

ABSTRACT
Toxoplasma gondii has become a model for studying the phylum Apicomplexa, in part due to the availability of excellent genetic tools. Although reverse genetic tools are available in a few widely utilized laboratory strains, they rely on special genetic backgrounds that are not easily implemented in natural isolates. Recent progress in modifying CRISPR (clustered regularly interspaced short palindromic repeats), a system of DNA recognition used as a defense mechanism in bacteria and archaea, has led to extremely efficient gene disruption in a variety of organisms. Here we utilized a CRISPR/CAS9-based system with single guide RNAs to disrupt genes in T. gondii. CRISPR/CAS9 provided an extremely efficient system for targeted gene disruption and for site-specific insertion of selectable markers through homologous recombination. CRISPR/CAS9 also facilitated site-specific insertion in the absence of homology, thus increasing the utility of this approach over existing technology. We then tested whether CRISPR/CAS9 would enable efficient transformation of a natural isolate. Using CRISPR/CAS9, we were able to rapidly generate both rop18 knockouts and complemented lines in the type I GT1 strain, which has been used for forward genetic crosses but which remains refractory to reverse genetic approaches. Assessment of their phenotypes in vivo revealed that ROP18 contributed a greater proportion to acute pathogenesis in GT1 than in the laboratory type I RH strain. Thus, CRISPR/CAS9 extends reverse genetic techniques to diverse isolates of T. gondii, allowing exploration of a much wider spectrum of biological diversity.
IMPORTANCE
Genetic approaches have proven very powerful for studying the biology of organisms, including microbes. However, ease of genetic manipulation varies widely among isolates, with common lab isolates often being the most amenable to such approaches. Unfortunately, such common lab isolates have also been passaged frequently in vitro and have thus lost many of the attributes of wild isolates, often affecting important traits, like virulence. On the other hand, wild isolates are often not amenable to standard genetic approaches, thus limiting inquiry about the genetic basis of biological diversity. Here we imported a new genetic system based on CRISPR/CAS9, which allows high efficiency of targeted gene disruption in natural isolates of T. gondii. This advance promises to bring the power of genetics to bear on the broad diversity of T. gondii strains that have been described recently.
INTRODUCTION
The protozoan parasite Toxoplasma gondii is an obligate intracellular pathogen belonging to the phylum Apicomplexa, which contains many other important pathogens (such as Plasmodium spp. and Cryptosporidium spp.) of humans and livestock (1). Due to the ease of growth in tissue culture and availability of tools for forward and reverse genetics, T. gondii has become the model organism for the study of apicomplexan parasites. In North American and Europe, Toxoplasma displays a highly clonal population structure (2), members of which differ greatly in their acute virulence in laboratory mice (2). Forward genetic approaches based on genetic crosses between these lineages have shown that a family of secreted serine threonine kinases largely controls differences in acute virulence of T. gondii in the mouse (3). Reverse genetic approaches have been used to validate the role of these effectors in selected laboratory strains (3). However, establishing to what extent these virulence factors contribute to pathogenesis in other related clonal strains, and in much more diverse strains that have been shown to exist in other regions of the world (4), has remained challenging.
Despite the availability of efficient systems for reverse genetics, homologous integration is not the preferred mechanism for insertion of foreign DNA into the genome of T. gondii. Instead, DNA is inserted into the genome randomly through nonhomologous-end joining (NHEJ). Inactivating the NHEJ pathway by knocking out the critical component KU80 has been used for increasing the efficiency of homologous recombination, thus facilitating gene knockouts and tagging endogenous loci in T. gondii (5, 6). Although this approach has clear advantages, it is limited to certain genetic backgrounds where KU80 has been deleted (5, 7). The lack of convenient genetic tools in many other T. gondii strains has hindered our understanding of the wider role that conserved genes play in diverse lineages. For example, the gene encoding the serine threonine kinase ROP18 was identified as the major gene responsible for differences between the highly virulent type I GT1 strain and the nonvirulent type III CTG strain (8). Confirmation of this role was based on a very strong gain of function when the type I allele of ROP18 from the RH strain was expressed in the type III CTG strain (8). In contrast, a rop18 deletion in the type I RH background causes only a modest loss of virulence (9). Although this apparent discrepancy may be due to underlying differences between type I strains, as described previously (10, 11), we have not been able to test this hypothesis due to difficulty in knocking out the ROP18 locus in the GT1 strain using conventional approaches.
