

Abstract
BACKGROUND
Volatile anesthetics cause widespread apoptosis in the developing brain. Carbon monoxide (CO) has antiapoptotic properties, and exhaled endogenous CO is commonly rebreathed during low-flow anesthesia in infants and children, resulting in subclinical CO exposure. Thus, we aimed to determine whether CO could limit isoflurane-induced apoptosis in the developing brain.
METHODS
Seven-day-old male CD-1 mouse pups underwent 1-hour exposure to 0 (air), 5, or 100 ppm CO in air with or without isoflurane (2%). We assessed carboxyhemoglobin levels, cytochrome c peroxidase activity, and cytochrome c release from forebrain mitochondria after exposure and quantified the number of activated caspase-3 positive cells and TUNEL positive nuclei in neocortex, hippocampus, and hypothalamus/thalamus.
RESULTS
Carboxyhemoglobin levels approximated those expected in humans after a similar time-weighted CO exposure. Isoflurane significantly increased cytochrome c peroxidase activity, cytochrome c release, the number of activated caspase-3 cells, and TUNEL positive nuclei in the forebrain of air-exposed mice. CO, however, abrogated isoflurane-induced cytochrome c peroxidase activation and cytochrome c release from forebrain mitochondria and decreased the number of activated caspase-3 positive cells and TUNEL positive nuclei after simultaneous exposure with isoflurane.
CONCLUSIONS
Taken together, the data indicate that CO can limit apoptosis after isoflurane exposure via inhibition of cytochrome c peroxidase depending on concentration. Although it is unknown whether CO directly inhibited isoflurane-induced apoptosis, it is possible that low-flow anesthesia designed to target rebreathing of specific concentrations of CO may be a desired strategy to develop in the future in an effort to prevent anesthesia-induced neurotoxicity in infants and children.
Anumber of commonly used anesthetic drugs cause widespread neuronal apoptosis in the developing mammalian brain.1–5 Vulnerability coincides with the period of synaptogenesis, and anesthesia-induced neurotoxicity has been shown to result in significant neuron loss, behavioral impairments, and cognitive deficits in a variety of newborn animal models.6,7 Although a causal relationship in humans has yet to be demonstrated, evidence indicating an association between anesthesia exposure and cognitive and behavioral disorders in young children continues to emerge.8–10 Thus, there is a need to develop protective strategies to prevent potential anesthesia-induced neurodegeneration in infants and children.
The exact upstream mechanisms that initiate anesthesia-induced neurotoxicity are not completely understood; however, downstream, the process is mediated by the mitochondrial pathway of apoptosis.6,11 After anesthetic exposure, Bax translocates to the outer mitochondrial membrane, resulting in mitochondrial permeabilization, release of cytochrome c, widespread caspase-3 activation, and DNA fragmentation.6 Upstream of this phenomenon, cytochrome c is bound to cardiolipin on the inner mitochondrial membrane via electrostatic and hydrophobic interactions.12 Cytochrome c has peroxidase activity and, in the presence of hydrogen peroxide, oxidizes cardiolipin to hydroperoxycardiolipin.12 This mobilizes cytochrome c from the inner membrane and permits it to be released after permeabilization of the outer mitochondrial membrane.
Carbon monoxide (CO) is a colorless and odorless gas that has antiapoptotic properties.13–18 CO prevents apoptosis by binding to the cytochrome c-cardiolipin complex and inhibiting cytochrome c peroxidase activity.12,19 This prevents oxidation of cardiolipin, mobilization and release of cytochrome c, and subsequent caspase activation. It has been demonstrated that brief exposure to low concentrations of CO inhibits developmental programmed cell death in vivo in the forebrain of newborn mice.19 It is important to note that infants and children are routinely exposed to CO during low-flow anesthesia when rebreathing is permitted.20,21 The source of CO in this setting is likely exhaled endogenous CO generated via heme catabolism.21 Because exhaled CO is not scavenged or removed from the anesthesia breathing circuit, during low-flow anesthesia, patients rebreathe exhaled CO and experience a subclinical CO exposure.21,22 In this work, we aimed to determine whether the antiapoptotic effects of subclinical concentrations of inspired CO could limit anesthesia-induced neuronal apoptosis. We demonstrated that CO exposure limits apoptosis in a variety of brain regions in newborn mice exposed to isoflurane by inhibiting cytochrome c peroxidase activity and subsequent cytochrome c release. These findings are clinically relevant and could have implications for the development of low-flow anesthesia as a standard paradigm to target low CO concentration exposures in infants and children to prevent anesthesia-induced neurotoxicity.
METHODS
Animal Exposures
The care of the animals in this study was in accordance with National Institutes of Health and Institutional Animal Care and Use Committee guidelines. Study approval was granted by the Children’s National Medical Center. Sixto 8-week-old CD-1 pregnant female mice (20–30 grams) were acquired (Charles River, Wilmington, MA) to yield newborn pups. CD-1 mice were chosen because pups have been shown to reliably demonstrate neuronal changes consistent with human neonatal injury in specific experimental models.23 On postnatal day 7 (P7), we exposed male CD-1 mouse pups to 0 ppm CO (air), 5 ppm CO in air, or 100 ppm CO in air with and without isoflurane (2%) for 1 hour in a 7-L Plexiglas chamber (25 × 20 × 14 cm). The 3 experimental CO cohorts represented: negative control (0 ppm CO), low concentration subclinical CO (5 ppm), and high concentration subclinical CO (100 ppm). The chamber had a port for fresh gas inlet and a port for gas outlet that was directed to a fume hood exhaust using standard suction tubing. Specific concentrations of CO in air (premixed gas H-cylinders, Air Products, Camden, NJ) were verified using an electrochemical sensing CO detector (Monoxor III, Bacharach, Anderson, CA). Designated CO mixtures were delivered through the variable bypass isoflurane vaporizer and exposure chamber at a flow rate of 8 to 12 L/min. Mice were kept warm with an infrared heating lamp (Cole-Parmer, Court Vernon Hills, IL). P7 was chosen because synaptogenesis peaks at day 7 in rodents and is completed by the second or third week of life.24,25 One hour exposure to 2% isoflurane has been shown to activate brain capsase-3 in 7-day-old mice and is a brief anesthetic exposure.26 After exposure, pups were placed with their respective dams. Eighty-four newborn mice were evaluated.
