

Abstract
Insulin receptor substrate 1 (IRS1) is a key mediator of insulin signal transduction. Perturbations involving IRS1 complexes may lead to the development of insulin resistance and type 2 diabetes (T2D). Surprisingly little is known about the proteins that interact with IRS1 in humans under health and disease conditions. We used a proteomic approach to assess IRS1 interaction partners in skeletal muscle from lean healthy control subjects (LCs), obese insulin-resistant nondiabetic control subjects (OCs), and participants with T2D before and after insulin infusion. We identified 113 novel endogenous IRS1 interaction partners, which represents the largest IRS1 interactome in humans and provides new targets for studies of IRS1 complexes in various diseases. Furthermore, we generated the first global picture of IRS1 interaction partners in LCs, and how they differ in OCs and T2D patients. Interestingly, dozens of proteins in OCs and/or T2D patients exhibited increased associations with IRS1 compared with LCs under the basal and/or insulin-stimulated conditions, revealing multiple new dysfunctional IRS1 pathways in OCs and T2D patients. This novel abnormality, increased interaction of multiple proteins with IRS1 in obesity and T2D in humans, provides new insights into the molecular mechanism of insulin resistance and identifies new targets for T2D drug development.
Introduction
Insulin resistance in skeletal muscle, the major site of insulin-stimulated glucose disposal, underlies a large number of aberrant conditions, such as the metabolic syndrome and type 2 diabetes (T2D) (1–4). Insulin receptor substrate 1 (IRS1) plays a central role in the insulin cascade, and its ability to form signaling complexes with the insulin receptor and intracellular signaling partners as a keystone, linking events at the plasma membrane with intracellular machinery. Abnormal protein-protein interactions involving IRS1 may interfere with proper insulin transduction and contribute to the development of insulin resistance and T2D. Most studies on IRS1 interaction partners have been carried out in cell culture or animal models, focusing on a few known targets (5–11). Whether these findings can be translated into humans is unclear. IRS1 contains a pleckstrin homology domain and a phosphotyrosine binding domain, through which it interacts with the insulin receptor and insulin-like growth factor 1 receptor. IRS1 also has several YXXM motifs. Upon tyrosine phosphorylation, IRS1 interacts with the p85 regulatory subunit of phosphatidylinositide 3-kinase (PI3K) which leads to the activation of this enzyme and subsequent activation of Akt, resulting in enhanced glucose uptake, and increased glycogen and protein synthesis (7,8). In addition, tyrosine phosphorylated IRS1 interacts with growth factor receptor-binding protein 2 (GRB2), leading to mitogen-activated protein kinase activation and subsequent promotion of cell survival and mitogenesis (10). Moreover, IRS1 interacts with negative regulators such as SH-protein tyrosine phosphatase-2, a protein tyrosine phosphatase that reduces levels insulin-stimulated tyrosine phosphorylation of IRS1 (11). Furthermore, IRS1 binds through its phosphotyrosine binding domain to tyrosine 14 of caveolin-1 (CAV1), and in CAV1-deficient cells IRS1 protein expression is reduced (9). Post-translational modifications, especially phosphorylation and glycosylation, of IRS1 have been implicated in insulin resistance and T2D. Reduced tyrosine phosphorylation of IRS1 is a common feature in insulin-resistant human skeletal muscle, while either increased or decreased site-specific serine/threonine phosphorylation of IRS1 have been reported in insulin resistance and T2D (12). Additionally, O-linked glycosylation of IRS1 is higher in insulin-resistant conditions (13). All of these insulin-signaling events involving IRS1 require the time-dependent formation of IRS1 complexes (14). Surprisingly, except for the p85α subunit, which has been shown to interact with IRS1 in human skeletal muscle (7,8), little is known about the proteins that interact with IRS1 in human skeletal muscle in health and disease conditions.
Proteomic approaches combining high-performance liquid chromatography (HPLC)-electrospray ionization (ESI)-tandem mass spectrometry (MS/MS) with coimmunoprecipitation (CO-IP) have been widely used to map protein-protein interaction networks (15,16). Nonetheless, most of these studies used protein overexpression and/or epitope-tagged bait proteins, and were performed in cell culture models. We developed a straightforward, label-free approach combining HPLC-ESI-MS/MS with CO-IP, without the use of protein overexpression or protein tags, to investigate changes in the abundance of endogenous proteins associated with a bait protein, and identified 11 novel endogenous insulin-stimulated IRS1 interaction partners in L6 myotubes (6). In the current study, we applied an improved proteomic approach to investigate IRS1 interaction partners in human skeletal muscle from lean control subjects (LCs), obese nondiabetic control subjects (OCs), and obese T2D patients. The goal of the study is to determine 1) whether the proteins shown to interact with IRS1 in cell culture or animal models indeed interact with IRS1 in human skeletal muscle, and whether there are new IRS1 interaction partners in human skeletal muscle; and 2) whether there are new abnormalities in protein-protein interactions involving IRS1 in skeletal muscle insulin resistance and T2D compared with the lean healthy condition.
Research Design and Methods
As illustrated in Supplementary Fig. 1, the approach we used included extensive clinical and proteomics data acquisition and data analysis. The clinical studies started with participant recruitment and were followed by comprehensive screening tests, and in-patient clinical procedures involving hyperinsulinemic-euglycemic clamp and muscle biopsies. The proteomics studies were conducted as follows: biopsy homogenization; immunoprecipitation of the “bait” protein (IRS1), at the endogenous level; followed by one-dimensional SDS-PAGE to separate cointeraction proteins; in-gel trypsin digestion to generate peptide fragments; and HPLC-ESI-MS/MS analysis to identify coimmunoprecipitating proteins. Multiple biological comparisons and immunoprecipitation of Normal IgG (NIgG; as nonspecific control) were used to minimize false-positive results. Extensive bioinformatics and literature searches were used to integrate clinical and proteomics data and to identify pathways/functional categories, in which identified IRS1 interaction partners were involved, that were impacted by obesity and T2D.
Reagents
The following suppliers were used: sequencing-grade modified trypsin (Promega, Madison, WI); protein A sepharose and iodoacetamide (Sigma-Aldrich, St. Louis, MO); C18 ZipTip (Millipore, Billerica, MA); and antibody to IRS1 (Millipore, Billerica, MA).
Subjects
A total of 22 volunteers—8 glucose-tolerant LCs; 7 glucose-tolerant OCs; and 7 T2D patients—were recruited and took part in the study at the C.S. Mott Center for Human Growth and Development at Wayne State University. The purpose, nature, and potential risks of the study were explained to all participants, and written consent was obtained before their participation. None of the participants had any significant medical problems (other than diabetes). None of the participants engaged in any heavy exercise, and they were instructed to stop any form of exercise for at least 2 days before the study. T2D patients were treated with metformin and/or sulfonylureas, which were withdrawn 3 days before the hyperinsulinemic-euglycemic clamp visit. The protocol was approved by the Institutional Review Board of Wayne State University.
