

Abstract
Microfibril-associated glycoprotein 1 (MAGP1) is a component of extracellular matrix microfibrils. Here we show that MAGP1 expression is significantly altered in obese humans, and inactivation of the MAGP1 gene (Mfap2−/−) in mice results in adipocyte hypertrophy and predisposition to metabolic dysfunction. Impaired thermoregulation was evident in Mfap2−/− mice prior to changes in adiposity, suggesting a causative role for MAGP1 in the increased adiposity and predisposition to diabetes. By 5 weeks of age, Mfap2−/− mice were maladaptive to cold challenge, uncoupling protein-1 expression was attenuated in the brown adipose tissue, and there was reduced browning of the subcutaneous white adipose tissue. Levels of transforming growth factor-β (TGF-β) activity were elevated in Mfap2−/− adipose tissue, and the treatment of Mfap2−/− mice with a TGF-β–neutralizing antibody improved their body temperature and prevented the increased adiposity phenotype. Together, these findings indicate that the regulation of TGF-β by MAGP1 is protective against the effects of metabolic stress, and its absence predisposes individuals to metabolic dysfunction.
Introduction
The extracellular matrix (ECM) has emerged as a pivotal component in cellular signaling either through direct interaction with cell-surface receptors or through the ability to regulate growth factor bioavailability. Microfibrils are abundant ECM components that impart strength to tissues and provide instructional signals that affect cellular differentiation and function (1–4). These multiprotein filaments appear in early development and are found in almost all tissues. The core components are the fibrillins, which are encoded by three genes in humans (FBN1, FBN2, and FBN3) but only two functional genes in mice (Fbn1 and Fbn2). In the mouse, combined deficiency of fibrillin-1 and fibrillin-2 results in embryonic death, demonstrating that microfibrils are required for normal development and survival (5). In vertebrates, microfibril-associated glycoprotein (MAGP) 1 and MAGP2 associate with fibrillin to create the functional form of the fiber (6,7). Unlike the fibrillins, which form the structural core of microfibrils, the MAGPs are modifiers of microfibril function, and not key structural elements.
A crucial function of microfibrils is their regulation of growth factor activity, particularly growth factors of the transforming growth factor-β (TGF-β) family (8). The fibrillins covalently bind the large latent complex form of TGF-β, and there is evidence that the pathomechanism associated with fibrillin-1 mutations (e.g., Marfan syndrome) is excess TGF-β activity that is due to an inability to sequester latent TGF-β in the ECM (8). The MAGPs also interact with TGF-β but bind the active, not latent, form of the growth factor (9). Mice lacking MAGP1 have phenotypes consistent with altered TGF-β activity, but they do not always overlap with those associated with fibrillin mutations, which is indicative of both overlapping and distinct functions of the two proteins (9–12). The generation of the MAGP1-deficient (Mfap2−/−) mouse, which has no defect in microfibril formation, provides a model to study the two distinct functions of the microfibril: providing mechanical stability to the tissue and regulation of cell signaling pathways.
One phenotype that shows complete penetrance in Mfap2−/− mice is increased adiposity. This characteristic is particularly interesting in light of studies associating obesity traits in humans to a locus on chromosome 1p36 that includes the gene for MAGP1 (MFAP2) (13–15). Further, it has been proposed that excess TGF-β is involved in the pathogenesis of metabolic diseases like obesity and diabetes (16). In humans, TGF-β1 levels in both adipose tissue and plasma positively correlate with BMI, and elevated TGF-β1 level is a risk factor for type 2 diabetes (17–19). TGF-β has pleiotropic effects on metabolic function, and, in mice, inhibiting TGF-β is protective against diet-induced obesity and diabetes (19). As reviewed by Tan et al. (16), TGF-β inhibits peroxisome proliferator–activated receptor γ coactivator (PGC-1α) and uncoupling protein-1 (UCP-1), which are necessary for uncoupling respiration for heat production, and inhibits the phenotypic transition of white adipocytes to brown. The result is reduced energy expenditure and excess lipid storage. Excess TGF-β in obesity facilitates the development of metabolic syndrome by promoting inflammation and fibrosis, which impairs adipose tissue, liver, and pancreatic function.
Given the importance of the microfibril in regulating TGF-β activity, the purpose of the current study was to explore whether the increased adiposity and changes in metabolic function in Mfap2−/− mice result from altered TGF-β activity. Here we show that Mfap2−/− mice have changes in metabolic function that lead to increased adipocyte size, ectopic lipid accumulation, and insulin resistance. These changes occur secondary to reduced energy expenditure (i.e., impaired thermogenesis) associated with TGF-β–mediated suppression of Ppargc1α (PGC-1α) and Ucp-1. Treating Mfap2−/− mice with a TGF-β neutralizing antibody improved body temperature and prevented excess adiposity. Our findings establish the importance of the ECM component MAGP1 in regulating metabolic pathways associated with obesity, and identify a mechanism whereby the potent effects of TGF-β on intracellular metabolic pathways are regulated by the microenvironment.
Research Design and Methods
Animals and Diets
Generation and genotyping of Mfap2−/− mice has been described (9). All mice used in this study were males on the C57BL/6 background (The Jackson Laboratory, Bar Harbor, ME), were housed in a pathogen-free animal facility, and were fed standard or high-fat chow ad libitum. For caloric intake studies, food consumption of individually housed mice was measured over a consecutive 7-day period. For high-fat diet (HFD) studies, mice were placed on high-fat or control chow for 16 weeks (D12492 [HFD, 60% fat] and D12450B [control 10% fat]; Research Diets, Inc., New Brunswick, NJ). For the neutralizing anti-TGF-β antibody (α-TGF-β) treatment study, mice received intraperitoneal injections of α-TGF-β (clone 1D11) or control IgG-I (clone 11711) three times per week for 5 weeks. Antibodies were purchased from R&D Systems (Minneapolis, MN), dissolved in sterile PBS, and delivered at a dose of 1.5 mg/kg. Mice described as “adults” were 5–6 months old, while “young” mice were 5–7 weeks old.