Carboxyhemoglobin (COHb) Levels
COHb levels were measured immediately after 1-hour exposure. At the time of euthanasia, after pentobarbital injection (150 mg/kg, intraperitoneal), 200 μL blood was sampled from the left ventricle and COHb measured via 6 wavelength co-oximetry (Radiometer Osm3 Hemoximeter, Copenhagen, Denmark, range 0–100 ± 0.2%). Five animals per cohort were evaluated.
Activated Caspase-3 Immunohistochemistry
Five hours after exposure, following euthanasia with pentobarbital injection (150 mg/kg, ip), the brain was perfused with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) via left ventricle injection for 30 minutes and then postfixed in additional fixative solution for 24 hours at 4°C. Serial sections were cut at a thickness of 6 μm in the coronal plane through the cerebral hemispheres beginning at −1.7 mm from bregma and 2.1 mm from interaural, and individual sections were slide mounted. Immunohistochemistry was performed on 3 to 4 nonserial nonadjacent sections using polyclonal antirabbit activated caspase-3 (Cell Signaling Technology, Beverly, MA), biotinylated secondary antibody (goat antirabbit, Cell Signaling Technology), and developed with diaminobenzidine. Nuclei were counterstained with hematoxylin. The number of activated caspase-3 positive cells per square millimeter was quantified at ×10 magnification in neocortex (primary and secondary somatosensory and auditory neocortices), hippocampus (dentate gyrus, CA1, CA2, and CA3 regions), and hypothalamic/thalamic region (laterodorsal, mediodorsal, ventromedial, ventrolateral, ventroposteromedial, ventroposterolateral thalamic nuclei, ventromedial hypothalamic nucleus, peduncular part of the lateral hypothalamus, and the central anterior hypothalamic area) of both hemispheres in 3 to 4 animals per group. Brain regions were defined in accordance with Mouse Brain Atlas.27,28
Terminal Deoxynucleotidyl Transferase-Mediated UTP Nick End-Labeling Staining
Five hours after exposure, following euthanasia, the brain was perfused with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) via left ventricle injection for 30 minutes and then postfixed in additional fixative solution for 24 hours at 4°C. Paraffin-embedded brain sections were cut into 6-μm sections in the coronal plane through the cerebral hemispheres beginning at −1.7 mm from bregma, 2.1 mm from interaural, slide mounted, and stained for terminal deoxynucleotidyl transferase-mediated UTP nick end-labeling (TUNEL). Sections were incubated in 0.5% Triton at room temperature, followed by proteinase K at 37°C, then immersed in terminal deoxynucleotidyl transferase (TdT) buffer (30 mmol/L Tris-HCl buffer, pH 7.2, 140 mmol/L sodium cacodylate, and 1 mmol/L cobalt chloride) at room temperature. This was followed by incubation with TdT and biotin-16-dUTP for 60 minutes at 37°C. The reaction was terminated with TB buffer (300 mmol/L sodium chloride with 30 mmol/L sodium citrate) at room temperature, followed by immersion in 3% hydrogen peroxide and 2% fetal bovine serum at room temperature. The sections were then covered with an Avidin Biotin Complex (1:200 dilution) for 30 minutes at room temperature, incubated with FITCAvidin D for detection, and counterstained with DAPI. The numbers of TUNEL positive nuclei in neocortex, hippocampus, and hypothalamic/thalamic region (identical regions as for activated caspase-3) were quantified at ×10 magnification in 3 to 4 nonserial sections per mouse, and 3 to 4 mice per cohort were evaluated. Brain regions were defined in accordance with Mouse Brain Atlas.27,28
Cytochrome C Peroxidase Activity
Immediately after 1-hour exposure, cytochrome c was extracted from fresh mitochondria as previously described.29 Isolated forebrain mitochondria (20 mg/mL) were suspended in a hypotonic 0.015 M KCl solution for 10 minutes on ice and then centrifuged at 105,000g for 15 minutes at 4°C. The pellet was resuspended in 0.15 M KCl solution for 10 minutes on ice and then centrifuged again at 105,000g for 15 minutes at 4°C. The supernatant was collected and cytochrome c content quantified with spectrophotometry. The peroxidase activity of 0.5 to 1 μM cytochrome c was determined by measuring the rate of oxidation of 50 μM 2,2′-azinobis-(2-ethylbenzthiazoline-6-sulfonate) (ABTS) in 10 mM potassium phosphate buffer (pH 7.4) at 415 nm (ε415 = 3.6 × 104 M−1 cm−1) after the addition of hydrogen peroxide.30 Five animals per cohort were evaluated.
Heme C Determination
Immediately after 1-hour exposure, forebrain mitochondria and cytosol were isolated by differential centrifugation.31 As previously described, forebrain was harvested and homogenized in ice-cold H medium (70 mM sucrose, 220 mM mannitol, 2.5 mM Hepes, pH 7.4 and 2 mM EDTA).31 The homogenate was spun at 1500g for 10 minutes at 4°C. Supernatant was removed and centrifuged at 10,000g for 10 minutes at 4°C. Cytosolic supernatant was collected, and pellet was resuspended in H medium and centrifuged again at 10,000g for 10 minutes at 4°C. Pellet was again resuspended in H medium, and mitochondrial and cytosolic protein concentrations subsequently determined using the method of Lowry.31
Mitochondrial and cytosolic heme c content were calculated from the difference in spectra (dithionate/ascorbate reduced minus air-oxidized) of mitochondria or cytosolic protein (0.5–1 mg) solubilized in 10% lauryl maltoside using an absorption coefficient of 20.5 mM−1 cm−1 at 550 to 535 nm.32,33 Five animals per cohort were evaluated.
Statistical Analysis
Sample sizes for each end point were chosen based on previous work.19 Our previous study used 8 animals per cohort for COHb and heme c determination, 3 to 4 animals per cohort for activated caspase-3 and TUNEL assessment, and 5 animals per cohort for measurement of cytochrome c peroxidase activity, and data followed normal probability distribution.19 For this work, sample sizes were based on the number of animals needed to detect a 30% difference from air-exposed control values with a power of 80 based on an α of 0.01. Data are presented as mean ± SE. To assess statistical significance, we performed pairwise comparisons in an analysis of variance design using Tukey test.
