

Abstract
Dopamine (DA) neurotransmission requires a complex series of enzymatic reactions that are tightly linked to catecholamine exocytosis and receptor interactions on pre- and postsynaptic neurons. Regulation of dopaminergic signalling is primarily achieved through reuptake of extracellular DA by the DA transporter (DAT) on presynaptic neurons. Aberrant regulation of DA signalling, and in particular hyperactivation, has been proposed as a key insult in the presentation of schizophrenia and related neuropsychiatric disorders. We recently identified 14-3-3ζ as an essential component of neurodevelopment and a central risk factor in the schizophrenia protein interaction network. Our analysis of 14-3-3ζ-deficient mice now shows that baseline hyperactivity of knockout (KO) mice is rescued by the antipsychotic drug clozapine. 14-3-3ζ KO mice displayed enhanced locomotor hyperactivity induced by the DA releaser amphetamine. Consistent with 14-3-3ζ having a role in DA signalling, we found increased levels of DA in the striatum of 14-3-3ζ KO mice. Although 14-3-3ζ is proposed to modulate activity of the rate-limiting DA biosynthesis enzyme, tyrosine hydroxylase (TH), we were unable to identify any differences in total TH levels, TH localization or TH activation in 14-3-3ζ KO mice. Rather, our analysis identified significantly reduced levels of DAT in the absence of notable differences in RNA or protein levels of DA receptors D1–D5. Providing insight into the mechanisms by which 14-3-3ζ controls DAT stability, we found a physical association between 14-3-3ζ and DAT by co-immunoprecipitation. Taken together, our results identify a novel role for 14-3-3ζ in DA neurotransmission and provide support to the hyperdopaminergic basis of pathologies associated with schizophrenia and related disorders.
Keywords: 14-3-3ζ, dopamine neurotransmission, dopamine transporter, schizophrenia, schizophrenia mouse model
Introduction
Schizophrenia and related neuropsychiatric disorders are widely believed to arise from neurodevelopmental defects that affect synaptic transmission.1 Indeed, neuropharmacological studies with antipsychotic drugs suggest that many of the positive symptoms associated with schizophrenia arise from increased dopamine (DA) signalling.2, 3 Neuroimaging studies following amphetamine treatment add strong support to this notion4, 5 and further implicate the mesolimbic pathway in the hyperdopaminergic hypothesis. Within the mesolimbic pathway, DA is produced by neurons predominantly located in the ventral tegmental area (VTA) and the substantia nigra (SN) of the midbrain. Central to the function of these neurons is the expression and activity of the rate-limiting catecholamine biosynthesis enzyme, tyrosine hydroxylase (TH), which catalyses the synthesis of DA from its precursor L-tyrosine. Following exocytosis from presynaptic neurons, DA binds to G-protein-coupled DA receptors D1–D5 to initiate signalling cascades in postsynaptic neurons. The activity of DA is tightly regulated by the DA transporter (DAT) that mediates reuptake of DA by presynaptic neurons where it is either recycled to the vesicular pool or degraded.6, 7
Recent studies have shown that the family of 14-3-3 regulatory proteins bind to TH to enhance phosphorylation of serine 31 (Ser-31) and Ser-40 to positively regulate its enzymatic activity.8, 9 Indeed, 14-3-3 proteins were originally identified as archetypical TH co-factors.10, 11 The 14-3-3 family comprises seven isoforms in mammals (β, ζ, ɛ, γ, η, τ and σ) that bind to phospho-serine/threonine residues on target proteins to modify their function and/or localization. In this manner, 14-3-3 proteins have been described to mediate a range of cell functions including cell cycle regulation, proliferation, migration, differentiation and apoptosis.10, 12, 13, 14 Although multiple 14-3-3 isoforms have the ability to bind TH in vitro, knockdown studies in midbrain-derived MN9D cells suggest that 14-3-3ζ is the major isoform involved in DA synthesis.15 In support of these findings, 14-3-3ζ is also the major isoform expressed in regions containing termini of dopaminergic neurons such as the striatum.16 However, the role of 14-3-3ζ in TH activity in vivo, or in other stages of DA neurotransmission, has not been explored.
We recently reported that 14-3-3ζ knockout (KO) mice have schizophrenia-like behavioural deficits such as hyperactivity and disrupted sensorimotor gating that are accompanied by aberrant neuronal migration and axonal guidance defects in the hippocampus.17 14-3-3ζ KO mice therefore represent a novel neurodevelopmental model of schizophrenia and associated disorders. In strong support of this notion, 14-3-3ζ is downregulated in post-mortem schizophrenia brain samples at the mRNA level18, 19 and is one of only 24 proteins downregulated across multiple neuroproteomic studies on schizophrenia patient samples.20, 21, 22 In addition, significant linkage to 14-3-3ζ has been identified through analysis of single-nucleotide polymorphisms from control and schizophrenia patient samples.23 Further support for a role in schizophrenia is derived from the recent finding that 14-3-3ζ is represented as a central hub within the schizophrenia-specific interaction network.24 At the molecular level, 14-3-3ζ interacts with several proteins essential for neuronal development that are also implicated in the pathogenesis of schizophrenia, including DISC1, NUDEL, LIS1 and TH.17, 25
Here we have explored the physiological and molecular basis of schizophrenia-like behavioural deficits by analyzing locomotor hyperactivity in 14-3-3ζ KO mice. We found that baseline hyperactivity of KO mice is rescued by the antipsychotic drug clozapine and that KO mice are hypersensitive to the DA releaser amphetamine. In strong support of DA underpinning some of the schizophrenia-like behavioural defects, in this model we found that total tissue DA levels were increased in KO mice. Our analysis of the dopaminergic signalling pathway indicates that 14-3-3ζ has an essential role in modulating protein levels of DAT. Our finding that 14-3-3ζ interacts with DAT provides insight into the molecular regulation of DAT stability. Unexpectedly, TH-positive neurons, TH expression and TH activation were unaffected in KO mice. Moreover, DA receptors were also expressed at similar levels to wild-type (WT) mice. Our results therefore implicate 14-3-3ζ as an essential component in the DA neurotransmission pathway by modulating the abundance of DAT.
Materials and methods
Mice
14-3-3ζGt(OST062)Lex (or 14-3-3ζ KO) mice on a SV129 background carrying a gene trap construct that contains the βGeo reporter gene disrupting 14-3-3ζ expression, have been described previously.17 14-3-3ζ Genotype was determined by PCR amplification of genomic tail DNA as described.17 Animal experiments were conducted in accordance with the guidelines of the Animal Ethics Committee of the Institute of Medical and Veterinary Sciences, the University of Adelaide and the Florey Institute for Neuroscience and Mental Health, University of Melbourne.
Behavioural assays
All procedures were carried out under normal light conditions (60–100 Lux) between 0800 and 1200 hours. Behavioural phenotyping was performed on the 14-3-3ζ KO line as previously described.26, 27, 28 One cohort of mice was used for the psychotropic drug-induced open field test at 30 weeks of age (11 WT, 5 females and 6 males; 11 KO, 5 females and 6 males). A separate cohort of mice was used at the age of 35 weeks for clozapine treatments and locomotor function tests (12 WT, 8 males and 4 females; 12 KO, 8 males and 4 females).
Clozapine treatment and locomotor function test
Clozapine was obtained from Sigma Aldrich (St Louis, MO, USA) and was dissolved in 10 mM HCl and diluted in sterile water.29 Vehicle was prepared in an identical manner without the addition of clozapine. Concentrated aliquots of both clozapine and vehicle were stored at −20 °C. Aliquots were thawed and diluted to their final concentration in sterile saline on the day of dosing. Solutions were buffered with NaOH to achieve a final pH of 6.5–7.5. Mice were given clozapine (5 mg kg–1) or vehicle daily for 14 days before behavioural testing. Dosing was continued for a further 11 days throughout the behavioural testing period, with dosing always conducted between 1530 and 1700 hours, following any behavioural testing. In all, 10 WT (3 males+7 females) and 9 KO (3 males+6 females) mice were given vehicle, whereas 10 WT (4 males+6 females) and 8 KO (4 males+4 females) mice were given clozapine. Mice were tested in a brightly lit square arena, 40 × 40 cm (Stoelting, Wood Dale, IL, USA), with clear walls 35 cm high for 5 min according to published protocols.30, 31 The floor was divided into inner and outer zones. Time spent in each zone was measured and total distance travelled was measured as an indication of baseline locomotor activity. An imaging program (ANY-maze, Wood Dale, IL, USA) was used to track movements and measure time in zones.