CRISPR (clustered regularly interspaced short palindromic repeats) is a naturally occurring adaptive immune system found in bacteria and archaea, where it operates to restrict mobile genetic elements, such as bacteriophages and plasmids (12). Recently, the CRISPR/CAS9 (CRISPR-associated gene 9) system was adapted for targeted genome editing in a variety of organisms (13). The type II CRISPR system from Streptococcus pyogenes is the most commonly used for this purpose due to its well-known mechanism of action (14). When in complex with CRISPR RNA and a transactivating RNA, the endonuclease CAS9 introduces double-strand DNA breaks in a target sequence that is homologous to CRISPR RNA, and these breaks can be repaired by NHEJ or homologous recombination (14). The CAS9 nuclease is also able to use a hybrid single guide RNA (sgRNA) made from the fusion of 20-nucleotide CRISPR RNA to the transactivating RNA scaffold to generate double-strand breaks in the target DNA (14). The utility of using sgRNAs designed to target specific genes combined with the high efficiency by disruption by CRISPR/CAS9 has proven useful for gene manipulation in many model organisms (15–20).
In the present study, we adapted the CRISPR/CAS9 system for efficient gene disruption in T. gondii. We used CAS9 combined with site-specific sgRNAs to disrupt the uracil phosphoribosyl transferase (UPRT) gene. Loss of UPRT results in resistance to fluorodeoxyribose (FUDR), thus allowing easy quantitation of the efficiency and providing a convenient site for monitoring the frequency of disruption. We extended the use of CRISPR/CAS9 to disrupt the ROP18 locus from the type I strain GT1 and to complement this loss at the UPRT locus, revealing that ROP18 plays a larger role in acute virulence than previously predicted (9). Our studies demonstrate that CAS9-mediated gene disruption provides an efficient and powerful means of testing the role of specific genes in diverse genetic backgrounds of T. gondii.
RESULTS
Introduction of site-specific mutations in the T. gondii genome using CRISPR/CAS9.
Previous studies have shown that combining CRISPR/CAS9 with sgRNAs can be used to generate site-specific double-strand breaks in the target DNA that are repaired by NHEJ, leading to short insertions and deletions that inactivate the gene (20). To adapt the CRISPR/CAS9 system to T. gondii, we constructed a plasmid expressing a nucleus-localized CAS9 fused to green fluorescent protein (CAS9-NLS-GFP) driven by the SAG1 promoter and a single guide RNA (sgRNA) driven by the T. gondii U6 (TgU6) promoter (Fig. 1A; also, see Table S1 in the supplemental material). The sgRNA contained a 20-nucleotide (nt) guide RNA that targeted a region in exon 5 of the UPRT gene immediately upstream of a protospacer-adjacent motif (PAM), which consists of NGG for the S. pyogenes CAS9 nuclease (14) (Fig. 1). We chose the UPRT locus to test the efficiency of the CRISPR/CAS9 system because its inactivation leads to FUDR resistance (21), providing a convenient assay for measuring gene disruption. To assess the frequency of CRISPR/CAS9-mediated gene inactivation in T. gondii, we transfected the UPRT-specific sgRNA (sgUPRT) CRISPR plasmid into the type I RH strain by electroporation. GFP expression was monitored to determine the transfection efficiency at 24 h after electroporation, revealing that ~20 to 30% of cells received the plasmid (Fig. 1B and Table 1). To check the frequency of UPRT inactivation, the pooled transfectants (collected 48 h after electroporation) were subjected to plaque assays with and without FUDR. Approximately 10% of the successful transfectants (defined as CAS9-GFP-positive cells) were FUDR resistant (Table 1). To confirm that FUDR-resistant parasites had mutations in the UPRT gene, we generated single clones and sequenced the targeted region. All the sequenced clones had mutations in UPRT, primarily consisting of short deletions or insertions (Fig. 1C). As a control for the above-described experiment, we replaced the UPRT-targeting sgRNA with one that targeted ROP18 (sgROP18). Transfection of the sgROP18 CRISPR plasmid resulted in a similar number of CAS9-GFP-positive cells but did not result in FUDR-resistant parasites (Table 1), indicating that CRISPR/CAS9-mediated mutagenesis is site specific. To further check the site specificity and potential off-target activity, we made two sgRNAs (OT1 and OT2) that each differed from the UPRT-targeting sgRNA by two nucleotides (Table 1). Transfection of these constructs produced similar levels of CAS9-GFP-positive cells but 200- to 3,000-fold fewer FUDR-resistant parasites (Table 1). Collectively, these results indicate that CRISPR/CAS9 efficiently and specifically introduces targeted mutations into T. gondii.