RESULTS
Brief Subclinical CO Exposure With and Without Isoflurane in Newborn Mouse Pups
To investigate the effects of subclinical CO on isoflurane-induced neuronal apoptosis, we exposed 7-day-old male CD-1 mouse pups to 0 ppm CO (air), 5 ppm CO (low concentration subclinical exposure), or 100 ppm CO (upper limit of subclinical exposure) for 1 hour with and without isoflurane (2%). P7 was chosen because synaptogenesis peaks on day 7 in rodents and is completed by the second or third week of life.24,25 The exposure time was chosen because inspiring isoflurane for 1 hour has been shown to activate brain capsase-3 in 7-day-old mice and is a clinically relevant anesthetic duration in infants and children.26
COHb levels increased significantly in the cohort exposed to 100 ppm CO without isoflurane with a trend toward significance in animals exposed to 100 ppm with isoflurane (Fig. 1). There was no significant difference in COHb levels in cohorts exposed to 5 ppm CO compared with air-exposed control values (Fig. 1). Isoflurane exposure had no independent effect on COHb levels (P = 0.34 [95% CI, 1.45 ± 0.05]) (Fig. 1). It is important to note that the resultant COHb levels measured after CO exposure in all groups approximated levels expected in humans after a similar time-weighted exposure (e.g., mean COHb of 3.27% in humans after 1-hour exposure to 100 ppm CO, 95% CI ± 0.12) (Fig. 1).34–36 Furthermore, these levels were well below values known to result in tissue hypoxia (70% COHb) and were markedly less than levels known to elicit signs and symptoms in humans (10% COHb).13,37 Thus, 1-hour exposure to 5 or 100 ppm CO in 7-day-old mouse pups was a subclinical CO exposure.
Figure 1.
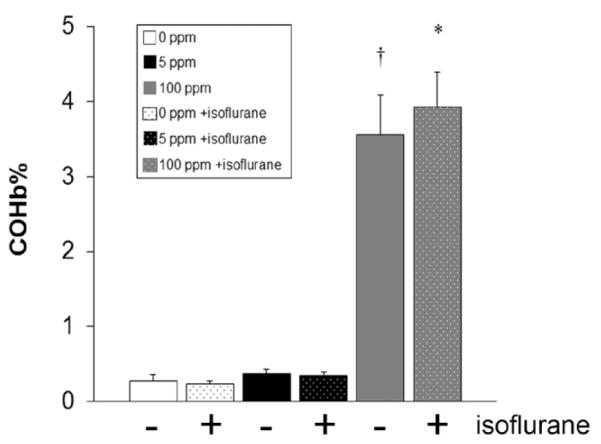
Carboxyhemoglobin (COHb) levels after carbon monoxide (CO) exposure with and without isoflurane. COHb levels were measured immediately after CO exposure with (+) and without (−) isoflurane. Values are expressed as percentage (%) COHb means plus standard error. N = 5 animals per cohort. *P < 0.05 vs air and 5 ppm CO cohorts. †P < 0.01 vs air and 5 ppm CO cohorts.
Brief Subclinical CO Exposure Inhibits Neuronal Apoptosis in Isoflurane-Exposed Mice
Commonly used anesthetics cause widespread apoptosis in the developing mammalian brain.1–5 Thus, we determined the effect of CO on isoflurane-induced neuronal apoptosis in the neocortex, hippocampus, and hypothalamic and thalamic regions by assessing for activated caspase-3 with immunohistochemistry and TdT-mediated TUNEL staining on slide-mounted brain sections. Pups were evaluated 5 hours after simultaneous exposure to either 0 ppm CO (air), 5 ppm CO, or 100 ppm CO with or without isoflurane on P7.
Consistent with previous work, 1-hour exposure to isoflurane in air significantly increased the number of activated caspase-3 positive cells and TUNEL positive nuclei in all brain regions examined compared with nonisoflurane air-exposed animals (Figs. 2 and 3).26 CO significantly decreased the number of activated caspase-3 positive cells and TUNEL positive nuclei in virtually every brain region of both cohorts exposed to CO with isoflurane compared with mice exposed to isoflurane in air (Figs. 2 and 3). It is important to note that activated caspase-3 and the number of TUNEL positive nuclei were at or below air-exposed levels in all brain regions evaluated after 100 ppm CO and isoflurane exposure. In addition, there appeared to be a concentration-dependent CO effect on the number of activated caspase-3 positive cells in the hypothalamus/thalamus and TUNEL positive nuclei in the neocortex and hippocampus of isoflurane-exposed mice.
Figure 2.

Activated caspase-3 after carbon monoxide (CO) exposure with and without isoflurane. Immunohistochemistry for activated caspase-3 was performed on coronal sections 5 hours after exposure. (A) Representative sections imaged at ×10 from somatosensory neocortex (NC), hippocampus (HC), and hypothalamic/thalamic region (H/T) obtained after 1-hour exposure to air (0 ppm CO), 5 ppm CO, or 100 ppm CO with (+) and without (−) isoflurane are depicted. Arrowheads indicate activated caspase-3 stained cells. Activated caspase-3 positive cells undergoing degeneration within the boxed area in each section are magnified in the inset. CA1, CA2, dentate gyrus (DG) regions of HC are labeled. Scale bars, 100 μm. Quantification of activated caspase-3 stained cells in (B) neocortex (C) hippocampus and (D) hypothalamic/thalamic region are demonstrated. Values are expressed as means plus standard error. N = 3–4 animals per cohort. *P < 0.05 vs 0 ppm CO − isoflurane. †P < 0.01 vs 0 ppm CO − isoflurane. ^P < 0.001 vs 0 ppm CO − isoflurane. @ P < 0.05 vs 0 ppm CO + isoflurane. ‡P < 0.01 vs 0 ppm CO + isoflurane. #P < 0.001 vs 0 ppm CO + isoflurane. $P < 0.05 vs 5 ppm CO + isoflurane.