Body Composition, Activity, Glucose Tolerance, and Blood Parameters
Lean (protein) and fat (lipid) mass was determined on mice and tissue samples using an EchoMRI 3-in-1 model instrument (Echo Medical Systems, Houston, TX). Whole-body fat was also determined by dual-energy X-ray absorptiometry (PIXImus; GE Lunar, Fitchburg, WI) as described by Craft et al. (11). The activity of individual mice was quantified over a 24-h period using an infrared motion sensor (InfraMot apparatus; TSE Systems, Midland, MI) as previously described (20). The first 4 h of data (acclimation period) were not included in the analysis. For glucose tolerance tests (GTTs), mice were fasted overnight prior to a 1–2 g/kg dextrose injection. For insulin sensitivity tests (ITTs), mice were fasted 6 h prior to 0.75 units/kg Humulin-R insulin injection (Lilly, Indianapolis, IN). Contour meters (Bayer, Whippany, NJ) measured tail blood glucose concentration. Serum insulin concentration following 1 g/kg dextrose injection was determined using an Erenna digital single molecule counting platform (Singulex, St. Louis, MO). Triglyceride level and cholesterol concentration were determined as previously described (21,22). Serum leptin was assayed by ELISA using commercial reagents (CrystalChem, Downers Grove, IL).
Adipocyte Size and Number
Adipocyte size and number was measured as previously described (23). Briefly, 50 mg of epididymal white adipose tissue (WAT) was fixed in a 0.2 mol/L collidine HCl/31 mg/mL osmium tetraoxide solution and dissociated in a solution containing 8 mol/L urea and 154 mmol/L NaCl. Samples were then analyzed on a Multisizer-3 (Beckman Coulter, Fullerton, CA) using a 400-m aperture (dynamic linear range 12–320 m). The total adipocyte number was determined for the amount of the sample analyzed (cells per milligram of tissue) then corrected for the total mass of the fat pad.
Lipolysis and Lipid Uptake
Assays were performed on explants of epididymal fat pads with intact ECM. Lipolysis was determined by glycerol release following 200 nmol/L isoproterenol treatment, using the Sigma free glycerol reagent, as described previously (24). For lipid uptake, fat pads were treated with 250 μmol/L tritiated oleic acid, with and without 10 nmol/L insulin. After incubation, fat pads were washed and homogenized in 0.1 mol/L NaOH, and radioactivity was determined using a Beckman LS6000SC scintillation counter (25).
Quantitative RT-PCR
RNA was extracted from tissue with TRIzol reagent (Invitrogen, Grand Island, NY) and RNeasy columns (Qiagen, Valencia, CA). For mouse analyses, RNA was reverse-transcribed using the Applied Biosystems (Grand Island, NY) cDNA-to-RNA reverse transcription kit, and then quantitative PCR (qPCR) was performed using a TaqMan Universal PCR Master Mix reagent kit (Applied Biosystems). For human studies, RNA was reverse transcribed using the Invitrogen VILO cDNA synthesis system, and qPCR was performed using SYBR green. For normalization, coamplification of the mRNA for the ribosomal protein 36B4 (mouse and human) and cyclophilin-A (mouse) was performed.
Histology
Tissue was fixed in 10% buffered formalin for 16 h, dehydrated via an ethanol gradient, and stored in 70% ethanol prior to paraffin embedding. MAGP1 expression was determined by incubating tissue sections with anti-MAGP1 antibody. Inflammation and ECM deposition were evaluated by incubating tissue sections with anti-MAC-3 antibody or trichrome stain.
Energy Expenditure
Mouse body temperature was determined by rectal probe thermometer. Adaptive thermogenesis was evaluated by 6- to 8-h cold challenge. Baseline body temperature was determined, then mice were placed in prechilled cages at 4°C. Metabolic rate was determined by indirect calorimetry. Mice were placed in a Columbus Instruments (Columbus, OH) Oxymax chamber for 24 h, with free access to food and water, at ambient temperature, and with a normal light/dark cycle. The first 4 h of data recording were considered acclimation time. Energy expenditure (Heat) was determined using the following equation: Heat = [3.815 + 1.232(VCO2/VO2)] × VO2 × body weight.
Mitochondrial Content
DNA was purified from brown adipose tissue (BAT) or subcutaneous WAT (scWAT) using DNeasy Blood and Tissue kit (Qiagen). qPCR was performed using Syber Green (Applied Biosystems) and primers specific for nuclear (H19) or mitochondrial DNA (CytB, ND1). Mitochondrial content was calculated as the ratio of mitochondrial DNA to nuclear DNA.
Human Subjects
Subcutaneous abdominal adipose tissue samples were obtained by percutaneous biopsy from 21 obese (BMI [mean ± SD] 40.9 ± 8.0 kg/m2) and 9 lean (BMI 22.7 ± 1.9 kg/m2) men and women, after subjects fasted for ∼12 h overnight, as described previously (26). No subject had any history or evidence of serious disease, took medications that can affect metabolism or the immune system, or had diabetes.
Study Approval
All animals were treated following animal protocols approved by the Washington University Animal Studies Committee. For human studies, subjects gave their written informed consent before participating in the study, which was approved by the Washington University Human Research Protection Office.
Results
MAGP1 Deficiency Causes Excess Adiposity and Metabolic Dysfunction
MAGP1 transcript was detectable in the adipose tissue of mice (Supplementary Fig. 1A), and deletion of MAGP1 in mice fed a standard chow diet resulted in significantly increased adipose tissue mass (Mfap2−/− relative to WT mice; Fig. 1A and B). Although lean mass was unchanged in adult Mfap2−/− animals, whole-body mass was proportionately increased relative to wild-type (WT) controls (Fig. 1B and C). Elevated whole-body adiposity in Mfap2−/− mice was detectable by ∼10 weeks of age (Fig. 1D). As expected with increased adiposity, serum leptin and WAT leptin expression were increased in Mfap2−/− mice (Table 1).
Figure 1.

MAGP1 deficiency causes excess adiposity in mice. A: Mfap2−/− mice have significantly more WAT. Mass and differential volume (photo) of epididymal fat pads from 5-month-old WT and Mfap2−/− mice (mean ± SEM; n = 9 and 8). B: Whole-body fat and lean content determined by EchoMRI on 5-month-old mice (mean ± SEM; n = 9 and 8). C: Body weight of 5-month-old mice (mean ± SEM.; n = 9 and 8). D: Increased adiposity due to MAGP1 deletion is apparent by 9–10 weeks of age. Longitudinal EchoMRI study of whole-body adiposity from 5 to 15 weeks of age (mean ± SEM; n = 10 and 8). E: Daily food consumption was measured in 7-week-old mice, prior to changes in adiposity. Food intake was determined per mouse over 7 days (mean ± SEM; n = 5 and 5). F: Infrared-based activity measured during light (day) and dark (night) cycles (mean ± SEM; n = 9 and 8). Student t test was used for single comparisons (*P ≤ 0.05). RU, relative units.
Table 1.
