

Abstract
The complete sequencing and annotation of the genomes of industrially-important Bacillus species has enhanced our understanding of their properties, and allowed advances in genetic manipulations in other Bacillus species. Post-genomic studies require simple and highly efficient tools to enable genetic manipulation. Here, we summarize the recent progress in genetic engineering strategies for Bacillus species. We review the available genetic tools that have been developed in Bacillus species, as well as methods developed in other species that may also be applicable in Bacillus. Furthermore, we address the limitations and challenges of the existing methods, and discuss the future research prospects in developing novel and useful tools for genetic modification of Bacillus species.
Keywords: Bacillus species, Genetic engineering strategies, Counter-selection marker, Operator-repressor system, Toxin gene, Site-specific recombination
Introduction
Bacillus subtilis and some related Bacillus species are non-pathogenic, free of exotoxins and endotoxins, and have a recognized history of safe use in foods. These species are also useful for fermentation and large-scale cultivation. Despite these natural advantages, protocols using Bacillus species lag far behind those for Escherichia coli and Saccharomyces cerevisiae, which are the most widely used cellular factories for producing industrially-important enzymes and biochemicals [1,2]. Therefore, efficient genetic manipulation and highly developed systems-level strategies for developing Bacillus species as microbial cell factories are needed. Recently, Bacillus species have received increased attention for their use in genetic engineering and production of heterologous proteins [3], valuable enzymes, vitamins, platform chemicals, and antibiotics [4-6].
The complete genome sequences of several B. subtilis strains have been determined and annotated, stimulating novel methods for interpreting metabolic pathways and supplying an overview of protein machinery [7]. The completion of the genome sequences of industrially-important Bacillus species (Bacillus licheniformis, GenBank accession number CP000560 and Bacillus amyloliquefaciens, GenBank accession number AE017333) revealed a high degree of homology to B. subtilis[8-10]. These complete genome sequences not only enhance our understanding of these strains, but also allow advances in genetic manipulations in other Bacillus species [6]. With rapid developments in post-genomic studies, simple and efficient genetic tools are required to conveniently enable multiple modifications of the genomes of Bacillus species.
Classical chromosomal modification is based on the insertion of a selectable marker, usually a drug resistance gene, into the chromosome of a bacterial strain [11]. Using this strategy, a second selective maker gene is required to introduce another chromosomal modification, so the number of available selection genes limits the feasibility of multiple chromosomal modifications. Moreover, the selectable gene should be removed by single-crossover recombination if the strain is to be used for further genetic manipulation. In addition, the chance of obtaining a positive strain is relatively low, and the selection process is laborious. To overcome these problems, methods that can eliminate marker cassettes in the primary transformants are desperately needed.
Recently, useful tools for genetic modification of Bacillus species have emerged from the fields of systems and synthetic biology. Thus, it is necessary to summarize recent progress, current obstacles, and future goals to inspire more research interest and advance studies in this field. First, we summarize the progress in genetic modification strategies of Bacillus species, including several kinds of marker-free genetic modification methods and site-specific recombination strategies. Next, we discuss some strategies developed in other species that could also be used in Bacillus species. Lastly, we compare current genetic engineering strategies, analyze their challenges and limitations, and discuss future research goals for developing novel and useful tools for genetic modification of Bacillus species.
Operator-repressor system-based genetic engineering strategies
The development of various inducible promoter systems has played an important role in the analysis of gene expression and function. Promoters responsive to an assortment of inducing agents, including heavy metals, hormones, and heat shock, as well as several viral, cellular, and bacterial regulatory factors, have been successfully used to manipulate gene expression. Some systems have been applied to genetically engineer Bacillus species, and the selection and counter-selection marker cassettes are shown in Figure 1.
Figure 1.
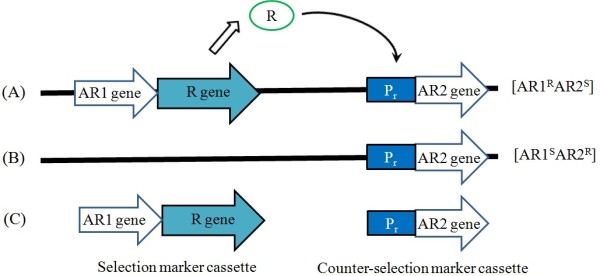
Operator-repressor system-based genetic engineering strategy. (A) In the presence of the R gene, the constitutively expressed R protein specifically binds to the Pr promoter and represses promoter activity. (B) When the R gene is excised from the genome, the Pr promoter is activated and mutants can be selected by antibiotic resistance. (C) Diagrams of selection and counter-selection marker cassettes. R: Repressor, AR: Antibiotic resistance, Pr: Promoter. [AR1R], [AR1S], [AR2R], and [AR2S] are AR1-resistant, AR1-sensitive, AR2-resistant, and AR2-sensitive bacterial phenotypes, respectively.
cI as a counter-selection marker
The CI repressor gene (cI857 or cI) from E. coli bacteriophage lambda encodes the CI repressor, which can bind to the Pr promoter of lambda phage to suppress its promoter activity. Itaya [12] developed a counter-selection method in which a neomycin-resistance gene is regulated by Pr. This system, controlled by the presence/absence of the CI repressor, allows precise selection for marker-free genetic modification of the chromosome [13-15]. Similarly, Uotsu-Tomita et al.[16] developed a double cI-Pr method for positive selection of B. subtilis recombinants, and Tsuge et al.[17] obtained markerless deletion of sfp, degQ, and ppsABCDE using the cI-Pr system. This method can be repeatedly used by changing the location of the cI gene [18].
araR as a counter-selection marker
araR encodes a negative regulator of the ara operon that can be induced by L-arabinose in B. subtilis[19]. Liu et al.[20] developed a method in which the chromosomal araR locus was replaced by P ara -neo, which confers neomycin resistance. By adding L-arabinose to the growth medium, the expression of P ara -neo can be induced based on the presence of araR in the chromosome. First, the selection marker cassette cat-araR is inserted upstream of the target gene via recombination, and then selected for with chloramphenicol. Next, single-crossover recombination between two downstream regions removes the cat-araR cassette, together with the target gene. The remaining P ara -neo in the genome can be used again for further genetic modification. The authors obtained 3.8-kb and 41.8-kb deletion strains using this system that left no selective marker gene in the targeted loci.
Pyrimidine metabolism-based genetic engineering strategies
Common approaches for counter-selection exploit genes involved in purine or pyrimidine metabolism and are based on the fact that purine or pyrimidine analogs can be converted to toxic compounds. Plating cells on media containing the analog leads to a strong selection for clones that have lost the chromosomally-integrated copy of the gene encoding the converting enzyme. Therefore, parental strains used for genome modification must lack the respective gene for purine or pyrimidine nucleotide biosynthesis. Recently, upp and pyrF, encoding uracil phosphoribosyltransferase (UPRTase) and orotidine 5′-phosphate decarboxylase (OMPdecase), respectively, have been used as counter-selection markers in Bacillus species. The relevant pyrimidine metabolism pathway and mechanisms of action of pyrimidine analogs are shown in Figure 2.
Figure 2.
