Vaccari et al., 2012). These reports support that probenecid and BBG have potential therapeutic value, and that pannexin 1, P2X4 and P2X7 receptors could be potential therapeutic targets.
Inflammasome is composed of a receptor such as NLPR1, NLPR2, NLPR3 and NLRC4, and caspase-1, and sometimes ASC. Therefore, targeting any of these components, such as the receptor, caspase-1 or ASC, could prevent inflammasome assembly and activation, and reduce IL-1β and IL-18 generation. In support, knockout of NLRP3 and caspase-1 have been shown to suppress amyloidogenesis and neuropathology, and improve cognition in AD transgenic mice (Heneka et al., 2013). Consequently, designing agents that control the activation or assembly of inflammasome could provide a promising approach to tackling neuroinflammasome. Reportedly a neutralizing antibody against NLRP1is ableto reduce cytokine levels after throboembolic stroke in mice, thereby reducing the detrimental consequences of post-ischemic inflammation (Abulafia et al., 2009). The administration of anti-ASC neutralizing antibodies in spinal cord injury in rats reducescaspase-1 activation and IL-1β and IL-18 levels, leading to better recovery after spinal cord trauma (de Rivero Vaccari et al., 2008). A tetrapeptide recognition sequence has been uncovered for caspase-1, WEHD (Trp-Glu-His-Asp), which can be inhibited by a compound, N-Acetyl-WEHD-aldehyde (Chakraborty et al., 2010; Garcia-Calvo et al., 1998). Thus far clinical trials of this promising application have not been reported. Therapies for treating inflammasome-mediated inflammation have not been fully investigated and would be an interesting area for future exploration.
6. Conclusion
To date, studies on the role of inflammasomes in regulating the maturation of interleukins have enhanced our understanding of inflammation in diseases, including AD. Blocking or neutralizing IL-1β reduces cognitive impairment and decreases AD-like pathological changes in AD mouse models (Gonzalez et al., 2009; Kitazawa et al., 2011). Similarly, knockout of IL-1β receptor antagonist in mice increases the neuronal damage induced by Aβ (Craft et al., 2005). These findings support a role of inflammasome in the pathogenesis of AD, however, the detail mechanism of inflammasome assembly and activation in the CNS is still emerging. AD is a complex neurodegenerative disease with an unclear etiology and thus far, there are no proven treatments to prevent or slow its progression. Evidence suggests that environmental factors, e.g. saturated fatty acids, can initiate the activation of glial cells to release inflammatory molecules, including IL-1β and IL-18 (Liu and Chan, 2014; Liu et al., 2013a; Wang et al., 2012). IL-1β and IL-18 can further induce the activation of glial cells in the CNS to secret more inflammatory molecules, creating a feedback loop to sustain the production of proinflamatory molecules and result in increased amyloidogenesis and neurofibrillary tangles (Figure 1). Therefore, studying interleukin generation through the inhibition of inflammasomes assembly and activation could prove crucial to gaining a better understanding of the role of inflammasomes in AD, and providing novel therapeutics for AD.
Figure 1. Scheme of inflammasome-mediated inflammatory response.
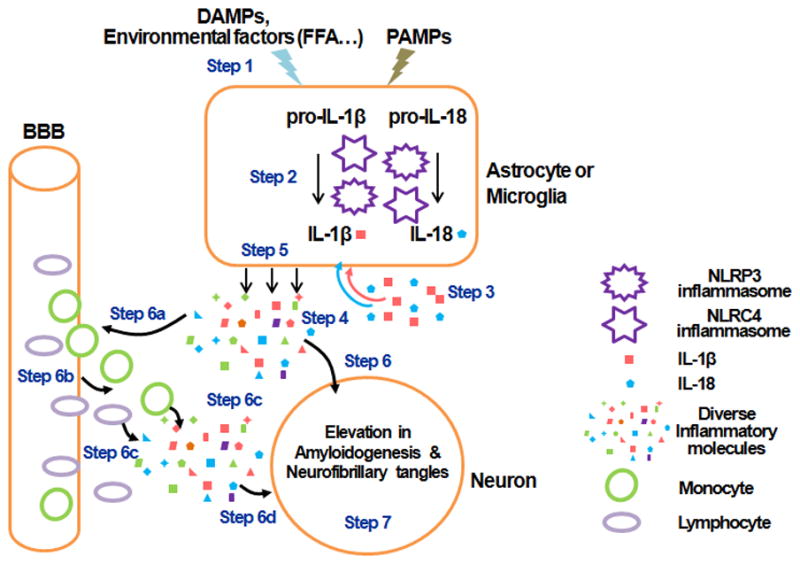
Damage associated molecular patterns (DAMPs), environmental factors such as elevated free fatty acids (FFA), or pathogen associated molecular patterns (PAMPs) initiate the activation of inflammasomes in astrocytes or microglial cells to release inflammatory molecules, IL-1β and IL-18 (Step1–3). IL-1β and IL-18 can further activate more astrocytes or microglial cells in the CNS to secret more diverse inflammatory molecules (Step4, 5). The inflammatory molecules induce the elevation of amyloidogenesis and neurofibrillary tangles in neurons (Step6, 7). Concomitantly, the diverse inflammatory molecules also recruit other cells such as monocytes and lymphocytes to cross the blood brain barrier (BBB) and to release more diverse inflammatory molecules, resulting in an increase in amyloidogenesis and neurofibrillary tangles in the neurons (Step6a–7).
Inflammasomes involved in IL-1β and IL-18 maturation contribute to AD progression
Environmental factors, i.e. fatty acids, can trigger the activation of inflammasome
Modulating inflammasome activation could be a potential strategy for treating AD
Acknowledgments
This study was supported in part by the National Institute of Health (R01GM079688 and R01GM089866), the National Science Foundation (CBET 0941055).