Hyperinsulinemic-Euglycemic Clamp With Muscle Biopsies
A hyperinsulinemic-euglycemic clamp was used to assess insulin sensitivity and expose skeletal muscle to insulin in vivo, as previously described (17,18). The study began at approximately 0830 h (time −60 min) after a minimum 10-h overnight fast. A catheter was placed in an antecubital vein and maintained throughout the study for infusions of insulin and glucose. A second catheter was placed in a vein in the contralateral arm, which was covered with a heating pad (60°C) for the sampling of arterialized venous blood. Baseline arterialized venous blood samples for determination of plasma glucose and insulin concentrations were drawn. At 0900 h (time −30 min), a percutaneous needle biopsy of the vastus lateralis muscle was performed under local anesthesia. Muscle biopsy specimens were immediately blotted free of blood, cleaned of connective tissue and fat (∼30 s), and then frozen in liquid nitrogen. At 0930 h (time 0 min), a primed, continuous infusion of human regular insulin (Humulin R; Eli Lilly, Indianapolis, IN) was started at a rate of 80 mU/m2/min, and continued for 120 min. Plasma glucose was measured at 5-min intervals throughout the clamp. Euglycemia was targeted for 90 mg/dL by variable infusion of 20% d-glucose. At 1130 h (time 120 min), another muscle biopsy sample was obtained from the contralateral leg.
Plasma Insulin Concentration Determinations
Plasma insulin concentration was measured by the ALPCO Insulin ELISA Jumbo (ALPCO Diagnostics, Salem, NH).
Proteomics Sample Preparation and Analysis
Biopsy samples were homogenized and processed as previously described (17,18). The lysate proteins were precleared with NIgG followed by IRS1 immunoprecipitation. The coimmunoprecipitates were resolved on one-dimensional SDS-PAGE, followed by in-gel trypsin digestion, peptide purification, and HPLC-ESI-MS/MS analysis using an LTQ Orbitrap Elite as described (6,19). Peptides/protein identification and quantification were performed using the MaxQuant, one of the popular quantitative proteomics software packages (20). Peak areas (PAs) for each protein were obtained by selecting the label-free quantification option in MaxQuant. Only proteins identified with a minimum of two unique peptides and with a false discovery rate (FDR) of 0.01 were considered.
To be considered as an IRS1 interaction partner, a protein has to further satisfy the following criteria: 1) the protein must be identified with label-free quantification PAs in more than half of the IRS1 immunoprecipitates (IPs) (i.e., >22 biopsy samples used), and 370 proteins met this criterion; and 2) those proteins must have an enrichment ratio of >10. The enrichment ratio was calculated as follows: first, the PA for a protein identified in a gel lane was normalized against the sum of the PAs for all proteins identified in the same gel lane to obtain normalized ratio for each protein, Norm:i:
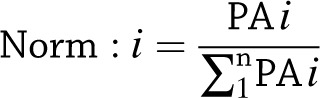 |
Then, the average of normalized ratio for each protein in the IRS1 coimmunoprecipitates, Average_Norm:i_IRS1, as well as the average of normalized ratio for the same protein in the NIgG coimmunoprecipitates, Average_Norm:i_NIgG, were obtained. Finally, Average_Norm:i_IRS1 was divided by Average_Norm:i_NIgG, to obtain the enrichment ratio for each protein:
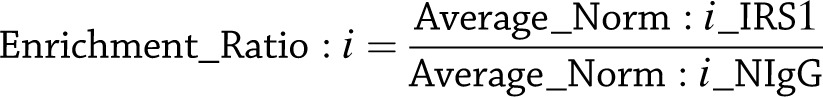 |
Since we used NIgG as a control, the first level of identification will be to search for proteins exclusively detected in the IRS1 IPs. However, this will result in false-negative results. Because of the high sensitivity of our approach, even if a trace amount of a protein was nonspecifically bound or adsorbed on the NIgG beads, it may be identified with a minimum of two unique peptides with an FDR of 0.01. Nonetheless, if this protein is a true component of the IRS1 complex, a higher PA will be assigned to this protein in the IRS1 sample than in the NIgG sample.
To determine the relative quantities of IRS1 interaction partners in lean, obese, and T2D subjects, the PA for each protein identified in a specific biopsy was normalized against the PA for IRS1 identified in the same biopsy sample, which results in Norm:j:
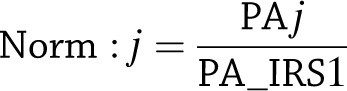 |
The normalization strategy is widely used in proteomics studies involving protein-protein interactions (15) and uses the same concept used in Western blotting, in which the Western blot signal for an interaction protein is normalized against that for the protein serving as the bait. The normalized PA for each IRS1 interaction partner, Norm:j, was log2-transformed and compared within the group to assess the effects of insulin or across the three groups to determine the effects of obesity or T2D on protein-protein interactions involving IRS1.
Statistical Analysis
Although a large number of proteins was assigned in at least 1 of 44 biopsy samples that were studied, a series of filters was used to narrow the number of proteins that were used in comparisons among groups as described above. This approach is diagrammed in Supplementary Fig. 1C. For within-group comparisons to assess the effects of insulin, statistical significance was assessed using paired t tests. For across-group comparisons, statistical significance was assessed using ANOVA with post hoc independent t tests. Differences were considered statistically significant at P < 0.05.
Bioinformatics Analysis
Pathway analysis on IRS1 interaction partners were performed using two bioinformatic software packages: Ingenuity Pathway Analysis (Ingenuity Systems, Redwood City, CA), which considers a pathway to be a set of genes; and Impact Analysis (21), which takes into account the topology of the pathway, in addition to the over-representation of genes. Both of these software packages are widely used, and contain biological and chemical interactions and functional annotations created by manual curation of the scientific literature (21,22). Ingenuity queries a proprietary database of Canonical Pathways; Impact Analysis uses the Kyoto Encyclopedia of Genes and Genomes (KEGG) (23) and Reactome Pathway (24) databases. Due to the fact that a gold standard pathway analysis tool or pathway database is currently unavailable, using multiple pathway analysis packages or databases may provide better results. A pathway was considered to be significantly enriched if both the FDR for the pathway was <0.01 and the pathway included at least four of the identified IRS1 partners.
Results
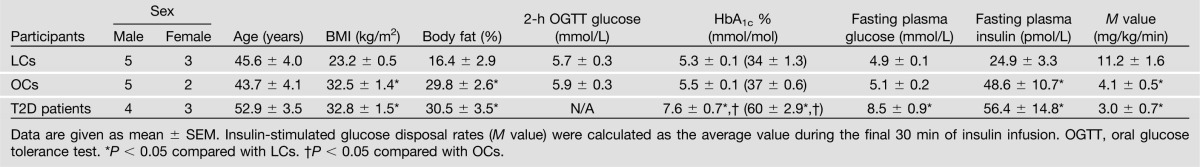
As can be seen from Table 1, no significant differences in sex and age were observed among the three groups. Compared with LCs, BMI, percentage of body fat, and fasting plasma insulin levels were significantly higher, whereas the M values, a measure of insulin sensitivity, were significantly lower in OCs and T2D patients.
Table 1.
Clinical characteristics of LC, OC, and T2D participants for the IRS1 interaction partner study
IRS1 was detected in IRS1 IPs from all 44 biopsy samples used for the study, but was not detected at all in the NIgG IPs. In total, 122 proteins met the criteria for the classification as IRS1 interaction partners (Table 2). Note that proteins may interact with IRS1 directly or indirectly through another protein that interacts with IRS1 directly. Among these 122 IRS1 interaction partners, 113 were previously unreported in any species. Of the nine proteins that were previously reported as IRS1 interaction partners (Table 2), only p85α was reported in human skeletal muscle (7,8). Pathway analysis of the 122 IRS1 interaction partners indicated that multiple pathways are significantly enriched, such as pathways related to mitochondrial function, insulin signaling, protein synthesis and degradation, and cytoskeleton dynamics (Fig. 1A and Supplementary Table 1). These results imply that IRS1 plays an important role in these biological processes or that these pathways regulate IRS1 function. A significantly enriched interaction network of IRS1 is shown in Fig. 1B. These novel IRS1 interaction partners in humans may help to understand the various roles that IRS1 plays in physiological and pathophysiological conditions in muscle and other tissues/organs.