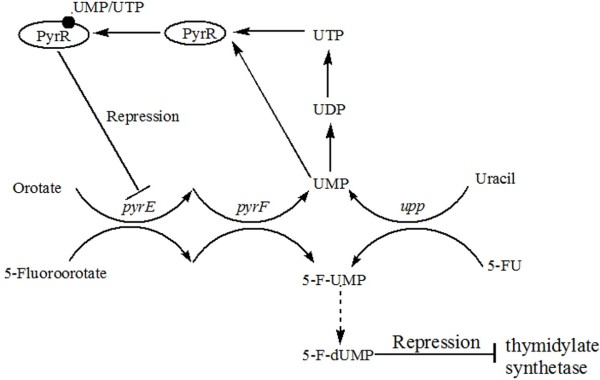
Summary of pyrimidine metabolism pathway and mechanisms of action of pyrimidine analogs. Orotate is metabolized to UMP by OPRTase and OMPdecase, encoded by pyrE and pyrF, respectively. These enzymes also convert 5-fluoroorotate to 5-fluoro-UMP. pyrR encodes an mRNA-binding attenuator (PyrR) that negatively regulates pyr expression by sensing UMP or UTP. UMP is also produced from uracil by UPRTase, encoded by upp. UPRTase converts 5-FU to 5-fluoro-UMP, which is further metabolized to the toxic metabolite 5-fluoro-dUMP. 5-fluoro-dUMP is a strong inhibitor of thymidylate synthetase, which provides the sole de novo source of dTMP for DNA biosynthesis.
upp as a counter-selection marker
UPRTase catalyzes the key reaction of the pyrimidine salvage pathway, from uracil to UMP, in many microorganisms. The toxic pyrimidine analog 5-fluorouracil (5-FU) can be converted to 5-fluoro-UMP by UPRTase. The latter compound can be further catalyzed into 5-fluoro-dUMP, which is a strong inhibitor of thymidylate synthetase. Deletion of upp endows the mutant strain with resistance to 5-FU.
Fabret et al.[21] developed a method using upp as a screening marker in B. subtilis. A PCR-generated DNA fragment, which consists of the target gene with a desired mutation linked to a upp cassette, was inserted into the genome by double-crossover recombination and selected for based on phleomycin resistance. The upp gene was excised through recombination of the direct repeats (DR) flanking the upp cassette in the linear DNA, and 5-FU-containing medium was used to select for strains that contained the desired chromosomal mutation.
The upp cassette has also been used to functionally analyze B. subtilis genes by constructing a mutant library [22], point mutations [23], and gene-null strains [24,25]. Morimoto et al.[26] generated a genome-reduced B. subtilis strain, MGB874, using the upp cassette to sequentially knock out genes. Compared with the parental B. subtilis 168 genome, the genome of strain MGB874 is depleted by 874 kb (20.7%), including 865 genes.
Tanaka et al.[27] developed an improved counter-selection system that combines the upp and cI-Pr systems. In this system, the master strain (MS) TF8A Δupp:: λPr-neo was constructed by replacing upp with a λPr-neo cassette, the counter-selection marker mentioned above. The upp-phleo-cI cassette undergoes homologous replacement with a targeted chromosome region after being introduced into the MS. Positive selection for cassette integration is based on resistance to phleomycin. The upp gene and the sak promoter construct Psak-λcI are used for counter-selection and cassette removal.
Recently, Shi et al.[28] developed a method combining upp and double-strand break (DSB) repair, which is caused by exogenous endonuclease I-SceI and comK overexpression, for fast preparation of competent cells. First, a foreign dsDNA fragment is integrated into the chromosome via double-crossover. The upp cassette can then be excised by a second intramolecular homologous recombination. The DSB repair potently induces the second intramolecular recombination, which enhances the frequency of resolution by one to two orders of magnitude.
A method based on upp has also been used in Bacillus species other than B. subtilis. Wemhoff et al. [29] developed an upp-based deletion method for Bacillus pumilus in which master strains used for gene deletion are generated by targeted deletion of a set of genes, including the essential sporulation gene yqfD, enabling rapid allelic exchange between upp and hsdR. The hsdR gene encodes the restrictase of a type I restriction modification system, and its deletion makes a strain readily transformable. The resultant B. pumilus mutant is easily transformable with plasmid DNA isolated from E. coli strains. In addition, direct gene disruption is possible, which enables relatively rapid genetic manipulations.
pyrF as a counter-selection marker
Orotate phosphoribosyltransferase (OPRTase) and OMPdecase, encoded by pyrE and pyrF respectively, are essential enzymes for metabolism of orotic acid to UMP and 5-fluoroorotate to 5-fluoro-UMP. 5-fluoro-UMP can be further converted into the toxic metabolite 5-fluoro-dUMP. The PyrR protein (encoded by pyrR) is an mRNA-binding attenuator, which regulates expression of pyrimidine biosynthetic (pyr) genes by sensing UMP or UTP.
Suzuki et al.[30] established a counter-selection system based on deletion of pyrR and pyrF in Geobacillus kaustophilus HTA426. The disruption of pyrF and pyrR makes the MS auxotrophic for uracil and resistant to 5-fluoroorotate. Heterologous β-galactosidase and α-amylase genes were integrated in the genome of G. kaustophilus by pyrF-based counter-selection using pGAM plasmids, without leaving the marker in the target loci. This system may be applied to other organisms harboring pyrR, such as Bacillus-related bacteria.
Auxotrophy-based genetic engineering strategies
A lysine-auxotrophic strain combined with an operator-repressor system
The lysA gene of B. subtilis 168 encodes diaminopimelate decarboxylase, which catalyzes the final step in the lysine biosynthetic pathway in which meso-diaminopimelate is converted into lysine. The lysA gene is essential for the strain to grow on minimal medium [31,32].
A lysine-auxotrophic strain combined with blaI
A conditional auxotrophy-based method for removing selection markers was developed by Brans et al. [33]. This method combines the use of blaI, a spectinomycin resistance gene, with a conditional lysine-auxotrophic B. subtilis strain (BS1541). The blaI gene of B. licheniformis encodes a cytoplasmic repressor (BlaI) that negatively regulates the expression of β-lactamase (encoded by blaP) in the absence of penicillin. The B. subtilis P lysA promoter was replaced with the B. licheniformis P blaP promoter to obtain strain BS1541, thus blaI can confer lysine auxotrophy to the strain. The blaI cassette, containing blaI flanked by two DR sequences, was inserted into the genome of strain BS1541 by homologous recombination and selected for with spectinomycin, and the cassette was removed by a single-crossover recombination between the two DRs. This strategy can be used consecutively to further modify the Bacillus chromosome.
A lysine-auxotrophic strain combined with lacI
A similar method was developed by Zhang et al.[34]. A conditionally lysine-auxotrophic B. subtilis strain (BS-PS) was generated by substituting the P lysA promoter with an isopropyl-β-D-thiogalactopyranoside (IPTG)-inducible P spac promoter. A vector containing lacI, which encodes a repressor of the P spac promoter, and a chloromycetin resistance gene was used to generate specific integrating vectors. The integration of these vectors into the BS-PS chromosome, and the excision of lacI and the chloromycetin resistance gene, were both achieved by a single crossover at the homologous arm.
A method based on a histidine-auxotrophic strain
hisF and hisI are the last two genes in the his operon of the B. subtilis 168 chromosome, and are followed by two genes of unknown function, yvcA and yvcB. An incomplete histidine biosynthesis operon leads to histidine auxotrophy in B. subtilis.
Motejadded et al.[35] developed a system based on two plasmids to obtain marker-free recombinant B. subtilis strains. One plasmid consists of the 3′-end of hisF, the 5′-end of hisI, a spectinomycin resistance gene, and a 1.3 kb fragment containing the 3′-end of yvcA and the 5′-end of yvcB. The other plasmid contains the same yvc-region and the 3′- end of hisF, but has a complete hisI and lacks the antibiotic marker. Spectinomycin-resistant His-auxotrophic mutants are obtained after integration of the first plasmid into the B. subtilis genome by double crossover. Insertion of the second plasmid into this mutant leads to a spectinomycin-sensitive His-prototrophic strain. These plasmids were successfully applied to integrate the lipase gene of Bacillus thermocatenulatus into a B. subtilis glucose-regulated system. Morabbi et al. also used this system to perform markerless integration of P mtlR -mtlR-H342D into the chromosome of B. subtilis[36].
Site-specific recombination-based genetic engineering strategies
Site-specific recombination (SSR) systems use recombinases that catalyze recombination between two site-specific recognition sites, which generates a desired DNA integration, deletion, or inversion. SSR systems that are used in bacterial genome engineering include Cre/loxP from bacteriophage P1 [37], Xis/attP from bacteriophage λ [38], and FLP/FRT[39] from S. cerevisiae. The recombination efficiency of SSR systems is much higher than that of native recombination systems, which makes them applicable for undertaking multiple sequential mutations of the same chromosome (Figure 3). Recently, Cre/loxP and Xer/dif systems have been used in B. subtilis species.