List of abbreviations
- AD
alzheimer’s disease
- ASC
apoptosis-associated speck-like protein containing a caspase recruitment domain
- Aβ
Aβ beta
- BACE1
beta site Aβ beta precursor protein cleaving enzyme 1
- Cdk5
cyclin dependent kinase 5
- CNS
central nervous system
- DAMPs
damage associated molecular patterns
- GSK-3β
glycogen synthase kinase 3β
- HepG2
hepatocellular carcinoma cells
- IFN-γ
interferon –γ
- IL-1β
interleukin-1β
- IL-18
interleukin-18
- IL-1RL1
IL-1 receptor-like 1 (also called ST2)
- IL-1RAcp
IL-1 receptor accessory protein
- Naip5
NLR family, apoptosis inhibitory protein 5
- NFκB
nuclear factor kappa B
- NFTs
neurofibrillary tangels
- PYD
pyrin domain
- BIR
baculovirus IAP repeat
- NLRP1
nucleotide-binding oligomerization domain (NOD)-like receptor (NLR) protein 1
- NLRP2
(NOD-like receptor protein 2)
- NLRP3
NOD-like receptor protein 3
- CARD domain
Caspase activation and recruitment domains
- NLRC4
CARD domain-containing protein 4
- IPAF
ICE-protease activating factor
- NSAIDs
non-steroidal anti-inflammatory drugs
- TNFα
tumor necrosis factor-α
- NFATc4
nuclear factor of activated T-cells, cytoplasmic 4
- NO
nitric oxide
- PA
palmitate
- SPT
serine palmitoyltransferase
- PAMPs
pathogen associated molecular patterns
- STAT3
Signal transducer and activator of transcription 3
Footnotes
The authors have declared no conflict interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abulafia DP, de Rivero Vaccari JP, Lozano JD, Lotocki G, Keane RW, Dietrich WD. Inhibition of the inflammasome complex reduces the inflammatory response after thromboembolic stroke in mice. J Cereb Blood Flow Metab. 2009;29:534–544. doi: 10.1038/jcbfm.2008.143. [DOI] [PubMed] [Google Scholar]
- Aizenstein HJ, Nebes RD, Saxton JA, Price JC, Mathis CA, Tsopelas ND, Ziolko SK, James JA, Snitz BE, Houck PR, Bi W, Cohen AD, Lopresti BJ, DeKosky ST, Halligan EM, Klunk WE. Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol. 2008;65:1509–1517. doi: 10.1001/archneur.65.11.1509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajuwon KM, Spurlock ME. Palmitate activates the NF-kappaB transcription factor and induces IL-6 and TNFalpha expression in 3T3-L1 adipocytes. J Nutr. 2005;135:1841–1846. doi: 10.1093/jn/135.8.1841. [DOI] [PubMed] [Google Scholar]
- Alboni S, Cervia D, Sugama S, Conti B. Interleukin 18 in the CNS. J Neuroinflammation. 2010;7:9. doi: 10.1186/1742-2094-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan SM, Rothwell NJ. Inflammation in central nervous system injury. Philos Trans R Soc Lond B Biol Sci. 2003;358:1669–1677. doi: 10.1098/rstb.2003.1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alzheimer’s A. Alzheimer’s disease facts and figures. Alzheimers Dement. 2012;8:131–168. doi: 10.1016/j.jalz.2012.02.001. [DOI] [PubMed] [Google Scholar]
- Annaert W, De Strooper B. A cell biological perspective on Alzheimer’s disease. Annu Rev Cell Dev Biol. 2002;18:25–51. doi: 10.1146/annurev.cellbio.18.020402.142302. [DOI] [PubMed] [Google Scholar]
- Bauer J, Strauss S, Schreiter-Gasser U, Ganter U, Schlegel P, Witt I, Yolk B, Berger M. Interleukin-6 and alpha-2-macroglobulin indicate an acute-phase state in Alzheimer’s disease cortices. FEBS Lett. 1991;285:111–114. doi: 10.1016/0014-5793(91)80737-n. [DOI] [PubMed] [Google Scholar]
- Bernoud N, Fenart L, Benistant C, Pageaux JF, Dehouck MP, Moliere P, Lagarde M, Cecchelli R, Lecerf J. Astrocytes are mainly responsible for the polyunsaturated fatty acid enrichment in blood-brain barrier endothelial cells in vitro. J Lipid Res. 1998;39:1816–1824. [PubMed] [Google Scholar]
- Blacker D, Bertram L, Saunders AJ, Moscarillo TJ, Albert MS, Wiener H, Perry RT, Collins JS, Harrell LE, Go RC, Mahoney A, Beaty T, Fallin MD, Avramopoulos D, Chase GA, Folstein MF, McInnis MG, Bassett SS, Doheny KJ, Pugh EW, Tanzi RE. Results of a high-resolution genome screen of 437 Alzheimer’s disease families. Hum Mol Genet. 2003;12:23–32. doi: 10.1093/hmg/ddg007. [DOI] [PubMed] [Google Scholar]
- Blasko I, Veerhuis R, Stampfer-Kountchev M, Saurwein-Teissl M, Eikelenboom P, Grubeck-Loebenstein B. Costimulatory effects of interferon-gamma and interleukin-1beta or tumor necrosis factor alpha on the synthesis of Abeta1-40 and Abeta1-42 by human astrocytes. Neurobiol Dis. 2000;7:682–689. doi: 10.1006/nbdi.2000.0321. [DOI] [PubMed] [Google Scholar]
- Blazquez C, Galve-Roperh I, Guzman M. De novo-synthesized ceramide signals apoptosis in astrocytes via extracellular signal-regulated kinase. FASEB J. 2000;14:2315–2322. doi: 10.1096/fj.00-0122com. [DOI] [PubMed] [Google Scholar]
- Blum-Degen D, Muller T, Kuhn W, Gerlach M, Przuntek H, Riederer P. Interleukin-1 beta and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci Lett. 1995;202:17–20. doi: 10.1016/0304-3940(95)12192-7. [DOI] [PubMed] [Google Scholar]
- Bonifati DM, Kishore U. Role of complement in neurodegeneration and neuroinflammation. Mol Immunol. 2007;44:999–1010. doi: 10.1016/j.molimm.2006.03.007. [DOI] [PubMed] [Google Scholar]
- Bossu P, Ciaramella A, Moro ML, Bellincampi L, Bernardini S, Federici G, Trequattrini A, Macciardi F, Spoletini I, Di Iulio F, Caltagirone C, Spalletta G. Interleukin 18 gene polymorphisms predict risk and outcome of Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2007;78:807–811. doi: 10.1136/jnnp.2006.103242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossu P, Ciaramella A, Salani F, Bizzoni F, Varsi E, Di Iulio F, Giubilei F, Gianni W, Trequattrini A, Moro ML, Bernardini S, Caltagirone C, Spalletta G. Interleukin-18 produced by peripheral blood cells is increased in Alzheimer’s disease and correlates with cognitive impairment. Brain Behav Immun. 2008;22:487–492. doi: 10.1016/j.bbi.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Breitner JC, Haneuse SJ, Walker R, Dublin S, Crane PK, Gray SL, Larson EB. Risk of dementia and AD with prior exposure to NSAIDs in an elderly community-based cohort. Neurology. 2009;72:1899–1905. doi: 10.1212/WNL.0b013e3181a18691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broussard GJ, Mytar J, Li RC, Klapstein GJ. The role of inflammatory processes in Alzheimer’s disease. Inflammopharmacology. 2012;20:109–126. doi: 10.1007/s10787-012-0130-z. [DOI] [PubMed] [Google Scholar]
- Cacabelos R, Franco-Maside A, Alvarez XA. Interleukin-1 in Alzheimer’s disease and multi-infarct dementia: neuropsychological correlations. Methods Find Exp Clin Pharmacol. 1991;13:703–708. [PubMed] [Google Scholar]
- Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, Jones T, Banati RB. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358:461–467. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- Chakraborty S, Kaushik DK, Gupta M, Basu A. Inflammasome signaling at the heart of central nervous system pathology. J Neurosci Res. 2010;88:1615–1631. doi: 10.1002/jnr.22343. [DOI] [PubMed] [Google Scholar]
- Chandrasekar B, Valente AJ, Freeman GL, Mahimainathan L, Mummidi S. Interleukin-18 induces human cardiac endothelial cell death via a novel signaling pathway involving NF-kappaB-dependent PTEN activation. Biochem Biophys Res Commun. 2006;339:956–963. doi: 10.1016/j.bbrc.2005.11.100. [DOI] [PubMed] [Google Scholar]
- Cho H, Wu M, Zhang L, Thompson R, Nath A, Chan C. Signaling dynamics of palmitate-induced ER stress responses mediated by ATF4 in HepG2 cells. BMC Syst Biol. 2013;7:9. doi: 10.1186/1752-0509-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colotta F, Re F, Muzio M, Bertini R, Polentarutti N, Sironi M, Giri JG, Dower SK, Sims JE, Mantovani A. Interleukin-1 type II receptor: a decoy target for IL-1 that is regulated by IL-4. Science. 1993;261:472–475. doi: 10.1126/science.8332913. [DOI] [PubMed] [Google Scholar]
- Comim CM, Reis PA, Frutuoso VS, Fries GR, Fraga DB, Kapczinski F, Zugno AI, Barichello T, Quevedo J, Castro-Faria-Neto HC. Effects of experimental cerebral malaria in memory, brain-derived neurotrophic factor and acetylcholinesterase activity [correction for acitivity] in the hippocampus of survivor mice. Neurosci Lett. 2012;523:104–107. doi: 10.1016/j.neulet.2012.06.051. [DOI] [PubMed] [Google Scholar]
- Conti B, Park LC, Calingasan NY, Kim Y, Kim H, Bae Y, Gibson GE, Joh TH. Cultures of astrocytes and microglia express interleukin 18. Brain Res Mol Brain Res. 1999;67:46–52. doi: 10.1016/s0169-328x(99)00034-0. [DOI] [PubMed] [Google Scholar]
- Craft JM, Watterson DM, Hirsch E, Van Eldik LJ. Interleukin 1 receptor antagonist knockout mice show enhanced microglial activation and neuronal damage induced by intracerebroventricular infusion of human beta-amyloid. J Neuroinflammation. 2005;2:15. doi: 10.1186/1742-2094-2-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craft JM, Watterson DM, Van Eldik LJ. Human amyloid beta-induced neuroinflammation is an early event in neurodegeneration. Glia. 2006;53:484–490. doi: 10.1002/glia.20306. [DOI] [PubMed] [Google Scholar]
- Culhane AC, Hall MD, Rothwell NJ, Luheshi GN. Cloning of rat brain interleukin-18 cDNA. Mol Psychiatry. 1998;3:362–366. doi: 10.1038/sj.mp.4000389. [DOI] [PubMed] [Google Scholar]
- Curran BP, O’Connor JJ. The inhibition of long-term potentiation in the rat dentate gyrus by pro-inflammatory cytokines is attenuated in the presence of nicotine. Neurosci Lett. 2003;344:103–106. doi: 10.1016/s0304-3940(03)00440-3. [DOI] [PubMed] [Google Scholar]
- D’Andrea MR, Cole GM, Ard MD. The microglial phagocytic role with specific plaque types in the Alzheimer disease brain. Neurobiol Aging. 2004;25:675–683. doi: 10.1016/j.neurobiolaging.2003.12.026. [DOI] [PubMed] [Google Scholar]
- Das S, Basu A. Inflammation: a new candidate in modulating adult neurogenesis. J Neurosci Res. 2008;86:1199–1208. doi: 10.1002/jnr.21585. [DOI] [PubMed] [Google Scholar]
- Das S, Mishra MK, Ghosh J, Basu A. Japanese Encephalitis Virus infection induces IL-18 and IL-1beta in microglia and astrocytes: correlation with in vitro cytokine responsiveness of glial cells and subsequent neuronal death. J Neuroimmunol. 2008;195:60–72. doi: 10.1016/j.jneuroim.2008.01.009. [DOI] [PubMed] [Google Scholar]
- de Rivero Vaccari JP, Bastien D, Yurcisin G, Pineau I, Dietrich WD, De Koninck Y, Keane RW, Lacroix S. P2X4 receptors influence inflammasome activation after spinal cord injury. J Neurosci. 2012;32:3058–3066. doi: 10.1523/JNEUROSCI.4930-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rivero Vaccari JP, Lotocki G, Alonso OF, Bramlett HM, Dietrich WD, Keane RW. Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. J Cereb Blood Flow Metab. 2009;29:1251–1261. doi: 10.1038/jcbfm.2009.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rivero Vaccari JP, Lotocki G, Marcillo AE, Dietrich WD, Keane RW. A molecular platform in neurons regulates inflammation after spinal cord injury. J Neurosci. 2008;28:3404–3414. doi: 10.1523/JNEUROSCI.0157-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deniz-Naranjo MC, Munoz-Fernandez C, Alemany-Rodriguez MJ, Perez-Vieitez MC, Aladro-Benito Y, Irurita-Latasa J, Sanchez-Garcia F. Cytokine IL-1 beta but not IL-1 alpha promoter polymorphism is associated with Alzheimer disease in a population from the Canary Islands, Spain. Eur J Neurol. 2008;15:1080–1084. doi: 10.1111/j.1468-1331.2008.02252.x. [DOI] [PubMed] [Google Scholar]
- Desikan RS, Cabral HJ, Hess CP, Dillon WP, Glastonbury CM, Weiner MW, Schmansky NJ, Greve DN, Salat DH, Buckner RL, Fischl B. Automated MRI measures identify individuals with mild cognitive impairment and Alzheimer’s disease. Brain. 2009;132:2048–2057. doi: 10.1093/brain/awp123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhopeshwarkar GA, Mead JF. Uptake and transport of fatty acids into the brain and the role of the blood-brain barrier system. Adv Lipid Res. 1973;11:109–142. doi: 10.1016/b978-0-12-024911-4.50010-6. [DOI] [PubMed] [Google Scholar]
- Di Paolo G, Kim TW. Linking lipids to Alzheimer’s disease: cholesterol and beyond. Nat Rev Neurosci. 2011;12:284–296. doi: 10.1038/nrn3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Rosa M, Dell’Ombra N, Zambito AM, Malaguarnera M, Nicoletti F, Malaguarnera L. Chitotriosidase and inflammatory mediator levels in Alzheimer’s disease and cerebrovascular dementia. Eur J Neurosci. 2006;23:2648–2656. doi: 10.1111/j.1460-9568.2006.04780.x. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. IL-1: discoveries, controversies and future directions. Eur J Immunol. 2010;40:599–606. doi: 10.1002/eji.201040319. [DOI] [PubMed] [Google Scholar]
- Dong Y, Benveniste EN. Immune function of astrocytes. Glia. 2001;36:180–190. doi: 10.1002/glia.1107. [DOI] [PubMed] [Google Scholar]
- Edison P, Archer HA, Gerhard A, Hinz R, Pavese N, Turkheimer FE, Hammers A, Tai YF, Fox N, Kennedy A, Rossor M, Brooks DJ. Microglia, amyloid, and cognition in Alzheimer’s disease: An [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis. 2008;32:412–419. doi: 10.1016/j.nbd.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Eisenberg SP, Evans RJ, Arend WP, Verderber E, Brewer MT, Hannum CH, Thompson RC. Primary structure and functional expression from complementary DNA of a human interleukin-1 receptor antagonist. Nature. 1990;343:341–346. doi: 10.1038/343341a0. [DOI] [PubMed] [Google Scholar]
