

Summary
It has long been known that apoptosis is vital to the generation and maintenance of proper adaptive immune function. An example of this is the vital requirement for apoptotic signaling during the generation of self-tolerant lymphocytes: the apoptotic death of B and T cells with overt autoreactivity is vital to central tolerance. More recently, the contributions of additional processes including cellular autophagy and programmed necrosis have been implicated in controlling both innate and adaptive immune functions. Evidence has been provided to demonstrate that the death of cells following ligation of death receptors (DR), a sub-family of cell surface molecules related to TNF receptor 1, is not exclusively the domain of caspase-dependent apoptosis. In cells lacking the capacity to activate caspase-8 following DR ligation, cell death instead occurs via programmed necrosis, or as it has been recently termed, “necroptosis.” This death process depends on RIP1 and RIP3, serine/threonine kinases that are recruited by DRs, and likely by other cellular signals including DNA damage and antigen receptor ligation. The generation of RIP1/RIP3 containing “necrosomes” activates downstream necroptotic signaling that ultimately targets cellular energetic metabolism. Also related to cellular metabolic regulation, cellular autophagy has also been found to play unique and important roles in immunity. In this review, we describe the roles of necroptosis and autophagy in innate and adaptive immunity, and speculate on the intriguing interplay between these two cellular processes.
Keywords: apoptosis, autophagy, necroptosis, immune tolerance, immune homeostasis, T cells, metabolism
Introduction
As this volume of reviews focuses on the roles of cellular metabolism in immune function, it might seem a discussion of autophagy and programmed cell death (PCD) might seem out of place. Yet, both of these cellular processes may be considered extreme examples of metabolism. Autophagic signaling is primarily engaged in preventing metabolic catastrophe, as it is required for the elimination of damaged organelles and macromolecules. In addition, the products of autophagic breakdown are often shunted toward the maintenance of cellular energy. In contrast, programmed cell death might be viewed as the terminal state of metabolism that cells must ultimate experience. It is not surprising that such cellular survival and death mechanisms are essential to vertebrate cellular physiology. Indeed, one may view these pathways as performing important but simple housekeeping functions in all cells, autophagy as a mechanism to deal with cellular stress and damage, and PCD as a means to initiate the conclusion of cellular metabolism under specific circumstances. Yet, as detailed in the following pages, autophagy and PCD are both are also vital to the development and proper functioning of vertebrate immunity, playing regulatory roles in these processes.
While numerous excellent reviews have addressed the myriad functions of apoptotic signaling in innate and adaptive immunity, here we focus specifically on programmed necrosis/necroptosis and autophagy. We start with an introduction regarding death receptor induced necroptosis, as this is probably the best understood non-apoptotic signaling pathway that involves the generation of RIP1/RIP3 containing complexes termed necrosomes. We then describe additional contexts under which necrosomal signaling promotes non-apoptotic cell death. Autophagy and its involvement in adaptive and innate immune function is addressed, followed by a treatise on the observed interplay between these two processes. As we shall describe, both programmed necrosis and autophagy are intricately involved in modulating immune cell metabolism, the major theme of this collection of essays.
Apoptosis vs. necrosis
For many years, the widely accepted form of cell death known to regulate embryonic development, immune homeostasis, and disease was apoptosis. Apoptotic death is described as an orderly, programmed event, characterized by an intact plasma membrane until the late stages, activation of caspases, DNA fragmentation, and membrane blebbing (1). Necrotic death is described as swelling of the endoplasmic reticulum, mitochondria, and cytoplasm, subsequently resulting in rupture of the plasma membrane and lysis of the cell, and is regarded as an unregulated and uncontrollable process (2). Necrosis has long been viewed as an accidental form of cell death caused by physical or chemical injury. Due to release of cellular contents, necrosis is thought to comprise an immunogenic death modality in contrast with apoptosis, which generally induces a tolerogenic response. While caspase 8-mediated apoptotic death has been well characterized, recent studies have now revealed that necrosis can also be regulated through specific intrinsic cellular programs.
Development of functional adaptive and innate immune responses requires strict regulation of programmed cell death signaling pathways. These signaling pathways are essential for shaping and maintaining the immune system, and ensuring functional immune responses. Programmed death is required for clonal deletion of lymphocytes during an infection, and dysregulation results in defective clearance of autoreactive T cells and autoimmune disease (3). Recent advancements have provided deeper insight into the molecular mechanisms that regulate the interplay between apoptosis and necroptosis, and uncovered additional, critical roles for programmed necrosis in immune function.
Death receptor/TNF receptor signaling
The tumor necrosis factor receptor (TNF) receptor superfamily is composed of a twenty-three cell surface receptors that potentiate various functions in vivo. Receptors belonging to the TNF receptor family play an important role in immune regulation by controlling various responses in mammalian cells. Of particular interest is a subset called the death receptors (DR), many of which are essential for immune homeostasis and tolerance and participate in the clonal deletion of activated lymphocytes (4). Death receptors share the feature of a “death domain” motif on their cytoplasmic tails, which promotes homotypic oligomerization and the recruitment of adaptor molecules such as FADD, TRADD, and RIPK1 (5–8) all of which bear death domains (DD) for interaction with DRs. Thus, the DD serves as the crucial bridge between adaptor proteins and death receptors to transmit downstream apoptotic signals.
The CD95/Fas/Apo-1 “DISC”
Ligation of the DR CD95/Fas/Apo-1 triggers formation of a cytosolic complex known as the “death-inducing signaling complex” (DISC) (9), which includes adaptor molecule FADD (Fas associated with death domain), RIPK1, caspase-8 (casp8), and a casp8-like molecule that lacks proteolytic activity called c-FLIP (10). Inactive pro-casp8 exists in a monomeric form in the cytosol: upon DR engagement, FADD is recruited to the DR via its DD, where it then recruits and activates monomeric casp8 via its death effector domain (DED) interactions (11, 12). Pro-casp8 homodimerization is accompanied by auto-cleavage and induction of catalytic activity (auto-activation) (13), ultimately leading to cleavage-mediated activation of downstream executioner caspases including casp3, 6 and 7 (14).
In response to various extracellular or intracellular stimuli, initiation of cellular death pathways is regulated by the activity of caspases. “Intrinsic” apoptosis resulting from mitochondrial outer membrane permeabilization (MOMP) is regulated by the “apoptosome” which includes Apaf-1, casp9, and Bcl-2 family members BAX and BAK (15). MOMP leads to the release of mitochondrial apoptotic-inducing factors cytochrome C and SMAC/DIABLO, thereby activating casp9 and downstream effector caspases. In contrast, “extrinsic” apoptosis is mediated by casp8 following DR ligation. Activated casp8 or casp9 induces Casp-3, -6 and -7 activity via cleavage of the short prodomain of these latter caspases. This cascade of inducer caspases acting on executioner caspases serves to amplify the apoptotic signal in a manner similar to the blood-clotting cascade. In certain cell types, active casp8 cleaves the pro-apoptotic Bcl-2 family member Bid. Truncated Bid (tBid) is then capable of interacting with mitochondrial membrane bound Bax and Bak, ultimately leading to the release of cytochrome C and the activation of casp9 via the “apoptosome.” (16–18). In this manner, there is a means for crosstalk between DR-mediated extrinsic cell death and cytochrome-C mediated intrinsic apoptotic cell death pathways.
TNFR1 promotes assembly of “complexes I and II”
Although analogous to death mediated by CD95/Fas/Apo-1, initiation of TNFR1 signaling by TNFα is followed by recruitment of a distinct set of adaptors. These include TNFR-associated death domain protein (TRADD) (19, 20), TRAF2 and ubiquitin ligases, TRAF2 and cIAP1 (21), to the membrane. RIPK1 is then recruited to form what is commonly referred to as “complex I” (22, 23). Following a sequence of ubiquitination events modulated by cIAP1/2 (24–26) and the linear ubiquitin assembly complex (LUBAC) (27–29), pro-survival complex I, containing ubiquitinated RIPK1 as a scaffolding protein, recruits NF-κB essential modulator (NEMO) and IκB kinase (IKK) complex resulting in NF-κB activation (30). NF-κB signaling drives cell survival and proliferation by upregulating synthesis of anti-apoptotic proteins, such as cFLIP and cIAP, and aids in the inflammatory response (25, 31). When NF-κB activation is blocked, and thus protein synthesis (cFLIP) is inhibited, assembly of complex II induces apoptosis.
The function of RIPK1 as a pro-survival or pro-death molecule is regulated by its ubiquitination status. Poly-ubiquitinated RIPK1 prevents the transition of the membrane-associated complex I to the cytosolic complex II (32, 33). Depending on the cellular conditions and posttranslational regulatory mechanisms, de-ubiquitylation of RIPK1 by enzyme cylindromatosis (CYLD) permits internalization of complex I (34) and generation of complex II, resulting in casp8 activation and triggering of apoptosis (35).
In response to TNFR1 engagement, adaptor protein TRADD recruits FADD to activate casp8, while following CD95/Fas/Apo-1 ligation, FADD binds directly to CD95 to activate casp8. Thus, the critical interaction for caspase activation is between FADD and Fas death domains, whereas the corresponding interaction responsible for TNFα-induced caspase activation is between FADD and TRADD death domains.