Body composition, biochemical, and hormonal characteristics (5-month-old male mice, n = 5–10)
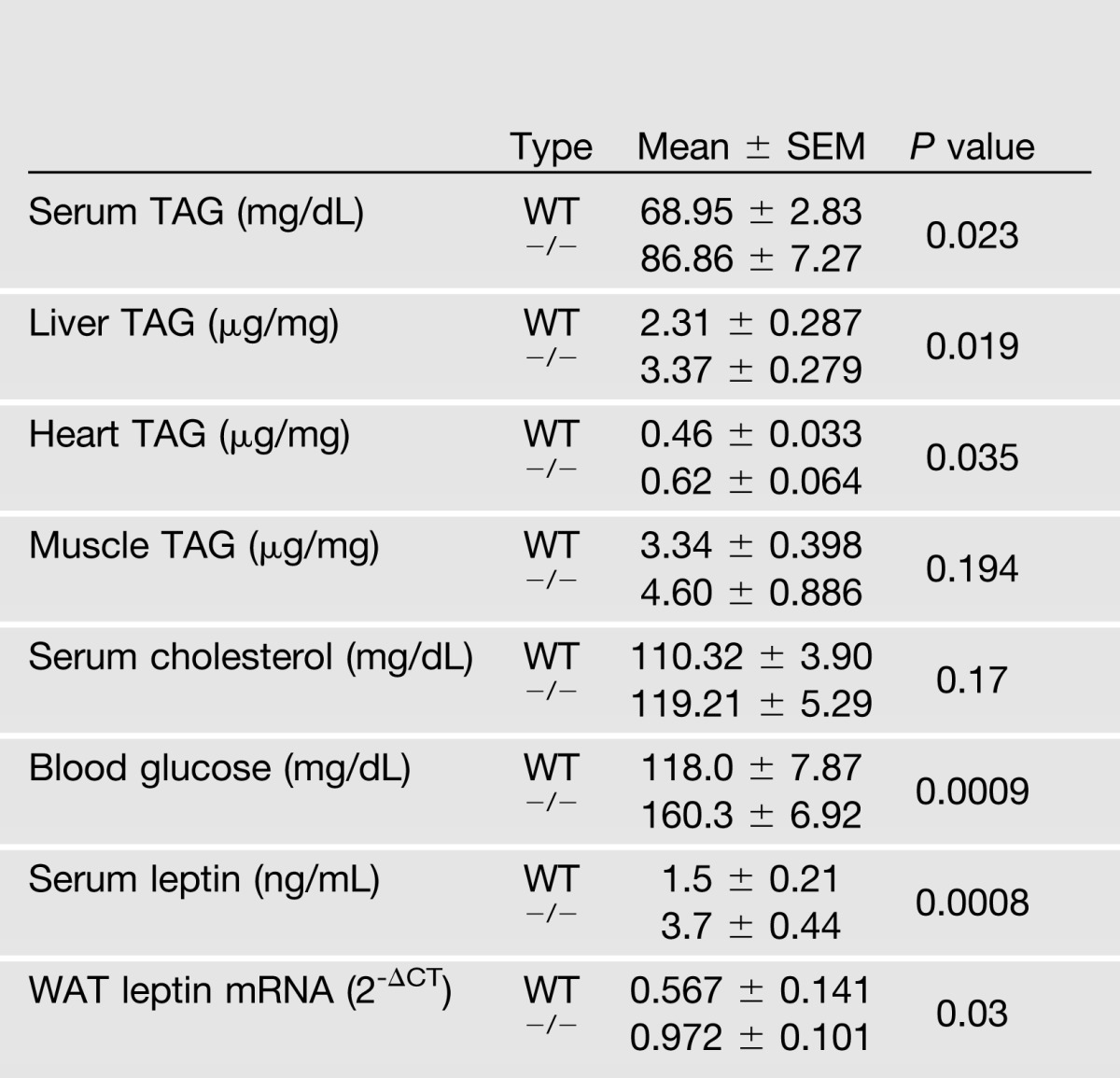
Excess adiposity in Mfap2−/− mice was the direct consequence of MAGP1 deficiency as MAGP2 and fibrillin-1 transcript expression was normal in the WAT of these animals (Supplementary Fig. 1B). MAGP1 deficiency in the outbred Black Swiss mouse strain also resulted in increased adiposity (Supplementary Fig. 1C), indicating that MAGP1 deficiency, and not genetic background, was the major determinant of fat overgrowth in these animals. Finally, excess adiposity in Mfap2−/− mice was not preceded by either increased caloric intake or reduced ambulatory activity (Fig. 1E and F).
Expansion of adipose tissue in Mfap2−/− animals was associated with poor metabolic health. Mfap2−/− mice had elevated serum triacylglycerol (TAG), tissue TAG, and serum cholesterol levels when compared with WT mice (Table 1). GTT results demonstrated that adult Mfap2−/− mice, when fed standard chow diet, had impaired glucose clearance leading to elevated blood glucose levels; and ITT results revealed reduced insulin sensitivity (Fig. 2A). Mfap2−/− mice were also more susceptible to the adverse metabolic effects of HFD. WT and Mfap2−/− mice show significant weight gain on the HFD; however, Mfap2−/− mice maintain their elevated adiposity compared with WT mice (Fig. 2B). Hyperglycemia, hyperinsulinemia, and insulin resistance were significantly accentuated in Mfap2−/− mice relative to WT animals fed HFD for 12 weeks (Fig. 2C–E). Further, ectopic lipid accumulation in the liver was substantially elevated in the knock-out animals compared with WT mice fed HFD for 14 weeks (Fig. 2F). These data demonstrate that the absence of MAGP1 predisposes mice to hyperlipidemia, hyperglycemia, hyperinsulinemia, ectopic lipid accumulation, and impaired glucose metabolism, suggesting that MAGP1 serves a protective role against metabolic disease.
Figure 2.

Adipose tissue expansion in Mfap2−/− mice is associated with metabolic dysfunction. A: GTT and ITT results in 5- to 6-month-old mice, following 12-h overnight fast and 1 g/kg dextrose injection or 6-h fast and 0.75 units/kg insulin injection (n = 7–10). B: High-fat feeding exacerbates the metabolic dysfunction associated with MAGP1 deficiency. Whole-body fat content in response to control chow and HFD (60% fat) was determined by EchoMRI (mean ± SEM; n = 8–10). C: 6-h fast blood glucose levels in WT and Mfap2−/− (−/−) mice fed chow or HFD for 12 weeks (mean ± SEM; n = 8–10). D: Serum insulin concentration in response to 1 g/kg dextrose injection; WT and Mfap2−/− mice were fed chow or HFD for 12 weeks (mean ± SEM; n = 7–10). E: ITT results following 6-h fast and 0.75 units/kg insulin injection in WT and Mfap2−/− mice consuming chow or HFD for 13 weeks (n = 8–10 per group). F: Ectopic lipid accumulation is enhanced in MAGP1-deficient tissue. Biopsy EchoMRI and histology (hematoxylin-eosin; scale bar 400 μm) was performed on liver from WT and Mfap2−/− mice that consumed HFD for 14 weeks (mean ± SEM; n = 5–6). Student t test was used for single comparisons, *P < 0.05.