Amphetamine-induced locomotor hyperactivity
Baseline locomotor activity and amphetamine-induced locomotor activity were assessed using a TruScan Photobeam Activity system (Coulbourn Instruments, Whitehall, PA, USA). This system consists of a mouse enclosure (25.4 × 25.4 × 40.6 cm) surrounded by a sensor-ring that included a 16 × 16 array of photobeams and a computerized data acquisition system. After 30 min of baseline locomotor activity and habituation to the test environment, the animals received either saline or 5 mg kg–1 of amphetamine by intraperitoneal injection and activity was monitored over a subsequent 90-min period.
Production of 14-3-3ζ monoclonal antibodies
Anti-14-3-3ζ monoclonal antibodies were generated in a female BALB/c 14.3.3ζ KO mouse, which was injected three times, each with 10 μg purified recombinant 14.3.3ζ protein, over a period of 6 weeks. Enzyme-linked immunoassay of serum was performed to verify immunoreactivity to 14-3-3ζ. Splenocytes were isolated and fused with NS1 myeloma cells. Hybridomas were selected by incubating the cells at 37 °C with humidified 5% CO2 atmosphere in hypoxanthine–aminopterin–thymidine containing media. Immunoreactivity of these hybridomas was determined using a modified enzyme-linked immunoassay in which recombinant 14-3-3ζ was adsorbed to the surface of 96-well tissue culture plates. Positive lines were then clonally expanded. From this screen, we identified 35 positive hybridoma cell lines. Monoclonal antibodies were purified from eight lines detailed in Supplementary Figure S1. Purified antibodies were tested by western blot against each 14-3-3 isoform obtained from overexpression of His-tagged pGEX-expression constructs in bacteria in comparison with a commercially available 14-3-3ζ polyclonal antibody (C-16, Santa Cruz, CA, USA). Antibodies M6 (for western blots) and G1-7 for immunoprecipitation (IPs) were grown up and purified by the Walter Eliza Hall Institute Antibody facility for use in this study.
Histology and immunohistochemistry
For all anatomical analyses, postnatal mice were perfuse fixed with fresh 4% paraformaldehyde dissolved in phosphate-buffered saline as previously described.32 Brains were rapidly dissected free from other tissue and post fixed in 4% paraformaldehyde for an additional 24 h at 4 °C. Tissue was cryopreserved in 20% sucrose at room temperature (RT) overnight and frozen in Tissue-Tek OCT (Sakura Finetek, Torrance, CA, USA). Sections were cut at a thickness of 10 μm on a CM1850 cryostat (Leica, North Ryde, Australia) and air-dried for 60 min before staining.
For immunohistochemistry, sections were blocked in 10% non-immune goat serum or 1% bovine serum albumin in PBST (0.1 M phosphate-buffered saline, 0.3% Triton X-100, 1% bovine serum albumin) for 1 h at RT and subsequently incubated with primary antibodies for 1 h at RT. Primary antibodies and dilutions: rabbit polyclonal to TH (1:200; Millipore, Billerica, MA, USA), rat monoclonal to DAT (1:20, Santa Cruz). Sections were washed several times with PBST and then incubated with 1:200 dilution of Alexa Fluor-labelled secondary antibodies (Molecular Probes, Mullgrave, VIC, Australia) or streptavidin-labelled secondary antibodies (Jackson Laboratories, Bar Harbor, ME, USA) for 1 h at RT. After three washes in PBST, fluorescent sections were mounted in Prolong Gold antifade reagent with 4,6-diamidino-2-phenylindole (Molecular Probes) and streptavidin-labelled sections were developed with DAB substrate (Sigma, Castle Hill, NSW, Australia).
Image analysis
Low-resolution images were recorded on an SZX10 stereo microscope (Olympus, Edwardstown, SA, Australia) equipped with a Micropublisher 3.3 digital camera (Q-Imaging, Waltham, MA, USA) and processed with OpenLab 2.2 software (Improvision, Waltham, MA, USA). High-resolution images were recorded on an IX81 inverted microscope (Olympus) equipped with an OCRA-ER digital CCD camera (Hamamatsu, Hamamatsu, Japan) and processed with CellR software (Olympus). DAT and TH immunofluorescence was captured on a LSM700 confocal microscope (Zeiss, North Ryde, NSW, Australia). All figures were constructed in Adobe Photoshop CS3 (Adobe Systems, San Jose, CA, USA). Quantitation of DAT and TH expression from confocal immunofluorescence images was completed as described previously.33 Briefly, images were split into separate channels for TH or DAT, converted to binary images and used for fluorescence intensity calculations with an Image J area calculator macro designed to detect staining in confocal image slices.
Cell culture
The dopaminergic neuronal progenitor cell line, SN4741,34 was maintained in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum (HyClone, South Logan, UT, USA), 1% glutamine and antibiotics. FLAG-His-DAT (generously provided by Alexander Sorkin) and Myc-14-3-3ζ (generously provided by Joanna Woodcock) were transiently transfected into cells with Lipofectamine 2000 (Life Sciences, Mullgrave, VIC, Australia) and allowed to grow for 48 h before extracting protein lysates.
Immunoprecipitation
All protein extracts were prepared by lysis in NP40 lysis buffer composed of 137 mM NaCl, 10 mM Tris-HCl (pH 7.4), 10% glycerol, 1% Nonidet P-40, and protease and phosphatase inhibitors (4.5 U of aprotinin per ml, 1 μg of leupeptin per ml, 1 mM phenylmethylsulfonyl fluoride, 10 mM sodium fluoride, 10 mM β-glycerol phosphate, 10 mM sodium pyrophosphate and 10 mM sodium vanadate). Samples were lysed for 60 min at 4 °C, then centrifuged at 10 000 g for 15 min. The supernatants were precleared with mouse Ig-coupled Sepharose beads for 30 min at 4 °C. The precleared lysates were incubated for 2 h at 4 °C with 2 μg ml–1 of anti-TH antibody (Millipore), monoclonal anti-1433ζ antibody (G1-7), anti-DAT (6-5G10, Santa Cruz; and MAB369, Millipore), anti-Flag (Sigma) and control immunoglobulin G (Sigma) absorbed to protein G-Sepharose (Amersham Biosciences, Amersham, UK). The sepharose beads were washed three times with lysis buffer before being boiled for 5 min in sodium dodecyl sulphate–polyacrylamide gel electrophoresis sample buffer. The immunoprecipitated proteins and lysates were separated by sodium dodecyl sulphate–polyacrylamide gel electrophoresis, electrophorectically transferred to a polyvinylidene difluoride (Hybond-P, Amersham, UK) membrane (GE Health, Rydalmere, NSW, Australia) and analysed by immunoblotting.
Immunoblotting
Polyvinylidene difluoride membranes were blocked with 5% skim milk powder in TBST and immunoblotted with polyclonal rabbit anti-14-3-3ζ C-16 (Santa Cruz) at 1:1000, rabbit anti-TH (Millipore) at 1:1000, rabbit anti-phospho-serine-31 TH (Cell Signalling Anibodies, Danvers, MA, USA) at 1:1000, rabbit anti-phospho-serine-40 TH (Cell Signalling Anibodies) at 1:1000, rat anti-DAT (6–5G10; Santa Cruz) at 1:200, mouse anti-Flag-M2 (Sigma-Aldrich) at 1:1000, mouse anti-Myc (9B11; Cell Signalling Technologies) at 1:1000 and mouse anti-HA (6E2; Cell Signalling Technologies) at 1:1000. Rabbit polyclonal against β-actin (1:5000, Millipore) was used as a loading control. Bound antibodies were detected with horseradish peroxidase-conjugated secondary antibody (1:5,000, Pierce-Thermo Scientific, Rockford, IL, USA). Immunoreactive proteins were visualized by ECL (Luminescent Image Analyzer LAS-4000, Fujifilm, Tokyo, Japan). The images were analysed with Multi Gauge Ver3.0 (Fujifilm).