The lack of FADD and casp8 reveals an alternative form of PCD
Many groups have now established that disabling casp8 dependent apoptotic cell death often leads to a form of cell death that has many hallmarks of cellular necrosis (36). Termed “programmed necrosis” or “necroptosis”, this form of death is induced by the formation of a necrosis-inducing complex comprised of receptor-interacting protein kinase 1, RIPK1, and RIPK3 (32, 33, 37). Recent findings have implicated necroptosis as a major cell death pathway of equal importance to apoptotic signaling in regulating development and immunity.
Although initial research supported a relatively straightforward apoptotic signaling cascade downstream of death receptors (14), it is now evident that casp8 induced apoptosis is not the sole pathway in DR-induced cell death (38). Perhaps the first evidence of this was provided by Nagata’s group (39), when an artificially multimerizing mutation of FADD unexpectedly induced a caspase-independent form of cell death (40). Whereas the apoptotic effect of TNFα is mediated by caspases, induction of necrotic death by TNFα or other members of the TNF family is enhanced by caspase inhibitors, as shown by Vandenabeele and colleagues (40). It was observed that L929 cells incubated with the pan-caspase inhibitor zVAD-FMK underwent an alternate form of cell death in response to TNFα. Further evidence of cells failing to follow this simple DR-signaling model was provided when Jürg Tschopp and coworkers revealed that this alternative form of death activated by DR ligation is dependent on the kinase activity of RIPK1 (41). Taken together, it is now established that disabling the casp8 apoptotic machinery results in cell death resembling necrosis, termed “necroptosis” or “programmed necrosis”, which is enhanced by zVAD-FMK.
RIP kinases are part of a family of seven serine-threonine kinases that have been shown to be involved in innate and adaptive immunity (42). RIP kinases are characterized by their effector or protein interaction domains, which allow them to participate in various signaling pathways. RIPK1 contains a C-terminal death domain motif, an intermediate domain known as a RIP homotypic interaction motif (RHIM) (43), and an N-terminal kinase domain. Using a small molecule library, Yuan and colleagues (44–46) have implicated the kinase activity of RIPK1 in elaboration of programmed necrosis by identifying a family of molecules termed “necrostatins”, specifically Necrostatin-1, that binds to and inhibits the catalytic activity of RIPK1. Addition of Nec-1 inhibited necroptotic death induced by DR signaling in several cell types.
In addition to RIPK1, RIPK3 was also found recruited to complex II (47) in response to DR-ligation or TCR stimulation, leading to formation of necroptotic-inducing complexes, “necrosomes” or “complex IIb” (42). When casp8 activity is compromised, RIPK3 is recruited to complex II by RIPK1 to form complex IIb. Association of RIPK1-RIPK3 complex promotes necroptosis, and necrosome assembly is blocked with RIPK1 inhibitor Nec-1 (48). Our work and that from others has shown RIPK1 kinase activity is required for elaboration of necrotic signaling, and casp8 negatively regulates necroptosis (49). Direct cleavage of RIPK1, and possibly RIPK3, by casp8 is one mechanism through which cells differentiate between apoptotic and necrotic death following DR ligation (Fig. 1). In addition, cleavage of CYLD by casp8 has also been shown to impact this decision (50).
Figure 1. Modulation of necroptotic vs. apoptotic cell death and TCR mediated proliferation by caspase-8.
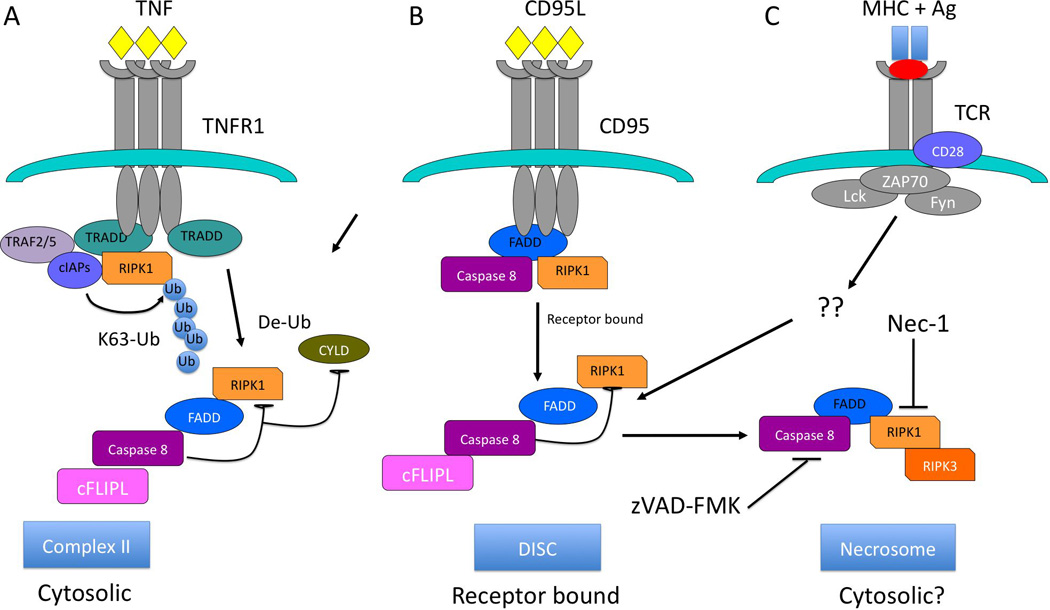
Schematic of A) TNF-induced cell death, B) CD95L/Fas induced cell death, and C) TCR-induced necroptosis. Following ligation of TNFR1 by TNFa, TRADD recruitment leads to assembly of a receptor bound complex containing RIP1, RIP3. In the presence of functional FADD and casp8, TNF or CD95L leads to assembly of complex I, or the DISC (A and B, respectively). For the TNFR1 complex 1, RIP1 is then deubiquinylated by CYLD, followed by assembly of complex II, which includes FADD, casp8 and cFLIPL. Casp8-mediated cleavage of CYLD and RIP1 prevents recruitment of RIP3 and activation of necroptosis. Similar signaling occurs following CD95L binding to CD95/Fas, except direct recruitment of FADD and casp8 to the receptor leads to activation of apoptosis via casp8 activity. For TCR stimulation (C), a RIP1/RIP3 containing necrosome occurs independent of death receptor ligation, possibly in the cytosol. In each case, casp8 activity is directed against RIP1 and CYLD, preventing the assembly of a necrosome containing RIP3.
RIPK3 is structurally similar to RIPK1; both contain a RHIM domain that facilitates their interaction, although RIPK3 lacks a DD motif, and thus plays no apparent role in the NF-κB survival pathway (51). RIPK3 is recruited to RIPK1-containing necrosomes via its RHIM where a series of phosphorylation events occurs (32). The kinase activity of RIPK1 is required to stabilize its interaction with RIPK3, and this interaction is blocked by Nec-1 (32, 33). Phosphorylation of RIPK1 and RIPK3 is mutually dependent as Nec-1 also suppresses RIPK3 phosphorylation.
Necroptosis in T cells
As a germline deficiency in FADD led to an early embryonic lethal phenotype, Winoto and colleagues made use of Rag1-deficient blastocyst chimerism to develop FADD deficient T cells (52) to investigate of the role of DR-mediated death in T cells. Other groups (53–55) used an alternative approach in which a mutant form of FADD (5) lacking a casp8 recruitment domain (FADDdd) was ectopically expressed in T cells. Both models unveiled the induction of RIPK1-dependent necroptosis in T cells following antigenic receptor signaling when casp8 activity is compromised. FADDdd and casp8−/− T cells unexpectedly displayed proliferative defects in vitro following TCR stimulation and in vivo in response to viral challenge, even though pro-survival NF-κB activation remained intact (56–58). Extracellular blockade of DR ligation failed to rescue casp8−/− or FADDdd-expressing T cells, suggesting that the nucleation of RIPK1/RIPK3 containing necrosomes occurs in a manner independent of DR signaling. Upon activation, FADDdd and Casp8−/− T cells exhibit excessive autophagy (59) before succumbing to necroptosis, supporting the role of RIPK1 in regulating both processes. Nec-1 treatment rescued the expansion defect and blocked the hyperautophagic phenotype in casp8−/− and FADDdd T cells (57, 59). Interestingly, naïve cells lacking functional DISC proteins did not display this hyperautophagic phenotype, demonstrating that increased autophagy requires TCR stimulation.
Similar results observed in mice conditionally lacking FADD, casp8 or c-FLIP in T cells established that these DISC proteins are also essential for T-cell clonal expansion (60–62). Research by Lenardo and colleagues (63) led to the observation that loss of casp8 function in humans leads to striking T cell defects. Taken together, these studies demonstrate that DISC proteins are required for T cell expansion following antigenic stimulation. Germline deletion of casp8, RIPK1, cFLIP, or FADD in mice results in embryonic or perinatal lethality, demonstrating a vital role for DISC proteins in multiple developmental processes. It is clear that casp8−/− or FADDdd T cells succumb to necroptosis when stimulated with antigens. The BCL-10-CARMA-1-MALT1 (BCM) complex, which activates NF-κB following TCR stimulation, has been proposed to also dictate DISC formation (64, 65). However, it remains to be determined if the BCM complex leads to the generation of necrosomes in cells lacking FADD, casp8 or cFLIP function.