- Emmanouilidou E, Elenis D, Papasilekas T, Stranjalis G, Gerozissis K, Ioannou PC, Vekrellis K. Assessment of alpha-synuclein secretion in mouse and human brain parenchyma. PLoS One. 2011;6:e22225. doi: 10.1371/journal.pone.0022225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillit H, Ding WH, Buee L, Kalman J, Altstiel L, Lawlor B, Wolf-Klein G. Elevated circulating tumor necrosis factor levels in Alzheimer’s disease. Neurosci Lett. 1991;129:318–320. doi: 10.1016/0304-3940(91)90490-k. [DOI] [PubMed] [Google Scholar]
- Galimberti D, Scarpini E. Disease-modifying treatments for Alzheimer’s disease. Ther Adv Neurol Disord. 2011;4:203–216. doi: 10.1177/1756285611404470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Calvo M, Peterson EP, Leiting B, Ruel R, Nicholson DW, Thornberry NA. Inhibition of human caspases by peptide-based and macromolecular inhibitors. J Biol Chem. 1998;273:32608–32613. doi: 10.1074/jbc.273.49.32608. [DOI] [PubMed] [Google Scholar]
- Geekiyanage H, Chan C. MicroRNA-137/181c regulates serine palmitoyltransferase and in turn amyloid beta, novel targets in sporadic Alzheimer’s disease. J Neurosci. 2011;31:14820–14830. doi: 10.1523/JNEUROSCI.3883-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geekiyanage H, Upadhye A, Chan C. Inhibition of serine palmitoyltransferase reduces Abeta and tau hyperphosphorylation in a murine model: a safe therapeutic strategy for Alzheimer’s disease. Neurobiol Aging. 2013 doi: 10.1016/j.neurobiolaging.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrmann J, Matsumoto Y, Kreutzberg GW. Microglia: intrinsic immuneffector cell of the brain. Brain Res Brain Res Rev. 1995;20:269–287. doi: 10.1016/0165-0173(94)00015-h. [DOI] [PubMed] [Google Scholar]
- Gemma C, Bickford PC. Interleukin-1beta and caspase-1: players in the regulation of age-related cognitive dysfunction. Rev Neurosci. 2007;18:137–148. doi: 10.1515/revneuro.2007.18.2.137. [DOI] [PubMed] [Google Scholar]
- Gonzalez PV, Schioth HB, Lasaga M, Scimonelli TN. Memory impairment induced by IL-1beta is reversed by alpha-MSH through central melanocortin-4 receptors. Brain Behav Immun. 2009;23:817–822. doi: 10.1016/j.bbi.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Gracie JA. Interleukin-18 as a potential target in inflammatory arthritis. Clin Exp Immunol. 2004;136:402–404. doi: 10.1111/j.1365-2249.2004.02475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin WS, Liu L, Li Y, Mrak RE, Barger SW. Interleukin-1 mediates Alzheimer and Lewy body pathologies. J Neuroinflammation. 2006;3:5. doi: 10.1186/1742-2094-3-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Wong S, Xie W, Lei T, Luo Z. Palmitate modulates intracellular signaling, induces endoplasmic reticulum stress, and causes apoptosis in mouse 3T3-L1 and rat primary preadipocytes. Am J Physiol Endocrinol Metab. 2007;293:E576–586. doi: 10.1152/ajpendo.00523.2006. [DOI] [PubMed] [Google Scholar]
- Gupta S, Knight AG, Keller JN, Bruce-Keller AJ. Saturated long-chain fatty acids activate inflammatory signaling in astrocytes. J Neurochem. 2012;120:1060–1071. doi: 10.1111/j.1471-4159.2012.07660.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halle A, Hornung V, Petzold GC, Stewart CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ, Golenbock DT. The NALP3 inflammasome is involved in the innate immune response to amyloid-beta. Nat Immunol. 2008;9:857–865. doi: 10.1038/ni.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haughey NJ, V, Bandaru V, Bae M, Mattson MP. Roles for dysfunctional sphingolipid metabolism in Alzheimer’s disease neuropathogenesis. Biochim Biophys Acta. 2010;1801:878–886. doi: 10.1016/j.bbalip.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins PN, Lachmann HJ, McDermott MF. Interleukin-1-receptor antagonist in the Muckle-Wells syndrome. N Engl J Med. 2003;348:2583–2584. doi: 10.1056/NEJM200306193482523. [DOI] [PubMed] [Google Scholar]
- Hayden KM, Zandi PP, Khachaturian AS, Szekely CA, Fotuhi M, Norton MC, Tschanz JT, Pieper CF, Corcoran C, Lyketsos CG, Breitner JC, Welsh-Bohmer KA. Does NSAID use modify cognitive trajectories in the elderly? The Cache County study. Neurology. 2007;69:275–282. doi: 10.1212/01.wnl.0000265223.25679.2a. [DOI] [PubMed] [Google Scholar]
- He X, Huang Y, Li B, Gong CX, Schuchman EH. Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol Aging. 2010;31:398–408. doi: 10.1016/j.neurobiolaging.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493:674–678. doi: 10.1038/nature11729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci. 2008;28:8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho GJ, Drego R, Hakimian E, Masliah E. Mechanisms of cell signaling and inflammation in Alzheimer’s disease. Curr Drug Targets Inflamm Allergy. 2005;4:247–256. doi: 10.2174/1568010053586237. [DOI] [PubMed] [Google Scholar]
- Hoffman HM, Rosengren S, Boyle DL, Cho JY, Nayar J, Mueller JL, Anderson JP, Wanderer AA, Firestein GS. Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist. Lancet. 2004;364:1779–1785. doi: 10.1016/S0140-6736(04)17401-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingworth P, Harold D, Jones L, Owen MJ, Williams J. Alzheimer’s disease genetics: current knowledge and future challenges. Int J Geriatr Psychiatry. 2011;26:793–802. doi: 10.1002/gps.2628. [DOI] [PubMed] [Google Scholar]
- Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372:216–223. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- Homayoun P, Rodriguez de Turco EB, Parkins NE, Lane DC, Soblosky J, Carey ME, Bazan NG. Delayed phospholipid degradation in rat brain after traumatic brain injury. J Neurochem. 1997;69:199–205. doi: 10.1046/j.1471-4159.1997.69010199.x. [DOI] [PubMed] [Google Scholar]
- Huang TT, Chong KY, Ojcius DM, Wu YH, Ko YF, Wu CY, Martel J, Lu CC, Lai HC, Young JD. Hirsutella sinensis mycelium suppresses interleukin-1beta and interleukin-18 secretion by inhibiting both canonical and non-canonical inflammasomes. Sci Rep. 2013;3:1374. doi: 10.1038/srep01374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana A, Pahan K. Fibrillar amyloid-beta-activated human astroglia kill primary human neurons via neutral sphingomyelinase: implications for Alzheimer’s disease. J Neurosci. 2010;30:12676–12689. doi: 10.1523/JNEUROSCI.1243-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jander S, Schroeter M, Stoll G. Interleukin-18 expression after focal ischemia of the rat brain: association with the late-stage inflammatory response. J Cereb Blood Flow Metab. 2002;22:62–70. doi: 10.1097/00004647-200201000-00008. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Patterson PH. Cytokine and growth factor involvement in long-term potentiation. Mol Cell Neurosci. 1999;14:273–286. [PubMed] [Google Scholar]