Table 2.
The 122 proteins that met the two rigorous criteria for classification as IRS1 interaction partners
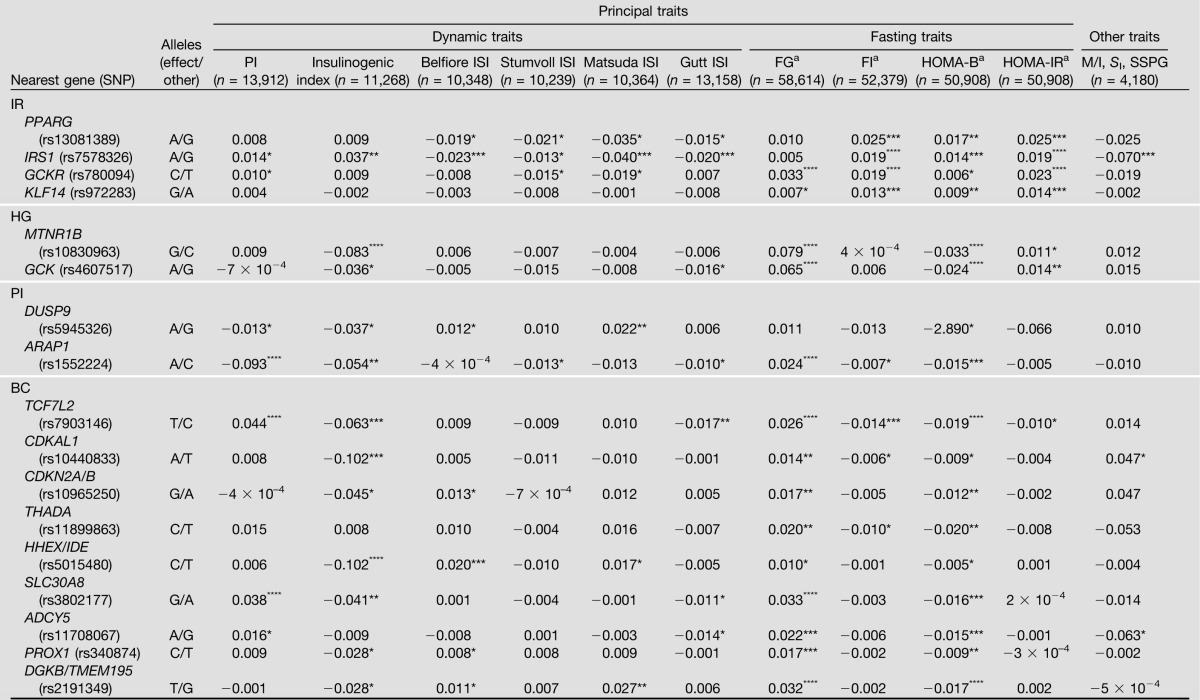
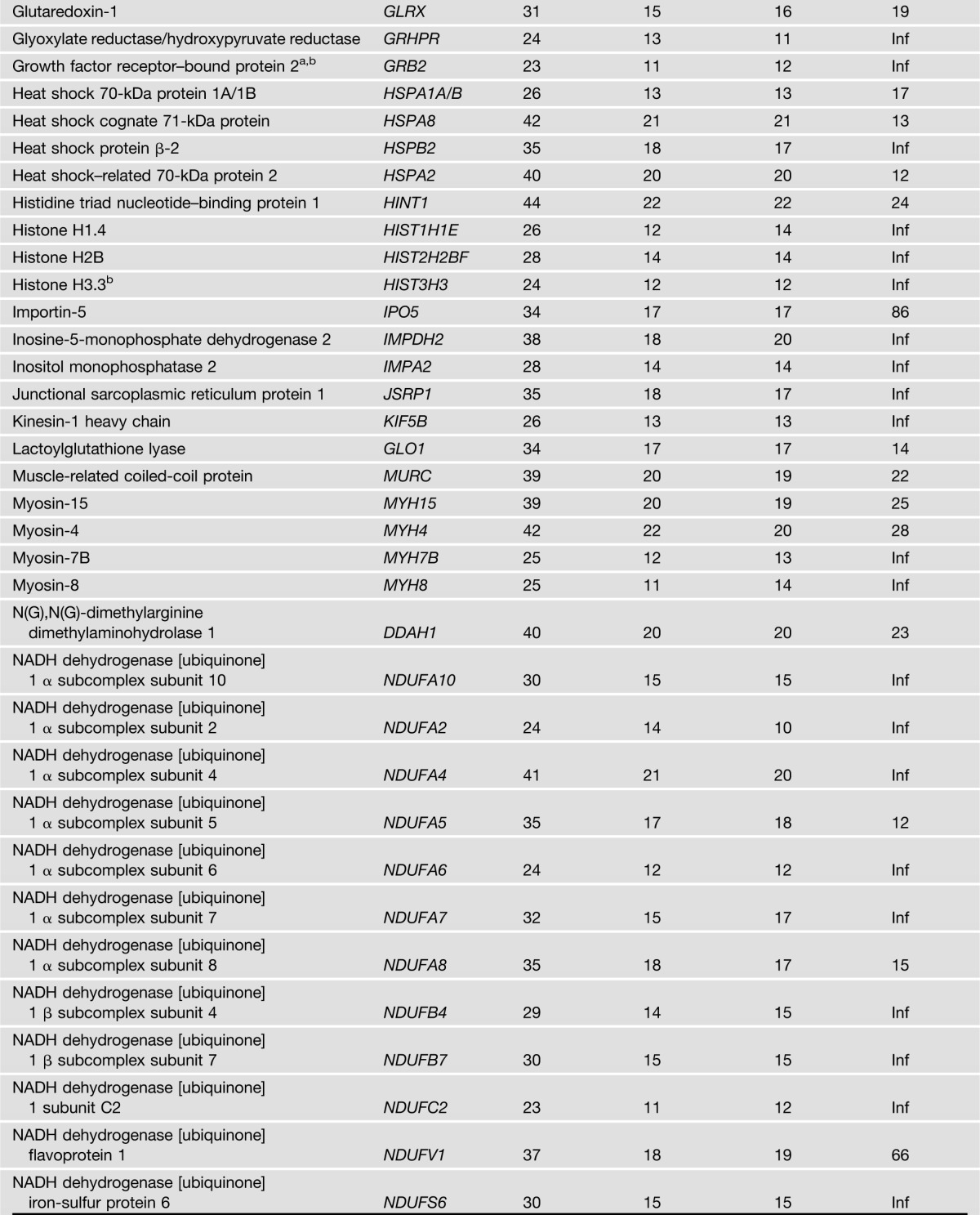
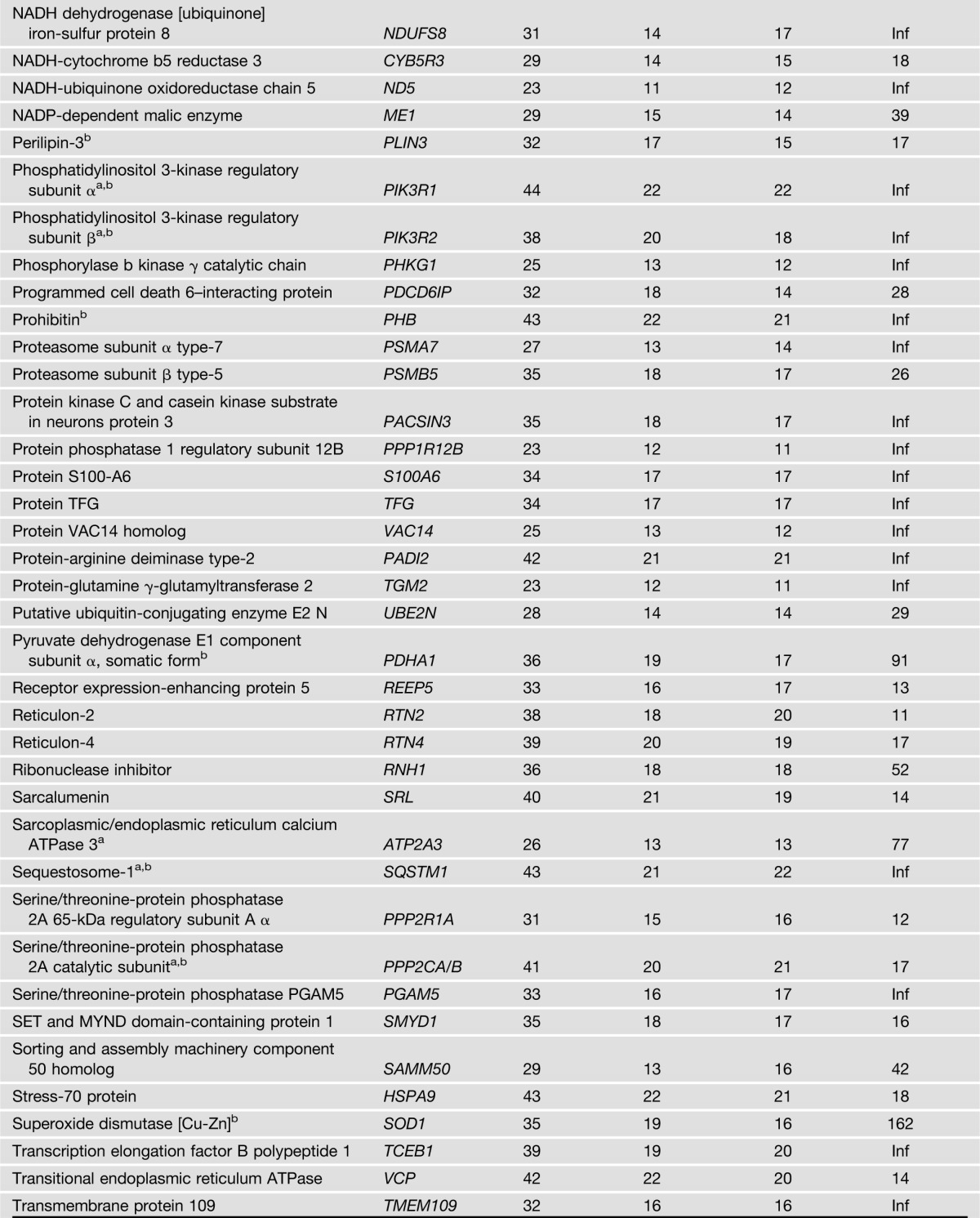
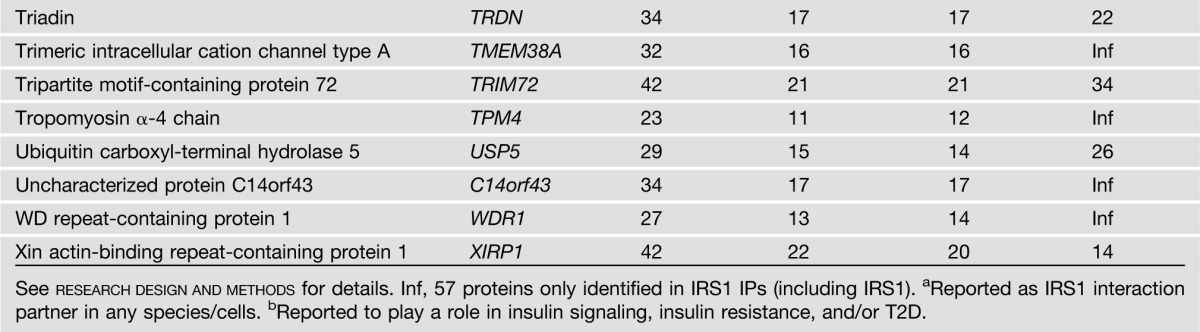
Figure 1.

Significantly enriched pathways and interaction network for IRS1 interaction partners. A: Significantly enriched pathways for the IRS1 interaction partners identified in this study revealed by Ingenuity Pathway Analysis and Impact Analysis. The pathways are grouped according to a common theme manually. The pathways were sorted by the database and then by pathway names. The total number of identified IRS1 interaction partners for a given pathway in this study are denoted above each bar. B: A significantly enriched IRS1 interaction network in human skeletal muscle revealed by proteomics and Ingenuity Pathway Analysis. All parameters were set as default, with the exception of the number of molecules per network, which was maximized to 140. The network with the top score (116), which is related to metabolic disorder, was shown, containing 62 molecules derived from the list of 122 IRS1 interaction partners identified in this study. IRS1 is highlighted in purple, and identified IRS1 interaction partners in this study are highlighted in green. Solid and dashed connecting lines indicate the presence of direct and indirect interactions in the Ingenuity database, respectively. eNOS, endothelial nitric oxide synthase; TCA, tricarboxylic acid.
Furthermore, 39 of the 122 proteins showed a significant difference in IRS1 interaction among the three groups or within a group in response to insulin (Table 3). Supplementary Fig. 2 provides a summary of IRS1 interaction partners in LCs, and how they differ in OCs and T2D patients with and without in vivo insulin infusion. Confirming the literature, both p85α (PIK3R1) and p85β (PIK3R2) showed a significant increase in the association with IRS1 in response to insulin infusion in LCs, but not in OCs and T2D patients. Since p85α and p85β are well-known insulin-stimulated interaction partners of IRS1 in skeletal muscle cells, and their interactions with IRS1 are impaired in insulin-resistant conditions (7,8), these results served as a positive control for our proteomics approach.
Table 3.