It is evident now that the death of FADDdd or casp8 deficient T cells is attributed to the loss of RIPK1-3 necroptotic regulation by casp8. Recent findings show that concurrent ablation of casp8 (or FADD function) and RIPK3 completely rescues the embryonic lethality resulting from developmental defects associated with a casp8 functional deficiency (60, 66, 67). As expected, these animals are resistant to the lethal effects of CD95 ligation in vivo. Importantly, deficiency of RIPK1 or RIPK3 rescues casp8−/−, FADDdd and FADD−/− T cells from TCR stimulation-induced necroptosis and restores proliferation of T cells lacking casp8 activity (49, 60, 66–68). Whether the dramatically similar phenotype of cFLIP−/− T cells (62, 69) may be rescued by a loss of RIPK1 and/or RIPK3, an issue likely complicated by cFLIP’s important anti-apoptotic properties, is an important question. In contrast, work by Winoto and colleagues (70) suggests RIPK3 signaling is not involved in the necroptotic demise of T cells lacking FADD based on the observation that RIPK3 failed to co-immunoprecipitate with RIPK1. Given the aforementioned genetic evidence provides a solid backing to support the hypothesis that RIPK3 does indeed facilitate the demise of T cells bearing defects in FADD and casp8 signaling, we surmise that these negative results are due to the highly transitory nature of the necrosome in the context of active casp8.
Although the immune defects associated with casp8−/− or FADDdd mice are restored with a RIPK3 deficiency, older casp8−/− × RIPK3−/− double-knockout animals display a lympho-accumulative disorder resembling that of lpr/lpr (Fas−/−) mice (71), characterized by enlarged lymphoid organs and accumulation of an aberrant population of B220+CD3+CD4−CD8− lymphoid cells (49, 66–68). Thus, RIPK1/3 mediated necroptosis appears to serve as a backup death pathway for elimination of excess T cells when casp8 dependent apoptosis is blocked. On the other hand, RIPK3 and casp8 play opposing roles during viral infection as FADDdd expressing mice failed to adequately respond to viral infection due to defective T cell proliferation (72). Double mutant mice display normal lymphoid compartments and restored T cell function and are capable of efficiently clearing mouse hepatitis virus (MHV) [41] and lymphocytic choriomeningitis (LCMV) (68). Our work has demonstrated that cleavage of both RIPK1 and RIPK3 by casp8 occurs in antigenically stimulated primary T cells and likely orchestrates the switch between apoptotic and necrotic cell death [41].
Negative regulation of necroptosis by casp8
While casp8 inhibition promotes necroptosis in response to DR engagement and T cell activation, an important question is how it exerts its pro-survival function without inducing apoptosis. The key targets of casp8 processing that prevent necroptosis remain a major subject of intense investigation. Once recruited and activated by FADD, casp8 homodimers transduce pro-apoptotic signals by cleaving and activating downstream executioner caspases. However, casp8 remains in an uncleaved, though catalytically active “single-chain” state during the assembly of necrosomes following TCR stimulation (73). This state is likely initiated by heterodimerization with the long form of cFLIP (67, 74), and such heterodimers may be retained proximal to the necrosome by virtue of casp8/cFLIPL pro-domain interactions with FADD. In such a tethered form, casp8 may be limited in diffusing to interact with executioner caspases such as casp3, thus preventing the elaboration of an apoptotic response.
By mutating the casp8 cleavage site within CYLD, Ting and coworkers (50) demonstrate that CYLD stabilizes and deubiquitinates RIPK1, disrupting its pro-survival interaction with NEMO, and permitting its relocation from complex I to complex II to trigger apoptosis. Neither RIPK1 nor RIPK3 were required for CYLD cleavage, and CYLD inactivation by casp8 is proposed to occur upstream of RIPK activity. Thus, RIPK1 recruitment to complex IIb is directly regulated by casp8 processing of CYLD. Several groups report a mechanism for necrosome inhibition by cFLIP in response to TCR stimulation and TLR signaling, respectively. cFLIP exists in two isoforms, cFLIP long (cFLIPL) and cFLIP short (c-FLIPs), and both can heterodimerize with caspase8 to negatively regulate its function by preventing auto-cleavage (74). When cellular RIPK3 expression is high, cFLIPs-casp8 heterodimers inhibit casp8 activity and RIPK1 necroptosis proceeds (67, 75, 76). When cFLIPL heterodimerizes with casp8, cFLIPL-caspase8 cannot induce apoptosis, but retains sufficient proteolytic activity to cleave RIPK1 and RIPK3, as well as CYLD and block necrosome formation. Thus, cFLIPL not only limits casp8 activation but also suppresses RIPK1-3 signaling.
Meier and colleagues addressed the role of cIAPs during death induced by genotoxic stress, and found that depletion of these proteins in etoposide treated cells promotes spontaneous ripoptosome formation (76). The mode of regulation is through cIAP-mediated ubiquitination, which inactivates the ripoptosome by targeting RIPK1 for proteasomal degradation. Another possible mechanism to account for this holds that ubiquitination of RIPK1 by cIAPs promotes NF-κB activation and prevents RIPK1 association with the ripoptosome (25). This scenario could serve as a potential overall model where a wide array of responses converge at regulation of cIAP levels to modulate cellular sensitivity to DISC proteins.
The Connection between Cellular Metabolism and Necroptotic signaling
Studies performed by Cho et al. and Zhang et al. (33, 37) in cell lines have implicated reactive oxygen species (ROS) in the mechanism of RIPK3 induced cell death, possibly bridging RIPK1-3-dependent necroptosis and autophagy (59, 77). However, ROS scavengers have been unsuccessful in completely preventing cell death in many cell lines, including Jurkat T cells lacking FADD expression (46). Several metabolic enzymes including glycogen phosphorylase (PYGL), glutamate-ammonia ligase (GLUL), glutamate dehydrogenase 1 (GLUD1), fructose-1,6-bisphosphatase 2 (FBP2), fumarate hydratase (FH), glycosyltransferase 25 domain-containing 1 (GLT25D1), and isocitrate dehydrogenase 1 (IDH1), were identified as RIPK3 substrates by Han and coworkers (37) using a mass spectrometric approach, but knockdown of many of these enzymes failed to convincingly block necroptosis. Cyclophilin D (CYPD), a mitochondrial matrix enzyme, was another potential candidate given its role in regulating mitochondrial permeability transition (78), but similarly, its inhibition was incapable of blocking RIPK-dependent necroptosis both in vitro (32) and in vivo (68).
Hitomi et al. (79) carried out an siRNA screen for genes required for TNFα-induced necroptosis and identified deubiquitylase CYLD. This is especially interesting because CYLD is recruited to TNFR1 to remove K63-ubiquitin chains from RIPK1. Other targets identified in the screen also include genes involved in ROS generation, glutathione peroxidase (gpx4), glutathione S-transferases (gsta3 and gsto2), as well as PARP-2 (80), linking RIPK1 necroptosis to increased ROS production and poly ADP-ribosylation. Bertrand and colleagues (81) discovered that polyubiquitination of RIPK1 allows it to bind to the pro-survival kinase Transforming Growth Factor-β-activated kinase 1 (TAK1) to prevent activation of apoptosis. Treatment with cIAP antagonist BV6 or inhibition of TAK1 sensitized L929 cells to TNFα-induced necroptosis. These authors note that this death is associated with increased RIPK1 recruitment to complex IIb, and augmentation of RIPK1-dependent ROS. Together, these results suggest a possible mechanism where necroptosis is driven by ROS levels, and cIAP1 and TAK1 play pro-survival roles by inhibiting ROS generation. Given that ROS are not absolutely required, we instead favor the notion that ROS instead amplify the necroptotic response. This may be due to the ability of ROS to inhibit the active site cysteine residues in cellular phosphatases (82), potentially leading to embellished signal transduction by RIPK1, RIPK3 or other kinases that participate in necroptotic signaling.
The events involved in the execution of necroptosis downstream of RIPK3 are still relatively unknown. Recent findings by Wang and colleagues (83) utilizing a chemical library screening approach identified a molecule, necrosulfonamide (NSA), that can block necroptosis downstream of RIPK3 activity. NSA blocks necrosis by modifying mixed lineage kinase like protein (MLKL1), itself identified as a functional substrate of RIPK3. Interaction of MLKL1 and the necrosome is shown to be necessary for propagating the necroptotic signal in Jurkats, HT-29, and HeLa cells. Wang et al. (84) simultaneously identified mitochondrial protein phosphatase, phosphoglycerate mutase 5 (PGAM5), as a downstream effector that binds the necrosome on the mitochondrial membrane. This PGAM5-MLKL1-RIPK1-3 interaction promotes Dynamin-Related Protein-1 (Drp1) activation, a GTPase necessary for mitochondrial fission, and Drp1 inhibition blocks necrosis. These findings suggest that the RIPK1-RIPK3 complex amplifies its necroptotic signal by recruiting mitochondrial fragmentation processes, and provides a potential mechanism of RIPK1-3 death regulation in the immune system.