Figure 3.
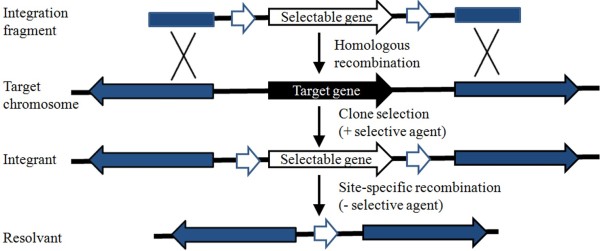
Site-specific recombination-based genetic engineering strategy. Selectable genes are often antibiotic resistance genes. White regions represent recognition sites of site-specific recombinases, and blue regions represent sequences homologous between the genome and the integration cassette.
The Cre/lox system
The Cre/lox system is a powerful genetic tool [40] that is widely used in eukaryotic and prokaryotic cells and consists of a Cre recombinase and a pair of loxP sites. Cre recombinase catalyzes the reciprocal site-specific recombination between the two loxP sites, which does not require any host cofactors or accessory proteins. A pair of modified lox sites, lox71 (L) and lox66 (R), are usually used [41] to minimize genetic instability, as microbial genomes can contain multiple native loxP sites that could be identified by Cre. A double mutant lox72 remnant site, which has weaker binding affinity for Cre, is obtained following recombination of lox71 and lox66, allowing for repeated mutations in a single genetic background [42,43].
Yan et al. [11] developed a genome engineering procedure for B. subtilis by combining the mutant Cre/lox system with a fusion PCR method. After the integration of a fusion PCR product (lox71-spc/zeo-lox66 cassette) into the genome, a thermosensitive vector containing cre is introduced to promote recombination between lox66 and lox71 sites. The PCR fusion product is only about 1.5 kb long, reducing the occurrence of PCR errors, which in turn reduces the probability of introducing errors into the chromosome. Three mutations have been successfully integrated into the same background strain using this method. Chromosomally-encoded tetR in B. subtilis was disrupted by insertion of a lox66-aphAIII-lox71 kanamycin resistance cassette, and subsequent marker excision by Cre recombinase led to the assembly of a novel tetR allele [44]. Phosphotransacetylase, encoded by pta, was ablated using the Cre/lox system in a strain of B. subtilis 168 engineered to produce L-malate [45]. Finally, five adjacent genes, nagP, gamP, gamA, nagA, nagB, were also deleted using the Cre/lox strategy and a PCR-based chromosome modification method for production of N-acetylglucosamine in B. subtilis[46].
Bacillus coagulans is a promising species for producing bulk chemicals from renewable resources in the industrial field, but there are few genetic tools for its modification. Kovacs et al. described a Cre-lox system that uses two plasmids for targeted gene modification in B. coagulans[47]. pMH77 is an integration vector that carries a lox66-cat-lox71 cassette flanked by restriction sites that can be used for cloning homologous regions. pMH66 is a Cre-recombinase plasmid that is used to promote recombination between the two lox sites. The authors used this technique to develop a LacZ reporter assay for measuring gene transcription and to express heterologous D-lactate dehydrogenase.
Although Cre-mediated recombination and excision of the chromosomal sequence between two lox sites is efficient, it does not occur in all cells. To address this, Wang et al.[48] developed a procedure combining Cre recombination and the hen egg white lysozyme gene (hewl) as a counter-selectable marker that eliminates the cells carrying the selection cassette. This procedure is based on the beta protein of lambda phage, a single-stranded annealing protein, and employs a single-stranded PCR product containing a lox71-ble-hewl-PR-lox66 cassette to modify a specific gene. The single-stranded DNA can be protected from exonuclease digestion by beta protein, and can be recombined via beta recombinase-catalyzed annealing at the replication fork [39,49]. The beta protein, regulated by promoter PRM in the lambda cI857 PRM–PR promoter system on the thermosensitive plasmid pWY121, promotes homologous recombination. The hewl gene is placed after promoter PR, which is effective in B. subtilis, and is precisely regulated by the CI857 repressor protein [50]. The efficiency of in-frame deletion using this method can reach 100%. As hen egg white lysozyme is active against Bacillus species, and its encoding gene is distantly related to Bacillus genes [51], it could also be effective in other Bacillus species.
Later, Enyeart et al.[52] combined retargetable mobile group II introns, or “targetrons”, and the Cre/lox system into a versatile platform known as Genome Editing via Targetrons and Recombinases (GETR). Targetrons can be inserted into desired DNA sites with such high efficiency that the inclusion of a selectable marker is not necessary. The lox sites are delivered to specific genomic loci by introns, which enable genomic manipulations, and the added flexibility of RNA hairpins formed by the lox sites enhances the efficiency. GETR is an efficient bacterial genetic engineering approach with broad host-applicability that can be used to generate insertions, deletions, inversions, and one-step cut-and-paste operations.
The Xer/dif system
Chromosome dimers, which are formed during the bacterial life cycle, must be resolved by the bacterial cell machinery for efficient chromosome segregation. The Xer/dif site-specific recombination system used by most bacteria resolves these chromosome dimers into monomers using two tyrosine recombinases to perform the recombination reaction at the dif site, which consists of 28–30 bp. Xer recombinases are represented by XerC and XerD in Gram-negative bacteria such as E. coli[53], and by CodV and RipX in B. subtilis and other Gram-positive bacteria [54]. Intramolecular Xer recombination can excise a dif-flanked DNA sequence from a chromosomally inserted cassette.
Bloor et al.[55] developed a simple and effective method for genetic modification in bacterial chromosomes based on the Xer/dif system. The insertion cassette used in this method consists of a selectable marker gene flanked by dif sites and homology arms that are homologous to the genomic target. After the insertion cassette integrates into the corresponding chromosomal region via double-crossover recombination, Xer recombinases can integrate the two dif sites into one site, thus removing the selectable marker gene. This method eliminates the requirement for an exogenous SSR system, as Xer recombinases are naturally present in bacteria. Furthermore, a counter-selectable gene is not necessary, as the frequency of Xer recombination is sufficient to allow detection of recombinant clones without antibiotic selection. It is worth noting that introducing multiple dif sites in close proximity can induce deletions of the intervening gene segments, so this method may not be suitable for modifying multiple adjacent genes. Pohl et al.[56] used the Xer system to generate B. subtilis mutants with precise deletions in ten extracytoplasmic proteases that affect recombinant protein secretion.
Toxin gene-based genetic engineering strategies
Toxin-antitoxin (TA) systems usually include a functional element consisting of a biologically active protein molecule and its corresponding inhibitor. TA loci are divided into three different types, but the major part of this monograph is devoted to describing type II loci because of the large numbers of known evolutionarily-independent type II gene families. mazF and ccdB are two well-described toxin genes in the mazEF and ccdAB type II TA systems, respectively, both of which have been applied in genetic modification systems. The toxin gene cassettes and commonly used genetic engineering strategies based on them are shown in Figure 4.
Figure 4.
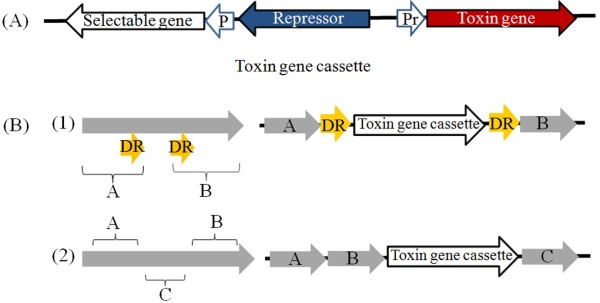
Toxin gene-based genetic engineering strategies. (A) Schematic representation of the toxin gene cassette. P: Commonly-used constitutive promoter. Pr: Promoter of operator-repressor system, which is repressed by the repressor and activated by an inducer e.g. Pspac, Pxyl. The repressor gene in the cassette can also be deleted in some methods. (B) Two different strategies based on toxin gene cassettes. A: upstream sequence; B: downstream sequence; C: sequence for integration of the toxin gene cassette, in combination with A in (2); DR: direct repeat sequence. After integration of the toxin gene cassette into a target chromosome locus via double-crossover recombination [A and B in (1) or A and C in (2)] and positive selection for antibiotic resistance, the cassette is removed by a single crossover event between two DR sequences in (1) or B sequences in (2).
mazF as a counter-selection marker
mazEF is one of the best-characterized TA systems. The MazF toxin of E. coli is an mRNA interferase that cleaves cellular free mRNAs specifically at ACA trinucleotides [57] to block protein synthesis, which inhibits growth. MazF expression in both bacterial and mammalian cells induces programmed cell death [58,59].