Mfap2−/− Adipocytes Are Hypertrophic
Adipocyte number and size were evaluated in standard chow-fed WT and Mfap2−/− mice. Adipocyte number was normal in Mfap2−/− WAT; however, adipocyte volume was increased. Cell number was calculated by normalizing the number of cells per milligram of digested tissue to total fat pad mass (Fig. 3A). Adipocyte sizing analysis of Mfap2−/− WAT revealed that there were more cells with diameters between 20 and 50 μm, fewer cells with diameters in the range of 60–100 μm, and more cells >100 μm in diameter compared with control animals (Fig. 3B). As a result, the separation between the populations of small (25–50 μm) and large (60–100 μm) adipocytes, which was well-delineated in WT tissue, was less so in Mfap2−/− WAT. Histological assessment supported larger adipocyte volume in the WAT of Mfap2−/− mice (Fig. 3C).
Figure 3.

Lipid storage is enhanced in Mfap2−/− WAT. A: Total adipocyte number in epididymal WAT (mean [±SEM] number of adipocytes per milligram of tissue multiplied by total fat pad mass; n = 5). B: White adipocyte size distribution was determined using a Beckman Multisizer, and lines represent the average of all samples (left). The distribution curves were used to determine the average volume of small and large adipocytes (right) (mean ± SEM; n = 10 and 8). C: A pictomicrograph of epididymal WAT from 5-month-old WT and Mfap2−/− mice (scale bar 100 μm; image color was inverted to accentuate adipocyte cell borders). D and F: FA uptake, not lipolysis, is enhanced in Mfap2−/− WAT. Assays were performed on intact WAT explants. D: FA uptake, determined by the uptake of tritiated oleic acid, in the presence/absence of 10 μmol/L insulin (mean ± SEM; n = 5–6). F: Lipolysis, determined by glycerol release following the addition of 10 μmol/L isoproterenol (mean ± SEM; n = 4–5). E and G: Differential expression of lipid metabolism–associated genes in Mfap2−/− muscle tissue. qPCR was performed on RNA extracted from the muscle of WT and Mfap2−/− mice fed control chow or HFD (mean ± SEM; n = 5 and 6). Gene targets included the following: Lpl (lipoprotein lipase), Cd36 (FA translocase), Plin5 (perilipin 5), Plin2 (perilipin 2, adipose differentiation-related protein [ADFP]), Dgat2 (diacylglycerol acyltransferase 2), Cpt1b (carnitine palmitoyl transferase 1b), Pparα, Pparδ, Ppargc1α (PGC-1α, PPARγ coactivator 1 α), and Ucp-3. The Student t test was used for single comparisons (*P ≤ 0.05). Lg, large; Sm, small; vol, volume.
To elucidate the underlying mechanisms for excess lipid content in chow-fed Mfap2−/− mice, the rates of fatty acid (FA) uptake and lipolysis were evaluated. Because MAGP1 is an ECM protein, these functional assays were performed on minced WAT with intact ECM. Stimulation of FA uptake by insulin was evident in WT WAT (Fig. 3D). Basal FA uptake in Mfap2−/− WAT was elevated and did not respond to stimulation by 10 μmol/L insulin as observed in WT tissue, indicating abnormal FA uptake in Mfap2−/− animals (Fig. 3D). Altered regulation of FA metabolism was then demonstrated in muscle, the major energy-using tissue, using qPCR analysis of key lipid uptake/storage genes. HFD feeding in mice induced the expression of Lpl, Cd36, Plin5, Plin2 (Adfp), and Dgat2 in both WT and Mfap2−/− cohorts. MAGP1 deficiency accentuates the expression of these genes (Fig. 3E). In contrast to FA uptake, lipolysis in response to isoproterenol was normal in Mfap2−/− WAT (Fig. 3F). Further, the expression of genes that support lipid catabolism (Cpt1b, Pparα, and Pparδ) was not different in Mfap2−/− tissue when compared with changes seen in WT controls (Fig. 3G).
Interestingly, expression of Ppargc1α (PGC-1α) and Ucp-3, genes important to mitochondrial uncoupling, was significantly decreased in Mfap2−/− muscle (Fig. 3G). These results suggest that MAGP1 deficiency results in a defect in energy utilization and that this defect leads to excess lipid storage and adipocyte hypertrophy.
Reduced Heat Production in Mfap2−/− Mice
WAT specializes in lipid storage, while the primary function of BAT is using FAs for heat production (27,28). To determine whether MAGP1 deficiency influences BAT function, we analyzed BAT composition and thermoregulation. On standard chow, Mfap2−/− mice had increased BAT mass due to increased lipid accumulation (Fig. 4A and B). The HFD increased both BAT mass and lipid content in WT animals, and accentuated the increased lipid content in Mfap2−/− BAT (Fig. 4A and B).
Figure 4.

Impaired heat production in Mfap2−/− mice. A: BAT mass is elevated in adult WT and Mfap2−/− mice fed either control chow or HFD for 16 weeks (mean ± SEM; n = 7 and 10). B: Increased BAT mass is due to increased lipid content. BAT lipid-to-protein ratio was determined by biopsy samples obtained using the EchoMRI (mean ± SEM; n = 5 and 6). C: Rectal temperatures of adult mice during the day (12:00 p.m.) and night (12:00 a.m.) (mean ± SEM; n = 5 and 6). D: Rectal body temperature in adult mice during a 4°C cold challenge (mean ± SEM; n = 4 and 5). E: qPCR of thermogenesis-related genes in BAT from adult mice kept at room temperature (RT) or 4°C for 6 h. Data represent the averages of samples from two independent studies (mean ± SEM; n = 7). *P ≤ 0.05.
Measurement of body temperature during light and dark cycles found that Mfap2−/− mice had reduced body temperature relative to control animals during both periods (Fig. 4C). To further characterize BAT functionality, adaptive thermogenesis was evaluated by exposing the mice to 4°C temperature for 6 h. Both WT and Mfap2−/− mice exhibited reduced body temperatures during cold exposure. However, Mfap2−/− animals had significantly greater body temperature loss (Fig. 4D). In agreement with the altered thermoregulation, Mfap2−/− mice were not as efficient as WT mice in upregulating expression of the BAT-associated genes Ppargc1α (PGC-1α) and Ucp-1 following cold exposure (Fig. 4E).