The 39 IRS1 interacting proteins showing differential interactions in muscle biopsies from LCs, OCs, and T2D patients revealed by proteomics
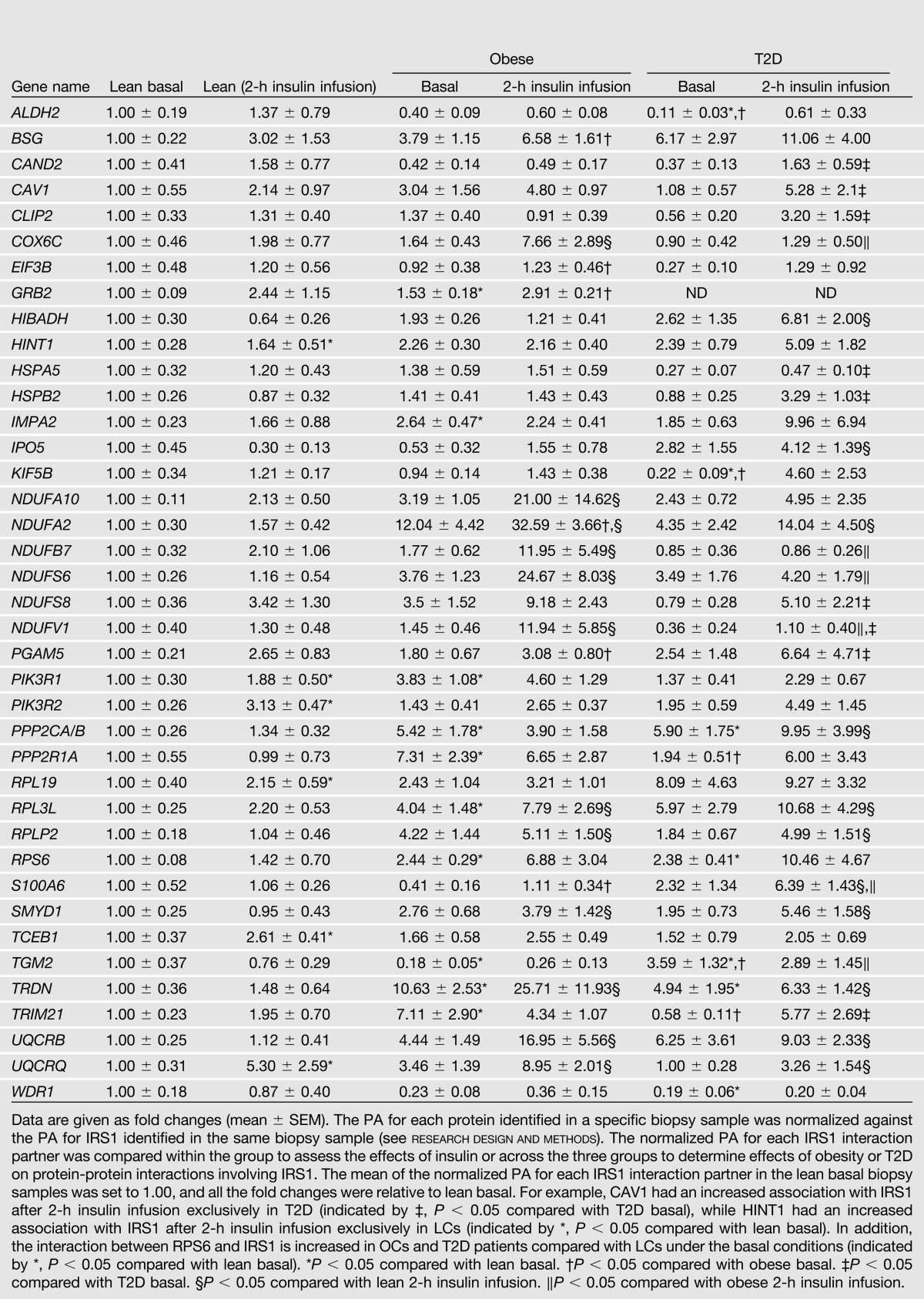
Of interest, we found nine insulin-stimulated interaction partners of IRS1 in T2D patients, and six in OCs that did not respond to insulin in LCs (Supplementary Fig. 2, highlighted in red and cyan, respectively). Moreover, 10 and 12 proteins in 2-h insulin-stimulated muscle in T2D patients and OCs, respectively, showed increased associations with IRS1 compared with 2-h insulin-stimulated LC muscle (Supplementary Fig. 2, highlighted in orange and brown, respectively). These results imply that in addition to reduced insulin response in classic signaling proteins, the increased interaction of multiple proteins with IRS1 in response to insulin also characterizes insulin resistance and T2D. Moreover, nine and four proteins in basal OC and T2D muscle, respectively, showed an increased association with IRS1 compared with basal LC muscle (Supplementary Fig. 2, highlighted in blue). The proteins that showed increased interaction with IRS1 in OC and T2D muscle are involved in multiple pathways and functional categories related to insulin signaling, such as PI3K/AKT signaling, mitochondrial function, inflammation, degradation/synthesis, and cytoskeletal dynamics. KEGG PI3K-AKT and KEGG Oxidative Phosphorylation metabolic pathways with IRS1 partners color-coded according to their differences in IRS1 interaction among LCs, OCs, and T2D patients are illustrated in Fig. 2A and Fig. 3, respectively.
Figure 2.

Abnormalities in IRS1 interaction in the KEGG PI3K-AKT signaling pathway (PI3K-AKT). GRB2, PI3KR1, PI3KR2, PPP2R1A, PPP2CA/B, and RPS6 were identified as IRS1 interaction partners in the study, and, except for PIK3R1, interactions between IRS1 and other identified interaction partners were previously unreported in human skeletal muscle. The KEGG PI3K-AKT pathway was exported from www.kegg.jp as a diagram. A: The exported diagram was color-coded for identified IRS1 interaction partners according to their differences in IRS1 interaction among LCs, OCs, and T2D patients. If a protein has altered interactions with IRS1 in more than one comparison, it may be highlighted in more than one color. The KEGG representation of this signaling pathway highlights the important processes that are regulated by insulin (e.g., protein synthesis, glucose uptake, and glycogen synthesis) and the relationship of identified IRS1 interaction partners within the KEGG PI3K-AKT insulin pathway and associated abnormalities in OCs and T2D patients. Please note that currently there is no comprehensive pathway database and some proteins known to be involved in the PI3K-AKT pathway may not be included in the KEGG PI3K-AKT pathway. B: The interaction between PP2Ac (also known as PPP2AC) and IRS1 was increased in OCs and T2D patients compared with LCs. Data are presented as the mean ± SEM. White bar, basal; black bar, 2-h insulin-stimulated. *P < 0.05 compared with lean basal; @P < 0.05 compared with lean 2-h insulin.
Figure 3.

Abnormalities in IRS1 interaction in the KEGG Oxidative Phosphorylation metabolic pathway. The figure for the KEGG Oxidative Phosphorylation metabolic pathway was downloaded from http://www.kegg.jp and modified to add color-coding to IRS1 partners according to their differences in IRS1 interaction among LCs, OCs, and T2D patients. If a protein has altered interactions with IRS1 in more than one comparison, it may be highlighted in more than one color. In total, 18 IRS1 interaction partners, all of which were novel, were assigned to the KEGG Oxidative Phosphorylation metabolic pathway: 14 in Complex I (shown in the NADH dehydrogenase block), 3 in Complex III (shown in the cytochrome c reductase block), and 1 in Complex IV (shown in the cytochrome c oxidase block). Among them, nine partners showed no differences in IRS1 interaction among LCs, OCs, and T2D patients (highlighted in green); one was insulin-stimulated in OCs, but not in LCs (NDUFA2, highlighted in light blue); two were insulin-stimulated in the muscle of T2D patients, but not in that of LCs or OCs (NDUFS8 and NDUFV1, highlighted in red); four had increased association with IRS1 in 2-h insulin-stimulated T2D patient muscle vs. 2-h insulin-stimulated LC muscle (NDUFA2, NDUFV1, QCR7, and QCR8, highlighted in orange); four had increased interaction with IRS1 in 2-h insulin-stimulated OC muscle vs. 2-h insulin-stimulated LC muscle (NDUFA10, NDUFB7, NDUFS6, and COX6C, highlighted in brown); and four exhibited altered interaction with IRS1 in 2-h insulin-stimulated T2D muscle vs. 2-h insulin-stimulated OC muscle (NDUFB7, NDUFS6, NDUFV1, and COX6C, highlighted in light green).
DISCUSSION
High-throughput technologies such as oligonucleotide microarrays are powerful tools for the study of physiological and pathological conditions with complex or multifactorial underlying mechanisms, such as insulin resistance and T2D. However, expression of muscle mRNA may not reflect the abundance of proteins (25) and can give no information regarding protein-protein interactions. MS/MS-based proteomics offers a powerful approach for studying the complex protein-protein networks in insulin resistance. IRS1 plays a central role in the insulin cascade, and abnormal IRS1 complexes may contribute to the development of insulin resistance. Most studies on IRS1 interaction partners have been carried out in cell culture or animal models, concentrating on a few known targets (5–11). The present project analyzed proteins isolated from muscle biopsies of LCs, OCs, and T2D patients using state-of-the-art HPLC-ESI-MS/MS to assess interacting partners of IRS1. Compared with the approach that we published previously (6), the main improvement includes the following: 1) use NIgG as a negative control to filter our nonspecific binders; 2) multiple biological comparisons to improve confidence; and 3) extensive bioinformatics analysis to reveal significantly enriched pathways. The strategy has the ability to detect endogenous protein complexes, without the use of labeling or protein overexpression/tags. This approach may be applicable to the study of other protein complexes in cells, animal models, and human tissue samples.