FIG 1 .

CRISPR/CAS9-directed mutagenesis in T. gondii. (A) (Top) Schematic illustration of the plasmid expressing CAS9 and a single guide RNA (sgRNA) targeting the UPRT gene in T. gondii. Detailed plasmid map and sequence files can be found in Fig. S1 in the supplemental material. CAS9 (from Streptococcus pyogenes) fused to GFP was expressed from the TgSAG1 promoter, and the sgRNA was expressed from the TgU6 promoter. (Bottom) Sequence of the sgRNA and the region targeted in UPRT (purple bar). The green arrowhead indicates the predicted cleavage site by CAS9. NLS, nuclear localization signal; PAM, protospacer-adjacent motif. (B) Expression of CAS9-NLS-GFP in T. gondii determined by immunofluorescence staining 24 h after transfection of the CAS9/sgUPRT plasmid. Parasites were stained with rabbit anti-TgALD (red), and CAS9-GFP was stained with mouse anti-GFP (green). Scale bar = 5 µm. (C) Mutations in the UPRT gene induced by CAS9 and the sgUPRT from FUDR-resistant clones (MUT 1, etc., represent different clones).
TABLE 1 .
Inactivation of UPRT by CRISPR/CAS9-mediated mutagenesis
| sgRNA target | Expt | % GFP- positive cellsa |
Plaque formationb |
% efficiencyc | |
|---|---|---|---|---|---|
| Without FUDR | With FUDR | ||||
| UPRT | 1 | 41.0 | 92/200 | 122/6,000 | 11.0 |
| 2 | 26.5 | 73/200 | 65/6,000 | 11.3 | |
| 3 | 21.6 | 70/200 | 39/6,000 | 8.6 | |
| ROP18 | 1 | 27.4 | 93/200 | 0/600,000 | 0.0 |
| 2 | 24.4 | 67/200 | 0/600,000 | 0.0 | |
| 3 | 19.8 | 71/200 | 0/600,000 | 0.0 | |
| UPRT-OT1d | 1 | 27.6 | 40/200 | 1/600,000 | 0.003 |
| 2 | 23.9 | 63/200 | 0/600,000 | 0.0 | |
| UPRT-OT2e | 1 | 16.7 | 57/200 | 6/600,000 | 0.021 |
| 2 | 20.0 | 67/200 | 12/600,000 | 0.050 | |
The CAS9-GFP expressing population was estimated by immunofluorescence staining 24 h posttransfection.
Number of plaques observed/number of parasites seeded. When applied, FUDR was used at 10 µM.
[(Number of plaques observed in the presence of FUDR/% GFP-positive cells)/number of plaques observed in the absence of FUDR] × 100.
The sgRNA sequence GGCGTCTCGATTGTGAGACG differs from the UPRT sgRNA sequence by two nucleotides (bold).
The gRNA sequence GGCGTCTCGATTGTGAAGGC differs from the UPRT sgRNA sequence by two nucleotides (bold).
Gene disruption based on CRISPR/CAS9-induced homologous recombination.