- Jeon GS, Park SK, Park SW, Kim DW, Chung CK, Cho SS. Glial expression of interleukin-18 and its receptor after excitotoxic damage in the mouse hippocampus. Neurochem Res. 2008;33:179–184. doi: 10.1007/s11064-007-9434-6. [DOI] [PubMed] [Google Scholar]
- Johnston H, Boutin H, Allan SM. Assessing the contribution of inflammation in models of Alzheimer’s disease. Biochem Soc Trans. 2011;39:886–890. doi: 10.1042/BST0390886. [DOI] [PubMed] [Google Scholar]
- Julien C, Tremblay C, Phivilay A, Berthiaume L, Emond V, Julien P, Calon F. High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse model. Neurobiol Aging. 2010;31:1516–1531. doi: 10.1016/j.neurobiolaging.2008.08.022. [DOI] [PubMed] [Google Scholar]
- Kanno T, Nagata T, Yamamoto S, Okamura H, Nishizaki T. Interleukin-18 stimulates synaptically released glutamate and enhances postsynaptic AMPA receptor responses in the CA1 region of mouse hippocampal slices. Brain Res. 2004;1012:190–193. doi: 10.1016/j.brainres.2004.03.065. [DOI] [PubMed] [Google Scholar]
- Karmi A, Iozzo P, Viljanen A, Hirvonen J, Fielding BA, Virtanen K, Oikonen V, Kemppainen J, Viljanen T, Guiducci L, Haaparanta-Solin M, Nagren K, Solin O, Nuutila P. Increased brain fatty acid uptake in metabolic syndrome. Diabetes. 2010;59:2171–2177. doi: 10.2337/db09-0138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsel P, Li C, Haroutunian V. Gene expression alterations in the sphingolipid metabolism pathways during progression of dementia and Alzheimer’s disease: a shift toward ceramide accumulation at the earliest recognizable stages of Alzheimer’s disease? Neurochem Res. 2007;32:845–856. doi: 10.1007/s11064-007-9297-x. [DOI] [PubMed] [Google Scholar]
- Kim NG, Lee H, Son E, Kwon OY, Park JY, Park JH, Cho GJ, Choi WS, Suk K. Hypoxic induction of caspase-11/caspase-1/interleukin-1beta in brain microglia. Brain Res Mol Brain Res. 2003;114:107–114. doi: 10.1016/s0169-328x(03)00135-9. [DOI] [PubMed] [Google Scholar]
- Kitazawa M, Cheng D, Tsukamoto MR, Koike MA, Wes PD, Vasilevko V, Cribbs DH, LaFerla FM. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal beta-catenin pathway function in an Alzheimer’s disease model. J Immunol. 2011;187:6539–6549. doi: 10.4049/jimmunol.1100620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kummer JA, Broekhuizen R, Everett H, Agostini L, Kuijk L, Martinon F, van Bruggen R, Tschopp J. Inflammasome components NALP 1 and 3 show distinct but separate expression profiles in human tissues suggesting a site-specific role in the inflammatory response. J Histochem Cytochem. 2007;55:443–452. doi: 10.1369/jhc.6A7101.2006. [DOI] [PubMed] [Google Scholar]
- Lachmann HJ, Quartier P, So A, Hawkins PN. The emerging role of interleukin-1beta in autoinflammatory diseases. Arthritis Rheum. 2011;63:314–324. doi: 10.1002/art.30105. [DOI] [PubMed] [Google Scholar]
- Lau LT, Yu AC. Astrocytes produce and release interleukin-1, interleukin-6, tumor necrosis factor alpha and interferon-gamma following traumatic and metabolic injury. J Neurotrauma. 2001;18:351–359. doi: 10.1089/08977150151071035. [DOI] [PubMed] [Google Scholar]
- Lawson LJ, V, Perry H, Gordon S. Turnover of resident microglia in the normal adult mouse brain. Neuroscience. 1992;48:405–415. doi: 10.1016/0306-4522(92)90500-2. [DOI] [PubMed] [Google Scholar]
- Lee YJ, Han SB, Nam SY, Oh KW, Hong JT. Inflammation and Alzheimer’s disease. Arch Pharm Res. 2010;33:1539–1556. doi: 10.1007/s12272-010-1006-7. [DOI] [PubMed] [Google Scholar]
- Levin-Allerhand JA, Lominska CE, Smith JD. Increased amyloid- levels in APPSWE transgenic mice treated chronically with a physiological high-fat high-cholesterol diet. J Nutr Health Aging. 2002;6:315–319. [PubMed] [Google Scholar]
- Li C, Zhao R, Gao K, Wei Z, Yin MY, Lau LT, Chui D, Hoi Yu AC. Astrocytes: implications for neuroinflammatory pathogenesis of Alzheimer’s disease. Curr Alzheimer Res. 2011;8:67–80. doi: 10.2174/156720511794604543. [DOI] [PubMed] [Google Scholar]
- Lightfield KL, Persson J, Trinidad NJ, Brubaker SW, Kofoed EM, Sauer JD, Dunipace EA, Warren SE, Miao EA, Vance RE. Differential requirements for NAIP5 in activation of the NLRC4 inflammasome. Infect Immun. 2011;79:1606–1614. doi: 10.1128/IAI.01187-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton P. Ischemic cell death in brain neurons. Physiol Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Little JP, Madeira JM, Klegeris A. The saturated fatty acid palmitate induces human monocytic cell toxicity toward neuronal cells: exploring a possible link between obesity-related metabolic impairments and neuroinflammation. J Alzheimers Dis. 2012;30(Suppl 2):S179–183. doi: 10.3233/JAD-2011-111262. [DOI] [PubMed] [Google Scholar]
- Liu L, Chan C. IPAF inflammasome is involved in interleukin-1beta production from astrocytes, induced by palmitate; implications for Alzheimer’s Disease. Neurobiol Aging. 2014;35:309–321. doi: 10.1016/j.neurobiolaging.2013.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Li Y, Van Eldik LJ, Griffin WS, Barger SW. S100B-induced microglial and neuronal IL-1 expression is mediated by cell type-specific transcription factors. J Neurochem. 2005;92:546–553. doi: 10.1111/j.1471-4159.2004.02909.x. [DOI] [PubMed] [Google Scholar]
- Liu L, Martin R, Chan C. Palmitate-activated astrocytes via serine palmitoyltransferase increase BACE1 in primary neurons by sphingomyelinases. Neurobiol Aging. 2013a;34:540–550. doi: 10.1016/j.neurobiolaging.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Martin R, Kohler G, Chan C. Palmitate induces transcriptional regulation of BACE1 and presenilin by STAT3 in neurons mediated by astrocytes. Exp Neurol. 2013b;248:482–490. doi: 10.1016/j.expneurol.2013.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo X, Yang Y, Shen T, Tang X, Xiao Y, Zou T, Xia M, Ling W. Docosahexaenoic acid ameliorates palmitate-induced lipid accumulation and inflammation through repressing NLRC4 inflammasome activation in HepG2 cells. Nutr Metab (Lond) 2012;9:34. doi: 10.1186/1743-7075-9-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maesako M, Uemura K, Kubota M, Kuzuya A, Sasaki K, Asada M, Watanabe K, Hayashida N, Ihara M, Ito H, Shimohama S, Kihara T, Kinoshita A. Environmental enrichment ameliorated high-fat diet-induced Abeta deposition and memory deficit in APP transgenic mice. Neurobiol Aging. 2012a;33:1011, e1011–1023. doi: 10.1016/j.neurobiolaging.2011.10.028. [DOI] [PubMed] [Google Scholar]