Detection of DA levels and metabolism
DA, and the metabolite 3,4-dihydroxyphenylacetic acid (DOPAC), levels in the striatum, prefrontal cortex and hippocampus of 7 WT and 7 KO animals were determined using reverse-phase high-performance liquid chromatography (HPLC) as previously described. For tissue preparation, small biopsies were dissected on a chilled plate, weighed, and stabilized in 200 μl 0.4 M perchloric acid (HClO4) containing 0.05% sodium metabisulphate (Na2S2O5) and 0.01% disodium EDTA. The sample tissue was then homogenized, cellular and vesicular membranes disrupted using a sonicator and finally stored at 70 °C. On the day of analysis, all samples were centrifuged at 10 500 g for 10 min and filtered though minispin filters for additional 3 min at 10 000 r.p.m. before being injected into the HPLC. For each sample, 10 μl was injected by a cooled autosampler (SIL 20A, Shimadzu, Rydalmere, NSW, Australia) and Shimadzu LC-AT pumpon to a reverse-phase C18 column (4.6 mm diameter, 150 mm length; CHROMPACK, Croydon, UK) coupled with an electrochemical detector (Decade II, Antec Leyden, Rydalmere, NSW, Australia). The mobile phase, comprises the following (mM): (KH2PO4, 70; EDTA di-sodium salt, 0.5; octane-sulphonic acid, sodium salt, 8; with 17% HPLC grade methanol, pH 3) was delivered at a flow rate of 500 μl min–1. The peaks were processed using LC solutions software (Antec Leyden). Concentrations of DA and its metabolite DOPAC were calculated for each sample.
Quantitative reverse transcriptase-PCR
Total RNA was isolated from cells using Trizol (Ambion, Austin, TX, USA) and single-stranded complementary DNA was synthesized using the QuantiTect Reverse transcription kit (Qiagen, Frankfurt, Germany). Quantitative PCR was performed with SYBR Green reagent (Qiagen) using the Rotor-Gene 6000 real-time PCR system (Corbett Life Science, Frankfurt, Germany). Primers used were: glyceraldehyde 3-phosphate dehydrogenase F: 5′-ACCCAGAAGACTGTGGATGG-3′, R: 5′-CAGTGAGCTTCCCGTTCA-3′ DA receptor D1 F: 5′-AACTGTATGGTGCCCTTCTGTGG-3′, R: 5′-CATTCGTAGTTGTTGTTGCCCCG-3′ DA receptor D2 F: 5′-CACTCCGCCACTTCTTGACATACA-3′, R: 5′-TCTCCTCCGACACCTACCCCGA-3′ DA receptor D3 F: 5′-GTCCTGCCCTCTCCTCTTTGGTTT-3′, R: 5′-AGTCTACGGTGCCCTGTTTAC-3′ DA receptor D4 F: 5′-TGCCCTCAACCCCATCATCTACAC-3′, R: 5′-AATACTTCCGACCCCCAACCCT-3′ DA receptor D5 F: 5′-GGGAGATCGCTGCTGCCTATGTC-3′, R: 5′-TTTTAGAGTGGTGAGTGGGGGTTA-3′ DAT F: 5′-ACGCTCAAAATACTCAGCAG-3′, R: 5′-TACCGAGAGGACAGCATTCC-3′. Relative mRNA levels were quantified using the comparative quantitation method in Rotor-Gene 6000 Series Software. Relative mRNAs levels were then normalized to glyceraldehyde 3-phosphate dehydrogenase. Each PCR was performed in technical triplicates, and each experiment was performed in at least three biological replicates. Error bars represent s.e.m. between biological replicates.
Statistical analysis
All data are presented as mean±s.e.m. Behavioural experiments were analysed using two-way analysis of variance (ANOVA) with repeated measures where appropriate (Systat, version 9.0, SPSS software; SPSS, Armonk, NY, USA). Neurochemical data were analysed using ANOVA and Student's t-test. In all studies, a P-value of <0.05 was considered to be statistically significant.
Results
Baseline hyperactivity of 14-3-3ζ KO mice is rescued with clozapine
Our previous studies identified several schizophrenia-like behavioural deficits in 14-3-3ζ KO mice, including a reduced capacity to learn and remember, hyperactivity and disrupted sensorimotor gating. To further test the relevance of this mouse model to schizophrenia and related disorders, we completed behavioural analyses with the antipsychotic drug clozapine, an antagonist of DA and serotonin receptors. Consistent with our previous report, we found that 14-3-3ζ KO mice have baseline hyperactivity over a 30-min test period. Following 2 weeks of daily intraperitoneal injections of 5 mg kg–1 clozapine, we found that baseline locomotor hyperactivity of 14-3-3ζ KO mice returned to levels similar to WT mice (Figure 1). ANOVA revealed main effects of genotype (F(1, 31)=7.2, P=0.012) and of clozapine treatment (F(1, 31)=4.3, P=0.046) and a trend towards a genotype × clozapine interaction (F(1, 31)=3.3, P=0.078). Further pairwise comparison confirmed the expected baseline locomotor hyperactivity in 14-3-3ζ KO mice compared with controls (F(1, 16)=9.9, P=0.006) but there was no difference between clozapine-treated 14-3-3ζ KO mice and WT controls (Figure 1). Distance moved was significantly reduced in clozapine-treated compared with saline-treated 14-3-3ζ KO mice (F(1, 14)=6.4, P=0.024) but there was no clozapine effect in WT controls (Figure 1). This functional rescue of hyperactivity was independent of sex.
Figure 1.

Clozapine rescues baseline hyperactivity of 14-3-3ζ knockout (KO) mice. 14-3-3ζ KO mice (white bar; n=8; 5 males and 3 females) have greater baseline exploratory behaviour than 14-3-3ζ wild-type (WT) mice (closed bar; n=10; 6 males and 4 females) in an open field test. Treatment with the antipsychotic clozapine has no effect on WT mice (dark grey hashed bar; n=10; 6 males and 4 females) but reduces baseline exploratory behaviour of 14-3-3ζ KO mice (light grey bar; n=8; 5 males and 3 females) to levels similar to WT.
14-3-3ζ KO mice are hypersensitive to amphetamine
A defining feature of human psychiatric conditions is enhanced behavioural effects of amphetamine.35, 36, 37 To further validate 14-3-3ζ KO mice as a robust schizophrenia-like mouse model, we completed analysis of amphetamine-induced hyperactivity. Amphetamine is a potent psychostimulant that enhances the release of DA from presynaptic dopaminergic terminals.38 Consistent with previous findings, we found that 14-3-3ζ KO mice showed hyperactivity relative to WT mice in the 20-min habituation phase before drug or after saline administration (Figure 2a; WT, n=12; KO, n=11). Indeed, ANOVA showed a significant main effect of genotype after injection of saline (F(1, 22)=11.0, P=0.003). WT and KO mice also demonstrated a decline in activity with habituation to the test arena. Subcutaneous injection of amphetamine-induced (5 mg kg–1) hyperactivity in both 14-3-3ζ WT and KO mice (WT, n=12; KO, n=11), however, this effect was significantly enhanced in the KO mice that had reduced time to become maximally hyperactive and also covered a greater distance than WT controls particularly in the first 45 min of the 90-min testing period (Figure 2b). The genotype-dependent difference in amphetamine time-course was reflected by a significant ANOVA amphetamine × genotype × time interaction (F(17, 374=2.8, P<0.001). During the first 45 min after amphetamine injection, there was also a genotype × amphetamine interaction (F(1, 22)=6.0, P=0.023), which was absent for the second 45 min after injection (Figure 2b). Induced hyperactivity was similar for both males and females with no sex bias (P>0.05).