To expand the CRISPR system to target genes for which no direct selection exists, we next sought to determine whether it could facilitate gene disruption by integration of a selectable marker. We again targeted the UPRT locus, as it provided a convenient means to test the efficacy of integration. For this experiment, we cotransfected the sgUPRT CRISPR plasmid with a homology template consisting of an amplicon (DNA fragments generated by PCR amplification) (see Tables S1 and S2 in the supplemental material) containing a pyrimethamine-resistant DHFR (DHFR*) cassette flanked by homology arms corresponding to the regions directly surrounding the sgUPRT target sequence (Fig. 2A; also, see Tables S1 and S2). Following cotransfection, parasites were selected with pyrimethamine, and the stably resistant pool was subjected to plaque assays with and without FUDR. We observed that the majority (70% to 100%) of the pyrimethamine-resistant parasites were also FUDR resistant, indicating a high frequency of UPRT disruption (Table 2). To confirm that FUDR resistance was due to DHFR* insertion at the UPRT locus, we isolated single clones of pyrimethamine-resistant parasites (without FUDR selection) and performed diagnostic PCR to check for integration (Fig. 2B). The majority (>45%) of these clones demonstrated correct integration of DHFR* into the UPRT locus (Fig. 2B), where both the 5′ and 3′ flanks were consistent with a double-crossover event by homologous recombination (Table 2). In other clones (15 to 20% of cases), the UPRT locus was disrupted but only one of the two flanking regions showed a pattern by PCR that was consistent with a simple homologous-crossover event (Table 2). Although the exact nature of these insertion events is unknown, >70% of clones showed targeted disruption of the UPRT gene, indicating a high degree of specific targeting (Table 2). In contrast, when parasites were transfected with only the UPRT homology template but not the sgUPRT plasmid, the pool of pyrimethamine-resistant parasites did not yield FUDR-resistant plaques (Table 2), consistent with the low frequency of homologous recombination in T. gondii. Likewise, although transfection of parasites with an unrelated gRNA (sgROP18) in combination with the UPRT-targeting DHFR* amplicon gave parasites that were resistant to pyrimethamine, it did not yield FUDR-resistant parasites (Table 2). Collectively, these results indicate that CRISPR/CAS9 enhances local recombination at the gRNA target region to introduce high-frequency insertion, resulting in gene disruption.
FIG 2 .
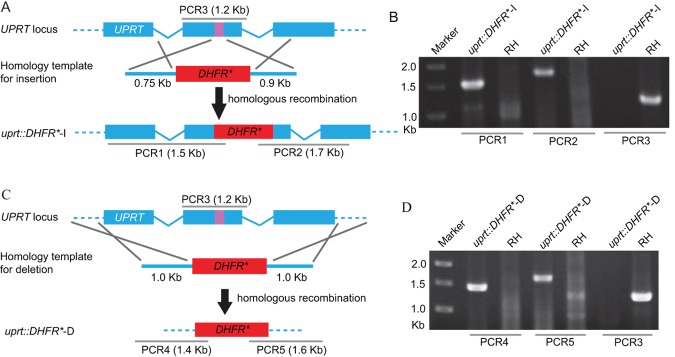
CRISPR/CAS9-mediated gene disruption and/or deletion of the UPRT locus. (A) Schematic of CRISPR/CAS9 strategy used to inactivate UPRT by inserting pyrimethamine-resistant DHFR (DHFR*). Transfection of the sgUPRT together with an amplicon containing a DHFR*-expressing cassette flanked by homology regions to UPRT was used to generate gene disruptions by insertion. The purple bar in UPRT gene represents the region targeted by the sgRNA. (B) Diagnostic PCR demonstrating homologous integration and gene disruption in a representative clone (uprt::DHFR*-I) compared with the parental line RH. PCR1 and PCR2 provide evidence of homologous integration based on products amplified between the DHFR* gene and regions in the UPRT locus that lie outside the targeting amplicon. PCR3 amplified a 1.2-kb fragment in wild-type cells that was lost due to the insertion of DHFR* (the larger fragment created by insertion of DHFR* [4.4 kb] does not amplify under conditions described here). The purple bar in UPRT gene represents the region targeted by the sgRNA. (C) Schematic of CRISPR/CAS9 strategy used to delete the entire coding region of UPRT by inserting DHFR*. Transfection of the sgUPRT together with the DHFR*-expressing amplicon shown was used to generate gene deletions. (D) Diagnostic PCR demonstrating homologous integration and gene deletion in a representative clone (uprt::DHFR*-D) compared with the parental line RH. PCR4 and PCR5 provide evidence of homologous integration based on products amplified between the DHFR* gene and regions in the UPRT locus that lie outside the targeting amplicon. PCR3 was as described above.