To investigate whether impaired thermoregulation accounts for the excess adiposity of Mfap2−/− mice, thermoregulation was evaluated in young Mfap2−/− mice at an age (4–6 weeks old) before excess lipid accumulation was detectable. At 1 month, Mfap2−/− mice had no change in whole-body adiposity, but at 5 months these mice had a significant increase in adiposity relative to WT mice (Fig. 5A). At 1 month, the same Mfap2−/− mice had reduced body temperature relative to WT mice, and this phenotype was maintained at 5 months (Fig. 5A). Data from indirect calorimetry demonstrated a trend toward reduced energy expenditure in Mfap2−/− mice compared with WT mice, but the differences were not statistically significant (Fig. 5B). Young Mfap2−/− mice were also maladaptive to 4°C cold challenge (Fig. 5C). Lipid content in Mfap2−/− BAT appeared slightly elevated by histology (Fig. 5D), and, similar to adult mice, young Mfap2−/− mice had reduced BAT expression of Ucp-1 (Fig. 5E). Interestingly, Mfap2−/− mice had reduced thermogenesis and Ucp-1 expression despite having no significant difference in mitochondrial content (Fig. 5F).
Figure 5.

Increased adiposity in Mfap2−/− mice is preceded by impaired energy expenditure (EE). A: Longitudinal comparison of adiposity (left) and temperature (right). Adiposity (EchoMRI) and rectal temperature were determined at 1 month, then at 5 months of age (mean ± SEM; n = 8–10). B: EE was determined by indirect calorimetry in mice that were ∼7 weeks old. B, Left: Heat production (presented as EE) curves represent the average of 3 WT or 4 Mfap2−/− experimental groups, where each group consisted of 3–4 mice. B, Right: EE is presented as the daytime (1:00–7:00 p.m.) and nighttime (8:00 p.m. to 7:00 a.m.) values. C: Rectal temperature in young (6 week) mice during 4°C cold challenge (mean ± SEM; n = 5). D: Hematoxylin-eosin–stained BAT sections from 6-week-old WT and Mfap2−/− mice (scale bar 100 μm). E: Transcript expression of Ucp-1 in the BAT of 6-week-old WT and Mfap2−/− mice (qPCR, mean ± SEM; n = 5). F: Normal mitochondrial copy number in Mfap2−/− BAT. Copy number is ratio of mitochondrial (Cytb, Nd1) to nuclear (H19) DNA (qPCR, mean ± SEM; n = 4). G: Hematoxylin-eosin–stained scWAT sections from 6-week-old WT and Mfap2−/− mice (scale bar 100 μm). H: PGC-1α and Ucp-1 transcript expression in scWAT from ∼6-week-old mice during an 8-h cold challenge (mean ± SEM; n = 3–5 per genotype per time point). I: Normal mitochondrial copy number in Mfap2−/− scWAT. The copy number is the ratio of mitochondrial (Cytb, cytochrome b; Nd1, NADH dehydrogenase subunit 1) to nuclear (H19) DNA (qPCR, mean ± SEM; n = 3–5). The Student t test was used for single comparisons (*P ≤ 0.05). Rel mito, relative mitochondrial; RT, room temperature.
While mouse gonadal WAT had little-to-no expression of thermogenic genes such as Ucp-1, scWAT has thermogenic potential through a process termed “adipocyte browning” (29). Because activating the PGC-1α/UCP-1 pathway in mouse scWAT is protective against diet-induced diabetes (29–31), adipocyte browning was explored in the scWAT of Mfap2−/− mice. By histology, Mfap2−/− scWAT appeared to have elevated lipid content and fewer clusters of multilocular adipocytes (Fig. 5G). Cold challenge induced adipocyte browning and expression of Ppargc1α (PGC-1α) and Ucp-1 in WT scWAT; however, this response was significantly blunted in Mfap2−/− scWAT (Fig. 5H). Reduced Ppargc1α (PGC-1α) and Ucp-1 expression was not due to a failure in mitochondrial biogenesis as mitochondrial content was increased appropriately in Mfap2−/− scWAT following cold exposure (Fig. 5I). These studies indicated that MAGP1 deficiency suppressed the acquisition of brown fat features in white fat, and further support a model where adiposity was increased in Mfap2−/− mice because of reduced energy expenditure in the form of heat production.
Neutralizing TGF-β Activity Resolves Mfap2−/− Phenotypes
Microfibrils facilitate storage of TGF-β family growth factors in the ECM (8,32), and we have shown that MAGP1 functionally interacts with TGF-β (9,10). Because TGF-β impairs thermogenesis while supporting WAT expansion and insulin resistance (16), aberrant TGF-β activity was investigated as the mechanism underlying the metabolic phenotypes in Mfap2−/− mice. Shown in Fig. 6A, loss of MAGP1 resulted in a significant increase in TGF-β activity in WAT, as assessed by Smad-2 phosphorylation. TGF-β contributes to the pathogenesis of obesity and metabolic syndrome by stimulating fibrosis and inflammation (16). Figure 6B shows that expression of the major collagens associated with the fibrotic response, collagen-1 and collagen-3, were elevated by HFD in both WT and Mfap2−/− WAT. However, this elevation was exaggerated in the Mfap2−/− tissue. Trichrome staining provided further support for increased collagen deposition in HFD-fed Mfap2−/− WAT versus WT cohorts. HFD feeding also induced expression of macrophage-associated genes in WT WAT (Fig. 6C). As predicted, the expression of these inflammatory genes was significantly higher in the Mfap2−/− WAT. Increased macrophage infiltration into Mfap2−/− WAT relative to WT WAT following HFD feeding was supported by anti-Mac3 immunohistochemistry (Fig. 6C). These data show that MAGP1 regulates TGF-β signaling and TGF-β–dependent processes in adipose tissue.
Figure 6.