Figure 2.

14-3-3ζ Knockout (KO) mice are hypersensitive to amphetamine. (a) 14-3-3ζ KO mice (closed square; n=11; 6 males and 5 females) have greater baseline exploratory behaviour than 14-3-3ζ wild-type (WT) mice (open square; n=12; 8 males and 4 females) in an open field test. (b) 14-3-3ζ KO mice (closed circle; n=11; 6 males and 5 females) have increased hyperactivity in response to amphetamine (5 mg kg–1) than 14-3-3ζ WT mice (open circle; n=12; 8 males and 4 females) in an open field test. Note difference in vertical scale.
DA levels and DA turnover are aberrant in 14-3-3ζ KO mice
Given the rescue of baseline hyperactivity with clozapine and the increased hyperactivity to the DA releaser amphetamine, we next investigated the levels of total tissue DA and DOPAC in the striatum, cortex and hippocampus by HPLC/EC. Our analysis found that tissue content of DA was significantly increased by 30% in the striatum of P100 14-3-3ζ KO mice when compared with WT controls (Figure 3a; WT, mean=135 pmol mg–1, n=6; KO, mean=178 pmol mg–1, n=6; P=0.038). Although not reaching levels of significance this trend was also observed in the cortex and hippocampus of 14-3-3ζ KO mice (Supplementary Figures S1a and d). We also observed an increase of DOPAC in the striatum of 14-3-3ζ KO mice when compared with WT controls (Figure 3b; WT, mean=14.7 pmol mg–1; KO, mean =20.9 pmol mg–1) that was not observed in the cortex (Supplementary Figure S1b). This increase of DOPAC resulted in a similar level of DA turnover (DOPAC/DA ratio) in 14-3-3ζ KO and WT mice (Figure 3c).
Figure 3.

Altered baseline dopamine (DA) in the striatum of 14-3-3ζ knockout (KO) mice. (a) Baseline DA and 3,4-dihydroxyphenylacetic acid (DOPAC) levels were measured in the striatum by high-performance liquid chromatography (HPLC)/EC. 14-3-3ζ KO mice (white bar; n=6; 4 males and 2 females) have increased DA compared with 14-3-3ζ wild-type (WT) mice (closed bar; n=6; 3 males and 3 females). (b) 14-3-3ζ KO mice (white bar) also have increased DOPAC compared with 14-3-3ζ WT mice (closed bar). (c) DA turnover (DOPAC/DA ratio) is conserved in 14-3-3ζ KO mice (white bar) compared with 14-3-3ζ WT mice (closed bar).
TH is preserved in 14-3-3ζ KO mice
Our previous findings raised the hypothesis that DA neurotransmission is affected in 14-3-3ζ KO mice. DA is produced by dopaminergic neurons that primarily reside in the VTA/SN and send their processes to the dorsal and ventral striatum, respectively. 14-3-3ζ Has previously been suggested to have an essential role in DA synthesis by interacting with TH to promote its phosphorylation and activity.15 To investigate the interactions and functions of 14-3-3ζ, we generated a suite of 14-3-3ζ monoclonal antibodies by immunizing 14-3-3ζ KO mice with recombinant 14-3-3ζ protein. Western blot analysis of recombinant 14-3-3 proteins expressed in bacteria shows that anti-14-3-3ζ M6, D1-4 and N4 predominantly interact with 14-3-3ζ while also recognizing 14-3-3τ (Supplementary Figure S2). However, as 14-3-3τ is not expressed in brain tissue16 these antibodies provide an ideal resource to specifically address the role of 14-3-3ζ in brain function. Using anti-14-3-3ζ M6 on a western blot of proteins co-immunoprecipitated with anti-TH from P35 mouse brain lysates, we were able to confirm the interaction of 14-3-3ζ and TH in vivo (Figure 4a).
Figure 4.

Tyrosine hydroxylase (TH) is activated normally in 14-3-3ζ knockout (KO) mice. (a) TH was precipitated from P35 whole-brain lysates with anti-TH antibody. TH immunoprecipitates were probed with anti-TH and monoclonal antibodies against 14-3-3ζ (M6). (b) Sagittal sections and (i, ii) coronal sections (iii iv) show that the abundance of TH-positive dopaminergic neurons in the substantia nigra (sn) and ventral tegmental area (vta), and their projections to the striatum (st) are similar in 14-3-3ζ KO and wild-type (WT) mice. Scale bars=500 μm. (c) Western blot analysis of brain lysates shows that total TH, phospho serine 31 (Ser-31) and phospho Ser-40 are present at similar levels in 14-3-3ζ KO and WT mice. Load control used for quantitating western blots was α tubulin. A representative blot of all four samples is shown in this figure that is quantitated in Supplementary Figure S3.
We next analysed the abundance of total TH in sagittal and coronal brain sections by immunohistochemistry. Our analysis identified similar levels of expression in the striatum, VTA and SN and further showed that 14-3-3ζ KO mice have an equivalent number of TH-positive dopaminergic neurons as WT controls (Figure 4b; WT, n=4; KO, n=4). As TH activity is controlled through the phosphorylation of Ser-31 and Ser-40, we next explored the abundance of active TH with phospho-specific antibodies. Our immunoblotting analysis was unable to identify any significant differences in the levels of total or active TH in 14-3-3ζ KO brains (Figure 4c and Supplementary Figure S3; WT, n=6; KO, n=6).
DAT density is reduced in 14-3-3ζ KO mice
Following release of DA into the synaptic cleft it initiates signalling cascades in postsynaptic neurons by interacting with the G-protein-coupled DA receptors D1–D5. Regulation of this interaction is primarily achieved through the reuptake of DA by presynaptic neurons with DAT. Given the distinct possibility of a DA signalling dysfunction in 14-3-3ζ KO mice, we therefore explored the abundance of DA receptors and DAT in 14-3-3ζ KO brain samples. Quantitative reverse transcriptase-PCR analysis from P35 whole-brain RNA shows that each of these receptors is expressed normally at the transcript level (Figure 5a). Furthermore, analysis of the DA receptors by immunoblotting of P35 whole-brain lysates shows that the major isoforms of D1–D5 are present at normal levels in 14-3-3ζ KO mice (Figure 5b and Supplementary Figure S4; WT, n=4; KO, n=4).
Figure 5.

Expression of dopamine (DA) receptors in 14-3-3ζ knockout (KO) mice. (a) Quantitative reverse transcriptase-PCR (qRT-PCR) of DA receptors D1–D5 and DA transporter (DAT) show that transcript levels of these genes are present at the same level in 14-3-3ζ KO (open bars; n=4) and 14-3-3ζ wild-type (WT) brains (closed bars; n=4). (b) Western blot analysis of whole-brain lysate shows that protein levels of DA receptors D1–D5 are present at the same level in 14-3-3ζ KO (n=4) and 14-3-3ζ WT brains (n=4). A representative blot of all four samples is shown in this figure that is quantitated in Supplementary Figure S4.
We next analysed the localization and abundance of DAT by co-labelling sagittal brain sections with anti-DAT and anti-TH antibodies. In comparison with 14-3-3ζ WT mice, we observed a reduction in DAT within the SN-VTA of KO mice (Figure 6a). Upon closer examination, 14-3-3ζ WT showed evenly distributed DAT staining throughout the cell body and neurites of dopaminergic neurons within the midbrain, whereas KO neurons had sparse staining in neuronal processes and irregular localization within the cell body (Figure 6b). Within these KO mice, DAT localization was predominantly polarized to one side of the nucleus (Figure 6b). Expression of DAT was quantified by measuring the fluorescence intensity of anti-DAT in SN-VTA neurons relative to that of anti-TH antibodies. Our analysis shows that DAT levels are reduced by approximately 30% in the SN-VTA of 14-3-3ζ KO mice (Figure 6c; WT, n=4; KO, n=4; P=0.043). Using the same immunostaining method, we also found a significant reduction of DAT in the striatum (Figure 7). Analysis of low magnification images shows that DAT is uniformly reduced across the entire striatum (Figure 7a). Higher magnification further identifies distinct TH-positive fibres within the striatum that lack DAT (arrowheads, Figure 7b). Quantitation of this staining shows that DAT levels are reduced by approximately 50% in the striatum of 14-3-3ζ KO mice (Figure 7c; WT, n=4; KO, n=4; P=0.009).