TABLE 2 .
Disruption of UPRT by DHFR through CRISPR/CAS9-induced homologous recombination
| Expt | Trial | sgRNA target |
Type of disruptiona |
Plaque formationb |
% efficiencyc |
Frequency of clones by PCR |
||
|---|---|---|---|---|---|---|---|---|
| Without FUDR |
With FUDR |
Complete homologous replacementd |
Partial homologous replacemente |
|||||
| 1 | 1 | none | —f | 49/200 | 0/200,000 | 0.0 | — | — |
| 2 | none | — | 43/200 | 0/200,000 | 0.0 | — | — | |
| 2 | 1 | UPRT | Insertion | 55/200 | 38/200 | 69.0 | 4/9 | 2/9 |
| 2 | UPRT | Insertion | 64/200 | 50/200 | 78.1 | 4/6 | 1/6 | |
| 3 | UPRT | Insertion | 34/200 | 51/200 | ≈100.0 | NDg | ND | |
| 3 | 1 | ROP18 | — | 75/200 | 0/200,000 | 0.0 | — | — |
| 2 | ROP18 | — | 47/200 | 0/200,000 | 0.0 | — | — | |
| 4 | 1 | UPRT | Deletion | 45/200 | 50/200 | ≈100.0 | 4/9 | 5/9 |
| 2 | UPRT | Deletion | 55/200 | 47/200 | 85.5 | 3/7 | 3/7 | |
Different homology templates were used for gene insertion or deletion as described for Fig. 2A and C.
Number of plaques observed/number of parasites seeded. When used, FUDR was added to a final concentration of 10 µM.
(Number of plaques observed in the presence of FUDR/number of plaques observed in the absence of FUDR) × 100.
Number of positive clones/number of total clones checked. Clones that showed correct 5′ integration (positive in PCR1 or PCR4), correct 3′ integration (positive in PCR2 or PCR5), and loss of the endogenous UPRT gene (negative in PCR3) were considered to have undergone complete homologous replacement events.
Number of positive clones/number of total clones checked. Clones that showed loss of the endogenous UPRT gene (negative in PCR3) and correct integration in at least one end (5′ or 3′ but not both) were considered to have undergone partial homologous replacement events.
—, not applicable.
ND, not determined.
Although gene disruption can be a powerful means of assessing gene function, it is limited by the possibility that regions upstream of the insertion may still be transcribed, potentially leading to truncated gene products being expressed. To test whether CRISPR/CAS9 could be used to delete the entire coding sequence of UPRT, we used the sgUPRT CRISPR plasmid in combination with a homology template that contained DHFR* flanked by 5′ and 3′ homology regions from outside the coding sequence of the UPRT gene (Fig. 2C; also, see Table S1 in the supplemental material). Following cotransfection of the sgUPRT plasmid with this DHFR* amplicon, we selected with pyrimethamine and then checked the frequency of FUDR resistance by plaque assay and homologous replacement by PCR (Table 2). We observed a high frequency of homologous recombination at the UPRT locus as described above (Fig. 2D and Table 2). These findings indicate that in addition to disruption of a gene by insertion, CRISPR/CAS9 can also be used to delete large genomic regions by homologous recombination, using a single guide RNA to create a double-strand break in the gene of interest.
CRISPR/CAS9 enhances site-specific nonhomologous integration of foreign DNA.