Abberrent TGF-β activity causes impaired thermoregulation and excess adiposity in Mfap2−/− mice. A: Immunoblot of WAT lysate using antibodies to phosphorylated Smad-2 (p-Smad2), total Smad2 (t-Smad2), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Shown are lysates from 2 mice per genotype. B and C: WAT from mice on chow or HFD for 16 weeks. B: ECM deposition in WAT was assessed by qPCR of collagen-1 (Col1) and collagen-3 (Col3) and by trichrome staining of tissue. C: WAT macrophage infiltration was assessed by qPCR of macrophage-associated genes (Tnfα [tumor necrosis factor-α], Il-6 [interleukin-6], Itgax [CD11c], Arg1 [arginase-1], and Il-10 [interleukin-10]), and immunohistochemistry using an anti-MAC-3 antibody. B and C: Bar graphs are presented as mean ± SEM (n = 5 and 6 for qPCR). Scale bars 100 μm. D and E: α-TGF-β treatment prevents excess adiposity and improves body temperature in Mfap2−/− (−/−) mice. Adiposity (EchoMRI) and rectal temperature were determined before (D) and after (E) 5 weeks of treatment with control IgG or α-TGF-β (mean ± SEM; n = 4 and 5 per treatment group). *P ≤ 0.05.
To determine whether dysregulation of TGF-β was responsible for the reduced thermogenesis and increased adiposity in standard chow–fed Mfap2−/− mice, WT and Mfap2−/− mice were treated with a neutralizing α-TGF-β antibody or control IgG. Before treatment, 8-week-old Mfap2−/− mice had reduced body temperature, but there was no difference in adiposity compared with WT mice (Fig. 6D). Five weeks of IgG treatment failed to prevent the increase in adiposity seen in Mfap2−/− mice. However, adiposity in Mfap2−/− mice treated with the α-TGF-β was not significantly different from WT mice (Fig. 6E). Importantly, treatment with the α-TGF-β antibody resulted in elevated body temperatures of Mfap2−/− mice to near WT levels (Fig. 6E). Thus, MAGP1 supports energy expenditure and protects against excess lipid accumulation by regulating the availability of TGF-β.
MAGP1 Expression in Obese Humans
To investigate the relationship among TGF-β, MAGP1, and obesity in humans, we measured TGF-β1 (TGF-B1) and MAGP1 (MFAP2) gene expression in scWAT from 30 individuals with varying BMI values. WAT expression of TGF-β1 positively correlated with obesity (Fig. 7A), a finding substantiated by other laboratories (17,19). MAGP1 expression was also elevated in individuals with excess adiposity, and statistical significance was reached when comparing individuals considered to be normal weight (BMI <25 kg/m2) to overweight-obese individuals (BMI >25 kg/m2) (Fig. 7B and C). Similar to human WAT, the MAGP1 transcript was significantly elevated in the WAT of obese mice, following HFD feeding (Fig. 7D). Immunohistochemistry using an anti-MAGP1 antibody supported increased MAGP1 expression in WAT during metabolic challenge. HFD feeding in mice did not alter the expression of the microfibril molecules MAGP2 or fibrillin-1.
Figure 7.

MAGP1 correlates with TGF-β1 expression in obese humans. A–C: TGF-β1 and MAGP1 transcript expression in human scWAT was determined and compared with each other and the individual’s BMI. qPCR was used to determine TGF-β1 (TGFB1) and MAGP1 (MFAP2) expression in human scWAT (n = 29). A and B: TGF-β1 expression in WAT positively correlates with BMI and MAGP1 expression. C: Human MFAP2 (MAGP1) transcript expression was plotted against the individual’s BMI (n = 29). D: Positive correlation between MAGP1 expression in WAT and obesity is confirmed in mice (top) MAGP1 (Mfap2), not MAGP2 (Mfap5) or fibrillin-1 (Fbn1), correlates with obesity in mice. Transcripts were measured in WAT of mice fed standard chow diet (chow) or HFD. Data were plotted as the mean ± SEM of HFD WAT expression relative to chow WAT expression (n = 5 and 5). D, Bottom: Immunohistochemistry with anti-MAGP1 antibody of WAT from WT mice fed control chow and HFD. E: Proposed model for the mechanism by which MAGP1 regulates energy expenditure in mice. MAGP1 is a sink for TGF-β thereby, supporting lipid catabolism and thermogenesis. When active TGF-β concentration becomes excessive (i.e., MAGP1 saturation or MAGP1 deficiency), free TGF-β reduces PPAR and PRDM-16 coactivation of PGC-1α, which in turn prevents activation of UCP-1 transcription and, thus, thermogenesis. The imbalance of TGF-β and MAGP1 not only leads to reduced thermogenesis but also to excess lipid accumulation and fibrosis/inflammation, and eventually to features of metabolic syndrome. RU, relative units; hWAT, human WAT. *P ≤ 0.05.
Collectively, the data in this manuscript demonstrate that MAGP1 supports energy expenditure by impeding TGF-β activity, excess fat accumulation is associated with increased expression of both TGF-β1 and MAGP1, and the absence of MAGP1 causes predisposition to obesity-associated metabolic dysfunction. Therefore, altered MAGP1 expression could be considered a protective-adaptive response to obesity.
Discussion
The adipose ECM provides a structural scaffold that defines the limits of tissue growth. Changing the physical properties of the ECM has functional consequences; fibrotic ECM restricts adipocyte expansion and function (33), while decreasing ECM rigidity results in a permissive environment that supports adipose tissue expansion (34–36). In this report, we demonstrated that ECM components contribute more than mechanical properties to adipose tissue, and identified a mechanism by which the ECM influences cellular processes involved in energy expenditure by restricting growth factor delivery. Specifically, we demonstrated that the microfibril-associated protein MAGP1 is involved in regulating thermogenesis and the browning of white adipocytes through a TGF-β–mediated pathway. Accordingly, these data provide evidence of a novel mechanism for regulating energy metabolism by ECM proteins in the adipose tissue microenvironment. Further, our findings suggest that induction of WAT MAGP1 expression is an adaptive response that protects against excess TGF-β associated with obesity.
Elevated TGF-β levels correlate with obesity in humans and mice (17,19), and suggest a mechanistic link between MAGP1 and metabolism. A study by Yadav et al. (19) demonstrated that, in mice, downregulation of the TGF-β signaling pathway through deletion of a TGF-β signaling mediator, Smad3, resulted in a phenotype nearly opposite to that of the Mfap2−/− mouse. Smad3 knock-out mice are lean, have improved glucose metabolism, and are protected from diet-induced obesity, and thermogenesis is improved. They also found that systemic blockade of TGF-β signaling protects mice from obesity, diabetes, and hepatic steatosis. MAGP1 binds active TGF-β and controls its bioavailability through sequestration in the ECM. In the absence of MAGP1, there is less sequestered and, hence, more active TGF-β. The mechanistic pathway whereby elevated TGF-β signaling associated with MAGP1 deficiency influences energy metabolism is through reduced energy dissipation and transcriptional regulation of PGC-1α and UCP-1. TGF-β–induced Smad phosphorylation allows Smad to bind to and inhibit PRDM-16 function. PRDM-16 is a transcription coregulator that is essential to BAT development and drives expression PGC-1α, which is a transcription coregulator of itself and the thermogenic gene Ucp-1. Smad also inhibits the function of peroxisome proliferator–activated receptor (PPAR) β/δ. PPARβ/δ supports energy dissipation through FA oxidation and PGC-1α expression. The metabolic phenotypes found in Mfap2−/− mice are supportive of excess TGF-β signaling. Mfap2−/− mice have increased Smad phosphorylation, impaired transcription of both PGC-1α and UCP-1, and reduced energy expenditure (thermogenesis). These changes contribute to an accumulation of body fat mass, adipose tissue inflammation, ectopic lipid accumulation, and predisposition to metabolic dysfunction.