Figure 6.

14–3-3ζ Regulates dopamine transporter (DAT )expression in the substantia nigra (SN)-ventral tegmental area (VTA). (a) Sagittal brain sections stained with anti-tyrosine hydroxylase (TH; red) and anti-DAT (green) show reduced levels of DAT in the SN-VTA of 14-3-3ζ knockout (KO) mice compared with wild-type (WT) littermates. Blue, 4,6-diamidino-2-phenylindole (DAPI); scale bars=50 um. (b) Higher magnification of anti-DAT immunostaining (green) in SN-VTA shows that DAT is mislocalized in the cell bodies of dopaminergic neurons (white arrowheads). Scale bar=20 um. (c) Quantitation of anti-DAT immunostaining normalized to anti-TH confirms that 14-3-3ζ KO (open bar) have an approximately 30% reduction of DAT compared with WT mice (closed bar).
Figure 7.

14–3-3ζ Regulates dopamine transporter (DAT) expression in the striatum. (a) Sagittal brain sections stained with anti-tyrosine hydroxylase (TH; red) and anti-DAT (green) show reduced levels of DAT in the striatum of 14-3-3ζ knockout (KO) mice compared with wild-type (WT) littermates. Blue, 4,6-diamidino-2-phenylindole (DAPI); scale bars=200 um. (b) Higher magnification of anti-DAT immunostaining (boxed region in a) shows that DAT is missing in the axonal terminals of dopaminergic neurons (white arrowheads). Blue, DAPI; scale bar=10 um. (c) Quantitation of anti-DAT immunostaining normalized to anti-TH confirms that 14-3-3ζ KO (open bar) have an approximately 50% reduction of DAT compared with WT mice (closed bar). (d) Western blot analysis of whole-brain lysate shows that proteins levels of unglycosylated DAT (50 kDa) and glycosylated DAT (80 kDa) are reduced in 14-3-3ζ KO (n=4) compared with 14-3-3ζ WT mice (n=4).
Analysis of P35 whole-brain lysates by immunoblotting further identified robust deficiency of the non-glycosylated form of DAT (approximately 50 kDa band) in KO mice and in around half the cases also deficiency of the glycosylated form (approximately 75 kDa band; Figure 7d). To confirm that 14-3-3ζ is expressed in dopaminergic neurons, and in appropriate regions to have a role in DAT function, we co-immunostained sagittal brain sections with anti-14-3-3ζ M6 and anti-TH antibodies. Our analysis found that 14-3-3ζ is widely expressed in the midbrain, and importantly, within TH-positive neurons in the SN/VTA and their terminals in the striatum (Supplementary Figure S5). The lack of staining in 14-3-3ζ KO brain sections further confirms the specificity of this antibody and the expression of 14-3-3ζ in dopaminergic neurons.
DAT associates with 14-3-3ζ
As DAT is phosphorylated on several serine and threonine residues by CAMKII/protein kinase C and contains putative 14-3-3 binding sites (data not shown), we next investigated if 14-3-3ζ interacts with this transporter. By transiently expressing Flag-His-DAT and Myc-14-3-3ζ in the SN-derived mouse neuronal cell line SN4741, we were able to identify an interaction between these proteins by co-immuoprecipitation. In these experiments, purification of 14-3-3ζ with anti-Myc antibodies also co-precipitated Flag-His-tagged DAT (Figure 8a). In addition, purification of DAT with anti-Flag antibodies co-precipitated Myc-tagged 14-3-3ζ (Figure 8b). In contrast, co-immunoprecipitation with an immunoglobulin G isotype control antibody was unable to purify either 14-3-3ζ or DAT from protein lysates in which purification of DAT with anti-Flag antibodies co-precipitated Myc-tagged 14-3-3ζ (Supplementary Figure S6). To confirm the interaction between these proteins in vivo, we next completed co-immunoprecipitation experiments with our G1-7 monoclonal antibody in striatal protein extracts from P35 WT mice. Consistent with our overexpression study, we found that antibodies recognizing 14-3-3ζ also precipitate DAT from these complex protein extracts (Figure 8c).
Figure 8.
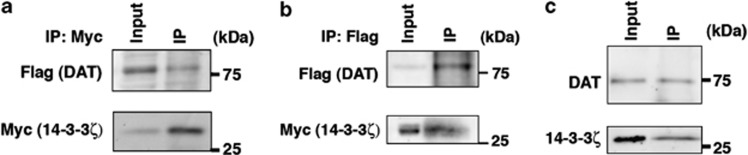
14–3-3ζ Associates with dopamine transporter (DAT). (a) SN4741 cells were transiently transfected with Flag-His-DAT and Myc-14-3-3ζ. 14-3-3ζ was precipitated from protein lysates with anti-Myc antibody. 14-3-3ζ Immunoprecipitates were probed with anti-Myc monoclonal antibody to recognise 14-3-3ζ and anti-Flag to recognize DAT. (b) DAT was precipitated from protein lysates as in (a) with anti-Flag antibody. DAT immunoprecipitates were probed with anti-Myc monoclonal antibody to recognise 14-3-3ζ and anti-Flag to recognize DAT (c) 14-3-3ζ Was precipitated from P35 striatum lysates with anti-14-3-3ζ G1-7 monoclonal antibody. 14-3-3ζ Immunoprecipitates were probed with anti-14-3-3ζ (C16) and antibodies against DAT.
Discussion
Over the past 50 years, there have been many clinical and pharmacological studies implicating aberrant DA signalling in the pathology of psychosis associated with schizophrenia and related disorders.5, 39, 40, 41, 42 Animal models with deficits in each component of the dopaminergic pathway, such as DA biosynthesis, DA receptors and DAT have provided valuable insight to the role of aberrant DA signalling in the presentation of psychosis-like behavioural defects.43 However, many of these models have also shown paradoxical effects when compared with the human condition. We recently described 14-3-3ζ KO mice as a novel neurodevelopmental schizophrenia-like mouse model that has many anatomical and behavioural defects associated with the human condition.17 This notion is supported by genetic linkage analysis23 and the observations that 14-3-3ζ is downregulated at the mRNA18 and protein20, 21, 44 levels in post-mortem schizophrenia brain samples. Further support for a role in schizophrenia is derived from the recent finding that 14-3-3ζ is represented as a central hub within the schizophrenia-specific protein interaction network.24 At the molecular level, 14-3-3ζ also interacts with several putative risk factors for schizophrenia that are essential for neuronal development, such as NdeL1, LIS1 and DISC1.17, 45 In this study, we have significantly advanced our understanding of the role of 14-3-3ζ in the presentation of schizophrenia-like deficits by identifying part of the molecular defects underpinning locomotor hyperactivity. First, we specifically identify dysregulated DA signalling as a cause of hyperactivity in 14-3-3ζ KO mice. Second, we have shown that DAT dysregulation occurs in the absence of any obvious change in abundance of TH or DA receptors. Finally, we found a reduction in the abundance of DAT in the brains of adult 14-3-3ζ KO mice, and show that in the WT setting, 14-3-3ζ and DAT interact.
A defining feature of schizophrenia is hypersensitivity to drugs that increase synaptic DA levels.35, 36, 37 To test the validity of 14-3-3ζ KO mice as a model for the human condition, we therefore analysed both this feature and the ability of antipsychotics to ameliorate psychosis-like symptoms in our mouse model in vivo.
In support of 14-3-3ζ KO mice modelling the behavioural deficits associated with schizophrenia and associated disorders, we found that the antipsychotic drug clozapine was able to rescue baseline hyperactivity. However, although a 2-week treatment regime with clozapine had dramatic pharmacological effects on hyperactivity it was unable to modify the neurodevelopmental and anatomical defects associated with the disorder, including lamination and mossy fibre navigation in the hippocampus (data not shown).