PI3K-AKT signaling is a well-defined insulin-signaling pathway regulating glucose metabolism (3,7). Multiple proteins involved in this pathway exhibited increased interaction with IRS1 in OCs and/or T2D patients (Fig. 2A), such as PI3KR1, GRB2, serine/threonine protein phosphatase 2A catalytic subunit (PPP2CA/B [also known as PP2Ac]), and 65-kDa regulatory subunit A α (PPP2R1A), and 40S ribosomal protein S6 (RPS6). Of interest, protein phosphatase 2A (PP2A) is involved in multiple aspects of insulin signal transduction, and is known to dephosphorylate several insulin-signaling kinases, such as S6K1 and Akt (26,27). Recently, PP2Ac was identified as an interaction partner of IRS1 in murine cardiomyocytes (5). Furthermore, emerging evidence suggests hyperactivation of PP2A in liver, muscle, and islets in diabetic cell/animal models (27). Our results provided the first evidence that PP2Ac interacted with IRS1 in LCs, and this interaction is increased in OCs and/or T2D patients (Fig. 2B). Correlation analysis of the combined LC, OC and T2D patient groups indicates that basal PP2Ac/IRS1 interaction has no significant correlation with sex, age, and 2-h oral glucose tolerance test. On the other hand, this interaction is positively correlated with BMI (r = 0.46, P < 0.05), percentage of body fat (r = 0.51, P < 0.05), HbA1c level (r = 0.46, P < 0.05), fasting plasma insulin level (r = 0.5, P < 0.05), and potentially fasting plasma glucose level (r = 0.45, P = 0.053). Moreover, this interaction is negatively correlated with M values (r = −0.52, P < 0.05), suggesting that increased PP2Ac/IRS1 interaction is associated with reduced insulin sensitivity.
Insulin resistance and T2D have been linked with mitochondrial dysfunction (4,28,29). Among the 39 IRS1 interaction proteins that showed a significant difference in interaction with IRS1 among the three groups, 12 are involved in mitochondrial processes (Fig. 3 and Supplementary Fig. 2). Eleven of them exhibited increased interaction with IRS1 in OCs/T2D patients, such as subunits of Complex I (NDUFS8, NDUFV1, NDUFA2, NDUFA10, NDUFB7, and NDUFS6), Complex III (UQCRB/Q), and Complex IV (COX6C), as well as 3-hydroxyisobutyrate dehydrogenase involved in branched-chain amino acid degradation (29). These results are rather intriguing since mitochondrial protein abundance is found to be down-regulated in OC and T2D in the basal state (30). A phosphotyrosine-dependent interaction between IRS1 and several mitochondrial proteins was previously reported in basal C2C12 mouse muscle cells (31). Our current findings further supported the association of insulin resistance and mitochondrial dysfunction, and more importantly, identified new protein-protein interactions involving IRS1 in humans that may contribute to mitochondrial dysfunction in insulin resistance and T2D.
Inflammation has been associated with insulin resistance (32,33). Three proteins involved in inflammation showed increased interaction with IRS1 in OCs and/or T2D: basigin (BSG [also known as EMMPRIN]), CAV1, and protein-glutamine γ-glutamyltransferase 2 (TGM2). Each of these proteins modulate nuclear factor-κB activation and are upregulated in inflammatory conditions (34–36). Our current findings suggest that IRS1 may play a role in inflammation-mediated insulin resistance.
Six proteins playing a role in protein degradation showed altered interaction with IRS1 in OCs and/or T2D patients compared with LCs, including cullin-associated NEDD8-dissociated protein 2 (CAND2), E3 ubiquitin-protein ligase TRIM21, histidine triad nucleotide-binding protein 1 (HINT1), transcription elongation factor B polypeptide 1 (TCEB1), heat shock protein β-2 (HSPB2), and 78-kDa glucose-regulated protein (HSPA5). CAND2, TRIM21, HINT1, and TCEB1 are members of ubiquitin ligases and may target IRS1 for degradation (37,38). Increased muscle protein degradation has been associated with insulin resistance due to overactivation of the ubiquitin proteasome (39). Additionally, upregulation of HSPB2 was observed in diabetic-induced rats (40), while HSPA5 heterozygous mice exhibited resistance to high-fat diet–induced obesity and T2D (41), suggesting a negative role of HSPB2 and HSPA5 in insulin signaling.
Multiple modulators of transcription and protein synthesis also had differential interaction with IRS1 in OCs and/or T2D patients compared with LCs, including SET and MYND domain–containing protein 1 (SMYD1), a transcriptional repressor (42); eukaryotic translation initiation factor 3 subunit B (EIF3B); ribosomal proteins (RPL3L, RPL19, RPLP2, and RPS6), key players in translation (43); and importin-5 (IPO5), a ribosomal protein transporter (44). Reduced mitochondrial protein synthesis was observed in T2D patients compared with matched control subjects (45). However, the mechanisms for these abnormalities are unclear. Our current findings indicated a role of IRS1 in reduced protein synthesis in T2D.
Cytoskeleton dynamics and muscle contraction regulate glucose uptake, and impairment in these processes may contribute to the development of insulin resistance (46,47). CAP-Gly domain–containing linker protein 2 (CLIP2) associates with the microtubule complex and regulates cytoskeletal organization. Clip-associating protein 2 (CLASP2), a known CLIP2 interaction partner, is an insulin-stimulated GLUT4 interaction partner in L6 myotubes (48). CLIP2 was insulin-responsive to IRS1 only in T2D patients. Interestingly, two other proteins involved in actin cytoskeleton dynamics showed decreased association with IRS1 under the basal condition, including kinesin-1 heavy chain (KIF5B) and WD repeat–containing protein 1 (WDR1). KIF5B is required for insulin-stimulated GLUT4 translocation, and also associates with mitochondria via its function as microtubule protein (49). WDR1 protects actin filaments from depolymerization and has been found to be upregulated by insulin in adipocytes (50). Our novel findings pinpoint the aberrant IRS1 interaction partners that may contribute to the defects in cytoskeleton dynamics and muscle contraction that are associated with glucose uptake in insulin resistance.
Although proteomic approaches using CO-IP have been widely used to map protein-protein interaction networks (15,16), one limitation of CO-IP exists: the proteins bound to the bait (in this case, IRS1) under the CO-IP condition may not necessarily bind to IRS1 in the cells, and vice versa. This limitation does raise the possibility of IRS1 interaction artifacts. IRS1, however, is distributed throughout the cytoplasm and nucleus, and binds the plasma membrane. It is interesting that a number of mitochondrial proteins were detected as IRS1 interaction partners. Currently, there is no evidence in the literature that IRS1 is localized to mitochondria. Nonetheless, all the mitochondrial proteins identified as IRS1 interaction partners in the study are encoded by nuclear DNA and translocated into mitochondria. Thus, before they are transported into mitochondria, they may interact with IRS1 within the cytoplasm.
It is noted that disease conditions may alter protein solubility. It is possible that the solubility of a protein in the CO-IP buffer was increased in OCs or T2D patients, and therefore more proteins were solubilized in the lysates, which might lead to the increased association of these proteins with IRS1 in OC/T2D. Additional experiments are warranted to explore this possibility.