Roles of necroptotic signaling in innate immunity
In addition to modulating T and B cell responses, upstream regulators of DR-induced apoptosis and necroptosis have been observed to contribute to innate immune responses. Although RIPK3−/− mice display no overt defects in DR-induced apoptosis, NF-kB activation, T cell function (53) or viral clearance (49, 68, 85), Francis Chan and colleagues (33) observed that RIPK3−/− mice fail to develop efficient inflammatory responses to vaccinia virus infection. These findings suggest that programmed necrosis may serve beneficial roles in vivo by promoting desirable inflammatory responses during pathogenic challenges. One hypothesis for the role of RIPK1/3 mediated necroptosis here is that infected necrotic cells induce a stronger inflammatory response and better antigen presentation than infected apoptotic cells (86), and is thus immunostimulatory. Upton et al. (85) demonstrate that RIPK3, but not RIPK1, is required for necrosis induced during viral infections. RIPK3−/− mice succumb to vaccinia virus by expressing a casp8 inhibitor, and jointly, viral replication is enhanced within infected RIPK3 deficient cells. Similarly, when mice were infected with cytomegalovirus (85), which expresses inhibitors of casp8 and a virally encoded inhibitor of RIPK3 m45/vIRA, RIPK3−/− mice were more susceptible to the M45/vIRA deficient virus. In these settings, necroptosis could function as a back-up mechanism against viruses encoding apoptotic inhibitors. Although casp8 and RIP kinases are both involved in Toll-like receptor (TLR3 and TLR4) signaling, described below, these findings support the hypothesis that both death pathways may act in parallel to coordinate a proper immune response.
Recent evidence from two groups (75, 76) has identified another cytosolic complex, which can activate both apoptosis and necroptosis through interactions between TLR domain-containing adaptor protein inducing interferon-B (TRIF) (87) and the RHIM of RIPK1. This ~2-megadalton signaling platform assembles in response to pattern recognition receptor (PRR) activation and genotoxic stress, and is comprised of complex IIb proteins casp8, FADD, cFLIP, and RIPK1 (Fig. 2a). These observations reveal this complex serves as a novel signaling platform that regulates casp8-dependent apoptosis or RIPK1-mediated necroptosis independent of TNFα, TRAIL, CD95L, and mitochondrial pathways (MOMP) and requires RIPK1 kinase activity for complex stabilization. Other groups have reported that casp8 and RIPK1 are recruited to the RNA sensor retinoic acid-inducible gene I (RIG1) complex (88). Additionally, the cytosolic DNA sensor DNA-dependent activator of interferon regulatory factors (DAI) (89), can also directly engage RIPK1 and RIPK3 through its RHIM motif to assemble a necrosome-like complex (Fig. 2b).
Figure 2. Induction of assembly of necrosomes following viral infection.
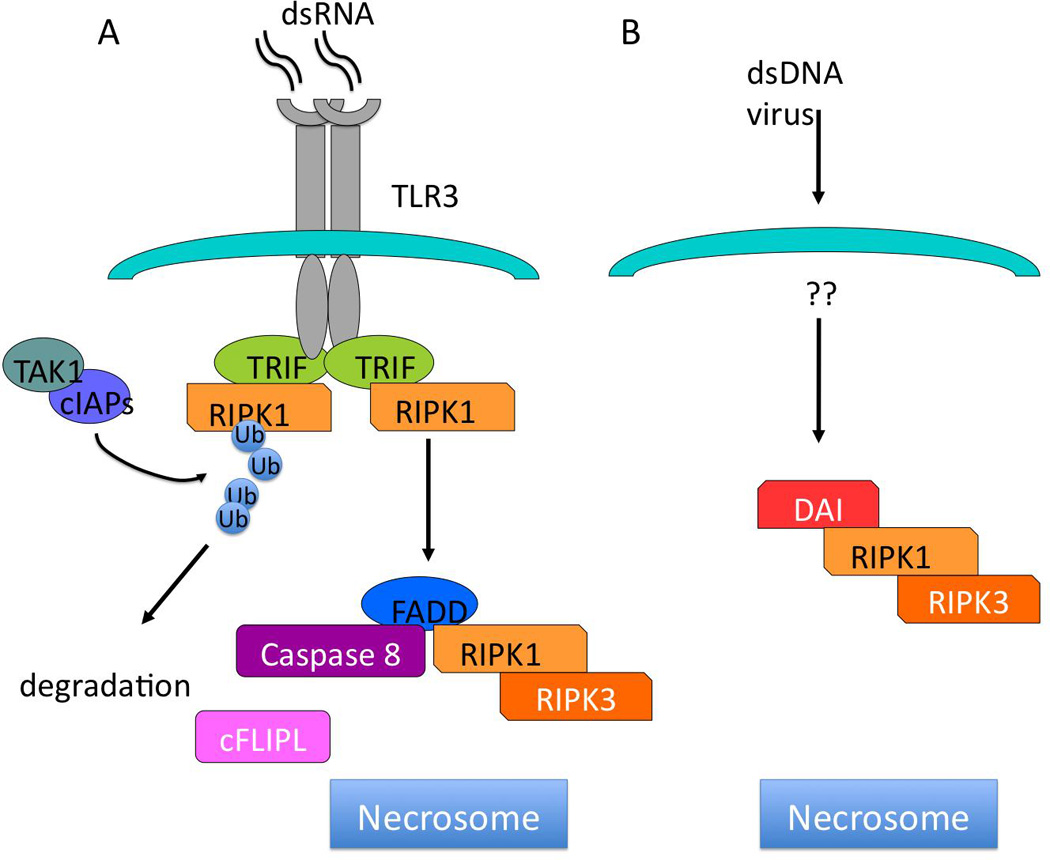
A) Binding of double-stranded RNA (dsRNA) to TLR3 leads to recruitment of TRIF, and RIPK1 via RHIM motif homotypic interactions. This leads to binding of FADD, casp8, RIP3 and possibly c-FLIPL. Cleavage of RIP1 (and RIP3?) by casp8 prevents recruitment of RIP3, and subsequent necroptosis. B) dsDNA viruses activate DAI, a RHIM domain containing factor that binds to RIP1 and RIP3, leading to the generation of necrosomes.
Overall, these reports describe additional stimuli where necroptosis is induced, thus expanding the number of intracellular and extracellular signals that are under caspase 8 regulation. Although various stimuli and different cell surface receptors (TNF, TCR, TLR3-4, genotoxic stress, RIG-I, DAI) activate death pathways and formation of necrotic complexes, the unifying mechanism of necroptosis is the recruitment of RIPK3 to these complexes. Importantly, ripoptosome assembly can occur independently of TNFR1 and TRADD/FADD, challenging the idea that DR ligation is necessary for RIPK1-RIPK3-containing complex formation. As embryonic developmental defects observed in FADD- and casp8-deficient mice are rescued by breeding onto RIPK1−/− and/or RIPK3−/− backgrounds, it is likely that additional stimuli that induce the formation of necrosome-like complexes will be discovered in future work.
Autophagy in T cells
Autophagy is a conserved process by which intracellular contents are degraded (90), and in the immune system, autophagy is important for clearing intracellular pathogens such as group A Streptococcus and Mycobacterium tuberculosis (91, 92). Interestingly, Li et al. (93) observed the formation of autophagosomes in activated CD4+ T cells indicating that autophagy does indeed occur in T cells, and can be intrinsically linked to T-cell activation and proliferation. Under starvation conditions, autophagy was also observed in T cells following TCR activation, implicating the process as a mode of regulating the metabolic pathways and energetic requirements associated with T cell activation and expansion.
Work by He and colleagues (94) first established an essential role for autophagy in activated T cells through the use of fetal liver reconstitution experiments using Atg5−/− T cells. Upon TCR stimulation, Atg5−/− T cells undergo inefficient clonal expansion accompanied by profound levels of cell death. Ch’en et al. (68) observed similar findings in T cells conditionally deficient for the autophagy gene Atg7. Following stimulation in vitro and in vivo with LCMV infection, Atg7−/− T cells exhibited defective proliferation similar to what is observed in activated caspa8−/− T cells. However, whereas the necroptosis inhibitor necrostatin-1 (Nec-1) rescues viability of casp8−/− cells, it had no effect in Atg7−/− T cells. Interestingly, the pan-caspase inhibitor qVD-FMK, was apparently capable of blocking Atg7−/− T cell death. Mice lacking either bearing T cell specific deletion of Atg5 or Atg7 displayed decreased thymic cellularity, though the percentages of double negative (DN), double positive (DP), and single positive (SP) subsets remained constant, suggesting that autophagy plays similar roles in these thymic subsets. Atg5-deficient and Atg7-deficient T cell numbers were also greatly reduced in the periphery compared to wildtype T cells, suggesting a developmental defect or an inability to maintain immune homeostasis. Together, these findings implicate autophagy as a critical process for T cell survival and maintenance.
To further investigate the relationship between Casp8-dependent death and autophagy, Ch’en et al. (68) crossed mice bearing a conditional Atg7 deletion with mice deficient in caspase 8. Deletion of Atg7 in combination with a caspase 8 deficiency exacerbated the loss of T cells, indicating the defects observed in T cells of mice lacking the capacity to induce autophagy are independent of RIPK1 activity. Given that autophagy is linked with RIPK1-mediated necroptotic death in activated T cells, it is surprising that autophagy plays a positive, homeostatic role for the catabolic process of rapidly proliferating, highly anabolic T cells.
Recent evidence suggests that autophagy may function as a critical process in mediating the events following TCR activation. Stimulation through the TCR results in an oxidative burst that may lead to generation of massive levels of toxic ROS. While ROS is beneficial for lowering T-cell activation thresholds through inhibition of phosphatase activity (82, 95), excess oxygen radicals can also be potentially toxic to T cells by inducing organelle damage through lipid peroxidation and other negative effects that promote T-cell death (96). Thus, it was proposed that selective autophagy of mitochondria, or mitophagy, may be used to compartmentalize mitochondria producing ROS and protect cells from damaged mitochondria that would further amplify ROS production.