Consistent with behavioural responses in patients with schizophrenia and related psychiatric disorders, we found that 14-3-3ζ KO mice are hypersensitive to the effects of amphetamine in a test of locomotor function. Taken together with our findings of baseline hyperactivity, disrupted senorimotor gating and developmental defects in the hippocampus,17 these analyses implicate 14-3-3ζ KO mice as a highly applicable neurodevelopmental model of schizophrenia and related disorders.
As amphetamine acts as a substrate for DAT to promote DA efflux from presynaptic neurons, and clozapine can antagonise DA receptors, our results further suggest that some of the psychosis-like behavioural defects of 14-3-3ζ KO mice may arise from defects in the dopaminergic pathway. Indeed, our analysis of total tissue DA levels found that DA is significantly increased in the striatum of KO mice and strongly supports a role for 14-3-3ζ in the DA signalling pathway. 14-3-3ζ Has been implicated as a regulator of DA signalling by interacting with TH through phosphorylation of Ser-19 to enhance activation of TH enzymatic activity by promoting further phosphorylation of Ser-31 and Ser-40.15 Our analysis of 14-3-3ζ KO mice not only shows that 14-3-3ζ is indispensable for the formation of dopaminergic neurons in the brain, it also shows that TH levels and TH activation are maintained in the absence of 14-3-3ζ. In contrast to the proposal that 14-3-3ζ is the major isoform controlling TH activity,15 our results suggest that 14-3-3 isoforms are likely to act redundantly in promoting TH activity in vivo. Indeed, multiple 14-3-3 isoforms have been shown to bind TH, and 14-3-3γ has recently been suggested to promote localization of TH to membrane fractions to preserve enzymatic activity.46 Although our data fit with the idea that DA production is normal in 14-3-3ζ KO mice, it will now be important to measure the rate of DA synthesis in vivo.
To determine the nature of the dopaminergic dysfunction in our schizophrenia-like mouse model, we therefore analysed later stages of signalling post-secretion of DA from presynaptic vesicles. DA interacts with receptors on postsynaptic neurons to initiate a myriad of downstream signalling events by modulating activity of adenylate cyclase. Our analysis of the abundance of DA receptor by quantitative reverse transcriptase-PCR and western blotting was unable to detect any differences in the levels of either total RNA or protein from whole-brain samples. This finding identifies important differences between our schizophrenia-like mouse model and that of the DISC1 mouse models. DISC1 mutant mice also display DA dysfunction, however, in these mice the primary defect has been attributed to excessive levels of DA receptor D2.47 Although the techniques used in our study provide a high level of quantitation, future experiments will be best directed at completing receptor binding studies to confirm our results and to analyse the levels and binding capacity of the DA receptors in more detail.
In the absence of gross defects in DA receptor levels, we next analysed the abundance of DAT that is expressed in presynaptic dopaminergic neurons to regulate levels of DA in the synaptic cleft. Our finding that DAT levels are reduced at the protein level and not at the RNA level suggests that the deficiency of DAT occurs via post-transcriptional mechanisms. In support of DAT dysfunction having a central role in the behavioural defects of our 14-3-3ζ mouse model, DAT KO mice have also been shown to have baseline hyperactivity.48 At steady-state levels DAT is constitutively internalized, recycled to the plasma membrane or degraded in the lysosome through the activity of the E3 ubiquitin ligase Nedd4-2.49 Although the precise mechanisms of this trafficking pathway are unclear it is reliant on amino acids 587–597 in the c-terminus of the protein50 and on the ubiquitination of three lysine residues in the N-terminus.51 DAT function is also heavily regulated by several kinases that primarily phosphorylate serine and threonine residues in its n-terminus.52 Upon pharmacological stimulation of DAT with amphetamine, several serine and threonine residues in this n-terminal region are phosphorylated by protein kinase C and CAMKII to induce DA efflux from presynaptic neurons.52 Our finding that DAT levels are reduced in 14-3-3ζ KO mice and also mislocalized in dopaminergic neurons therefore raises the hypothesis that 14-3-3ζ may have a role in modulating DAT phosphorylation, trafficking and/or degradation. In support of this notion, 14-3-3ζ has previously been found to bind to and inhibit the function of Nedd4-2 to positively regulate the abundance of other substrates such as amiloride-sensitive epithelial Na+ channel.53 Whether 14-3-3ζ also binds Nedd4-2 to regulate levels of DAT is currently unknown, however, as Nedd4-2 is proposed to modulate membrane associated DAT (that is, the glycosylated form) our finding of reduced non-glycosylated DAT may argue against a primary role in regulating DAT levels through this pathway. A role for 14-3-3ζ in modulating DAT phosphorylation or function may also help to explain the ability of amphetamine to increase hyperactivity of KO mice over and above that of WT controls albeit in the presence of a 30% reduction of total DAT levels. Thus, dysregulation of receptor phosphorylation and/or activity may promote degradation in the resting state but retain DAT in a primed state for activation upon pharmacological stimulation. In support of this notion, phosphorylation of DAT Ser-7 has previously been found to hold DAT in a primed state for amphetamine-induced DA efflux.52 It will now be of interest to examine the response of 14-3-3ζ KO mice to alternative stimulants of DAT such as cocaine and to test the ability of 14-3-3ζ to reduce amphetamine-induced DA efflux, for example by micro-dialysis.
In this study, we also identified an interaction between DAT and 14-3-3ζ in SN4741 cells and striatum brain extracts. However, as we were only able to co-purify a small proportion of total input protein in our co-immunoprecipitation experiments this further suggests that the interaction between DAT and 14-3-3ζ is quite weak and/or unstable. Whether this interaction is direct or mediated by an intermediate adaptor protein and whether this has a role in DAT function is currently unknown. Our in silico analysis of DAT identifies several high stringency phospho-serine/threonine 14-3-3 binding motifs in the c-terminus of the protein. Interestingly, these sites are located near the regions predicted to act as interaction sites for CAMKII and in the regions predicted to be essential for DAT internalization. It will now be of interest to determine the interaction dynamics of 14-3-3ζ and DAT and to determine if this has any role in kinase binding and receptor phosphorylation.
Finally, consistent with a functional abnormality in DAT physiology we found that total tissue DA levels are increased in 14-3-3ζ KO mice. Our study also found an increase in the DA degradation by-product, DOPAC. Although a 30% reduction in DAT may be expected to result in reduced DA degradation, our finding is in agreement with that observed in DAT KO mice that also have abundant levels of DOPAC.54 Our findings therefore add strong support to the notion that DOPAC represents a by-product of newly synthesized DA rather than a by-product of DA recycled from the synaptic cleft.54 Thus, although TH levels and activation are normal in 14-3-3ζ KO mice it will be of interest to determine the rate of DA synthesis in this model.
In conclusion, our data supports a model in which 14-3-3ζ interacts with DAT to modulate its activity and stability and thereby control the availability of DA in the synaptic cleft. This finding has important implications to the physiological basis of schizophrenia-like behavioural defects, the mechanisms controlling DAT function and the potential modulation of this pathway in the treatment of disorders with a hyperdopaminergic basis.