Figure 3.

Apoptosis after carbon monoxide (CO) exposure with and without isoflurane. TUNEL assays were performed on coronal sections 5 hours after exposure. (A) Representative sections imaged at ×10 from somatosensory neocortex (NC), hippocampus (HC), and hypothalamic/thalamic region (H/T) obtained after 1-hour exposure to air (0 ppm CO), 5 ppm CO, or 100 ppm CO with (+) and without (−) isoflurane are depicted. Green TUNEL positive nuclei are visible. CA1, CA2, dentate gyrus (DG) regions of HC are labeled. Scale bars, 100 μm. Quantification of total TUNEL positive nuclei from NC, HC, and H/T in 3–4 nonserial coronal sections are demonstrated in (B) neocortex (C) hippocampus and (D) hypothalamic/thalamic region. Values are expressed as means plus standard error. N = 3–4 animals per cohort. *P < 0.05 vs 0 ppm CO − isoflurane. †P < 0.01 vs 0 ppm CO − isoflurane. ^P < 0.001 vs 0 ppm CO − isoflurane. @ P < 0.05 vs 0 ppm CO + isoflurane. ‡P < 0.01 vs 0 ppm CO + isoflurane. % P < 0.05 vs 5 ppm CO − isoflurane. &P < 0.01 vs 5 ppm CO − isoflurane. $P < 0.05 vs 5 ppm CO + isoflurane.?P < 0.05 vs 100 ppm CO − isoflurane.
Exposure to CO without isoflurane had variable effects on activated caspase-3 and the number of TUNEL positive nuclei compared with air-exposed controls (Figs. 2 and 3). Differences trended toward significance between CO-exposed cohorts without isoflurane in the number of TUNEL positive nuclei in the hippocampus and hypothalamus/thalamus of 100 ppm CO exposed mice vs animals exposed to 5 ppm CO (Fig. 3).
CO Inhibits Forebrain Cytochrome C Peroxidase Activity and Isoflurane-Induced Cytochrome C Release from Mitochondria
Peroxidation of cardiolipin is an upstream event that is important for cytochrome c release and initiation of the mitochondrial apoptosis pathway.12 CO can bind to the cytochrome c-cardiolipin complex and inhibit cytochrome c peroxidase activity, thereby preventing oxidation of cardiolipin and mobilization and release of cytochrome c.12,19 Thus, we extracted cytochrome c from forebrain mitochondria immediately after 1-hour exposure to either 0 ppm CO (air), 5 ppm CO, or 100 ppm CO with or without isoflurane on P7 and measured peroxidase activity of cytochrome c using spectrophotometry. To assess for cytochrome c release, we measured the amount of heme c (the heme moiety of cytochrome c) in forebrain mitochondrial and cytosolic fractions immediately after exposure.
Isoflurane either significantly increased or trended toward a significant increase in forebrain cytochrome c peroxidase activity after 1-hour exposure in air compared with all nonisoflurane-exposed cohorts (Fig. 4). All isoflurane-exposed cohorts demonstrated significantly higher or a trend toward significantly higher cytochrome c peroxidase activity compared with nonisoflurane CO matched cohorts (Fig. 4). CO exposure significantly decreased forebrain cytochrome c peroxidase activity in isoflurane and nonisoflurane-exposed cohorts in a concentration-dependent manner (Fig. 4). CO-mediated inhibition of cytochrome c peroxidase resulted in enzyme activities that were below that of air-exposed control values in all CO-exposed cohorts (Fig. 4).
Figure 4.
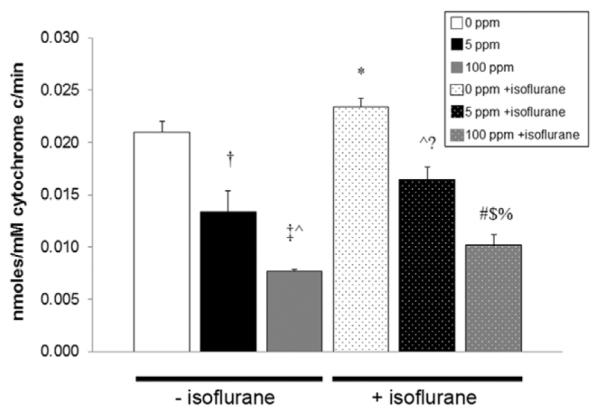
Cytochrome c peroxidase activity after carbon monoxide (CO) exposure with and without isoflurane. Steady-state cytochrome c peroxidase activity immediately after 1-hour exposure is shown. Values are expressed as means plus standard error. N = 5 animals per cohort. *P < 0.05 vs 0 ppm CO − isoflurane, P < 0.001 vs 5 ppm CO − isoflurane, vs 100 ppm CO − isoflurane. †P < 0.01 vs 0 ppm CO − isoflurane. ‡P < 0.001 vs 0 ppm CO − isoflurane. ^P < 0.05 vs 5 ppm CO − isoflurane. @P < 0.01 vs 5 ppm CO − isoflurane. #P < 0.05 vs 100 ppm CO − isoflurane.?P < 0.01 vs 0 ppm CO + isoflurane. $P < 0.01 vs 5 ppm CO + isoflurane. %P < 0.001 vs 0 ppm CO + isoflurane.
After isoflurane exposure for 1 hour in air, heme c levels decreased significantly in the mitochondrial fraction and increased significantly in the cytosolic fraction compared with nonisoflurane air-exposed controls, suggesting increased cytochrome c release in the forebrain after exposure (Fig. 5). CO exposure without isoflurane trended toward a significant increase in the amount of heme c in forebrain mitochondria in 5 ppm exposed animals compared with air-exposed controls (Fig. 5A). Cytosolic heme c content in forebrain decreased significantly in both cohorts exposed to CO without isoflurane compared with air-exposed controls, indicating inhibition of cytochrome c release (Fig. 5B). Exposure to CO with isoflurane resulted in significantly increased heme c levels within forebrain mitochondria of 100 ppm exposed mice and significantly decreased heme c in cytosol of both CO-exposed cohorts compared with animals exposed to isoflurane alone (Fig. 5). Decreases in cytosolic heme c after CO exposure with isoflurane were dose-dependent, and levels were at or below air-exposed control values (Fig. 5B). Mitochondrial heme c levels in animals exposed to isoflurane with 100 ppm CO were equivalent to levels seen in forebrain mitochondria of controls exposed to air alone (Fig. 5A). Taken together, the data suggest that CO inhibits forebrain cytochrome c peroxidase and, depending on concentration, can decrease isoflurane-induced cytochrome c release.