This hypothesis was tested by Pua et al. (94, 97) using mice conditionally deficient in autophagy protein, Atg7. T-cell-specific deletion of the Atg7 gene resulted in defective proliferation, similar to studies involving Atg5−/− fetal liver transplants. Interestingly, high levels of ROS and increased mitochondrial content were observed in naive Atg7−/− T cells, presumably due to the failure to eliminate mitochondria as thymocytes develop into mature T cells. Concurrent studies by Swat and colleagues (98) found that Atg5 deficient T cells displayed a similar increase in mitochondrial mass and enhanced biosynthesis of mitochondria-specific genes. These observations are consistent with the idea that mitophagy plays a crucial role during T cell development and activation. As T cells switch from oxidative phosphorylation to an aerobic glycolytic metabolism following their activation through the TCR, they become less reliant on mitochondria for their source of energy (99). Therefore, the ability of T cells to induce autophagy is necessary for the removal of nonfunctional or unnecessary mitochondria that may release excess ROS or apoptosis-inducing mitochondrial proteins (e.g. cytochrome C) that may trigger apoptotic or necrotic cell death.
Calcium signaling is also a consequence of T cell activation, characterized by the opening of CRAC channels and subsequent entry of millimolar levels of calcium (100). After TCR stimulation, ER calcium channels release calcium stores into the cytosol, leading to opening of CRAC channels and calcium influx from the extracellular environment into intracellular spaces. Thus, the role of autophagy in ER homeostasis may indirectly impact calcium mobilization in T lymphocytes (77). Consistent with the increased ER content observed in Atg5−/− and Atg7−/− T cells, He and colleagues (101) found that activated Atg7−/− T cells display impaired calcium influx and increased ER calcium stores. These authors proposed that this increased ER content leads to excessive calcium uptake, and the inability to deplete these stores results in impaired calcium influx. These observations suggest the that the function of autophagy in proliferating T cells is not only to maintain organelle homeostasis, but also to regulate ER content and ensure calcium homeostasis.
Another explanation for the profound requirement for autophagy in activated T cells is based on the known function of autophagy in degradation of cytosolic components to ensure bioenergetic and metabolic output (102). After antigenic stimulation, naïve T cells rapidly transition to an activated state to respond to antigen and proliferate. T cell activation imposes significant bioenergetic demands required for transcriptional remodeling and activation of biosynthetic pathways (103). As described, resting T cells switch from dependence on oxidative phosphorylation to aerobic glycolysis during the activated state as a potential method to accommodate their biosynthetic needs (104, 105). Nutrient transporter proteins are dramatically upregulated in T cells during the first 48 hours of activation, and autophagy may be required during the early stages when access to extracellular nutrients is limited but metabolic reprogramming to support proliferation has already begun. Using the conditional Atg7 KO model, Macian and colleagues (106) noted that blockade of autophagy inhibits Atg7−/− T cell activation, but in contrast to Pua et al. [82, 86], they did not observe an increase in cell death. The contents of basal autophagosomes in resting cells is largely composed of mitochondria, whereas in activated T cells, autophagosomes contain mostly cytosolic components and less inclusion of mitochondria was observed. It is proposed that in naïve T cells, autophagy functions to renew the cell proteome and organelle composition, whereas in activated T cells, it plays an energy-supplying role. Activated Atg7−/− T cells were defective in ATP production, and this defect could be partially rescued with addition of methyl pyruvate, an end product of glycolysis. Thus, it was proposed that, in response to antigen, T cells upregulate autophagy for selective breakdown of intracellular macromolecules and recycling of amino acids, particularly glutamine which plays a critical role in biosynthetic reactions, to generate sufficient energy to sustain clonal expansion.
In support of the critical role of autophagy in T lymphocytes and to eliminate the possibility that the results observed were due to autophagy-independent functions of Atg5 and Atg7, He and colleagues (107) developed Atg3−/− mice. Similar to the in vivo and in vitro phenotypes observed in Atg5−/− and Atg7−/− mice, Atg3−/− mice displayed a significantly decreased number of peripheral T cells, and these were incapable of proliferating efficiently upon stimulation. Additionally, T cells lacking Atg3 contained increased mitochondrial and ER content. An increased fraction of memory-like T lymphocytes was observed, consistent with lymphopoenia-driven proliferation in mice lacking T cells.
The impaired survival of T cells lacking autophagy was characterized by the spontaneous death observed in freshly isolated splenocytes of Atg3−/− mice, corresponding with increased casp9 activity. It is hypothesized that apoptotic pathways may mediate the death of Atg3−/− T cells, as incubation with the pan-caspase inhibitor zVAD-FMK lead to partial rescue in cell viability. Jia et al. suggested increased ROS production is a result of expanded mitochondrial and ER content, thus autophagy is critical for regulating organelle homeostasis (107). When Atg3 was acutely deleted, no change in mitochondria and ER content was observed in vitro until 10 days post deletion, indicating the accumulation of organelles over time may be responsible for the death of naïve T cells incapable of autophagy.
Lastly, it is possible that autophagy is required for efficient transitions through cell cycle stages based on the observation of increased LC3-II levels (authors’ unpublished observations) when G1/S and G2/M stages of cell cycle are blocked using hydroxyurea and nocodazole, respectively. This result suggests that autophagic flux may be enhanced as a means to accomplish cytoplasmic remodeling as a T cell transitions through distinct cell cycle stages. Autophagy has also been shown to be associated with necroptotic death in T cells lacking casp8 (59). However, as described above, deletion of autophagy genes impaired T cell activation and proliferation. Although several potential functions of autophagy have been put forward, perhaps the most vexing issue is that the loss of this vital cellular process leads to lymphopoenia. As a result, immune mechanisms cause the few remaining T cells to proliferate in an effort to regain T cell homeostasis (108). Given this caveat, a clearer picture of the function(s) of autophagy in activated T cells awaits more acute deletion of autophagy genes in T cells following their antigenic stimulation.
Crosstalk between necroptosis and autophagy
While there are likely a number of unique cell death pathways, perhaps dependent both on the death stimulus as well as the cellular developmental stage and gene expression pattern, three major forms of cell death have been described based on the cellular morphologies they induce (109). These include a) type-I, apoptotic cell death, b) type-II, autophagic cell death, and type-III, necrotic cell death. While type-I cell death has long been appreciated to be a programmed process, type-III cell death has, until very recently, been thought to occur simply by accident. Perhaps the most controversial of these is type-II cell death, as autophagy is primarily thought to be involved in promoting survival under high levels of cellular stress (110). Nevertheless, many have observed a strong concordance between necroptosis and autophagy. Lenardo and colleagues observed a significant enhancement in basal autophagy in cells following casp8 knockdown, and this was associated with significantly diminished cell survival (111, 112). At the time, it was proposed as a model of autophagic, type-II cell death. Importantly, this death process was highly dependent on RIPK1, as its knockdown led to diminished autophagic hallmarks (including LC3-II levels). Similarly our group (59), and that of Stephen Hedrick (57) observed hyper-autophagic phenotypes in FADDdd and casp8−/− T cells following antigenic stimulation that was associated with defective T cell survival and clonal expansion. We also observed high levels of basal autophagy in FADD−/− murine embryonic fibroblasts that was rescued by expression of full-length FADD (59). In addition, Yuan and colleagues also described high levels of basal and induced autophagy in Jurkat T cells lacking FADD (46). Significantly, knockdown of RIPK1 or treatment with Nec-1 not only prevented necroptotic cell death in these various cell systems, but also diminished the hyper-autophagic hallmarks. These results demonstrate that autophagy itself may be induced by death, and suggest that autophagy itself does not promote cell death (113–115).
How might necroptosis promote autophagy? From a very basic standpoint, one might assume that the very cellular stresses that autophagosomes mitigate may be heightened during apoptotic and necrotic cell death. Indeed, cellular ROS have been associated with both necroptosis and autophagy (38, 116, 117). Thus, the high level of ROS produced during necroptosis may lead to the induction of autophagy, the latter serving to eliminate radical-damaged organelles and proteins. Similarly, it is possible that the alteration in cellular energetics, a significant feature of necroptotic and necrotic cell death (2), may play a role. For example, the release of AIF from mitochondria, and subsequent translocation to nuclei is thought to promote the activation of PARPs. As a result, highly activated PARP consumes cellular NAD+, ultimately leading to diminished pools of ATP, and an increase in AMP (36). Under such artificial cellular starvation, high levels of AMP would activate AMPK, itself phosphorylating Ulk1, the mammalian Atg1 homolog (118–120). Since AMPK phosphorylation of Ulk1 would lead to induction of autophagy, the depletion of cellular energy pools induced by autophagy alone might be sufficient to cause the induction of autophagy in necroptotic cells. An alternative is that RIPK1 itself plays a significant and necroptosis-independent role in modulating autophagic signaling, a result potentially consistent with the observation of diminished autophagy with RIPK1 knockdown or inhibition. However, the means by which autophagic and necroptotic signaling pathways interact mechanistically will require further exploration.