Acknowledgments
We thank Rebecca Krake for genotyping mice and maintaining the mouse colonies. This work was funded by grants from the National Health and Medical Research Council of Australia (NHMRC) and Medvet Laboratories. HR is the recipient of the Peter Nelson Leukemia Research Fellowship and QS is the recipient of a NHMRC Career Development Award. MvdB is a NHMRC Senior Research Fellow. CLP is the recipient of a Viertel charitable foundation senior medical research fellowship.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies the paper on the Translational Psychiatry website (http://www.nature.com/tp)
Supplementary Material
References
- Tamminga CA, Stan AD, Wagner AD. The hippocampal formation in schizophrenia. Am J Psychiatry. 2010;167:1178–1193. doi: 10.1176/appi.ajp.2010.09081187. [DOI] [PubMed] [Google Scholar]
- Carpenter WT, Koenig JI. The evolution of drug development in schizophrenia: past issues and future opportunities. Neuropsychopharmacology. 2008;33:2061–2079. doi: 10.1038/sj.npp.1301639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leucht S, Wahlbeck K, Hamann J, Kissling W. New generation antipsychotics versus low-potency conventional antipsychotics: a systematic review and meta-analysis. Lancet. 2003;361:1581–1589. doi: 10.1016/S0140-6736(03)13306-5. [DOI] [PubMed] [Google Scholar]
- Drevets WC, Gautier C, Price JC, Kupfer DJ, Kinahan PE, Grace AA, et al. Amphetamine-induced dopamine release in human ventral striatum correlates with euphoria. Biol Psychiatry. 2001;49:81–96. doi: 10.1016/s0006-3223(00)01038-6. [DOI] [PubMed] [Google Scholar]
- Laruelle M, Abi-Dargham A, Gil R, Kegeles L, Innis R. Increased dopamine transmission in schizophrenia: relationship to illness phases. Biol Psychiatry. 1999;46:56–72. doi: 10.1016/s0006-3223(99)00067-0. [DOI] [PubMed] [Google Scholar]
- Amara SG, Kuhar MJ. Neurotransmitter transporters: recent progress. Annu Rev Neurosci. 1993;16:73–93. doi: 10.1146/annurev.ne.16.030193.000445. [DOI] [PubMed] [Google Scholar]
- Giros B, Caron MG. Molecular characterization of the dopamine transporter. Trends Pharmacol Sci. 1993;14:43–49. doi: 10.1016/0165-6147(93)90029-j. [DOI] [PubMed] [Google Scholar]
- Itagaki C, Isobe T, Taoka M, Natsume T, Nomura N, Horigome T, et al. Stimulus-coupled interaction of tyrosine hydroxylase with 14-3-3 proteins. Biochemistry. 1999;38:15673–15680. doi: 10.1021/bi9914255. [DOI] [PubMed] [Google Scholar]
- Toska K, Kleppe R, Armstrong CG, Morrice NA, Cohen P, Haavik J. Regulation of tyrosine hydroxylase by stress-activated protein kinases. J Neurochem. 2002;83:775–783. doi: 10.1046/j.1471-4159.2002.01172.x. [DOI] [PubMed] [Google Scholar]
- Aitken A. 14-3-3 proteins: a historic overview. Semin Cancer Biol. 2006;16:162–172. doi: 10.1016/j.semcancer.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Ichimura T, Isobe T, Okuyama T, Yamauchi T, Fujisawa H. Brain 14-3-3 protein is an activator protein that activates tryptophan 5-monooxygenase and tyrosine 3-monooxygenase in the presence of Ca2+,calmodulin-dependent protein kinase II. FEBS Lett. 1987;219:79–82. doi: 10.1016/0014-5793(87)81194-8. [DOI] [PubMed] [Google Scholar]
- Berg D, Holzmann C, Riess O. 14-3-3 proteins in the nervous system. Nat Rev Neurosci. 2003;4:752–762. doi: 10.1038/nrn1197. [DOI] [PubMed] [Google Scholar]
- Fu H, Subramanian RR, Masters SC. 14-3-3 Proteins: structure, function, and regulation. Annu Rev Pharmacol Toxicol. 2000;40:617–647. doi: 10.1146/annurev.pharmtox.40.1.617. [DOI] [PubMed] [Google Scholar]
- Rosner M, Hengstschlager M. 14-3-3 Proteins are involved in the regulation of mammalian cell proliferation. Amino Acids. 2006;30:105–109. doi: 10.1007/s00726-005-0240-7. [DOI] [PubMed] [Google Scholar]
- Wang J, Lou H, Pedersen CJ, Smith AD, Perez RG. 14-3-3Zeta contributes to tyrosine hydroxylase activity in MN9D cells: localization of dopamine regulatory proteins to mitochondria. J Biol Chem. 2009;284:14011–14019. doi: 10.1074/jbc.M901310200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxter HC, Liu WG, Forster JL, Aitken A, Fraser JR. Immunolocalisation of 14-3-3 isoforms in normal and scrapie-infected murine brain. Neuroscience. 2002;109:5–14. doi: 10.1016/s0306-4522(01)00492-4. [DOI] [PubMed] [Google Scholar]
- Cheah PS, Ramshaw HS, Thomas PQ, Toyo-Oka K, Xu X, Martin S, et al. Neurodevelopmental and neuropsychiatric behaviour defects arise from 14-3-3zeta deficiency Mol Psychiatry 2012. Picture solicited for cover17451–466. [DOI] [PubMed] [Google Scholar]
- Middleton FA, Peng L, Lewis DA, Levitt P, Mirnics K. Altered expression of 14-3-3 genes in the prefrontal cortex of subjects with schizophrenia. Neuropsychopharmacology. 2005;30:974–983. doi: 10.1038/sj.npp.1300674. [DOI] [PubMed] [Google Scholar]
- Wong AH, Likhodi O, Trakalo J, Yusuf M, Sinha A, Pato CN, et al. Genetic and post-mortem mRNA analysis of the 14-3-3 genes that encode phosphoserine/threonine-binding regulatory proteins in schizophrenia and bipolar disorder. Schizophr Res. 2005;78:137–146. doi: 10.1016/j.schres.2005.06.009. [DOI] [PubMed] [Google Scholar]
- English JA, Dicker P, Focking M, Dunn MJ, Cotter DR. 2-D DIGE analysis implicates cytoskeletal abnormalities in psychiatric disease. Proteomics. 2009;9:3368–3382. doi: 10.1002/pmic.200900015. [DOI] [PubMed] [Google Scholar]
- English JA, Pennington K, Dunn MJ, Cotter DR. The neuroproteomics of schizophrenia. Biol Psychiatry. 2011;69:163–172. doi: 10.1016/j.biopsych.2010.06.031. [DOI] [PubMed] [Google Scholar]
- Sivagnanasundaram S, Crossett B, Dedova I, Cordwell S, Matsumoto I. Abnormal pathways in the genu of the corpus callosum in schizophrenia pathogenesis: a proteome study. Proteomics Clin Appl. 2007;1:1291–1305. doi: 10.1002/prca.200700230. [DOI] [PubMed] [Google Scholar]
- Jia Y, Yu X, Zhang B, Yuan Y, Xu Q, Shen Y. An association study between polymorphisms in three genes of 14-3-3 (tyrosine 3-monooxygenase/tryptophan 5-monooxygenase activation protein) family and paranoid schizophrenia in northern Chinese population. Eur Psychiatry. 2004;19:377–379. doi: 10.1016/j.eurpsy.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Sun J, Jia P, Fanous AH, van den Oord E, Chen X, Riley BP, et al. Schizophrenia gene networks and pathways and their applications for novel candidate gene selection. PLoS One. 2010;5:e11351. doi: 10.1371/journal.pone.0011351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyo-oka K, Shionoya A, Gambello MJ, Cardoso C, Leventer R, Ward HL, et al. 14-3-3epsilon is important for neuronal migration by binding to NUDEL: a molecular explanation for Miller-Dieker syndrome. Nat Genet. 2003;34:274–285. doi: 10.1038/ng1169. [DOI] [PubMed] [Google Scholar]
- Coyle P, Tran N, Fung JN, Summers BL, Rofe AM. Maternal dietary zinc supplementation prevents aberrant behaviour in an object recognition task in mice offspring exposed to LPS in early pregnancy. Behav Brain Res. 2009;197:210–218. doi: 10.1016/j.bbr.2008.08.022. [DOI] [PubMed] [Google Scholar]
- Summers BL, Rofe AM, Coyle P. Prenatal zinc treatment at the time of acute ethanol exposure limits spatial memory impairments in mouse offspring. Pediatr Res. 2006;59:66–71. doi: 10.1203/01.pdr.0000190573.23893.13. [DOI] [PubMed] [Google Scholar]