Figure 5.

Cytochrome c release after carbon monoxide (CO) exposure with and without isoflurane. Heme c content within (A) mitochondria and (B) cytosol is demonstrated. Values are expressed as means plus standard error. N = 5 animals per cohort. *P < 0.05 vs 0 ppm CO − isoflurane. †P < 0.01 vs 0 ppm CO − isoflurane. #P < 0.05 vs 5 ppm CO + isoflurane. ‡P < 0.001 vs 0 ppm CO − isoflurane. ^P < 0.01 vs 0 ppm CO and 5 ppm + isoflurane. @P < 0.01 vs 5 ppm CO − isoflurane.
DISCUSSION
Our findings are consistent with previous work and support the concept that isoflurane causes neurotoxicity in the developing mammalian brain via activation of the intrinsic apoptosis pathway.6,11 In addition, we demonstrate for the first time that, upstream from cytochrome c release, isoflurane increases forebrain cytochrome c peroxidase activity. It is important to note that subclinical concentrations of CO inhibited isoflurane-induced cytochrome c peroxidase activation in a dose-dependent manner and decreased the release of cytochrome c, limiting apoptosis in the developing brain after exposure to isoflurane.
Because hydrogen peroxide is required for induction of cytochrome c peroxidase, increased peroxidase activity likely resulted from isoflurane-induced oxidative stress.38,39 Although we did not measure free radicals as part of this study, isoflurane and other anesthetic drugs generate reactive oxygen, nitrogen species, and hydrogen peroxide in developing neurons, hippocampus, subiculum, and thalamus.40–42 In addition, cytochrome c peroxidase activity has been shown to increase in the forebrain of mice during oxidative stress, and cytochrome c has been developed as a biosensor for hydrogen peroxide detection.43,44 Thus, isoflurane exposure likely increased hydrogen peroxide production that, in turn, enhanced the peroxidase activity of cytochrome c.38
Finding increased cytochrome c peroxidase activity after isoflurane exposure is significant because it uncovers a potential target for therapeutic intervention. Peroxidase activation is necessary and critical for mobilization and release of cytochrome c from mitochondria during apoptosis.12 Release of cytochrome c is often considered the “point of no return” in the pathway.45 Thus, targeting the peroxidase activity of cytochrome c is logical and inhibiting cytochrome c peroxidase could prevent cytochrome c release during proapoptotic stimuli such as with anesthetic exposure. In support of this concept, low concentrations of inhaled CO (50–500 ppm) have been shown to prevent apoptosis in endothelium, vascular smooth muscle, liver, lung tissue during hyperoxia, sepsis, and ischemia-reperfusion.14–18 In previous work, we demonstrated that brief exposure to low CO concentrations can inhibit cytochrome c peroxidase in vivo and impair programmed cell death in the developing forebrain of newborn mice.19 Here we demonstrate that CO, inspired at subclinical concentrations, inhibits activation of cytochrome c peroxidase during isoflurane exposure and impairs release of cytochrome c.
CO prevents apoptosis by binding to the cytochrome c-cardiolipin complex and inhibiting cytochrome c peroxidase activity.12 This prevents oxidation of cardiolipin and subsequent release of cytochrome c.12 Although it is possible that CO may exert its prosurvival response via a variety of other mechanisms, our data suggest that abrogation of isoflurane-induced apoptosis may be due to CO-mediated inhibition of cytochrome c peroxidase activation. This is supported by finding concentration-dependent responses to subclinical CO exposure within different aspects of the intrinsic apoptosis pathway that we assayed (cytochrome c peroxidase, cytochrome c release, activation of caspase-3, and DNA breakage [TUNEL]).
Although the data indicate that low concentrations of CO can inhibit apoptosis in the developing brain, we have not conclusively shown that CO directly prevents isoflurane-induced neuronal apoptosis. This is important because the CO effect could simply be a generalized, nonspecific phenomenon. Thus, it is possible that CO-mediated inhibition of apoptosis and anesthetic-induced apoptosis are 2 distinct processes that occur simultaneously and independently within the developing brain but not necessarily in the same cells. If these 2 processes are mutually exclusive, then certain cell populations would undergo apoptosis after exposure to isoflurane while totally different cell populations, destined to die via developmental programmed cell death, would survive due to CO-mediated inhibition of the intrinsic apoptosis pathway. Although the total number of neurons undergoing apoptosis in this scenario would be relatively decreased compared with isoflurane exposure alone, the impact on neurodevelopment could be devastating. This is because we have previously demonstrated that exposure to low concentrations of CO in the absence of an anesthetic prevents natural programmed cell death in the neocortex and hippocampus of 10-day-old mice and impairs neurocognitive development.19 Thus, although CO has antiapoptotic properties, its unchecked effects in the developing brain could be equally as deleterious as the effects of anesthetics alone.
Furthermore, brief, low concentration CO exposures can also cause oxidative stress.46 Such undesired effects could enhance apoptosis during development and could explain our findings regarding activated caspase-3 and TUNEL in the cohorts exposed to CO for 1 hour without isoflurane. So it is possible that CO has the potential to exert proapoptotic effects that could act synergistically with isoflurane. Thus, before implementing subclinical CO exposure in everyday clinical practice, its safety and efficacy must be established given the fact that its pro- and antiapoptotic effects could have adverse consequences in the developing brain.
The only way to prove that CO directly prevents the proapoptotic effect of isoflurane is to evaluate neurocognitive function and behavior after exposure. The fact that we have not included such an assessment in this work is a major limitation of the study. However, this will be the focus of future work. Thus, until we determine neurodevelopmental outcome after combined exposure to CO with isoflurane, we cannot draw definitive conclusions about the benefit of CO during an anesthetic.