Conclusions
Cellular death pathways are induced in response to various stimuli, both intrinsic and extrinsic, and as we know now, can also be triggered independent of receptor ligation. Although a broad range of cellular processes can activate necrosis-inducing signals, many groups have established RIPK3 as a critical mediator in these pathways. Recent work has defined necrosome suppression through direct cleavage of RIPK1 and RIPK3 as a non-apoptotic role for caspase 8 and FADD. However, the signals that induce RIPK1 and RIPK3 assembly and the execution of necrosis downstream of RIPK3 still remain to be answered.
Identification of CYLD as a caspase 8 target provides insight into the key determinants of cell survival or death. Further discovery of molecules involved in necroptosis as well as the physiological contexts that lead to necroptosis will expand our overall view of this alternate death pathway.
Necroptosis has been regarded possibly as a back-up pathway in the event of caspase 8 inactivation. However, the development of an LPR phenotype in casp8−/− or FADDdd × RIPK3−/− double mutants indicates both pathways (apoptosis and necrosis) play active, opposing roles, and stringent regulation is essential for maintaining immune homeostasis. The recently uncovered mechanism of caspase 8 regulation by cIAP and cFLIP provides a better understanding of how proliferating T cells can possess caspase 8 activity, yet are not sensitized to apoptosis. A major question that has yet to be resolved is the significance of necroptosis in immune homeostasis and tolerance. Situations where necroptosis is induced directly and not as a secondary response have yet to be defined.
Increasing evidence that necroptosis is ROS-mediated requires more work in linking cellular bioenergetics as a stress signal that promotes necroptotic sensitivity, possibly through differential regulation of cIAP levels. The identification of mitochondrial fission proteins downstream of RIPK3 targets requires further investigation to elucidate the potential involvement of metabolism/bioenergetics that links autophagy with necroptosis in lymphocytes. Autophagy is clearly important in activated T cells; whether for cytoplamic remodeling, mitophagy, or to supply energy, this process has been shown to be critical for T cell survival.
As we probe deeper into characterizing the exact paradigms of necroptosis and uncover the switch between apoptosis and necrosis, we will be able to manipulate death pathways as a therapeutic approach in modulating desired immune responses to various diseases and infections.
Abbreviations
- FADD
Fas-associated death domain
- TRADD
TNF receptor associated death domain
- Nec-1
necrostatin-1
- TCR
T cell antigen receptor
- TNF
tumor necrosis factor
- PCD
programmed cell death
- casp8
caspase-8
- RIP
receptor interacting protein
- cFLIP
FLICE-like inhibitory protein
- DR
death receptor
- DD
death domain
- Ub
ubiquitin
- cIAP
cellular inhibitor of apoptosis protein
- NEMO
NF-κB essential modulator
- IKK
I kappa B kinase
- NF-kB
nuclear factor kappa B
- CYLD
cylindromatosis
- RHIM
RIP kinase homotypic interaction motif
References
- 1.Strasser A, O'Connor L, Dixit VM. Apoptosis signaling. Annu Rev Biochem. 2000;69:217–245. doi: 10.1146/annurev.biochem.69.1.217. [DOI] [PubMed] [Google Scholar]
- 2.Yuan J, Kroemer G. Alternative cell death mechanisms in development and beyond. Genes Dev. 2010;24:2592–2602. doi: 10.1101/gad.1984410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gatzka M, Walsh CM. Apoptotic signal transduction and T cell tolerance. Autoimmunity. 2007;40:442–452. doi: 10.1080/08916930701464962. [DOI] [PubMed] [Google Scholar]
- 4.Siegel RM. Caspases at the crossroads of immune-cell life and death. Nat Rev Immunol. 2006;6:308–317. doi: 10.1038/nri1809. [DOI] [PubMed] [Google Scholar]
- 5.Chinnaiyan AM, O'Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 6.Boldin M, Varfolomeev E, Pancer Z, Mett I, Camonis J, Wallach D. A novel protein that interacts with the death domain of Fas/APO1 contains a sequence motif related to the death domain. J Biol Chem. 1995;270(14):7795–7798. doi: 10.1074/jbc.270.14.7795. [DOI] [PubMed] [Google Scholar]
- 7.Hsu H, Xiong J, Goeddel DV. The TNF receptor 1-associated protein TRADD signals cell death and NF-kappa B activation. Cell. 1995;81:495–504. doi: 10.1016/0092-8674(95)90070-5. [DOI] [PubMed] [Google Scholar]
- 8.Stanger BZ, Leder P, Lee TH, Kim E, Seed B. RIP: a novel protein containing a death domain that interacts with Fas/APO-1 (CD95) in yeast and causes cell death. Cell. 1995;81:513–523. doi: 10.1016/0092-8674(95)90072-1. [DOI] [PubMed] [Google Scholar]
- 9.Kischkel F, et al. Cytotoxicity-dependent APO-1 Fas/CD95 -associated proteins form a death-inducing signaling complex DISC with the receptor. EMBO J. 1995;14:5579–5588. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Irmler M, et al. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- 11.Donepudi M, Mac Sweeney A, Briand C, Grutter MG. Insights into the regulatory mechanism for caspase-8 activation. Mol Cell. 2003;11:543–549. doi: 10.1016/s1097-2765(03)00059-5. [DOI] [PubMed] [Google Scholar]
- 12.Oberst A, et al. Inducible dimerization and inducible cleavage reveal a requirement for both processes in caspase-8 activation. J Biol Chem. 2010;285:16632–16642. doi: 10.1074/jbc.M109.095083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pop C, Fitzgerald P, Green DR, Salvesen GS. Role of proteolysis in caspase-8 activation and stabilization. Biochemistry. 2007;46:4398–4407. doi: 10.1021/bi602623b. [DOI] [PubMed] [Google Scholar]
- 14.Ashkenazi A, Dixit V. Death receptors: signaling and modulation. Science. 1998;281(5381):1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 15.Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol Cell. 2010;37:299–310. doi: 10.1016/j.molcel.2010.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/s0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 17.Luo X, Budihardjo I, Zou H, Slaughter C, Wang X. Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell. 1998;94:481–490. doi: 10.1016/s0092-8674(00)81589-5. [DOI] [PubMed] [Google Scholar]
- 18.Grinberg M, et al. tBID homooligomerizes in the mitochondrial membrane to induce apoptosis. J Biol Chem. 2002 doi: 10.1074/jbc.M104893200. [DOI] [PubMed] [Google Scholar]
- 19.Ermolaeva MA, et al. Function of TRADD in tumor necrosis factor receptor 1 signaling and in TRIF-dependent inflammatory responses. Nat Immunol. 2008;9:1037–1046. doi: 10.1038/ni.1638. [DOI] [PubMed] [Google Scholar]
- 20.Harper N, Hughes M, MacFarlane M, Cohen GM. Fas-associated death domain protein and caspase-8 are not recruited to the tumor necrosis factor receptor 1 signaling complex during tumor necrosis factor-induced apoptosis. J Biol Chem. 2003;278:25534–25541. doi: 10.1074/jbc.M303399200. [DOI] [PubMed] [Google Scholar]
- 21.Wajant H, Pfizenmaier K, Scheurich P. Tumor necrosis factor signaling. Cell Death Differ. 2003;10:45–65. doi: 10.1038/sj.cdd.4401189. [DOI] [PubMed] [Google Scholar]
- 22.Ting A, Pimentel-Muinos F, Seed B. RIP mediates tumor necrosis factor receptor 1 activation of NF-kappaB but not Fas/APO-1-initiated apoptosis. EMBO J. 1996;15(22):6189–6196. [PMC free article] [PubMed] [Google Scholar]
- 23.Hsu H, Huang J, Shu H, Baichwal V, Goeddel D. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996;4:387–396. doi: 10.1016/s1074-7613(00)80252-6. [DOI] [PubMed] [Google Scholar]
- 24.Vince JE, et al. TRAF2 must bind to cellular inhibitors of apoptosis for tumor necrosis factor (tnf) to efficiently activate nf-{kappa}b and to prevent tnf-induced apoptosis. J Biol Chem. 2009;284:35906–35915. doi: 10.1074/jbc.M109.072256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bertrand MJ, et al. cIAP1 and cIAP2 facilitate cancer cell survival by functioning as E3 ligases that promote RIP1 ubiquitination. Mol Cell. 2008;30:689–700. doi: 10.1016/j.molcel.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 26.Varfolomeev E, Vucic D. (Un)expected roles of c-IAPs in apoptotic and NFkappaB signaling pathways. Cell Cycle. 2008;7:1511–1521. doi: 10.4161/cc.7.11.5959. [DOI] [PubMed] [Google Scholar]
- 27.Haas TL, et al. Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell. 2009;36:831–844. doi: 10.1016/j.molcel.2009.10.013. [DOI] [PubMed] [Google Scholar]
- 28.Rahighi S, et al. Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell. 2009;136:1098–1109. doi: 10.1016/j.cell.2009.03.007. [DOI] [PubMed] [Google Scholar]
- 29.Gerlach B, et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature. 2011;471:591–596. doi: 10.1038/nature09816. [DOI] [PubMed] [Google Scholar]
- 30.Ea CK, Deng L, Xia ZP, Pineda G, Chen ZJ. Activation of IKK by TNFalpha requires site-specific ubiquitination of RIP1 and polyubiquitin binding by NEMO. Mol Cell. 2006;22:245–257. doi: 10.1016/j.molcel.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 31.Hacker G, Bauer A, Villunger A. Apoptosis in activated T cells: what are the triggers, and what the signal transducers? Cell Cycle. 2006;5:2421–2424. doi: 10.4161/cc.5.21.3397. [DOI] [PubMed] [Google Scholar]