Zhang et al. presented a simple method, using E. coli mazF as a counter-selection marker, that can be applied to many Bacillus species without prior genetic modification of the host [60]. This method uses a mazF cassette containing mazF under the control of IPTG-inducible P spac promoter, a spectinomycin resistance gene, and two flanking DR sequences. After the mazF cassette from the linearized delivery vector is integrated into a target chromosome locus via double-crossover recombination, the mazF cassette is removed by a single-crossover event between the two DR sequences. This method requires cloning and takes approximately 2 weeks. In addition, mazF is regulated by the P spac promoter, which has a low induction rate and can have leaky expression in the absence of an inducer in B. subtilis[61-63]. Such leaky expression of mazF increases the frequency of spontaneous mazF-resistant mutants, which decreases the likelihood of isolating colonies with the designed mutation.
Morimoto et al.[64] combined the IPTG-inducible expression system with a high-fidelity fusion PCR method to generate marker-free deletion mutants of B. subtilis. In this method, sequences were designed for integration and excision of the mazF cassette by double- and single-crossover, respectively. This procedure is quicker than the method developed by Zhang et al.[60]. However, it still has some limitations, including possible DNA mutations introduced by the 4.0-kb PCR fusion fragment, the difficulty of assembling different DNA fragments, and some leakiness.
Yu et al.[65] replaced the IPTG-inducible spac expression system with the xyl expression system from Bacillus megaterium, which has tighter transcriptional regulation and a higher induction rate than the spac expression system [62,66]. In this system, mazF is placed under the control of the P xyl promoter, which is repressed by the xylose-responsive repressor XylR in the absence of xylose. However, the long PCR-fusion fragment-generated mutations and spontaneously-generated mazF-resistant mutants should also be taken into consideration when using the above method.
Like its counterpart in B. megaterium, the inducible P xyl promoter of B. subtilis also has strict transcriptional regulation [67] and, in fact, P xyl from B. subtilis W23 shows tighter regulation than P xyl from B. megaterium[61,62]. Lin et al.[68] constructed a mini-mazF cassette containing P xyl from B. subtilis, mazF, and a zeocin resistance gene. The mini-mazF cassette is about 2 kb long, which can somewhat reduce the possibility of PCR-induced mutations. The transformation frequency of this cassette is three-fold higher than the above-mentioned mazF cassettes, and the rate of spontaneous mazF-resistant mutants is low.
ccdB as a counter-selection marker
ccdAB is often used for positive selection of transformants, primarily in E. coli strains. Commercially-available systems (e.g. StabyCloning™ and StabyExpress™, Delphi Genetics SA, Charleroi, Belgium) are based on CcdB toxicity against gyrase and allow one-step selection of transformants, ensuring stable vector plasmid maintenance.
Recently, the plasmid F toxin gene ccdB was used as a counter-selection marker to construct markerless mutants of Vibrio splendidus[69]. A suicide vector carrying ccdB under the control of the arabinose P BAD promoter, which can be transferred to any Vibrio strain by RP4-based conjugation, was developed. The genetic modification system based on this suicide vector requires a two-step allelic exchange procedure. In the presence of arabinose, the counter-selection provided by the integrated vector enabled efficient markerless gene replacement in both V. splendidus and Vibrio cholerae.
Although ccdB has been used as a positive selection marker for a long time, its use for counter-selection in markerless genetic engineering is limited to a few strains and it has not yet been used in Bacillus species. Further analysis of ccdB and toxin genes from other TA systems is needed to determine whether they may be widely applicable for counter-selection in other species.
Thermosensitive plasmid-based genetic engineering strategies
Zakataeva et al.[70] developed a simple method based on a thermosensitive replication plasmid to introduce markerless mutations into the chromosomes of B. amyloliquefaciens. In this method, a delivery plasmid is efficiently introduced into cells for gene replacement, and a two-step replacement procedure mediated by single-crossover events is used. The procedure is efficient and fast and no counter-selection marker or special strain is required. Although this method is designed for B. amyloliquefaciens, it has also been successfully adapted to B. subtilis. Using this method, Sheremet et al. constructed a series of markerless B. amyloliquefaciens strains to produce inosine and 5-aminoimidazole-4- carboxamide ribonucleoside [71].
Transconjugation-based genetic engineering strategies
In some applications, B. licheniformis and B. megaterium outperform the better-studied microbiological model, B. subtilis. However, commonly-used methods for genetic modification of these strains, such as protoplast transformation, are time-consuming and complicated. Recently, some easy markerless deletion methods based on transconjugation have been developed.
A sacB-based transconjugation system for B. megaterium
B. megaterium is an industrially-important species, as it has been used to produce heterologous proteins and valuable enzymes. However, genetic manipulation of B. megaterium is difficult, primarily because of low transformation efficiency. Richhardt et al.[72] developed a simple and efficient transconjugation method for B. megaterium, combining a known transconjugation method [73] and B. subtilis sacB, which encodes levansucrase. The activity of this enzyme in the presence of sucrose is lethal to E. coli[74], so it is used as a counter-selection marker to eliminate the E. coli donor cells after mating. The transfer efficiency of this method is approximately 5 × 10−5 transconjugants/recipient, which is sufficient to allow direct selection of mutants in a one-step procedure.
A transconjugative plasmid-system in B. licheniformis
Rachinger et al.[75] established a markerless gene modification method for Bacilli species without natural competence, such as B. licheniformis. Chromosomal gene deletion is accomplished by the pKVM series of conjugative shuttle vectors, which contain regions flanking the target gene. These shuttle vectors carry the temperature-sensitive origin of replication from pE194ts and a thermostable β-galactosidase, allowing blue/white screening of recombinant clones on X-gal-containing agar plates, and can be conjugated to B. licheniformis and B. subtilis strains. Integration of the vector at the target locus, and its subsequent excision, are both mediated by homologous recombination and identified based on appropriate selection markers. These pKVM vectors can be used to efficiently generate deletions and insertions in B. licheniformis and other Bacillus strains.
Proposed methods for genetic engineering of Bacillus species
Tetracycline-dependent conditional gene knockout in B. subtilis
The tetracycline repressor (TetR) and its reverse mutant (revTetR) can be used for reversible, tetracycline-dependent induction and silencing of gene expression, respectively. Kamionka et al.[76] used both of these approaches in B. subtilis, as an example of a Gram-positive bacteria. In this system, the genomic spoVG-lacZ fusion gene is regulated by one or two tet operators, and either TetR or revTetR is controlled by different promoters, allowing precise adjustment of regulatory windows. TetR or revTetR turn expression on or off, respectively, when anhydrotetracycline is added, which means these two components can be used to construct conditional knockouts in B. subtilis and many other Gram-positive bacteria.
oroP from Lactococcus lactis as a counter-selection marker
The orotate transporter of L. lactis, encoded by oroP, mediates 5-fluoroorotate sensitivity in B. subtilis 168, E. coli XL1-Blue, and 5-fluoroorotate-sensitive lactococci. oroP is necessary for pyrimidine-auxotrophic derivative strains to use orotate as a sole pyrimidine source [77].