- Maesako M, Uemura K, Kubota M, Kuzuya A, Sasaki K, Hayashida N, Asada-Utsugi M, Watanabe K, Uemura M, Kihara T, Takahashi R, Shimohama S, Kinoshita A. Exercise is more effective than diet control in preventing high fat diet-induced beta-amyloid deposition and memory deficit in amyloid precursor protein transgenic mice. J Biol Chem. 2012b;287:23024–23033. doi: 10.1074/jbc.M112.367011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar A, Cruz D, Asamoah N, Buxbaum A, Sohar I, Lobel P, Maxfield FR. Activation of microglia acidifies lysosomes and leads to degradation of Alzheimer amyloid fibrils. Mol Biol Cell. 2007;18:1490–1496. doi: 10.1091/mbc.E06-10-0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaguarnera L, Motta M, Di Rosa M, Anzaldi M, Malaguarnera M. Interleukin-18 and transforming growth factor-beta 1 plasma levels in Alzheimer’s disease and vascular dementia. Neuropathology. 2006;26:307–312. doi: 10.1111/j.1440-1789.2006.00701.x. [DOI] [PubMed] [Google Scholar]
- Prince Martin, Bryce Renata, Ferri Cleusa. World Alzheimer report 2012. ALzheimer Disease International; 2012. [Google Scholar]
- Martinon F, Mayor A, Tschopp J. The inflammasomes: guardians of the body. Annu Rev Immunol. 2009;27:229–265. doi: 10.1146/annurev.immunol.021908.132715. [DOI] [PubMed] [Google Scholar]
- Mathew A, Lindsley TA, Sheridan A, Bhoiwala DL, Hushmendy SF, Yager EJ, Ruggiero EA, Crawford DR. Degraded mitochondrial DNA is a newly identified subtype of the damage associated molecular pattern (DAMP) family and possible trigger of neurodegeneration. J Alzheimers Dis. 2012;30:617–627. doi: 10.3233/JAD-2012-120145. [DOI] [PubMed] [Google Scholar]
- Mawhinney LJ, de Rivero Vaccari JP, Dale GA, Keane RW, Bramlett HM. Heightened inflammasome activation is linked to age-related cognitive impairment in Fischer 344 rats. BMC Neurosci. 2011;12:123. doi: 10.1186/1471-2202-12-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng XF, Wang XL, Tian XJ, Yang ZH, Chu GP, Zhang J, Li M, Shi J, Zhang C. Nod-Like Receptor Protein 1 Inflammasome Mediates Neuron Injury under High Glucose. Mol Neurobiol. 2013 doi: 10.1007/s12035-013-8551-2. [DOI] [PubMed] [Google Scholar]
- Minkiewicz J, de Rivero Vaccari JP, Keane RW. Human astrocytes express a novel NLRP2 inflammasome. Glia. 2013;61:1113–1121. doi: 10.1002/glia.22499. [DOI] [PubMed] [Google Scholar]
- Moll M, Kuemmerle-Deschner JB. Inflammasome and cytokine blocking strategies in autoinflammatory disorders. Clin Immunol. 2013;147:242–275. doi: 10.1016/j.clim.2013.04.008. [DOI] [PubMed] [Google Scholar]
- Monti LD, Landoni C, Setola E, Galluccio E, Lucotti P, Sandoli EP, Origgi A, Lucignani G, Piatti P, Fazio F. Myocardial insulin resistance associated with chronic hypertriglyceridemia and increased FFA levels in Type 2 diabetic patients. Am J Physiol Heart Circ Physiol. 2004;287:H1225–1231. doi: 10.1152/ajpheart.00629.2003. [DOI] [PubMed] [Google Scholar]
- Morales I, Farias G, Maccioni RB. Neuroimmunomodulation in the pathogenesis of Alzheimer’s disease. Neuroimmunomodulation. 2010;17:202–204. doi: 10.1159/000258724. [DOI] [PubMed] [Google Scholar]
- Morand O, Baumann N, Bourre JM. In vivo incorporation of exogenous [1–14C]stearic acid into neurons and astrocytes. Neurosci Lett. 1979;13:177–181. doi: 10.1016/0304-3940(79)90038-7. [DOI] [PubMed] [Google Scholar]
- Morris MC, Tangney CC. Diet and prevention of Alzheimer disease. JAMA. 2010;303:2519–2520. doi: 10.1001/jama.2010.844. [DOI] [PubMed] [Google Scholar]
- Motta M, Imbesi R, Di Rosa M, Stivala F, Malaguarnera L. Altered plasma cytokine levels in Alzheimer’s disease: correlation with the disease progression. Immunol Lett. 2007;114:46–51. doi: 10.1016/j.imlet.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Nelson PT, Kryscio RJ, Abner EL, Schmitt FA, Jicha GA, Mendiondo MS, Cooper G, Smith CB, Markesbery WR. Acetylcholinesterase inhibitor treatment is associated with relatively slow cognitive decline in patients with Alzheimer’s disease and AD + DLB. J Alzheimers Dis. 2009;16:29–34. doi: 10.3233/JAD-2009-0926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ojala J, Alafuzoff I, Herukka SK, van Groen T, Tanila H, Pirttila T. Expression of interleukin-18 is increased in the brains of Alzheimer’s disease patients. Neurobiol Aging. 2009;30:198–209. doi: 10.1016/j.neurobiolaging.2007.06.006. [DOI] [PubMed] [Google Scholar]
- Ojala JO, Sutinen EM, Salminen A, Pirttila T. Interleukin-18 increases expression of kinases involved in tau phosphorylation in SH-SY5Y neuroblastoma cells. J Neuroimmunol. 2008;205:86–93. doi: 10.1016/j.jneuroim.2008.09.012. [DOI] [PubMed] [Google Scholar]
- Oksman M, Iivonen H, Hogyes E, Amtul Z, Penke B, Leenders I, Broersen L, Lutjohann D, Hartmann T, Tanila H. Impact of different saturated fatty acid, polyunsaturated fatty acid and cholesterol containing diets on beta-amyloid accumulation in APP/PS1 transgenic mice. Neurobiol Dis. 2006;23:563–572. doi: 10.1016/j.nbd.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Otth C, Concha, Arendt T, Stieler J, Schliebs R, Gonzalez-Billault C, Maccioni RB. AbetaPP induces cdk5-dependent tau hyperphosphorylation in transgenic mice Tg2576. J Alzheimers Dis. 2002;4:417–430. doi: 10.3233/jad-2002-4508. [DOI] [PubMed] [Google Scholar]
- Ozturk C, Ozge A, Yalin OO, Yilmaz IA, Delialioglu N, Yildiz C, Tesdelen B, Kudiaki C. The diagnostic role of serum inflammatory and soluble proteins on dementia subtypes: correlation with cognitive and functional decline. Behav Neurol. 2007;18:207–215. doi: 10.1155/2007/432190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan XD, Zhu YG, Lin N, Zhang J, Ye QY, Huang HP, Chen XC. Microglial phagocytosis induced by fibrillar beta-amyloid is attenuated by oligomeric beta-amyloid: implications for Alzheimer’s disease. Mol Neurodegener. 2011;6:45. doi: 10.1186/1750-1326-6-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil S, Chan C. Palmitic and stearic fatty acids induce Alzheimer-like hyperphosphorylation of tau in primary rat cortical neurons. Neurosci Lett. 2005;384:288–293. doi: 10.1016/j.neulet.2005.05.003. [DOI] [PubMed] [Google Scholar]