- 32.He SW, Lai, Miao Lin, Wang Tao, Du Fenghe, Zhao Liping, Wang Xiaodong. Receptor Interacting Protein Kinase-3 Determines Cellular Necrotic Response to TNF-alpha. Cell. 2009;137:1100–1111. doi: 10.1016/j.cell.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 33.Cho YS, et al. Phosphorylation-driven assembly of the RIP1-RIP3 complex regulates programmed necrosis and virus-induced inflammation. Cell. 2009;137:1112–1123. doi: 10.1016/j.cell.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schneider-Brachert W, et al. Compartmentalization of TNF receptor 1 signaling: internalized TNF receptosomes as death signaling vesicles. Immunity. 2004;21:415–428. doi: 10.1016/j.immuni.2004.08.017. [DOI] [PubMed] [Google Scholar]
- 35.Wang L, Du F, Wang X. TNF-alpha induces two distinct caspase-8 activation pathways. Cell. 2008;133:693–703. doi: 10.1016/j.cell.2008.03.036. [DOI] [PubMed] [Google Scholar]
- 36.Penninger JM, Kroemer G. Mitochondria, AIF and caspases--rivaling for cell death execution. Nat Cell Biol. 2003;5:97–99. doi: 10.1038/ncb0203-97. [DOI] [PubMed] [Google Scholar]
- 37.Zhang DW, et al. RIP3, an Energy Metabolism Regulator that Switches TNF-Induced Cell Death from Apoptosis to Necrosis. Science. 2009 doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 38.Walsh CM, Luhrs KA, Arechiga AF. The "fuzzy logic" of the death-inducing signaling complex in lymphocytes. J Clin Immunol. 2003;23:333–353. doi: 10.1023/a:1025313415487. [DOI] [PubMed] [Google Scholar]
- 39.Kawahara A, Ohsawa Y, Matsumura H, Uchiyama Y, Nagata S. Caspase-independent cell killing by Fas-associated protein with death domain. J Cell Biol. 1998;143:1353–1360. doi: 10.1083/jcb.143.5.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vercammen D, et al. Inhibition of caspases increases the sensitivity of L929 cells to necrosis mediated by tumor necrosis factor. J Exp Med. 1998;187:1477–1485. doi: 10.1084/jem.187.9.1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Holler N, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 42.Declercq W, Vanden Berghe T, Vandenabeele P. RIP kinases at the crossroads of cell death and survival. Cell. 2009;138:229–232. doi: 10.1016/j.cell.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 43.Sun X, Yin J, Starovasnik MA, Fairbrother WJ, Dixit VM. Identification of a novel homotypic interaction motif required for the phosphorylation of receptor-interacting protein (RIP) by RIP3. J Biol Chem. 2002;277:9505–9511. doi: 10.1074/jbc.M109488200. [DOI] [PubMed] [Google Scholar]
- 44.Degterev A, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Degterev A, Yuan J. Expansion and evolution of cell death programmes. Nat Rev Mol Cell Biol. 2008;9:378–390. doi: 10.1038/nrm2393. [DOI] [PubMed] [Google Scholar]
- 46.Degterev A, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 47.Feng S, et al. Cleavage of RIP3 inactivates its caspase-independent apoptosis pathway by removal of kinase domain. Cell Signal. 2007;19:2056–2067. doi: 10.1016/j.cellsig.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 48.Moquin D, Chan FK. The molecular regulation of programmed necrotic cell injury. Trends Biochem Sci. 2010;35:434–441. doi: 10.1016/j.tibs.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu JV, et al. Complementary roles of Fas-associated death domain (FADD) and receptor interacting protein kinase-3 (RIPK3) in T-cell homeostasis and antiviral immunity. Proc Natl Acad Sci U S A. 2011;108:15312–15317. doi: 10.1073/pnas.1102779108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O'Donnell MA, et al. Caspase 8 inhibits programmed necrosis by processing CYLD. Nat Cell Biol. 2011;13:1437–1442. doi: 10.1038/ncb2362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Newton K, Sun X, Dixit VM. Kinase RIP3 is dispensable for normal NF-kappa Bs, signaling by the B-cell and T-cell receptors, tumor necrosis factor receptor 1, and Toll-like receptors 2 and 4. Mol Cell Biol. 2004;24:1464–1469. doi: 10.1128/MCB.24.4.1464-1469.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang J, Cado D, Chen A, Kabra N, Winoto A. Fas-mediated apoptosis and activation-induced T-cell proliferation are defective in mice lacking FADD/Mort1. Nature. 1998;392(6673):296–300. doi: 10.1038/32681. [DOI] [PubMed] [Google Scholar]
- 53.Newton K, Harris A, Bath M, Smith K, Strasser A. A dominant interfering mutant of FADD/MORT1 enhances deletion of autoreactive thymocytes and inhibits proliferation of mature T lymphocytes. EMBO J. 1998;17(3):706–718. doi: 10.1093/emboj/17.3.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Walsh C, Wen B, Chinnaiyan A, O'Rourke K, Dixit V, Hedrick S. A role for FADD in T cell activation and development. Immunity. 1998;8(4):439–449. doi: 10.1016/s1074-7613(00)80549-x. [DOI] [PubMed] [Google Scholar]
- 55.Zornig M, Hueber AO, Evan G. p53-dependent impairment of T-cell proliferation in FADD dominant-negative transgenic mice. Curr Biol. 1998;8:467–470. doi: 10.1016/s0960-9822(98)70182-4. [DOI] [PubMed] [Google Scholar]
- 56.Arechiga AF, et al. Cutting Edge: FADD Is Not Required for Antigen Receptor-Mediated NF-{kappa}B Activation. J Immunol. 2005;175:7800–7804. doi: 10.4049/jimmunol.175.12.7800. [DOI] [PubMed] [Google Scholar]
- 57.Ch'en IL, et al. Antigen-mediated T cell expansion regulated by parallel pathways of death. Proc Natl Acad Sci U S A. 2008;105:17463–17468. doi: 10.1073/pnas.0808043105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ch'en IL, Hedrick SM, Hoffmann A. NF-kappaB as a determinant of distinct cell death pathways. Methods Enzymol. 2008;446:175–187. doi: 10.1016/S0076-6879(08)01610-8. [DOI] [PubMed] [Google Scholar]
- 59.Bell BD, et al. FADD and caspase-8 control the outcome of autophagic signaling in proliferating T cells. Proc Natl Acad Sci U S A. 2008;105:16677–16682. doi: 10.1073/pnas.0808597105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang H, Zhou X, McQuade T, Li J, Chan FK, Zhang J. Functional complementation between FADD and RIP1 in embryos and lymphocytes. Nature. 2011;471:373–376. doi: 10.1038/nature09878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Salmena L, et al. Essential role for caspase 8 in T-cell homeostasis and T-cell-mediated immunity. Genes Dev. 2003;17:883–895. doi: 10.1101/gad.1063703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chau H, et al. Cellular FLICE-inhibitory protein is required for T cell survival and cycling. J Exp Med. 2005;202:405–413. doi: 10.1084/jem.20050118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Su H, et al. Requirement for caspase-8 in NF-kappaB activation by antigen receptor. Science. 2005;307:1465–1468. doi: 10.1126/science.1104765. [DOI] [PubMed] [Google Scholar]
- 64.Kawadler H, Gantz MA, Riley JL, Yang X. The paracaspase MALT1 controls caspase-8 activation during lymphocyte proliferation. Mol Cell. 2008;31:415–421. doi: 10.1016/j.molcel.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Misra RS, et al. Caspase-8 and c-FLIPL associate in lipid rafts with NF-kappaB adaptors during T cell activation. J Biol Chem. 2007;282:19365–19374. doi: 10.1074/jbc.M610610200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kaiser WJ, et al. RIP3 mediates the embryonic lethality of caspase-8-deficient mice. Nature. 2011;471:368–372. doi: 10.1038/nature09857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Oberst A, et al. Catalytic activity of the caspase-8-FLIP(L) complex inhibits RIPK3-dependent necrosis. Nature. 2011;471:363–367. doi: 10.1038/nature09852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ch'en IL, Tsau JS, Molkentin JD, Komatsu M, Hedrick SM. Mechanisms of necroptosis in T cells. J Exp Med. 2011;208:633–641. doi: 10.1084/jem.20110251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang N, He YW. An essential role for c-FLIP in the efficient development of mature T lymphocytes. J Exp Med. 2005;202:395–404. doi: 10.1084/jem.20050117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Osborn SL, et al. Fas-associated death domain (FADD) is a negative regulator of T-cell receptor-mediated necroptosis. Proc Natl Acad Sci U S A. 2010;107:13034–13039. doi: 10.1073/pnas.1005997107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Takahashi T, et al. Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell. 1994;76(6):969–976. doi: 10.1016/0092-8674(94)90375-1. [DOI] [PubMed] [Google Scholar]
- 72.Beisner DR, Chu IH, Arechiga AF, Hedrick SM, Walsh CM. The requirements for fas-associated death domain signaling in mature T cell activation and survival. J Immunol. 2003;171:247–256. doi: 10.4049/jimmunol.171.1.247. [DOI] [PubMed] [Google Scholar]