Solem et al.[78] developed a selection/counter-selection vector, pCS1966, which harbors oroP and can only replicate in E. coli. This plasmid can be used for homologous recombination at a specific site, and for integration at bacteriophage attachment sites. The plasmid contains an erythromycin-resistance gene for positive selection of cassette integration, and orotate utilization can be used for counter-selection and cassette excision in a pyrimidine auxotrophic mutant. As oroP can be functionally expressed in B. subtilis, its use for counter-selection can potentially be exploited in this species.
bgl/lacZ as counter-selection markers
β-glucosidase, encoded by bgl, can cleave 5-bromo-4-chloro-3-indolyl (BCI) to produce an indoxyl derivative that is toxic to bacteria. Angelov et al.[79] described a markerless mutation method that uses bgl and lacZ as counter-selection markers, and demonstrated the method in the thermophile Thermus thermophilus HB27 and in Micrococcus luteus ATCC 27141. This method uses a delivery plasmid containing the counter-selection markers and flanking regions of the target gene for efficient gene replacement in a two-step replacement process mediated by single crossover events. As Bacillus species are also sensitive to BCI substrate cleavage, this approach could be used to generate markerless chromosomal mutations in Bacillus.
Strategy comparison and prospects
Genome engineering strategies usually require two steps: the integration of a disruption cassette into the genome, and the excision of a selectable marker. Examples of these gene modification strategies, classified based on the different procedures used, are shown in Figure 5. Plasmid-borne disruption cassettes can integrate into the genome by single- or double-crossover, whereas those in PCR fragments usually integrate via the latter mechanism. Restriction endonuclease/ligase-dependent methods are not compatible with large scale approaches, and it generally takes about 2 weeks to complete a marker-free modification. In these methods, single-crossover events have a higher rate of positive recombinants in the cassette integration step than in double-crossover events, although the number of false-positive recombinants will increase in the selection marker excision step. In comparison, methods that use fusion PCR or long-flanking homology PCR techniques to generate the disruption cassette can modify a target gene more rapidly; however, these methods are prone to point mutations and may have difficulties in assembling different DNA fragments. High-fidelity PCR would reduce the incidence of point mutations in short fragments (<4 kb) [80].
Figure 5.
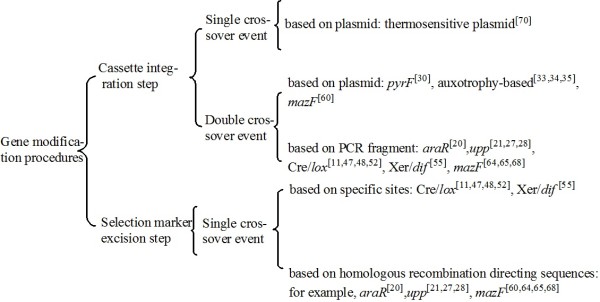
Classification of genetic modification strategies according to different procedures.
Marker excision is usually mediated by a single-crossover event, and the efficiency of this step largely determines the overall success of the genetic modification process. SSR can be used to efficiently eliminate the selection marker from the mutated locus, but it leaves remnant sequences (scars) at the targeted site. Some methods using counter-selection markers are scarless, but they are less efficient than methods based on SSR. Thus, methods combining SSR with counter-selection markers have emerged.
Many methods, such as those derived from upp deletion, can only be used in strains that have a specific gene mutated, which limits their application. Hence, methods that require no such prior modifications in the host and can be applied to some Bacillus species to satisfy the strong demand for a universal unmarked delivery system will receive much attention.
In general, an innovative method to modify genomes of Bacillus species should have certain characteristics: 1) it allows markerless or scarless genome manipulation that is both efficient and precise; 2) it has no requirement for any prior mutation prior to genetic modification; 3) it can be used for system-level genetic modifications or can generate multiple genomic mutations simultaneously.
Based on the above analysis, efforts to improve genetic modification technology for Bacillus species should focus on: 1) optimizing existing homologous-recombination-based genetic modification methods; 2) introducing more advanced technologies from other species into Bacillus species; 3) developing new combinatorial engineering tools for genome-wide modification, such as global transcription machinery engineering [81], tractable multiplex recombineering [82], and multiplex automated genome engineering [83]; 4) adapting novel genome editing engineering technology, such as clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated sequences and similar multi-loci editing systems, to Bacillus species [84,85].
Conclusions
Bacillus species have become important platforms for producing various enzymes and chemicals. A vast range of cellular phenotypes can be obtained in Bacillus species by regulating and modifying the corresponding metabolic pathways at global and gene-specific levels. Advances in genetic engineering strategies have helped realize the potential of Bacillus species as production hosts for manufacturing commodities, and make Bacillus species competitive with the traditional industrial microbes E. coli and S. cerevisiae.
Many useful tools for genetic modification of Bacillus species have been developed in recent years. In this review, we summarized and compared the design principles of current genetic engineering strategies and their recent progress. These strategies still have their own challenges and limitations, so comprehensive and efficient tools for systems-level genetic modifications are still required. We also detailed future research prospects for developing novel genetic modification systems for Bacillus species, which are expected to inspire further interest and advance studies in related fields.
Abbreviations
UPRTase: Uracil-phosphoribosyltransferase; 5-FU: 5-fluorouracil; DR: Direct repeats; MS: Master strain; DSB: Double-strand break; OPRTase: Orotate phosphoribosyltransferase; OMPdecase: Orotidine 5′-phosphate decarboxylase; IPTG: Isopropyl-β-Dthiogalactopyranoside; SSR: Site-specific recombination; GETR: Genome Editing via Targetrons and Recombinases; TA: Toxin-antitoxin; TetR: Tetracycline repressor; BCI: 5-bromo-4-chloro-3-indolyl; CRISPR: Clustered regularly interspaced short palindromic repeats.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
All authors defined the topic of the review and wrote, read and approved the manuscript. Both authors read and approved the final manuscript.
Contributor Information
Huina Dong, Email: dong_hn@tib.cas.cn.
Dawei Zhang, Email: zhang_dw@tib.cas.cn.
Acknowledgements
Authors would like to express their thanks to financial supports from State Key Development Program for Basic Research of China (973 Program, 2013CB733600), National Nature Science Foundation of China (31200036, 31370089), the Key Projects in the Tianjin Science & Technology Pillar Program (12ZCZDSY12700, 11ZCZDSY08500, 11ZCZDSY08400). The authors gratefully acknowledge the support of K. C. Wong Education Foundation, Hong Kong.