Regarding clinical toxicity, elevated COHb levels are formed after exposure to high concentrations of CO and can interfere with tissue oxygen delivery by impairing oxygen binding to and dissociation from hemoglobin.37 Overt clinical toxicity manifests from tissue hypoxia when COHb levels are higher than 70%, and signs and symptoms first appear when COHb levels are higher than 10%.13,37 Acute exposure to CO concentrations larger than 800 ppm can rapidly cause brain injury, cerebral edema, coma, and death, while brief exposure to 220 ppm results in headache, dizziness, and impaired judgment.34 Exposure to concentrations <120 ppm does not elicit any appreciable clinical symptoms.34 Thus, short-term exposure to 5 or 100 ppm CO is a non–life-threatening subclinical exposure, and the resultant COHb levels are well below values that cause tissue hypoxia and clinical signs and symptoms.
CO is endogenously produced, and infants and children routinely inspire subclinical concentrations of CO when rebreathing is permitted during low-flow general anesthesia.20,21,47 In previous work, we found that children inspire an average of 2 to 3 ppm CO and as much as 18 ppm CO during low-flow anesthesia.20,21 This resulted in an increase in COHb by an average of 0.2% up to 0.5% from baseline after 1 hour of low-flow anesthesia.20 In the current work, we found a similar increase in COHb after 1-hour exposure to 5 ppm CO and a 3% to 4% increase after exposure to 100 ppm CO. Thus, exposing newborn mice to 5 ppm CO with isoflurane mimics a subclinical CO exposure during lowflow anesthesia at a time point in late infancy.
Here we found that 5 ppm CO limited apoptosis after isoflurane exposure while 100 ppm CO maintained levels of apoptosis at or below control values. These findings suggest that higher concentrations of subclinical CO may be necessary to completely offset the proapoptotic response to isoflurane. Thus, it is possible that levels of CO encountered with rebreathing during routine lowflow anesthesia may limit isoflurane-induced neurotoxicity but may not be adequate to completely prevent it. However, more investigation is necessary before such conclusions can be made.
Targeting cytochrome c peroxidase to offset the oxidative stress of an anesthetic exposure is a novel and intriguing concept. Given that CO is commonly rebreathed during lowflow anesthesia and readily diffuses across the blood-brain barrier to gain access to the mitochondrial inner membrane, it has the potential to be developed as an antiapoptotic agent to prevent anesthesia-induced neurotoxicity.48 However, before routine application of CO as such a therapeutic agent, neurodegeneration needs to be definitively shown to occur in children after anesthetic exposure, CO must be shown to directly inhibit isoflurane-induced apoptosis, and the safety of subclinical CO exposure during development needs to be established. With further work, however, it is possible that low-flow anesthesia, intended to result in low concentration CO exposure, may be established as a standard paradigm designed to protect the developing brain of the infants and children we care for.
Acknowledgments
Funding: None.
Footnotes
DISCLOSURES
Name: Ying Cheng
Contribution: This author helped collect data, analyzed the data, and contributed in study design.
Attestation: Ms. Cheng attests to the integrity of the data and the analysis and approves the final manuscript. She is the archival author.
Name: Richard J. Levy, MD.
Contribution: This author designed the study, analyzed the data, and prepared the manuscript.
Attestation: Dr. Levy attests to the integrity of the data and the analysis and approves the final manuscript.
This manuscript was handled by: Gregory J. Crosby, MD.
The authors declare no conflicts of interest.
Reprints will not be available from the authors.
REFERENCES
- 1.Jevtovic-Todorovic V, Hartman RE, Izumi Y, Benshoff ND, Dikranian K, Zorumski CF, Olney JW, Wozniak DF. Early exposure to common anesthetic agents causes widespread neurode-generation in the developing rat brain and persistent learning deficits. J Neurosci. 2003;23:876–82. doi: 10.1523/JNEUROSCI.23-03-00876.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stefovska VG, Uckermann O, Czuczwar M, Smitka M, Czuczwar P, Kis J, Kaindl AM, Turski L, Turski WA, Ikonomidou C. Sedative and anticonvulsant drugs suppress postnatal neurogenesis. Ann Neurol. 2008;64:434–45. doi: 10.1002/ana.21463. [DOI] [PubMed] [Google Scholar]
- 3.Istaphanous GK, Loepke AW. General anesthetics and the developing brain. Curr Opin Anaesthesiol. 2009;22:368–73. doi: 10.1097/aco.0b013e3283294c9e. [DOI] [PubMed] [Google Scholar]
- 4.Brambrink AM, Evers AS, Avidan MS, Farber NB, Smith DJ, Zhang X, Dissen GA, Creeley CE, Olney JW. Isoflurane-induced neuroapoptosis in the neonatal rhesus macaque brain. Anesthesiology. 2010;112:834–41. doi: 10.1097/ALN.0b013e3181d049cd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Istaphanous GK, Howard J, Nan X, Hughes EA, McCann JC, McAuliffe JJ, Danzer SC, Loepke AW. Comparison of the neuroapoptotic properties of equipotent anesthetic concentrations of desflurane, isoflurane, or sevoflurane in neonatal mice. Anesthesiology. 2011;114:578–87. doi: 10.1097/ALN.0b013e3182084a70. [DOI] [PubMed] [Google Scholar]