- van den Buuse M, Wischhof L, Lee RX, Martin S, Karl T. Neuregulin 1 hypomorphic mutant mice: enhanced baseline locomotor activity but normal psychotropic drug-induced hyperlocomotion and prepulse inhibition regulation. Int J Neuropsychopharmacol. 2009;12:1383–1393. doi: 10.1017/S1461145709000388. [DOI] [PubMed] [Google Scholar]
- Williams AA, Ingram WM, Levine S, Resnik J, Kamel CM, Lish JR, et al. Reduced levels of serotonin 2A receptors underlie resistance of Egr3-deficient mice to locomotor suppression by clozapine. Neuropsychopharmacology. 2012;37:2285–2298. doi: 10.1038/npp.2012.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baune BT, Wiede F, Braun A, Golledge J, Arolt V, Koerner H. Cognitive dysfunction in mice deficient for TNF- and its receptors. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:1056–1064. doi: 10.1002/ajmg.b.30712. [DOI] [PubMed] [Google Scholar]
- Hart PC, Bergner CL, Smolinsky AN, Dufour BD, Egan RJ, Laporte JL, et al. Experimental models of anxiety for drug discovery and brain research. Methods Mol Biol. 2010;602:299–321. doi: 10.1007/978-1-60761-058-8_18. [DOI] [PubMed] [Google Scholar]
- Koike H, Arguello PA, Kvajo M, Karayiorgou M, Gogos JA. Disc1 is mutated in the 129S6/SvEv strain and modulates working memory in mice. Proc Natl Acad Sci USA. 2006;103:3693–3697. doi: 10.1073/pnas.0511189103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel MS, Lopez JI, McGhee EJ, Croft DR, Strachan D, Timpson P, et al. Actomyosin-mediated cellular tension drives increased tissue stiffness and beta-catenin activation to induce epidermal hyperplasia and tumor growth. Cancer Cell. 2011;19:776–791. doi: 10.1016/j.ccr.2011.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son JH, Chun HS, Joh TH, Cho S, Conti B, Lee JW. Neuroprotection and neuronal differentiation studies using substantia nigra dopaminergic cells derived from transgenic mouse embryos. J Neurosci. 1999;19:10–20. doi: 10.1523/JNEUROSCI.19-01-00010.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DL, Smith M, Robinson D, Jody D, Lerner G, Alvir J, et al. Methylphenidate increases thought disorder in recent onset schizophrenics, but not in normal controls. Biol Psychiatry. 1993;34:507–514. doi: 10.1016/0006-3223(93)90192-g. [DOI] [PubMed] [Google Scholar]
- Lieberman JA, Alvir J, Geisler S, Ramos-Lorenzi J, Woerner M, Novacenko H, et al. Methylphenidate response, psychopathology and tardive dyskinesia as predictors of relapse in schizophrenia. Neuropsychopharmacology. 1994;11:107–118. doi: 10.1038/npp.1994.40. [DOI] [PubMed] [Google Scholar]
- Lieberman JA, Kane JM, Alvir J. Provocative tests with psychostimulant drugs in schizophrenia. Psychopharmacology (Berl) 1987;91:415–433. doi: 10.1007/BF00216006. [DOI] [PubMed] [Google Scholar]
- Sulzer D. How addictive drugs disrupt presynaptic dopamine neurotransmission. Neuron. 2011;69:628–649. doi: 10.1016/j.neuron.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abi-Dargham A, Gil R, Krystal J, Baldwin RM, Seibyl JP, Bowers M, et al. Increased striatal dopamine transmission in schizophrenia: confirmation in a second cohort. Am J Psychiatry. 1998;155:761–767. doi: 10.1176/ajp.155.6.761. [DOI] [PubMed] [Google Scholar]
- Breier A, Su TP, Saunders R, Carson RE, Kolachana BS, de Bartolomeis A, et al. Schizophrenia is associated with elevated amphetamine-induced synaptic dopamine concentrations: evidence from a novel positron emission tomography method. Proc Natl Acad Sci USA. 1997;94:2569–2574. doi: 10.1073/pnas.94.6.2569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laruelle M. Imaging dopamine transmission in schizophrenia. A review and meta-analysis. Q J Nucl Med. 1998;42:211–221. [PubMed] [Google Scholar]
- van Rossum JM. The significance of dopamine-receptor blockadefor the mechanism of action of neuroleptic drugs. Arch Int de Pharmacodynamie et de Thérapie. 1966;160:492–494. [PubMed] [Google Scholar]
- van den Buuse M. Modeling the positive symptoms of schizophrenia in genetically modified mice: pharmacology and methodology aspects. Schizophr Bull. 2010;36:246–270. doi: 10.1093/schbul/sbp132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivagnanasundaram S, Fletcher D, Hubank M, Illingworth E, Skuse D, Scambler P. Differential gene expression in the hippocampus of the Df1/+ mice: a model for 22q11.2 deletion syndrome and schizophrenia. Brain Res. 2007;1139:48–59. doi: 10.1016/j.brainres.2007.01.014. [DOI] [PubMed] [Google Scholar]
- Toyo-Oka K, Sasaki S, Yano Y, Mori D, Kobayashi T, Toyoshima YY, et al. Recruitment of katanin p60 by phosphorylated NDEL1, an LIS1 interacting protein, is essential for mitotic cell division and neuronal migration. Hum Mol Genet. 2005;14:3113–3128. doi: 10.1093/hmg/ddi339. [DOI] [PubMed] [Google Scholar]
- Halskau O, Jr., Ying M, Baumann A, Kleppe R, Rodriguez-Larrea D, Almas B, et al. Three-way interaction between 14-3-3 proteins, the N-terminal region of tyrosine hydroxylase, and negatively charged membranes. J Biol Chem. 2009;284:32758–32769. doi: 10.1074/jbc.M109.027706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaaro-Peled H, Niwa M, Foss CA, Murai R, de Los Reyes S, Kamiya A, et al. Subcortical dopaminergic deficits in a DISC1 mutant model: a study in direct reference to human molecular brain imaging. Hum Mol Genet. 2013;22:1574–1580. doi: 10.1093/hmg/ddt007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, Caron MG. Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature. 1996;379:606–612. doi: 10.1038/379606a0. [DOI] [PubMed] [Google Scholar]
- Vina-Vilaseca A, Sorkin A. Lysine 63-linked polyubiquitination of the dopamine transporter requires WW3 and WW4 domains of Nedd4-2 and UBE2D ubiquitin-conjugating enzymes. J Biol Chem. 2010;285:7645–7656. doi: 10.1074/jbc.M109.058990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holton KL, Loder MK, Melikian HE. Nonclassical distinct endocytic signals dictate constitutive and PKC-regulated neurotransmitter transporter internalization. Nat Neurosci. 2005;8:881–888. doi: 10.1038/nn1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda M, Dionne KR, Sorkina T, Sorkin A. Three ubiquitin conjugation sites in the amino terminus of the dopamine transporter mediate protein kinase C-dependent endocytosis of the transporter. Mol Biol Cell. 2007;18:313–323. doi: 10.1091/mbc.E06-08-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoshbouei H, Sen N, Guptaroy B, Johnson L, Lund D, Gnegy ME, et al. N-terminal phosphorylation of the dopamine transporter is required for amphetamine-induced efflux. PLoS Biol. 2004;2:E78. doi: 10.1371/journal.pbio.0020078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura T, Yamamura H, Sasamoto K, Tominaga Y, Taoka M, Kakiuchi K, et al. 14-3-3 proteins modulate the expression of epithelial Na+ channels by phosphorylation-dependent interaction with Nedd4-2 ubiquitin ligase. J Biol Chem. 2005;280:13187–13194. doi: 10.1074/jbc.M412884200. [DOI] [PubMed] [Google Scholar]
- Jones SR, Gainetdinov RR, Jaber M, Giros B, Wightman RM, Caron MG. Profound neuronal plasticity in response to inactivation of the dopamine transporter. Proc Natl Acad Sci USA. 1998;95:4029–4034. doi: 10.1073/pnas.95.7.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.