- 73.Leverrier S, Salvesen GS, Walsh CM. Enzymatically active single chain caspase-8 maintains T-cell survival during clonal expansion. Cell Death Differ. 2011;18:90–98. doi: 10.1038/cdd.2010.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pop C, et al. FLIP(L) induces caspase 8 activity in the absence of interdomain caspase 8 cleavage and alters substrate specificity. Biochem J. 2011;433:447–457. doi: 10.1042/BJ20101738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Feoktistova M, et al. cIAPs block Ripoptosome formation, a RIP1/caspase-8 containing intracellular cell death complex differentially regulated by cFLIP isoforms. Mol Cell. 2011;43:449–463. doi: 10.1016/j.molcel.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tenev T, et al. The Ripoptosome, a signaling platform that assembles in response to genotoxic stress and loss of IAPs. Mol Cell. 2011;43:432–448. doi: 10.1016/j.molcel.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 77.Bell BD, Walsh CM. Coordinate regulation of autophagy and apoptosis in T cells by death effectors: FADD or foundation. Autophagy. 2009;5:238–240. doi: 10.4161/auto.5.2.7512. [DOI] [PubMed] [Google Scholar]
- 78.Baines CP, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature. 2005;434:658–662. doi: 10.1038/nature03434. [DOI] [PubMed] [Google Scholar]
- 79.Hitomi J, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135:1311–1323. doi: 10.1016/j.cell.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cande C, Cecconi F, Dessen P, Kroemer G. Apoptosis-inducing factor (AIF): key to the conserved caspase-independent pathways of cell death? J Cell Sci. 2002;115:4727–4734. doi: 10.1242/jcs.00210. [DOI] [PubMed] [Google Scholar]
- 81.Vanlangenakker N, et al. cIAP1 and TAK1 protect cells from TNF-induced necrosis by preventing RIP1/RIP3-dependent reactive oxygen species production. Cell Death Differ. 2011;18:656–665. doi: 10.1038/cdd.2010.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 83.Sun L, et al. Mixed Lineage Kinase Domain-like Protein Mediates Necrosis Signaling Downstream of RIP3 Kinase. Cell. 2012;148:213–227. doi: 10.1016/j.cell.2011.11.031. [DOI] [PubMed] [Google Scholar]
- 84.Wang Z, Jiang H, Chen S, Du F, Wang X. The Mitochondrial Phosphatase PGAM5 Functions at the Convergence Point of Multiple Necrotic Death Pathways. Cell. 2012;148:228–243. doi: 10.1016/j.cell.2011.11.030. [DOI] [PubMed] [Google Scholar]
- 85.Upton JW, Kaiser WJ, Mocarski ES. Virus inhibition of RIP3-dependent necrosis. Cell Host Microbe. 2010;7:302–313. doi: 10.1016/j.chom.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Weinlich R, Dillon CP, Green DR. Ripped to death. Trends Cell Biol. 2011;21:630–637. doi: 10.1016/j.tcb.2011.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Weber A, Kirejczyk Z, Besch R, Potthoff S, Leverkus M, Hacker G. Proapoptotic signalling through Toll-like receptor-3 involves TRIF-dependent activation of caspase-8 and is under the control of inhibitor of apoptosis proteins in melanoma cells. Cell Death Differ. 2010;17:942–951. doi: 10.1038/cdd.2009.190. [DOI] [PubMed] [Google Scholar]
- 88.Rajput A, et al. RIG-I RNA helicase activation of IRF3 transcription factor is negatively regulated by caspase-8-mediated cleavage of the RIP1 protein. Immunity. 2011;34:340–351. doi: 10.1016/j.immuni.2010.12.018. [DOI] [PubMed] [Google Scholar]
- 89.Upton JW, Kaiser WJ, Mocarski ES. DAI/ZBP1/DLM-1 Complexes with RIP3 to Mediate Virus-Induced Programmed Necrosis that Is Targeted by Murine Cytomegalovirus vIRA. Cell Host Microbe. 2012;11:290–297. doi: 10.1016/j.chom.2012.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. 2000;290:1717–1721. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nakagawa I, et al. Autophagy defends cells against invading group A Streptococcus. Science. 2004;306:1037–1040. doi: 10.1126/science.1103966. [DOI] [PubMed] [Google Scholar]
- 92.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–766. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 93.Li C, et al. Autophagy is induced in CD4+ T cells and important for the growth factor-withdrawal cell death. J Immunol. 2006;177:5163–5168. doi: 10.4049/jimmunol.177.8.5163. [DOI] [PubMed] [Google Scholar]
- 94.Pua HH, Dzhagalov I, Chuck M, Mizushima N, He YW. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med. 2007;204:25–31. doi: 10.1084/jem.20061303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Reyes BM, Danese S, Sans M, Fiocchi C, Levine AD. Redox equilibrium in mucosal T cells tunes the intestinal TCR signaling threshold. J Immunol. 2005;175:2158–2166. doi: 10.4049/jimmunol.175.4.2158. [DOI] [PubMed] [Google Scholar]
- 96.Hildeman DA, et al. Reactive oxygen species regulate activation-induced T cell apoptosis. Immunity. 1999;10:735–744. doi: 10.1016/s1074-7613(00)80072-2. [DOI] [PubMed] [Google Scholar]
- 97.Pua HH, Guo J, Komatsu M, He YW. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol. 2009;182:4046–4055. doi: 10.4049/jimmunol.0801143. [DOI] [PubMed] [Google Scholar]
- 98.Stephenson LM, et al. Identification of Atg5-dependent transcriptional changes and increases in mitochondrial mass in Atg5-deficient T lymphocytes. Autophagy. 2009;5:625–635. doi: 10.4161/auto.5.5.8133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Michalek RD, Rathmell JC. The metabolic life and times of a T-cell. Immunol Rev. 2010;236:190–202. doi: 10.1111/j.1600-065X.2010.00911.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cahalan MD, Zhang SL, Yeromin AV, Ohlsen K, Roos J, Stauderman KA. Molecular basis of the CRAC channel. Cell Calcium. 2007;42:133–144. doi: 10.1016/j.ceca.2007.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jia W, Pua HH, Li QJ, He YW. Autophagy regulates endoplasmic reticulum homeostasis and calcium mobilization in T lymphocytes. J Immunol. 2011;186:1564–1574. doi: 10.4049/jimmunol.1001822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cuervo AM, Macian F. Autophagy, nutrition and immunology. Mol Aspects Med. 2012;33:2–13. doi: 10.1016/j.mam.2011.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Jones RG, Thompson CB. Revving the engine: signal transduction fuels T cell activation. Immunity. 2007;27:173–178. doi: 10.1016/j.immuni.2007.07.008. [DOI] [PubMed] [Google Scholar]
- 104.Frauwirth KA, et al. The CD28 signaling pathway regulates glucose metabolism. Immunity. 2002;16:769–777. doi: 10.1016/s1074-7613(02)00323-0. [DOI] [PubMed] [Google Scholar]
- 105.Jones RG, et al. CD28-dependent activation of protein kinase B/Akt blocks Fas-mediated apoptosis by preventing death-inducing signaling complex assembly. J Exp Med. 2002;196:335–348. doi: 10.1084/jem.20020307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hubbard VM, Valdor R, Patel B, Singh R, Cuervo AM, Macian F. Macroautophagy regulates energy metabolism during effector T cell activation. J Immunol. 2010;185:7349–7357. doi: 10.4049/jimmunol.1000576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jia W, He YW. Temporal regulation of intracellular organelle homeostasis in T lymphocytes by autophagy. J Immunol. 2011;186:5313–5322. doi: 10.4049/jimmunol.1002404. [DOI] [PubMed] [Google Scholar]
- 108.Surh CD, Sprent J. Regulation of mature T cell homeostasis. Semin Immunol. 2005;17:183–191. doi: 10.1016/j.smim.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 109.Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16:663–669. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 110.Klionsky DJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4:151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yu L, Lenardo MJ, Baehrecke EH. Autophagy and caspases: a new cell death program. Cell Cycle. 2004;3:1124–1126. [PubMed] [Google Scholar]
- 112.Yu L, et al. Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science. 2004;304:1500–1502. doi: 10.1126/science.1096645. [DOI] [PubMed] [Google Scholar]
- 113.Klionsky DJ, et al. A comprehensive glossary of autophagy-related molecules and processes (2nd edition) Autophagy. 2011;7:1273–1294. doi: 10.4161/auto.7.11.17661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Thorburn A. I think autophagy controls the death of my cells: what do I do to get my paper published? Autophagy. 2011;7:455–456. doi: 10.4161/auto.7.5.14797. [DOI] [PubMed] [Google Scholar]
- 115.Gump JM, Thorburn A. Autophagy and apoptosis: what is the connection? Trends Cell Biol. 2011;21:387–392. doi: 10.1016/j.tcb.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. Embo J. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Berghe TV, et al. Necroptosis, necrosis and secondary necrosis converge on similar cellular disintegration features. Cell Death Differ. 2010;17:922–930. doi: 10.1038/cdd.2009.184. [DOI] [PubMed] [Google Scholar]
- 118.Jung CH, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20:1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hosokawa N, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20:1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]