References
- Ajikumar PK, Xiao WH, Tyo KE, Wang Y, Simeon F, Leonard E, Mucha O, Phon TH, Pfeifer B, Stephanopoulos G. Isoprenoid pathway optimization for Taxol precursor overproduction in Escherichia coli. Science. 2010;330(6000):70–74. doi: 10.1126/science.1191652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jong B, Siewers V, Nielsen J. Systems biology of yeast: enabling technology for development of cell factories for production of advanced biofuels. Curr Opin Biotechnol. 2012;23(4):624–630. doi: 10.1016/j.copbio.2011.11.021. [DOI] [PubMed] [Google Scholar]
- Schumann W. Production of recombinant proteins in Bacillus subtilis. Adv Appl Microbiol. 2007;62:137–189. doi: 10.1016/S0065-2164(07)62006-1. [DOI] [PubMed] [Google Scholar]
- Schallmey M, Singh A, Ward OP. Developments in the use of Bacillus species for industrial production. Can J Microbiol. 2004;50(1):1–17. doi: 10.1139/w03-076. [DOI] [PubMed] [Google Scholar]
- Perkins JWM, Sauer U, Hohmann HP. In: Metabolic pathway engineering handbook. Smolke CD, Nielsen J, editor. Texas: CRC press; 2009. Metabolic engineering of B. subtilis. [Google Scholar]
- Liu L, Liu Y, Shin H-d, Chen RR, Wang NS, Li J, Du G, Chen J. Developing Bacillus spp. as a cell factory for production of microbial enzymes and industrially important biochemicals in the context of systems and synthetic biology. Appl Microbiol Biotechnol. 2013;97(14):6113–6127. doi: 10.1007/s00253-013-4960-4. [DOI] [PubMed] [Google Scholar]
- Kunst F, Ogasawara N, Moszer I, Albertini AM, Alloni G, Azevedo V, Bertero MG, Bessieres P, Bolotin A, Borchert S, Borriss R, Boursier L, Brans A, Braun M, Brignell SC, Bron S, Brouillet S, Bruschi CV, Caldwell B, Capuano V, Carter NM, Choi SK, Codani JJ, Connerton IF, Cummings NJ, Daniel RA, Denizot F, Devine KM, Düsterhöft A, Ehrlich SD. et al. The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature. 1997;390(6657):249–256. doi: 10.1038/36786. [DOI] [PubMed] [Google Scholar]
- Chen XH, Koumoutsi A, Scholz R, Eisenreich A, Schneider K, Heinemeyer I, Morgenstern B, Voss B, Hess WR, Reva O, Junge H, Voigt B, Jungblut PR, Vater J, Süssmuth R, Liesegang H, Strittmatter A, Gottschalk G, Borriss R. Comparative analysis of the complete genome sequence of the plant growth-promoting bacterium Bacillus amyloliquefaciens FZB42. Nat Biotechnol. 2007;25(9):1007–1014. doi: 10.1038/nbt1325. [DOI] [PubMed] [Google Scholar]
- Ruckert C, Blom J, Chen X, Reva O, Borriss R. Genome sequence of B. amyloliquefaciens type strain DSM7(T) reveals differences to plant-associated B. amyloliquefaciens FZB42. J Biotechnol. 2011;155(1):78–85. doi: 10.1016/j.jbiotec.2011.01.006. [DOI] [PubMed] [Google Scholar]
- Veith B, Herzberg C, Steckel S, Feesche J, Ouml RG, Maurer KH, Ehrenreich P, Auml, Umer S, Bäumer S, Henne A, Liesegang H, Merkl R, Ehrenreich A, Gottschalk G. The complete genome sequence of Bacillus licheniformis DSM13, an organism with great industrial potential. J Mol Microbiol Biotechnol. 2004;7(4):204–211. doi: 10.1159/000079829. [DOI] [PubMed] [Google Scholar]
- Yan X, Yu HJ, Hong Q, Li SP. Cre/lox system and PCR-based genome engineering in Bacillus subtilis. Appl Environ Microbiol. 2008;74(17):5556–5562. doi: 10.1128/AEM.01156-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itaya M. Effective cloning of unmarked DNA fragments in the Bacillus subtilis 168 Genome. Biosci Biotechnol Biochem. 1999;63(3):602–604. doi: 10.1271/bbb.63.602. [DOI] [PubMed] [Google Scholar]
- Kaneko S, Tsuge K, Takeuchi T, Itaya M. Conversion of sub-megasized DNA to desired structures using a novel Bacillus subtilis genome vector. Nucleic Acids Res. 2003;31(18):e112. doi: 10.1093/nar/gng114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itaya M, Fujita K, Ikeuchi M, Koizumi M, Tsuge K. Stable positional cloning of long continuous DNA in the Bacillus subtilis genome vector. J Biochem. 2003;134(4):513–519. doi: 10.1093/jb/mvg168. [DOI] [PubMed] [Google Scholar]
- Itaya M, Nagata T, Shiroishi T, Fujita K, Tsuge K. Efficient cloning and engineering of giant DNAs in a novel Bacillus subtilis genome vector. J Biochem. 2000;128(5):869–875. doi: 10.1093/oxfordjournals.jbchem.a022825. [DOI] [PubMed] [Google Scholar]
- Uotsu-Tomita R, Kaneko S, Tsuge K, Itaya M. Insertion of unmarked DNA sequences in multiple loci of the Bacillus subtilis 168 genome: an efficient selection method. Biosci Biotechnol Biochem. 2005;69(5):1036–1039. doi: 10.1271/bbb.69.1036. [DOI] [PubMed] [Google Scholar]
- Tsuge K, Matsui K, Itaya M. Production of the non-ribosomal peptide plipastatin in Bacillus subtilis regulated by three relevant gene blocks assembled in a single movable DNA segment. J Biotechnol. 2007;129(4):592–603. doi: 10.1016/j.jbiotec.2007.01.033. [DOI] [PubMed] [Google Scholar]
- Tsuge K, Inoue S, Ano T, Itaya M, Shoda M. Horizontal transfer of iturin A operon, itu, to Bacillus subtilis 168 and conversion into an iturin A producer. Antimicrob Agents Chemother. 2005;49(11):4641–4648. doi: 10.1128/AAC.49.11.4641-4648.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sa-Nogueira I, Mota LJ. Negative regulation of L-arabinose metabolism in Bacillus subtilis: characterization of the araR (araC) gene. J Bacteriol. 1997;179(5):1598–1608. doi: 10.1128/jb.179.5.1598-1608.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Endo K, Ara K, Ozaki K, Ogasawara N. Introduction of marker-free deletions in Bacillus subtilis using the AraR repressor and the ara promoter. Microbiology. 2008;154(Pt 9):2562–2570. doi: 10.1099/mic.0.2008/016881-0. [DOI] [PubMed] [Google Scholar]
- Fabret C, Ehrlich SD, Noirot P. A new mutation delivery system for genome-scale approaches in Bacillus subtilis. Mol Microbiol. 2002;46(1):25–36. doi: 10.1046/j.1365-2958.2002.03140.x. [DOI] [PubMed] [Google Scholar]
- Dervyn E, Noirot-Gros M-F, Mervelet P, McGovern S, Ehrlich SD, Polard P, Noirot P. The bacterial condensin/cohesin-like protein complex acts in DNA repair and regulation of gene expression. Mol Microbiol. 2004;51(6):1629–1640. doi: 10.1111/j.1365-2958.2003.03951.x. [DOI] [PubMed] [Google Scholar]
- Noirot-Gros MF. Functional dissection of YabA, a negative regulator of DNA replication initiation in Bacillus subtilis. Proc Natl Acad Sci U S A. 2006;103(7):2368–2373. doi: 10.1073/pnas.0506914103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen SO, Thompson LS, Harry EJ. Cell division in Bacillus subtilis: FtsZ and FtsA Association Is Z-Ring Independent, and FtsA Is Required for Efficient Midcell Z-Ring Assembly. J Bacteriol. 2005;187(18):6536–6544. doi: 10.1128/JB.187.18.6536-6544.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt A. Functional analysis of 11 putative essential genes in Bacillus subtilis. Microbiology. 2006;152(10):2895–2907. doi: 10.1099/mic.0.29152-0. [DOI] [PubMed] [Google Scholar]