- Patil S, Melrose J, Chan C. Involvement of astroglial ceramide in palmitic acid-induced Alzheimer-like changes in primary neurons. Eur J Neurosci. 2007;26:2131–2141. doi: 10.1111/j.1460-9568.2007.05797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil S, Sheng L, Masserang A, Chan C. Palmitic acid-treated astrocytes induce BACE1 upregulation and accumulation of C-terminal fragment of APP in primary cortical neurons. Neurosci Lett. 2006;406:55–59. doi: 10.1016/j.neulet.2006.07.015. [DOI] [PubMed] [Google Scholar]
- Permanne B, Adessi C, Saborio GP, Fraga S, Frossard MJ, Van Dorpe J, Dewachter I, Banks WA, Van Leuven F, Soto C. Reduction of amyloid load and cerebral damage in a transgenic mouse model of Alzheimer’s disease by treatment with a beta-sheet breaker peptide. FASEB J. 2002;16:860–862. doi: 10.1096/fj.01-0841fje. [DOI] [PubMed] [Google Scholar]
- Pizza V, Agresta A, D’Acunto CW, Festa M, Capasso A. Neuroinflamm-aging and neurodegenerative diseases: an overview. CNS Neurol Disord Drug Targets. 2011;10:621–634. doi: 10.2174/187152711796235014. [DOI] [PubMed] [Google Scholar]
- Price DL, Sisodia SS. Cellular and molecular biology of Alzheimer’s disease and animal models. Annu Rev Med. 1994;45:435–446. doi: 10.1146/annurev.med.45.1.435. [DOI] [PubMed] [Google Scholar]
- Puglielli L, Ellis BC, Saunders AJ, Kovacs DM. Ceramide stabilizes beta-site amyloid precursor protein-cleaving enzyme 1 and promotes amyloid beta-peptide biogenesis. J Biol Chem. 2003;278:19777–19783. doi: 10.1074/jbc.M300466200. [DOI] [PubMed] [Google Scholar]
- Qin J, Berdyshev E, Goya J, Natarajan V, Dawson G. Neurons and oligodendrocytes recycle sphingosine 1-phosphate to ceramide: significance for apoptosis and multiple sclerosis. J Biol Chem. 2010;285:14134–14143. doi: 10.1074/jbc.M109.076810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- Roher AE, Weiss N, Kokjohn TA, Kuo YM, Kalback W, Anthony J, Watson D, Luehrs DC, Sue L, Walker D, Emmerling M, Goux W, Beach T. Increased A beta peptides and reduced cholesterol and myelin proteins characterize white matter degeneration in Alzheimer’s disease. Biochemistry. 2002;41:11080–11090. doi: 10.1021/bi026173d. [DOI] [PubMed] [Google Scholar]
- Rossi D, Volterra A. Astrocytic dysfunction: insights on the role in neurodegeneration. Brain Res Bull. 2009;80:224–232. doi: 10.1016/j.brainresbull.2009.07.012. [DOI] [PubMed] [Google Scholar]
- Rossi F, Bianchini E. Synergistic induction of nitric oxide by beta-amyloid and cytokines in astrocytes. Biochem Biophys Res Commun. 1996;225:474–478. doi: 10.1006/bbrc.1996.1197. [DOI] [PubMed] [Google Scholar]
- Rothwell NJ. Annual review prize lecture cytokines - killers in the brain? J Physiol. 1999;514(Pt 1):3–17. doi: 10.1111/j.1469-7793.1999.003af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-Perez JM, Morillas-Ruiz JM. A review: inflammatory process in Alzheimer’s disease, role of cytokines. Scientific World Journal. 2012;2012:756357. doi: 10.1100/2012/756357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saddoughi SA, Ogretmen B. Diverse functions of ceramide in cancer cell death and proliferation. Adv Cancer Res. 2013;117:37–58. doi: 10.1016/B978-0-12-394274-6.00002-9. [DOI] [PubMed] [Google Scholar]
- Salminen A, Ojala J, Suuronen T, Kaarniranta K, Kauppinen A. Amyloid-beta oligomers set fire to inflammasomes and induce Alzheimer’s pathology. J Cell Mol Med. 2008;12:2255–2262. doi: 10.1111/j.1582-4934.2008.00496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarmeas N, Stern Y, Tang MX, Mayeux R, Luchsinger JA. Mediterranean diet and risk for Alzheimer’s disease. Ann Neurol. 2006;59:912–921. doi: 10.1002/ana.20854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt RL, Lenz LL. Distinct licensing of IL-18 and IL-1beta secretion in response to NLRP3 inflammasome activation. PLoS One. 2012;7:e45186. doi: 10.1371/journal.pone.0045186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- Shaftel SS, Griffin WS, O’Banion MK. The role of interleukin-1 in neuroinflammation and Alzheimer disease: an evolving perspective. J Neuroinflammation. 2008;5:7. doi: 10.1186/1742-2094-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng JG, Ito K, Skinner RD, Mrak RE, Rovnaghi CR, Van Eldik LJ, Griffin WS. In vivo and in vitro evidence supporting a role for the inflammatory cytokine interleukin-1 as a driving force in Alzheimer pathogenesis. Neurobiol Aging. 1996;17:761–766. doi: 10.1016/0197-4580(96)00104-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi JQ, Zhang CC, Sun XL, Cheng XX, Wang JB, Zhang YD, Xu J, Zou HQ. Antimalarial Drug Artemisinin Extenuates Amyloidogenesis and Neuroinflammation in APPswe/PS1dE9 Transgenic Mice via Inhibition of Nuclear Factor-kappaB and NLRP3 Inflammasome Activation. CNS Neurosci Ther. 2013 doi: 10.1111/cns.12066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada IS, LeComte MD, Granger JC, Quinlan NJ, Spees JL. Self-renewal and differentiation of reactive astrocyte-derived neural stem/progenitor cells isolated from the cortical peri-infarct area after stroke. J Neurosci. 2012;32:7926–7940. doi: 10.1523/JNEUROSCI.4303-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman WR, de Rivero Vaccari JP, Locovei S, Qiu F, Carlsson SK, Scemes E, Keane RW, Dahl G. The pannexin 1 channel activates the inflammasome in neurons and astrocytes. J Biol Chem. 2009;284:18143–18151. doi: 10.1074/jbc.M109.004804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith QR, Nagura H. Fatty acid uptake and incorporation in brain: studies with the perfusion model. J Mol Neurosci. 2001;16:167–172. doi: 10.1385/JMN:16:2-3:167. discussion 215–121. [DOI] [PubMed] [Google Scholar]
- So A, Ives A, Joosten LA, Busso N. Targeting inflammasomes in rheumatic diseases. Nat Rev Rheumatol. 2013 doi: 10.1038/nrrheum.2013.61. [DOI] [PubMed] [Google Scholar]
- Solfrizzi V, D’Introno A, Colacicco AM, Capurso C, Del Parigi A, Capurso S, Gadaleta A, Capurso A, Panza F. Dietary fatty acids intake: possible role in cognitive decline and dementia. Exp Gerontol. 2005;40:257–270. doi: 10.1016/j.exger.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Srinivasan D, Yen JH, Joseph DJ, Friedman W. Cell type-specific interleukin-1beta signaling in the CNS. J Neurosci. 2004;24:6482–6488. doi: 10.1523/JNEUROSCI.5712-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudduth TL, Schmitt FA, Nelson PT, Wilcock DM. Neuroinflammatory phenotype in early Alzheimer’s disease. Neurobiol Aging. 2013;34:1051–1059. doi: 10.1016/j.neurobiolaging.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugama S, Cho BP, Baker H, Joh TH, Lucero J, Conti B. Neurons of the superior nucleus of the medial habenula and ependymal cells express IL-18 in rat CNS. Brain Res. 2002;958:1–9. doi: 10.1016/s0006-8993(02)03363-2. [DOI] [PubMed] [Google Scholar]