Several studies have associated obesity traits in humans to a locus around chromosome 1p36 that includes the gene MFAP2 (13–15). In a study of human WAT from normal weight, overweight, and obese individuals, we found MAGP1 levels to be elevated in the WAT from individuals with BMI values >25 kg/m2. While it remains to be determined in humans what role the increase in MAGP1 plays in obesity-associated metabolic dysfunction, we demonstrate in this report that the inability to increase MAGP1 expression during diet-induced obesity results in exacerbated metabolic disease in mice, suggesting that the modulation of MAGP1 expression is a protective-adaptive response to metabolic challenge. Figure 7E is a graphic representation of the proposed function of MAGP1. In adipose tissue, MAGP1 supports the sequestration of active TGF-β in the ECM. The capacity of MAGP1 to limit free TGF-β facilitates homeothermy and adaptive thermogenesis by allowing the transcription of Ppargc1α (PGC-1α) and Ucp-1. In pathologic conditions where TGF-β secretion is excessive, such as obesity, we hypothesize that MAGP1 expression is induced as a protective-adaptive response to sequester excess active TGF-β, thereby facilitating energy dissipation and protecting against inflammation and fibrosis.
Exemplifying the distinct functions of the ECM (mechanical vs. cell signaling) is the change in adiposity associated with Marfan syndrome (fibrillin-1 mutation). Marfan syndrome has been attributed to the disrupted assembly of microfibrils, and subsequently to the inability to sequester the latent TGF-β complex (8,32). Given the positive effect of TGF-β on adipocyte hypertrophy and the negative effect on thermoregulation, it would be expected that individuals with Marfan syndrome would be predisposed to obesity and diabetes, similar to Mfap2−/− mice. However, most individuals with Marfan syndrome have reduced adiposity (32,37,38). It is somewhat surprising that mutations in two proteins of the same extracellular fiber can result in such contrasting phenotypes. However, MAGP1 is not a structural protein of the microfibril but a modifier of fibrillin function. The MAGP1 deletion leaves the core microfibril intact. In contrast, mutation of fibrillin, the structural backbone of microfibrils, can disrupt the mechanical integrity of microfibrils and thus alter the physical properties of the adipose ECM. It is plausible that individuals with mutant fibrillin-1 have adipose ECM that constrains adipocyte growth, resulting in smaller adipocytes.
In summary, this study demonstrates that the ECM component MAGP1 has the capacity to regulate growth factor availability that is important for maintaining normal metabolic function, and provides further support for the role of TGF-β in the etiology of obesity-associated metabolic disease. Our results also highlight the contribution that accessory proteins, like MAGP1, provide to overall microfibril function and tissue homeostasis.
Article Information
Acknowledgments. The authors thank present and former members of the Mecham laboratory at Washington University, especially Daniel Abernathy.
Funding. This work was supported by National Institutes of Health grants DK-37948 to S.K., DK-033301 to N.A.A., HL-053325 to R.P.M., HL-105314 to R.P.M., HL-074138 to R.P.M., and UL1-RR-024992 (Clinical and Translation Science Award). Additional support was received from American Diabetes Association grant 7-13-JF-16 to C.S.C. and an award from the Washington University Nutrition and Obesity Research Center, P30-DK-056341 to C.S.C. Technical support was provided by the Mouse Phenotyping Core, the Adipocyte Biology and Molecular Nutrition Core of the Nutrition and Obesity Research Center (grant P30-DK-056341), and the Morphology Core of the Digestive Disease Research Core Center (grant P30-DK-52574).
Duality of Interest. S.K. is a shareholder of Aspire Bariatrics. No other potential conflicts of interest relevant to this article were reported.
Author Contributions. C.S.C. designed and performed the experiments, analyzed data, and wrote the manuscript. T.A.P. and T.C. provided technical support and gave conceptual advice. T.S. and M.D.C. provided technical support. S.K. provided valuable research materials and contributed to editing of the manuscript. N.A.A. and R.P.M. gave conceptual advice, provided valuable research materials, and contributed to editing of the manuscript. C.S.C. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db13-1604/-/DC1.
See accompanying article, p. 1858.