- Morimoto T, Kadoya R, Endo K, Tohata M, Sawada K, Liu S, Ozawa T, Kodama T, Kakeshita H, Kageyama Y, Manabe K, Kanaya S, Ara K, Ozaki K, Ogasawara N. Enhanced recombinant protein productivity by genome reduction in Bacillus subtilis. DNA Res. 2008;15(2):73–81. doi: 10.1093/dnares/dsn002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Henry CS, Zinner JF, Jolivet E, Cohoon MP, Xia F, Bidnenko V, Ehrlich SD, Stevens RL, Noirot P. Building the repertoire of dispensable chromosome regions in Bacillus subtilis entails major refinement of cognate large-scale metabolic model. Nucleic Acids Res. 2012;41(1):687–699. doi: 10.1093/nar/gks963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi T, Wang G, Wang Z, Fu J, Chen T, Zhao X. Establishment of a markerless mutation delivery system in Bacillus subtilis stimulated by a double-strand break in the chromosome. PLoS ONE. 2013;8(11):e81370. doi: 10.1371/journal.pone.0081370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wemhoff S, Meinhardt F. Generation of biologically contained, readily transformable, and genetically manageable mutants of the biotechnologically important Bacillus pumilus. Appl Microbiol Biotechnol. 2013;97(17):7805–7819. doi: 10.1007/s00253-013-4935-5. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Murakami A, Yoshida KI. Counterselection system for Geobacillus kaustophilus HTA426 through disruption of pyrF and pyrR. Appl Environ Microbiol. 2012;78(20):7376–7383. doi: 10.1128/AEM.01669-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Ehrlich SD, Albertini A, Amati G, Andersen KK, Arnaud M, Asai K, Ashikaga S, Aymerich S, Bessieres P, Boland F, Brignell SC, Bron S, Bunai K, Chapuis J, Christiansen LC, Danchin A, Débarbouille M, Dervyn E, Deuerling E, Devine K, Devine SK, Dreesen O, Errington J, Fillinger S, Foster SJ, Fujita Y, Galizzi A, Gardan R, Eschevins C. Essential Bacillus subtilis genes. Proc Natl Acad Sci U S A. 2003;100(8):4678–4683. doi: 10.1073/pnas.0730515100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosner A. Control of Lysine biosynthesis in Bacillus subtilis: Inhibition of dianminopimelate decarboxylase by Lysine. J Biotechnol. 1975;121(1):20–28. doi: 10.1128/jb.121.1.20-28.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brans A, Filee P, Chevigne A, Claessens A, Joris B. New integrative method to generate Bacillus subtilis recombinant strains free of selection markers. Appl Environ Microbiol. 2004;70(12):7241–7250. doi: 10.1128/AEM.70.12.7241-7250.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Zhang X, Yao Z, Lu Y, Lu F, Lu Z. A new method for multiple gene inactivations in Bacillus subtilis168, producing a strain free of selectable markers. Can J Microbiol. 2011;57(5):427–436. doi: 10.1139/w11-035. [DOI] [PubMed] [Google Scholar]
- Motejadded H, Altenbuchner J. Integration of a lipase gene into the Bacillus subtilis chromosome: Recombinant strains without antibiotic resistance marker. Iran J Biotechnol. 2007;5(2):105–109. [Google Scholar]
- Morabbi Heravi K, Wenzel M, Altenbuchner J. Regulation of mtl operon promoter of Bacillus subtilis: requirements of its use in expression vectors. Microb Cell Fact. 2011;10(1):83. doi: 10.1186/1475-2859-10-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Borgne S, Bolivar F, Gosset G. Plasmid vectors for marker-free chromosomal insertion of genetic material in Escherichia coli. Methods Mol Biol. 2004;267:135–143. doi: 10.1385/1-59259-774-2:135. [DOI] [PubMed] [Google Scholar]
- Zubko E, Scutt C, Meyer P. Intrachromosomal recombination between attP regions as a tool to remove selectable marker genes from tobacco transgenes. Nat Biotechnol. 2000;18(4):442–445. doi: 10.1038/74515. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A. 2000;97(12):6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn R, Torres RM. Cre/loxP recombination system and gene targeting. Methods Mol Biol. 2002;180:175–204. doi: 10.1385/1-59259-178-7:175. [DOI] [PubMed] [Google Scholar]
- Albert H, Dale EC, Lee E, Ow DW. Site-specific integration of DNA into wild-type and mutant lox sites placed in the plant genome. Plant J. 1995;7(4):649–659. doi: 10.1046/j.1365-313X.1995.7040649.x. [DOI] [PubMed] [Google Scholar]
- Lambert JM, Bongers RS, Kleerebezem M. Cre-lox-based system for multiple gene deletions and selectable-marker removal in Lactobacillus plantarum. Appl Environ Microbiol. 2006;73(4):1126–1135. doi: 10.1128/AEM.01473-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki N, Nonaka H, Tsuge Y, Inui M, Yukawa H. New multiple-deletion method for the Corynebacterium glutamicum genome, using a mutant lox sequence. Appl Environ Microbiol. 2005;71(12):8472–8480. doi: 10.1128/AEM.71.12.8472-8480.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram R, Kolb M, Hillen W. In vivo Activation of Tetracycline Rep ressor by Cre/lox-Mediated Gene Assembly. J Mol Microbiol Biotechnol. 2009;17(3):136–145. doi: 10.1159/000229606. [DOI] [PubMed] [Google Scholar]
- Mu L, Wen J. Engineered Bacillus subtilis 168 produces l-malate by heterologous biosynthesis pathway construction and lactate dehydrogenase deletion. World J Microbiol Biotechnol. 2012;29(1):33–41. doi: 10.1007/s11274-012-1155-6. [DOI] [PubMed] [Google Scholar]
- Liu Y, Liu L, Shin H-d, Chen RR, Li J, Du G, Chen J. Pathway engineering of Bacillus subtilis for microbial production of N-acetylglucosamine. Metab Eng. 2013;19:107–115. doi: 10.1016/j.ymben.2013.07.002. [DOI] [PubMed] [Google Scholar]
- Kovacs AT, Van Hartskamp M, Kuipers OP, Van Kranenburg R. Genetic tool development for a new host for biotechnology, the thermotolerant bacterium Bacillus coagulans. Appl Environ Microbiol. 2010;76(12):4085–4088. doi: 10.1128/AEM.03060-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Weng J, Waseem R, Yin X, Zhang R, Shen Q. Bacillus subtilis genome editing using ssDNA with short homology regions. Nucleic Acids Res. 2012;40(12):e91. doi: 10.1093/nar/gks248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maresca M, Erler A, Fu J, Friedrich A, Zhang Y, Stewart AF. Single-stranded heteroduplex intermediates in lambda Red homologous recombination. BMC Mol Biol. 2010;11:54. doi: 10.1186/1471-2199-11-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitling R, Sorokin AV, Behnke D. Temperature-inducible gene expression in Bacillus subtilis mediated by the cI857-encoded repressor of bacteriophage lambda. Gene. 1990;93(1):35–40. doi: 10.1016/0378-1119(90)90132-B. [DOI] [PubMed] [Google Scholar]
- Abdou AM, Higashiguchi S, Aboueleinin AM, Kim M, Ibrahim HR. Antimicrobial peptides derived from hen egg lysozyme with inhibitory effect against Bacillus species. Food Control. 2007;18(2):173–178. doi: 10.1016/j.foodcont.2005.09.010. [DOI] [Google Scholar]
- Enyeart PJ, Chirieleison SM, Dao MN, Perutka J, Quandt EM, Yao J, Whitt JT, Keatinge-Clay AT, Lambowitz AM, Ellington AD. Generalized bacterial genome editing using mobile group II introns and Cre-lox. Mol Syst Biol. 2013;9(685):685. doi: 10.1038/msb.2013.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakely G, May G, McCulloch R, Arciszewska LK, Burke M, Lovett ST, Sherratt DJ. Two related recombinases are required for site-specific recombination at dif and cer in E. coli K12. Cell. 1993;75(2):351–361. doi: 10.1016/0092-8674(93)80076-Q. [DOI] [PubMed] [Google Scholar]
- Sciochetti SA, Piggot PJ, Sherratt DJ, Blakely G. The ripX locus of Bacillus subtilis encodes a site-specific recombinase involved in proper chromosome partitioning. J Bacteriol. 1999;181(19):6053–6062. doi: 10.1128/jb.181.19.6053-6062.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloor AE, Cranenburgh RM. An efficient method of selectable marker gene excision by Xer recombination for gene replacement in bacterial chromosomes. Appl Environ Microbiol. 2006;72(4):2520–2525. doi: 10.1128/AEM.72.4.2520-2525.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl S, Bhavsar G, Hulme J, Bloor AE, Misirli G, Leckenby MW, Radford DS, Smith W, Wipat A, Williamson ED, Harwood CR, Cranenburgh RM. Proteomic analysis of Bacillus subtilis strains engineered for improved production of heterologous proteins. Proteomics. 2013;13(22):3298–3308. doi: 10.1002/pmic.201300183. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhang J, Hara H, Kato I, Inouye M. Insights into the mRNA cleavage mechanism by MazF, an mRNA interferase. J Biol Chem. 2005;280(5):3143–3150. doi: 10.1074/jbc.M411811200. [DOI] [PubMed] [Google Scholar]