Multiple reported IRS1 interaction partners were not identified as IRS1 interaction partners in human skeletal muscle in the current study (Supplementary Table 2); all of them were identified only in cell culture or animal models, but not in human skeletal muscle tissue, which might be due to the low abundance of these proteins in human skeletal muscle or difference in protein isoforms in cell culture/animal tissue/human tissue.
As mentioned earlier in the article, insulin-signaling events involving IRS1 require the time-dependent formation of IRS1 complexes (14), which might be altered in OCs/T2D patients compared with LCs. Therefore, it is possible that some of the observed differences in the interaction of IRS1 with several proteins among LCs, OCs, and T2D patients could be explained by the differential time of interaction among these groups.
In summary, we have identified 122 IRS1 interaction partners in human skeletal muscle, the majority of which are novel. Moreover, 39 of the 122 proteins showed a significant difference in IRS1 interaction among the three groups or within a group in response to insulin. It is well-known that in insulin-resistant and T2D states, reduced response to insulin exists, such as reduced insulin-stimulated association of IRS1 with p85α, decreased insulin-stimulated AKT phosphorylation, as well as impaired insulin-stimulated GLUT4 translocation and glucose uptake (3,7). Here, we did observe that both p85α and p85β along with three other proteins had a significant increase in their association with IRS1 in LCs, but not in OCs or T2D patients. On the other hand, surprisingly, we observed for the first time that dozens of other proteins, in multiple pathways related to insulin signaling, exhibited an augmented interaction with IRS1 in skeletal muscle from OCs and/or T2D patients in either basal or insulin-stimulated conditions. These proteins may be negative regulators of insulin action and possibly over-react to IRS1 in insulin resistance and T2D, resulting in the early termination of insulin signaling. These novel abnormalities in IRS1 interaction partners in obesity and T2D not only provide new insights into the molecular mechanism of insulin resistance, but also identify new targets for the development of drugs to treat metabolic diseases.
Article Information
Acknowledgments. The authors thank Roy Collins, RN (Wayne State University, Department of Clinical and Translational Science, School of Medicine); Ardella Magee, RN (Wayne State University, Department of Clinical and Translational Science, School of Medicine); Ginger Steinhilber, RN (Wayne State University, Department of Clinical and Translational Science, School of Medicine); Julie Mcqueeney (Wayne State University, Department of Clinical and Translational Science, School of Medicine); Mohammed Hammoude, MD (Wayne State University, Department of Clinical and Translational Science, School of Medicine); Samar Hafida, MD (Wayne State University, Department of Clinical and Translational Science, School of Medicine); Jackie Parker (Wayne State University, Department of Clinical and Translational Science, School of Medicine); Amy Stolinski (Wayne State University, Department of Clinical and Translational Science, School of Medicine); Nour Daboul, MD (Wayne State University, Department of Clinical and Translational Science, School of Medicine); Mindy Perez (Wayne State University, Department of Clinical and Translational Science, School of Medicine); Lisa Palmer (Wayne State University, Department of Clinical and Translational Science, School of Medicine); Evette Garcia (Wayne State University, Department of Clinical and Translational Science, School of Medicine); Rodney O. Berry (Wayne State University, Pharmaceutical Sciences); Hollilyne Drury, BS (Wayne State University, Pharmaceutical Sciences); Raluca Buceschi (Wayne State University, Pharmaceutical Sciences); Patrick Stevens (Wayne State University, Pharmaceutical Sciences); Akilah Franklin, BS (Wayne State University, Pharmaceutical Sciences); Rani N. Kattoula (Wayne State University, Pharmaceutical Sciences); Nazmine K. Sohi (Wayne State University, Pharmaceutical Sciences); and Jesse Sami Haddad (Wayne State University, Pharmaceutical Sciences) for their participation with the clinical studies; and Zhao Yang (Wayne State University, Pharmaceutical Sciences) for his technical support with insulin measurement.
Funding. This work was supported by Wayne State University faculty competition for postdoctoral fellow award (M.C. and Z.Y.); National Institutes of Health/National Institute of Diabetes and Digestive and Kidney Diseases grants T32-DK-080657 (Z.M. and A.B.A.-S.), R01-DK-081750-04S1 (M.L. and Z.Y.), and R01-DK-081750 (Z.Y.); and Wayne State University faculty start-up grant (Z.Y.).
Duality of Interest. No potential conflicts of interest relevant to this article were reported.
Author Contributions. M.C. and D.M. designed and performed the clinical and proteomic experiments, analyzed the data, generated figures, and wrote the manuscript. Z.M., M.L., B.S., and W.A.-j. carried out muscle biopsy and hyperinsulinemic-euglycemic clamp experiments and performed other clinically related tasks. M.D., A.B.A.-S., K.H., and J.F.H. helped with the clinical study design, muscle biopsy and hyperinsulinemic-euglycemic clamp setup, and data interpretation. R.T. and S.D. assisted with computer-aided data analysis. X.Z. helped with the experimental design. Z.Y. supervised the project, designed the clinical and proteomic experiments, analyzed the data, helped with data interpretation, and wrote the manuscript. Z.Y. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db13-1872/-/DC1.
References
- 1.Rask-Madsen C, Kahn CR. Tissue-specific insulin signaling, metabolic syndrome, and cardiovascular disease. Arterioscler Thromb Vasc Biol 2012;32:2052–2059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abdul-Ghani MA, DeFronzo RA. Pathogenesis of insulin resistance in skeletal muscle. J Biomed Biotechnol 2010;2010:476279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hojlund K, Beck-Nielsen H. Impaired glycogen synthase activity and mitochondrial dysfunction in skeletal muscle: markers or mediators in type 2 diabetes? Curr Diabetes Rev 2006;2:375–395 [DOI] [PubMed] [Google Scholar]
- 4.Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science 2005;307:384–387 [DOI] [PubMed] [Google Scholar]
- 5.Mandavia C, Sowers JR. Phosphoprotein phosphatase PP2A regulation of insulin receptor substrate 1 and insulin metabolic signaling. Cardiorenal Med 2012;2:308–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geetha T, Langlais P, Luo M, et al. Label-free proteomic identification of endogenous, insulin-stimulated interaction partners of insulin receptor substrate-1. J Am Soc Mass Spectrom 2011;22:457–466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cusi K, Maezono K, Osman A, et al. Insulin resistance differentially affects the PI 3-kinase- and MAP kinase-mediated signaling in human muscle. J Clin Invest 2000;105:311–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grimmsmann T, Levin K, Meyer MM, Beck-Nielsen H, Klein HH. Delays in insulin signaling towards glucose disposal in human skeletal muscle. J Endocrinol 2002;172:645–651 [DOI] [PubMed] [Google Scholar]
- 9.Chen J, Capozza F, Wu A, et al. Regulation of insulin receptor substrate-1 expression levels by caveolin-1. J Cell Physiol 2008;217:281–289 [DOI] [PubMed] [Google Scholar]