Previous studies have shown that homologous recombination in T. gondii requires relatively long flanking regions that match the genomic locus in order to ensure efficient integration (5). To examine whether this requirement might also limit the utility of CRISPR/CAS9, we explored the length of homology needed to drive efficient integration at the UPRT locus. We compared the efficiency of targeting with long homology regions (i.e., 750 to 900 bp) to that of using flanking regions of 20 bp, in each case surrounding the DHFR* cassette (Fig. 3A). As a control, we transfected the sgUPRT plasmid along with a construct containing the DHFR* selectable marker that lacked any homology to the UPRT gene (Fig. 3A). Transfected parasites were selected with pyrimethamine and then tested for FUDR resistance by plaque assay. Regardless of the targeting construct used, we observed high frequencies of parasites that were resistant to both drugs, suggesting that the DHFR* cassette was integrated into the UPRT gene. To further test the efficiency of integration, we obtained pyrimethamine-resistant single clones (without FUDR selection) and checked for integration into the UPRT locus by PCR (Fig. 3B). We used the size of the PCR3 product instead of PCR1 and PCR2 (Fig. 2A) to examine DHFR* insertion, because DHFR* may be incorporated at either orientation when short or no homology arms are used. For both the long (i.e., 750- to 900-bp) and short (i.e., 20-bp) homology flanks, as well as for the DHFR* cassette that lacked any homology, a high percentage (≥50%) of clones demonstrated correct integration into the UPRT locus (Table 3). These results indicate that CRISPR/CAS9 can also be used to facilitate nonhomologous integration into specific sites that are targeted for double-strand breaks using single guide RNAs, making targeted gene disruption much easier.
FIG 3 .
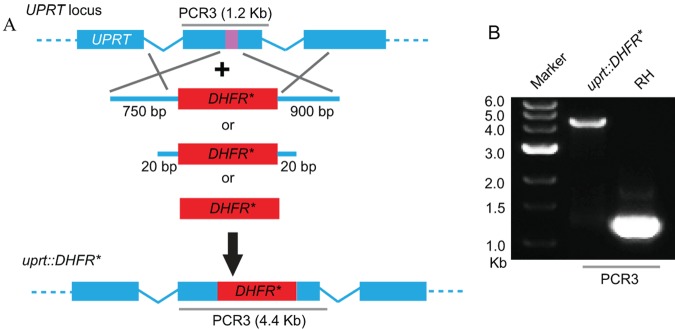
Role of flanking regions in CRISPR/CAS9-mediated gene disruption at the UPRT locus. (A) Schematic of CRISPR/CAS9 strategy used to inactivate UPRT by inserting DHFR*. Transfection of the sgUPRT plasmid together with DHFR*-expressing amplicons that contained variable-length flanks with homology to UPRT, or with no homology, were used to generate gene disruptions by insertion. The purple bar in UPRT gene represents the region targeted by the sgRNA. (B) PCR3 analysis of the UPRT locus. Amplification of the endogenous locus (RH) yielded a band of 1.2 kb, while insertion of a single copy of DHFR* resulted in a band of 4.4 kb (uprt::DHFR*). The clone shown was derived by cotransfection of an amplicon of DHFR* with no homology flanks.
TABLE 3 .
Site-specific integration of DHFR into the UPRT locus mediated by CRISPR/CAS9
| Expt | Trial | sgRNA target |
Type of disruption |
Size (bp) of homology armsa |
Plaque formationb |
% efficiencyc |
Frequency of clones by PCR3 |
||
|---|---|---|---|---|---|---|---|---|---|
| Without FUDR |
With FUDR |
Integrationd | Disruptione | ||||||
| 1 | 1 | UPRT | Insertion | 750, 900 | 72/200 | 65/200 | 90.3 | 3/5 | 2/5 |
| 2 | UPRT | Insertion | 750, 900 | 86/200 | 76/200 | 88.4 | NDf | ND | |
| 2 | 1 | UPRT | Insertion | 20, 20 | 25/200 | 22/200 | 88.0 | 3/5 | 2/5 |
| 2 | UPRT | Insertion | 20, 20 | 53/200 | 49/200 | 92.5 | 4/6 | 1/6 | |
| 3 | 1 | UPRT | Insertion | 0, 0 | 32/200 | 24/200 | 75.0 | 3/8 | 2/8 |
| 2 | UPRT | Insertion | 0, 0 | 52/200 | 37/200 | 71.2 | 4/5 | 1/5 | |
The sizes of the fragments flanking the 5′ and 3′ ends of DHFR that are homologous to UPRT, as indicated in Fig. 3A.