- Sugama S, Conti B. Interleukin-18 and stress. Brain Res Rev. 2008;58:85–95. doi: 10.1016/j.brainresrev.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Sugawara S, Uehara A, Nochi T, Yamaguchi T, Ueda H, Sugiyama A, Hanzawa K, Kumagai K, Okamura H, Takada H. Neutrophil proteinase 3-mediated induction of bioactive IL-18 secretion by human oral epithelial cells. J Immunol. 2001;167:6568–6575. doi: 10.4049/jimmunol.167.11.6568. [DOI] [PubMed] [Google Scholar]
- Suk K, Yeou Kim S, Kim H. Regulation of IL-18 production by IFN gamma and PGE2 in mouse microglial cells: involvement of NF-kB pathway in the regulatory processes. Immunol Lett. 2001;77:79–85. doi: 10.1016/s0165-2478(01)00209-7. [DOI] [PubMed] [Google Scholar]
- Sun A, Liu M, Nguyen XV, Bing G. P38 MAP kinase is activated at early stages in Alzheimer’s disease brain. Exp Neurol. 2003;183:394–405. doi: 10.1016/s0014-4886(03)00180-8. [DOI] [PubMed] [Google Scholar]
- Sutinen EM, Pirttila T, Anderson G, Salminen A, Ojala JO. Pro-inflammatory interleukin-18 increases Alzheimer’s disease-associated amyloid-beta production in human neuron-like cells. J Neuroinflammation. 2012;9:199. doi: 10.1186/1742-2094-9-199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szekely CA, Zandi PP. Non-steroidal anti-inflammatory drugs and Alzheimer’s disease: the epidemiological evidence. CNS Neurol Disord Drug Targets. 2010;9:132–139. doi: 10.2174/187152710791012026. [DOI] [PubMed] [Google Scholar]
- Takechi R, Galloway S, Pallebage-Gamarallage MM, Lam V, Mamo JC. Dietary fats, cerebrovasculature integrity and Alzheimer’s disease risk. Prog Lipid Res. 2010;49:159–170. doi: 10.1016/j.plipres.2009.10.004. [DOI] [PubMed] [Google Scholar]
- Tang BL. Neuronal protein trafficking associated with Alzheimer disease: from APP and BACE1 to glutamate receptors. Cell Adh Migr. 2009;3:118–128. doi: 10.4161/cam.3.1.7254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey MG, McCoy MK, Frank-Cannon TC. Neuroinflammatory mechanisms in Parkinson’s disease: potential environmental triggers, pathways, and targets for early therapeutic intervention. Exp Neurol. 2007;208:1–25. doi: 10.1016/j.expneurol.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trendelenburg G. Acute neurodegeneration and the inflammasome: central processor for danger signals and the inflammatory response? J Cereb Blood Flow Metab. 2008;28:867–881. doi: 10.1038/sj.jcbfm.9600609. [DOI] [PubMed] [Google Scholar]
- van de Veerdonk FL, Netea MG, Dinarello CA, Joosten LA. Inflammasome activation and IL-1beta and IL-18 processing during infection. Trends Immunol. 2011;32:110–116. doi: 10.1016/j.it.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Varnum MM, Ikezu T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer’s disease brain. Arch Immunol Ther Exp (Warsz) 2012;60:251–266. doi: 10.1007/s00005-012-0181-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker DG, Dalsing-Hernandez JE, Campbell NA, Lue LF. Decreased expression of CD200 and CD200 receptor in Alzheimer’s disease: a potential mechanism leading to chronic inflammation. Exp Neurol. 2009;215:5–19. doi: 10.1016/j.expneurol.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SW, Wang M, Grossman BM, Martin RJ. Effects of dietary fat on food intake and brain uptake and oxidation of fatty acids. Physiol Behav. 1994;56:517–522. doi: 10.1016/0031-9384(94)90295-x. [DOI] [PubMed] [Google Scholar]
- Wang Z, Liu D, Wang F, Liu S, Zhao S, Ling EA, Hao A. Saturated fatty acids activate microglia via Toll-like receptor 4/NF-kappaB signalling. Br J Nutr. 2012;107:229–241. doi: 10.1017/S0007114511002868. [DOI] [PubMed] [Google Scholar]
- Webb AC, Collins KL, Auron PE, Eddy RL, Nakai H, Byers MG, Haley LL, Henry WM, Shows TB. Interleukin-1 gene (IL1) assigned to long arm of human chromosome 2. Lymphokine Res. 1986;5:77–85. [PubMed] [Google Scholar]
- Weber A, Wasiliew P, Kracht M. Interleukin-1beta (IL-1beta) processing pathway. Sci Signal. 2010;3:cm2. doi: 10.1126/scisignal.3105cm2. [DOI] [PubMed] [Google Scholar]
- Wenk GL. Neuropathologic changes in Alzheimer’s disease. J Clin Psychiatry. 2003;64(Suppl 9):7–10. [PubMed] [Google Scholar]
- Whitmer RA, Gunderson EP, Barrett-Connor E, Quesenberry CP, Jr, Yaffe K. Obesity in middle age and future risk of dementia: a 27 year longitudinal population based study. BMJ. 2005;330:1360. doi: 10.1136/bmj.38446.466238.E0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RW, Herrup K. The control of neuron number. Annu Rev Neurosci. 1988;11:423–453. doi: 10.1146/annurev.ne.11.030188.002231. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Kiyota T, Horiba M, Buescher JL, Walsh SM, Gendelman HE, Ikezu T. Interferon-gamma and tumor necrosis factor-alpha regulate amyloid-beta plaque deposition and beta-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol. 2007;170:680–692. doi: 10.2353/ajpath.2007.060378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki Y, Matsuura N, Shozuhara H, Onodera H, Itoyama Y, Kogure K. Interleukin-1 as a pathogenetic mediator of ischemic brain damage in rats. Stroke. 1995;26:676–680. doi: 10.1161/01.str.26.4.676. discussion 681. [DOI] [PubMed] [Google Scholar]
- Yang-Wei Fann D, Lee SY, Manzanero S, Tang SC, Gelderblom M, Chunduri P, Bernreuther C, Glatzel M, Cheng YL, Thundyil J, Widiapradja A, Lok KZ, Foo SL, Wang YC, Li YI, Drummond GR, Basta M, Magnus T, Jo DG, Mattson MP, Sobey CG, Arumugam TV. Intravenous immunoglobulin suppresses NLRP1 and NLRP3 inflammasome-mediated neuronal death in ischemic stroke. Cell Death Dis. 2013;4:e790. doi: 10.1038/cddis.2013.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JT, Tan L, Song JH, Sun YP, Chen W, Miao D, Tian Y. Interleukin-18 promoter polymorphisms and risk of late onset Alzheimer’s disease. Brain Res. 2009a;1253:169–175. doi: 10.1016/j.brainres.2008.11.083. [DOI] [PubMed] [Google Scholar]
- Yu Y, He J, Zhang Y, Luo H, Zhu S, Yang Y, Zhao T, Wu J, Huang Y, Kong J, Tan Q, Li XM. Increased hippocampal neurogenesis in the progressive stage of Alzheimer’s disease phenotype in an APP/PS1 double transgenic mouse model. Hippocampus. 2009b;19:1247–1253. doi: 10.1002/hipo.20587. [DOI] [PubMed] [Google Scholar]
- Zou J, Crews FT. Inflammasome-IL-1beta Signaling Mediates Ethanol Inhibition of Hippocampal Neurogenesis. Front Neurosci. 2012;6:77. doi: 10.3389/fnins.2012.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]