- 6.Olney JW, Young C, Wozniak DF, Ikonomidou C, Jevtovic-Todorovic V. Anesthesia-induced developmental neuroapoptosis. Does it happen in humans? Anesthesiology. 2004;101:273–5. doi: 10.1097/00000542-200408000-00004. [DOI] [PubMed] [Google Scholar]
- 7.Rizzi S, Ori C, Jevtovic-Todorovic V. Timing versus duration: determinants of anesthesia-induced developmental apoptosis in the young mammalian brain. Ann N Y Acad Sci. 2010;1199:43–51. doi: 10.1111/j.1749-6632.2009.05173.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilder RT, Flick RP, Sprung J, Katusic SK, Barbaresi WJ, Mickelson C, Gleich SJ, Schroeder DR, Weaver AL, Warner DO. Early exposure to anesthesia and learning disabilities in a population-based birth cohort. Anesthesiology. 2009;110:796–804. doi: 10.1097/01.anes.0000344728.34332.5d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DiMaggio C, Sun LS, Kakavouli A, Byrne MW, Li G. A retrospective cohort study of the association of anesthesia and hernia repair surgery with behavioral and developmental disorders in young children. J Neurosurg Anesthesiol. 2009;21:286–91. doi: 10.1097/ANA.0b013e3181a71f11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Flick RP, Katusic SK, Colligan RC, Wilder RT, Voigt RG, Olson MD, Sprung J, Weaver AL, Schroeder DR, Warner DO. Cognitive and behavioral outcomes after early exposure to anesthesia and surgery. Pediatrics. 2011;128:e1053–61. doi: 10.1542/peds.2011-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yon JH, Daniel-Johnson J, Carter LB, Jevtovic-Todorovic V. Anesthesia induces neuronal cell death in the developing rat brain via the intrinsic and extrinsic apoptotic pathways. Neuroscience. 2005;135:815–27. doi: 10.1016/j.neuroscience.2005.03.064. [DOI] [PubMed] [Google Scholar]
- 12.Kapetanaki SM, Silkstone G, Husu I, Liebl U, Wilson MT, Vos MH. Interaction of carbon monoxide with the apoptosis-inducing cytochrome c-cardiolipin complex. Biochemistry. 2009;48:1613–9. doi: 10.1021/bi801817v. [DOI] [PubMed] [Google Scholar]
- 13.Kao LW, Nañagas KA. Carbon monoxide poisoning. Emerg Med Clin North Am. 2004;22:985–1018. doi: 10.1016/j.emc.2004.05.003. [DOI] [PubMed] [Google Scholar]
- 14.Zhou H, Liu J, Pan P, Jin D, Ding W, Li W. Carbon monoxide inhalation decreased lung injury via anti-inflammatory and anti-apoptotic effects in brain death rats. Exp Biol Med (Maywood) 2010;235:1236–43. doi: 10.1258/ebm.2010.010147. [DOI] [PubMed] [Google Scholar]
- 15.Song R, Kubo M, Morse D, Zhou Z, Zhang X, Dauber JH, Fabisiak J, Alber SM, Watkins SC, Zuckerbraun BS, Otterbein LE, Ning W, Oury TD, Lee PJ, McCurry KR, Choi AM. Carbon monoxide induces cytoprotection in rat orthotopic lung transplantation via anti-inflammatory and anti-apoptotic effects. Am J Pathol. 2003;163:231–42. doi: 10.1016/S0002-9440(10)63646-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bernardini C, Zannoni A, Bacci ML, Forni M. Protective effect of carbon monoxide pre-conditioning on LPS-induced endothelial cell stress. Cell Stress Chaperones. 2010;15:219–24. doi: 10.1007/s12192-009-0136-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sarady JK, Zuckerbraun BS, Bilban M, Wagner O, Usheva A, Liu F, Ifedigbo E, Zamora R, Choi AM, Otterbein LE. Carbon monoxide protection against endotoxic shock involves reciprocal effects on iNOS in the lung and liver. FASEB J. 2004;18:854–6. doi: 10.1096/fj.03-0643fje. [DOI] [PubMed] [Google Scholar]
- 18.Wang X, Wang Y, Kim HP, Nakahira K, Ryter SW, Choi AM. Carbon monoxide protects against hyperoxia-induced endothelial cell apoptosis by inhibiting reactive oxygen species formation. J Biol Chem. 2007;282:1718–26. doi: 10.1074/jbc.M607610200. [DOI] [PubMed] [Google Scholar]
- 19.Cheng Y, Thomas A, Mardini F, Bianchi SL, Tang JX, Peng J, Wei H, Eckenhoff MF, Eckenhoff RG, Levy RJ. Neurodevelopmental consequences of sub-clinical carbon monoxide exposure in newborn mice. PLoS One. 2012;7:e32029. doi: 10.1371/journal.pone.0032029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Levy RJ, Nasr VG, Rivera O, Roberts R, Slack M, Kanter JP, Ratnayaka K, Kaplan RF, McGowan FX., Jr Detection of carbon monoxide during routine anesthetics in infants and children. Anesth Analg. 2010;110:747–53. doi: 10.1213/ANE.0b013e3181cc4b9f. [DOI] [PubMed] [Google Scholar]
- 21.Nasr V, Emmanuel J, Deutsch N, Slack M, Kanter J, Ratnayaka K, Levy R. Carbon monoxide re-breathing during low-flow anaesthesia in infants and children. Br J Anaesth. 2010;105:836–41. doi: 10.1093/bja/aeq271. [DOI] [PubMed] [Google Scholar]
- 22.Woehlck HJ. Carbon monoxide rebreathing during low flow anesthesia. Anesth Analg. 2001;93:516–7. doi: 10.1097/00000539-200108000-00058. [DOI] [PubMed] [Google Scholar]
- 23.Farahani R, Kanaan A, Gavrialov O, Brunnert S, Douglas RM, Morcillo P, Haddad GG. Differential effects of chronic intermittent and chronic constant hypoxia on postnatal growth and development. Pediatr Pulmonol. 2008;43:20–8. doi: 10.1002/ppul.20729. [DOI] [PubMed] [Google Scholar]
- 24.Rice D, Barone S., Jr Critical periods of vulnerability for the developing nervous system: evidence from humans and animal models. Environ Health Perspect. 2000;108(Suppl 3):511–33. doi: 10.1289/ehp.00108s3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sanno H, Shen X, Kuru N, Bormuth I, Bobsin K, Gardner HA, Komljenovic D, Tarabykin V, Erzurumlu RS, Tucker KL. Control of postnatal apoptosis in the neocortex by RhoA-subfamily GTPases determines neuronal density. J Neurosci. 2010;30:4221–31. doi: 10.1523/JNEUROSCI.3318-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Johnson SA, Young C, Olney JW. Isoflurane-induced neuroapoptosis in the developing brain of nonhypoglycemic mice. J Neurosurg Anesthesiol. 2008;20:21–8. doi: 10.1097/ANA.0b013e3181271850. [DOI] [PubMed] [Google Scholar]