- 10.Valverde AM, Mur C, Pons S, et al. Association of insulin receptor substrate 1 (IRS-1) y895 with Grb-2 mediates the insulin signaling involved in IRS-1-deficient brown adipocyte mitogenesis. Mol Cell Biol 2001;21:2269–2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Myers MG, Jr, Mendez R, Shi P, Pierce JH, Rhoads R, White MF. The COOH-terminal tyrosine phosphorylation sites on IRS-1 bind SHP-2 and negatively regulate insulin signaling. J Biol Chem 1998;273:26908–26914 [DOI] [PubMed] [Google Scholar]
- 12.Copps KD, White MF. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia 2012;55:2565–2582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ball LE, Berkaw MN, Buse MG. Identification of the major site of O-linked beta-N-acetylglucosamine modification in the C terminus of insulin receptor substrate-1. Mol Cell Proteomics 2006;5:313–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saltiel AR, Pessin JE. Insulin signaling pathways in time and space. Trends Cell Biol 2002;12:65–71 [DOI] [PubMed] [Google Scholar]
- 15.Wepf A, Glatter T, Schmidt A, Aebersold R, Gstaiger M. Quantitative interaction proteomics using mass spectrometry. Nat Methods 2009;6:203–205 [DOI] [PubMed] [Google Scholar]
- 16.Marcilla M, Albar JP. Quantitative proteomics: a strategic ally to map protein interaction networks. IUBMB Life 2013;65:9–16 [DOI] [PubMed] [Google Scholar]
- 17.Yi Z, Langlais P, De Filippis EA, et al. Global assessment of regulation of phosphorylation of insulin receptor substrate-1 by insulin in vivo in human muscle. Diabetes 2007;56:1508–1516 [DOI] [PubMed] [Google Scholar]
- 18.Langlais P, Yi Z, Finlayson J, et al. Global IRS-1 phosphorylation analysis in insulin resistance. Diabetologia 2011;54:2878–2889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen XW, Wang H, Bajaj K, et al. SEC24A deficiency lowers plasma cholesterol through reduced PCSK9 secretion. eLife 2013;2:e00444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol 2008;26:1367–1372 [DOI] [PubMed] [Google Scholar]
- 21.Donato M, Xu Z, Tomoiaga A, et al. Analysis and correction of crosstalk effects in pathway analysis. Genome Res 2013;23:1885–1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thomas S, Bonchev D. A survey of current software for network analysis in molecular biology. Hum Genomics 2010;4:353–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kanehisa M, Goto S. KEGG: Kyoto Encyclopedia of Genes And Genomes. Nucleic Acids Res 2000;28:27–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Croft D, O’Kelly G, Wu G, et al. Reactome: a database of reactions, pathways and biological processes. Nucleic Acids Res 2011;39(Database issue):D691–D697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gygi SP, Rochon Y, Franza BR, Aebersold R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol 1999;19:1720–1730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ni YG, Wang N, Cao DJ, et al. FoxO transcription factors activate Akt and attenuate insulin signaling in heart by inhibiting protein phosphatases. Proc Natl Acad Sci U S A 2007;104:20517–20522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kowluru A, Matti A. Hyperactivation of protein phosphatase 2A in models of glucolipotoxicity and diabetes: potential mechanisms and functional consequences. Biochem Pharmacol 2012;84:591–597 [DOI] [PubMed] [Google Scholar]
- 28.Jelenik T, Roden M. Mitochondrial plasticity in obesity and diabetes mellitus. Antioxid Redox Signal 2013;19:258–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newgard CB. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab 2012;15:606–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hwang H, Bowen BP, Lefort N, et al. Proteomics analysis of human skeletal muscle reveals novel abnormalities in obesity and type 2 diabetes. Diabetes 2010;59:33–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hanke S, Mann M. The phosphotyrosine interactome of the insulin receptor family and its substrates IRS-1 and IRS-2. Mol Cell Proteomics 2009;8:519–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Odegaard JI, Chawla A. Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science 2013;339:172–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dali-Youcef N, Mecili M, Ricci R, Andrès E. Metabolic inflammation: connecting obesity and insulin resistance. Ann Med 2013;45:242–253 [DOI] [PubMed] [Google Scholar]
- 34.Garrean S, Gao XP, Brovkovych V, et al. Caveolin-1 regulates NF-kappaB activation and lung inflammatory response to sepsis induced by lipopolysaccharide. J Immunol 2006;177:4853–4860 [DOI] [PubMed] [Google Scholar]
- 35.Kim SY. Transglutaminase 2 in inflammation. Front Biosci 2006;11:3026–3035 [DOI] [PubMed] [Google Scholar]
- 36.Reddy VS, Prabhu SD, Mummidi S, et al. Interleukin-18 induces EMMPRIN expression in primary cardiomyocytes via JNK/Sp1 signaling and MMP-9 in part via EMMPRIN and through AP-1 and NF-kappaB activation. Am J Physiol Heart Circ Physiol 2010;299:H1242–H1254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shi J, Luo L, Eash J, Ibebunjo C, Glass DJ. The SCF-Fbxo40 complex induces IRS1 ubiquitination in skeletal muscle, limiting IGF1 signaling. Dev Cell 2011;21:835–847 [DOI] [PubMed] [Google Scholar]
- 38.Rui L, Yuan M, Frantz D, Shoelson S, White MF. SOCS-1 and SOCS-3 block insulin signaling by ubiquitin-mediated degradation of IRS1 and IRS2. J Biol Chem 2002;277:42394–42398 [DOI] [PubMed] [Google Scholar]
- 39.Wang X, Hu Z, Hu J, Du J, Mitch WE. Insulin resistance accelerates muscle protein degradation: activation of the ubiquitin-proteasome pathway by defects in muscle cell signaling. Endocrinology 2006;147:4160–4168 [DOI] [PubMed] [Google Scholar]
- 40.Sharma AK, Bharti S, Ojha S, et al. Up-regulation of PPARγ, heat shock protein-27 and -72 by naringin attenuates insulin resistance, β-cell dysfunction, hepatic steatosis and kidney damage in a rat model of type 2 diabetes. Br J Nutr 2011;106:1713–1723 [DOI] [PubMed] [Google Scholar]
- 41.Ye R, Jung DY, Jun JY, et al. Grp78 heterozygosity promotes adaptive unfolded protein response and attenuates diet-induced obesity and insulin resistance. Diabetes 2010;59:6–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gottlieb PD, Pierce SA, Sims RJ, et al. Bop encodes a muscle-restricted protein containing MYND and SET domains and is essential for cardiac differentiation and morphogenesis. Nat Genet 2002;31:25–32 [DOI] [PubMed] [Google Scholar]
- 43.Proud CG, Denton RM. Molecular mechanisms for the control of translation by insulin. Biochem J 1997;328:329–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jäkel S, Görlich D. Importin beta, transportin, RanBP5 and RanBP7 mediate nuclear import of ribosomal proteins in mammalian cells. EMBO J 1998;17:4491–4502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Halvatsiotis P, Short KR, Bigelow M, Nair KS. Synthesis rate of muscle proteins, muscle functions, and amino acid kinetics in type 2 diabetes. Diabetes 2002;51:2395–2404 [DOI] [PubMed] [Google Scholar]
- 46.Hoffman NJ, Elmendorf JS. Signaling, cytoskeletal and membrane mechanisms regulating GLUT4 exocytosis. Trends Endocrinol Metab 2011;22:110–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stöckli J, Fazakerley DJ, James DE. GLUT4 exocytosis. J Cell Sci 2011;124:4147–4159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Langlais P, Dillon JL, Mengos A, et al. Identification of a role for CLASP2 in insulin action. J Biol Chem 2012;287:39245–39253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Semiz S, Park JG, Nicoloro SM, et al. Conventional kinesin KIF5B mediates insulin-stimulated GLUT4 movements on microtubules. EMBO J 2003;22:2387–2399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rodgers BD, Levine MA, Bernier M, Montrose-Rafizadeh C. Insulin regulation of a novel WD-40 repeat protein in adipocytes. J Endocrinol 2001;168:325–332 [DOI] [PubMed] [Google Scholar]