References
- 1.Corson GM, Charbonneau NL, Keene DR, Sakai LY. Differential expression of fibrillin-3 adds to microfibril variety in human and avian, but not rodent, connective tissues. Genomics 2004;83:461–472 [DOI] [PubMed] [Google Scholar]
- 2.Sakai LY, Keene DR, Engvall E. Fibrillin, a new 350-kD glycoprotein, is a component of extracellular microfibrils. J Cell Biol 1986;103:2499–2509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang H, Apfelroth SD, Hu W, et al. Structure and expression of fibrillin-2, a novel microfibrillar component preferentially located in elastic matrices. J Cell Biol 1994;124:855–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang H, Hu W, Ramirez F. Developmental expression of fibrillin genes suggests heterogeneity of extracellular microfibrils. J Cell Biol 1995;129:1165–1176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carta L, Pereira L, Arteaga-Solis E, et al. Fibrillins 1 and 2 perform partially overlapping functions during aortic development. J Biol Chem 2006;281:8016–8023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cleary EG, Gibson MA. Elastin-associated microfibrils and microfibrillar proteins. Int Rev Connect Tissue Res 1983;10:97–209 [DOI] [PubMed] [Google Scholar]
- 7.Segade F. Functional evolution of the microfibril-associated glycoproteins. Gene 2009;439:43–54 [DOI] [PubMed] [Google Scholar]
- 8.Ramirez F, Carta L, Lee-Arteaga S, Liu C, Nistala H, Smaldone S. Fibrillin-rich microfibrils - structural and instructive determinants of mammalian development and physiology. Connect Tissue Res 2008;49:1–6 [DOI] [PubMed] [Google Scholar]
- 9.Weinbaum JS, Broekelmann TJ, Pierce RA, et al. Deficiency in microfibril-associated glycoprotein-1 leads to complex phenotypes in multiple organ systems. J Biol Chem 2008;283:25533–25543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Craft CS, Broekelmann TJ, Zou W, Chappel JC, Teitelbaum SL, Mecham RP. Oophorectomy-induced bone loss is attenuated in MAGP1-deficient mice. J Cell Biochem 2012;113:93–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Craft CS, Zou W, Watkins M, et al. Microfibril-associated glycoprotein-1, an extracellular matrix regulator of bone remodeling. J Biol Chem 2010;285:23858–23867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Werneck CC, Vicente CP, Weinberg JS, et al. Mice lacking the extracellular matrix protein MAGP1 display delayed thrombotic occlusion following vessel injury. Blood 2008;111:4137–4144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoffmann K, Mattheisen M, Dahm S, et al. A German genome-wide linkage scan for type 2 diabetes supports the existence of a metabolic syndrome locus on chromosome 1p36.13 and a type 2 diabetes locus on chromosome 16p12.2. Diabetologia 2007;50:1418–1422 [DOI] [PubMed] [Google Scholar]
- 14.Liu YJ, Xu FH, Shen H, et al. A follow-up linkage study for quantitative trait loci contributing to obesity-related phenotypes. J Clin Endocrinol Metab 2004;89:875–882 [DOI] [PubMed] [Google Scholar]
- 15.Pausova Z, Gaudet D, Gossard F, et al. Genome-wide scan for linkage to obesity-associated hypertension in French Canadians. Hypertension 2005;46:1280–1285 [DOI] [PubMed] [Google Scholar]
- 16.Tan CK, Chong HC, Tan EHP, Tan NS. Getting “Smad” about obesity and diabetes. Nutr Diabetes 2012;2:e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Alessi MC, Bastelica D, Morange P, et al. Plasminogen activator inhibitor 1, transforming growth factor-beta1, and BMI are closely associated in human adipose tissue during morbid obesity. Diabetes 2000;49:1374–1380 [DOI] [PubMed] [Google Scholar]
- 18.Herder C, Zierer A, Koenig W, Roden M, Meisinger C, Thorand B. Transforming growth factor-beta1 and incident type 2 diabetes: results from the MONICA/KORA case-cohort study, 1984-2002. Diabetes Care 2009;32:1921–1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yadav H, Quijano C, Kamaraju AK, et al. Protection from obesity and diabetes by blockade of TGF-β/Smad3 signaling. Cell Metab 2011;14:67–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chakravarthy MV, Zhu Y, López M, et al. Brain fatty acid synthase activates PPARalpha to maintain energy homeostasis. J Clin Invest 2007;117:2539–2552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li B, Nolte LA, Ju JS, et al. Skeletal muscle respiratory uncoupling prevents diet-induced obesity and insulin resistance in mice. Nat Med 2000;6:1115–1120 [DOI] [PubMed] [Google Scholar]
- 22.Marshall BA, Tordjman K, Host HH, et al. Relative hypoglycemia and hyperinsulinemia in mice with heterozygous lipoprotein lipase (LPL) deficiency. Islet LPL regulates insulin secretion. J Biol Chem 1999;274:27426–27432 [DOI] [PubMed] [Google Scholar]
- 23.Harris RB, Martin RJ. Metabolic response to a specific lipid-depleting factor in parabiotic rats. Am J Physiol 1986;250:R276–R286 [DOI] [PubMed] [Google Scholar]
- 24.Liu Y, Zhou D, Abumrad NA, Su X. ADP-ribosylation factor 6 modulates adrenergic stimulated lipolysis in adipocytes. Am J Physiol Cell Physiol 2010;298:C921–C928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nassir F, Wilson B, Han X, Gross RW, Abumrad NA. CD36 is important for fatty acid and cholesterol uptake by the proximal but not distal intestine. J Biol Chem 2007;282:19493–19501 [DOI] [PubMed] [Google Scholar]
- 26.Bradley D, Conte C, Mittendorfer B, et al. Gastric bypass and banding equally improve insulin sensitivity and β cell function. J Clin Invest 2012;122:4667–4674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bartelt A, Bruns OT, Reimer R, et al. Brown adipose tissue activity controls triglyceride clearance. Nat Med 2011;17:200–205 [DOI] [PubMed] [Google Scholar]
- 28.Saely CH, Geiger K, Drexel H. Brown versus white adipose tissue: a mini-review. Gerontology 2012;58:15–23 [DOI] [PubMed] [Google Scholar]
- 29.Wu J, Boström P, Sparks LM, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012;150:366–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fisher FM, Kleiner S, Douris N, et al. FGF21 regulates PGC-1α and browning of white adipose tissues in adaptive thermogenesis. Genes Dev 2012;26:271–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koncarevic A, Kajimura S, Cornwall-Brady M, et al. A novel therapeutic approach to treating obesity through modulation of TGFβ signaling. Endocrinology 2012;153:3133–3146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Judge DP, Dietz HC. Marfan’s syndrome. Lancet 2005;366:1965–1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chun TH, Hotary KB, Sabeh F, Saltiel AR, Allen ED, Weiss SJ. A pericellular collagenase directs the 3-dimensional development of white adipose tissue. Cell 2006;125:577–591 [DOI] [PubMed] [Google Scholar]
- 34.Bradshaw AD, Graves DC, Motamed K, Sage EH. SPARC-null mice exhibit increased adiposity without significant differences in overall body weight. Proc Natl Acad Sci USA 2003;100:6045–6050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khan T, Muise ES, Iyengar P, et al. Metabolic dysregulation and adipose tissue fibrosis: role of collagen VI. Mol Cell Biol 2009;29:1575–1591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kos K, Wilding JP. SPARC: a key player in the pathologies associated with obesity and diabetes. Nat Rev Endocrinol 2010;6:225–235 [DOI] [PubMed] [Google Scholar]
- 37.Graul-Neumann LM, Kienitz T, Robinson PN, et al. Marfan syndrome with neonatal progeroid syndrome-like lipodystrophy associated with a novel frameshift mutation at the 3′ terminus of the FBN1-gene. Am J Med Genet A 2010;152A:2749–2755 [DOI] [PubMed] [Google Scholar]
- 38.Summers KM, Nataatmadja M, Xu D, et al. Histopathology and fibrillin-1 distribution in severe early onset Marfan syndrome. Am J Med Genet A 2005;139:2–8 [DOI] [PubMed] [Google Scholar]