- 27.Franklin KBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. Academic Press; San Diego, CA: 1997. [Google Scholar]
- 28.Paxinos G, Watson C. Atlas of the Developing Mouse Brain: At E17.5, PO, and P6. Academic Press; San Diego, CA: 2007. [Google Scholar]
- 29.Jacobs EE, Sanadi DR. The reversible removal of cytochrome c from mitochondria. J Biol Chem. 1960;235:531–4. [PubMed] [Google Scholar]
- 30.Kim NH, Jeong MS, Choi SY, Kang JH. Peroxidase activity of cytochrome c. bull. Korean Chem Soc. 2004;25:1889–92. [Google Scholar]
- 31.Piel DA, Gruber PJ, Weinheimer CJ, Courtois MR, Robertson CM, Coopersmith CM, Deutschman CS, Levy RJ. Mitochondrial resuscitation with exogenous cytochrome c in the septic heart. Crit Care Med. 2007;35:2120–7. doi: 10.1097/01.ccm.0000278914.85340.fe. [DOI] [PubMed] [Google Scholar]
- 32.Ozawa T, Tanaka M, Shimomura Y. Crystallization of the middle part of the mitochondrial electron transfer chain: cyto-chrome bc1-cytochrome c complex. Proc Natl Acad Sci U S A. 1980;77:5084–6. doi: 10.1073/pnas.77.9.5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tanaka M, Ogawa N, Ihara K, Sugiyama Y, Mukohata Y. Cytochrome aa(3) in Haloferax volcanii. J Bacteriol. 2002;184:840–5. doi: 10.1128/JB.184.3.840-845.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Winter PM, Miller JN. Carbon monoxide poisoning. JAMA. 1976;236:1502. [PubMed] [Google Scholar]
- 35.Peterson JE, Stewart RD. Absorption and elimination of carbon monoxide by inactive young men. Arch Environ Health. 1970;21:165–71. doi: 10.1080/00039896.1970.10667215. [DOI] [PubMed] [Google Scholar]
- 36.Stewart RD, Peterson JE, Baretta ED, Bachand RT, Hosko MJ, Herrmann AA. Experimental human exposure to carbon mon-oxide. Arch Environ Health. 1970;21:154–64. doi: 10.1080/00039896.1970.10667214. [DOI] [PubMed] [Google Scholar]
- 37.Gorman D, Drewry A, Huang YL, Sames C. The clinical toxicology of carbon monoxide. Toxicology. 2003;187:25–38. doi: 10.1016/s0300-483x(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 38.Puchkov MN, Vassarais RA, Korepanova EA, Osipov AN. Cytochrome c produces pores in cardiolipin-containing planar bilayer lipid membranes in the presence of hydrogen peroxide. Biochim Biophys Acta. 2013;1828:208–12. doi: 10.1016/j.bbamem.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 39.Kim H, Oh E, Im H, Mun J, Yang M, Khim JY, Lee E, Lim SH, Kong MH, Lee M, Sul D. Oxidative damages in the DNA, lipids, and proteins of rats exposed to isofluranes and alcohols. Toxicology. 2006;220:169–78. doi: 10.1016/j.tox.2005.12.010. [DOI] [PubMed] [Google Scholar]
- 40.Bai X, Yan Y, Canfield S, Muravyeva MY, Kikuchi C, Zaja I, Corbett JA, Bosnjak ZJ. Ketamine enhances human neural stem cell proliferation and induces neuronal apoptosis via reactive oxygen species-mediated mitochondrial pathway. Anesth Analg. 2013;116:869–80. doi: 10.1213/ANE.0b013e3182860fc9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Matsuoka H, Watanabe Y, Isshiki A, Quock RM. Increased production of nitric oxide metabolites in the hippocampus under isoflurane anaesthesia in rats. Eur J Anaesthesiol. 1999;16:216–24. doi: 10.1046/j.1365-2346.1999.00459.x. [DOI] [PubMed] [Google Scholar]
- 42.Boscolo A, Milanovic D, Starr JA, Sanchez V, Oklopcic A, Moy L, Ori CC, Erisir A, Jevtovic-Todorovic V. Early exposure to general anesthesia disturbs mitochondrial fission and fusion in the developing rat brain. Anesthesiology. 2013;118:1086–97. doi: 10.1097/ALN.0b013e318289bc9b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheng Y, Corbin JG, Levy RJ. Programmed Cell Death is Impaired in Fmr1mutant mice. Dev Neuroscience. 2013;35:347–58. doi: 10.1159/000353248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Wael K, Bashir Q, Van Vlierberghe S, Dubruel P, Heering HA, Adriaens A. Electrochemical determination of hydrogen peroxide with cytochrome c peroxidase and horse heart cytochrome c entrapped in a gelatin hydrogel. Bioelectrochemistry. 2012;83:15–8. doi: 10.1016/j.bioelechem.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 45.Ferraro E, Pulicati A, Cencioni MT, Cozzolino M, Navoni F, di Martino S, Nardacci R, Carrì MT, Cecconi F. Apoptosomedeficient cells lose cytochrome c through proteasomal degradation but survive by autophagy-dependent glycolysis. Mol Biol Cell. 2008;19:3576–88. doi: 10.1091/mbc.E07-09-0858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Thom SR, Ischiropoulos H. Mechanism of oxidative stress from low levels of carbon monoxide. Res Rep Health Eff Inst. 1997;80:1–19. [PubMed] [Google Scholar]
- 47.Hayashi M, Takahashi T, Morimatsu H, Fujii H, Taga N, Mizobuchi S, Matsumi M, Katayama H, Yokoyama M, Taniguchi M, Morita K. Increased carbon monoxide concentration in exhaled air after surgery and anesthesia. Anesth Analg. 2004;99:444–8. doi: 10.1213/01.ANE.0000123821.51802.F3. [DOI] [PubMed] [Google Scholar]
- 48.Sutherland BA, Harrison JC, Nair SM, Sammut IA. Inhalation gases or gaseous mediators as neuroprotectants for cerebral ischaemia. Curr Drug Targets. 2013;14:56–73. doi: 10.2174/138945013804806433. [DOI] [PubMed] [Google Scholar]