- Yang J, Jiang W, Yang S. mazF as a counter-selectable marker for unmarked genetic modification of Pichia pastoris. FEMS Yeast Res. 2009;9(4):600–609. doi: 10.1111/j.1567-1364.2009.00503.x. [DOI] [PubMed] [Google Scholar]
- Kolodkin-Gal I, Hazan R, Gaathon A, Carmeli S, Engelberg-Kulka H. A linear pentapeptide is a quorum-sensing factor required for mazEF-mediated cell death in Escherichia coli. Science. 2007;318(5850):652–655. doi: 10.1126/science.1147248. [DOI] [PubMed] [Google Scholar]
- Zhang XZ. mazF, a novel counter-selectable marker for unmarked chromosomal manipulation in Bacillus subtilis. Nucleic Acids Res. 2006;34(9):e71. doi: 10.1093/nar/gkl358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhavsar AP, Zhao X, Brown ED. Development and characterization of a xylose-dependent system for expression of cloned genes in Bacillus subtilis: conditional complementation of a teichoic acid mutant. Appl Environ Microbiol. 2001;67(1):403–410. doi: 10.1128/AEM.67.1.403-410.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl B, Wehrl W, Wiegert T, Homuth G, Schumann W. Development of a new integration site within the Bacillus subtilis chromosome and construction of compatible expression cassettes. J Bacteriol. 2001;183(8):2696–2699. doi: 10.1128/JB.183.8.2696-2699.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagner V, Dervyn E, Ehrlich SD. A vector for systematic gene inactivation in Bacillus subtilis. Microbiology. 1998;144:3097–3104. doi: 10.1099/00221287-144-11-3097. [DOI] [PubMed] [Google Scholar]
- Morimoto T, Ara K, Ozaki K, Ogasawara N. A new simple method to introduce marker-free deletions in the Bacillus subtilis genome. Genes Genet Syst. 2009;84:315–318. doi: 10.1266/ggs.84.315. [DOI] [PubMed] [Google Scholar]
- Yu H. Efficient and precise construction of markerless manipulations in the Bacillus subtilis genome. J Microbiol Biotechnol. 2010;20(1):45–53. [PubMed] [Google Scholar]
- Kim L, Mogk A, Schumann W. A xylose-inducible Bacillus subtilis integration vector and its application. Gene. 1996;181:71–76. doi: 10.1016/S0378-1119(96)00466-0. [DOI] [PubMed] [Google Scholar]
- Lewis PJ, Marston AL. GFP vectors for controlled expression and dual labelling of protein fusions in Bacillus subtilis. Gene. 1999;227(1):101–110. doi: 10.1016/S0378-1119(98)00580-0. [DOI] [PubMed] [Google Scholar]
- Lin Z, Deng B, Jiao Z, Wu B, Xu X, Yu D, Li W. A versatile mini-mazF-cassette for marker-free targeted genetic modification in Bacillus subtilis. J Microbiol Methods. 2013;95(2):207–214. doi: 10.1016/j.mimet.2013.07.020. [DOI] [PubMed] [Google Scholar]
- Le Roux F, Binesse J, Saulnier D, Mazel D. Construction of a vibrio splendidus mutant lacking the metalloprotease gene vsm by use of a novel counterselectable suicide vector. Appl Environ Microbiol. 2006;73(3):777–784. doi: 10.1128/AEM.02147-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakataeva NP, Nikitina OV, Gronskiy SV, Romanenkov DV, Livshits VA. A simple method to introduce marker-free genetic modifications into the chromosome of naturally nontransformable Bacillus amyloliquefaciens strains. Appl Microbiol Biotechnol. 2009;85(4):1201–1209. doi: 10.1007/s00253-009-2276-1. [DOI] [PubMed] [Google Scholar]
- Sheremet AS, Gronskiy SV, Akhmadyshin RA, Novikova AE, Livshits VA, Shakulov RS, Zakataeva NP. Enhancement of extracellular purine nucleoside accumulation by Bacillus strains through genetic modifications of genes involved in nucleoside export. J Ind Microbiol Biotechnol. 2010;38(1):65–70. doi: 10.1007/s10295-010-0829-z. [DOI] [PubMed] [Google Scholar]
- Richhardt J, Larsen M, Meinhardt F. An improved transconjugation protocol for Bacillus megaterium facilitating a direct genetic knockout. Appl Microbiol Biotechnol. 2010;86(6):1959–1965. doi: 10.1007/s00253-010-2503-9. [DOI] [PubMed] [Google Scholar]
- Muroa MA, Priest FG. Construction of chromosomal integrants of Bacillus sphaericus 2362 by conjugation with Escherichia coli. Res Microbiol. 2000;151:547–555. doi: 10.1016/S0923-2508(00)00224-2. [DOI] [PubMed] [Google Scholar]
- Gay P, Cqq DL, Steinmetz M, Berkelman T, Kado CI. Positive selection procedure for entrapment of insertion sequence elements in Gram-negative bacteria. J Bacteriol. 1985;164(2):918–921. doi: 10.1128/jb.164.2.918-921.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachinger M, Bauch M, Strittmatter A, Bongaerts J, Evers S, Maurer K-H, Daniel R, Liebl W, Liesegang H, Ehrenreich A. Size unlimited markerless deletions by a transconjugative plasmid-system in Bacillus licheniformis. J Biotechnol. 2013;167(4):365–369. doi: 10.1016/j.jbiotec.2013.07.026. [DOI] [PubMed] [Google Scholar]
- Kamionka A, Bertram R, Hillen W. Tetracycline-dependent conditional gene knockout in Bacillus subtilis. Appl Environ Microbiol. 2005;71(2):728–733. doi: 10.1128/AEM.71.2.728-733.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defoor E, Kryger MB, Martinussen J. The orotate transporter encoded by oroP from Lactococcus lactis is required for orotate utilization and has utility as a food-grade selectable marker. Microbiology. 2007;153(11):3645–3659. doi: 10.1099/mic.0.2007/005959-0. [DOI] [PubMed] [Google Scholar]
- Solem C, Defoor E, Jensen PR, Martinussen J. Plasmid pCS1966, a new selection/counterselection tool for lactic acid bacterium strain construction based on the oroP gene, encoding an orotate transporter from Lactococcus lactis. Appl Environ Microbiol. 2008;74(15):4772–4775. doi: 10.1128/AEM.00134-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelov A, Li H, Geissler A, Leis B, Liebl W. Toxicity of indoxyl derivative accumulation in bacteria and its use as a new counterselection principle. Syst Appl Microbiol. 2013;36(8):585–592. doi: 10.1016/j.syapm.2013.06.001. [DOI] [PubMed] [Google Scholar]
- Shevchuk NA, Bryksin AV, Nusinovich YA, Cabello FC, Sutherland M, Ladisch S. Construction of long DNA molecules using long PCR-based fusion of several fragments simultaneously. Nucleic Acids Res. 2004;32(2):e19. doi: 10.1093/nar/gnh014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alper H, Stephanopoulos G. Global transcription machinery engineering: a new approach for improving cellular phenotype. Metab Eng. 2007;9(3):258–267. doi: 10.1016/j.ymben.2006.12.002. [DOI] [PubMed] [Google Scholar]
- Warner JR, Reeder PJ, Karimpour-Fard A, Woodruff LB, Gill RT. Rapid profiling of a microbial genome using mixtures of barcoded oligonucleotides. Nat Biotechnol. 2010;28(8):856–862. doi: 10.1038/nbt.1653. [DOI] [PubMed] [Google Scholar]
- Wang HH, Isaacs FJ, Carr PA, Sun ZZ, Xu G, Forest CR, Church GM. Programming cells by multiplex genome engineering and accelerated evolution. Nature. 2009;460(7257):894–898. doi: 10.1038/nature08187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nat Biotechnol. 2013;31(3):233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpentier E, Doudna JA. Biotechnology: Rewriting a genome. Nature. 2013;495(7439):50–51. doi: 10.1038/495050a. [DOI] [PubMed] [Google Scholar]