Number of plaques observed/number of parasites seeded. When used, FUDR was added to a final concentration of 10 µM.
(Number of plaques observed in the presence of FUDR [10 µM]/number of plaques observed in the absence of FUDR) × 100.
Clones that showed the presence of a 4.4-kb band and absence of a 1.2-kb band in PCR3 were considered integration events. Number of positive clones/number of total clones checked.
Clones that lacked the endogenous 1.2-kb band for the UPRT locus but which did not give the expected band of 4.4 kb, corresponding to insertion of DHFR*, were considered disruptions. Number of positive clones/number of total clones checked.
ND, not determined.
CRISPR/CAS9-mediated gene disruption in diverse type I strains.
All the preceding experiments were done with the type I RH strain, which is highly amenable to genetic manipulation but not entirely representative of natural type I variants (11, 22). To test whether CRISPR/CAS9 would allow broader application of reverse genetic approaches, we attempted to knock out out a gene in the type I strain GT1, which is a low-passage-number isolate that retains the entire life cycle. For this test, we chose the ROP18 gene, which had previously been implicated in the virulence of the type I strain GT1 (8). To disrupt ROP18, we designed a guide RNA to target the 5′ coding region of ROP18 (sgROP18) and paired this with a PCR amplicon consisting of the DHFR* selectable marker flanked with regions of homology to ROP18 (Fig. 4A). After cotransfection of the sgROP18 CRISPR plasmid and homology template into GT1, parasites were selected with pyrimethamine and cloned by limiting dilutions. All six randomly selected pyrimethamine-resistant clones were positive for correct integration of DHFR* at the ROP18 locus by PCR (Fig. 4B) and subsequent Western blot analysis confirmed the loss of ROP18 expression (Fig. 4C). All of the CRISPR/CAS9 plasmids used here are transiently expressed and lack selection for retention in T. gondii, and such plasmids are normally lost over the first few days in culture. However, we performed immunofluorescence assays and Western blot analysis to check the CAS9-GFP expression to see whether any clones retained the sgROP18 CRISPR plasmid (which would interfere with the complementation described below). We did not observe any GFP expression in any of the three clones checked (data not shown), indicating loss of the CRISPR plasmid. We then generated a complemented line in the GT1 knockout by expressing ROP18 with a Ty epitope tag driven by the IMC1 promoter at the UPRT locus, as described previously (23). The sgUPRT CRISPR plasmid was used to introduce double-strand breaks, while the ROP18 expression cassette was flanked by homology regions to drive homologous integration into the UPRT locus (Fig. 4D). Following cotransfection, parasites were selected with FUDR, and single clones were screened for ROP18 expression by immunofluorescence staining. Approximately 20 to 30% of FUDR-resistant parasites expressed ROP18 (data not shown), a number that is probably limited by the efficiency of cotransfection. A representative clone showing a level of ROP18 expression similar to that of wild-type GT1 (Fig. 4C) cells and correct integration into the UPRT locus (Fig. 4E) was chosen for further analysis. These results indicate that the CRISPR/CAS9 system also works efficiently in genetic backgrounds that have previously been refractory to genetic manipulations.
FIG 4 .

CRISPR/CAS9-mediated disruption of ROP18 in the type I strain GT1. (A) Schematic of CRISPR/CAS9-mediated disruption of ROP18 by insertion of DHFR*. Transfection of sgROP18 together with the DHFR* amplicon shown was used to disrupt the ROP18 coding region. The purple bar indicates the sgROP18 target region. (B) Diagnostic PCR demonstrating homologous integration in a representative rop18::DHFR* clone compared with the parental line GT1. (C) Expression of ROP18 in a rop18-disrupted line (rop18::DHFR*) and a complemented line (rop18::DHFR*/uprt::ROP18) determined by Western blotting. ROP18 was detected by rabbit anti-TgROP18, and GRA7 was detected by mouse anti-TgGRA7 as a loading control. Primary antibodies were detected with Li-Cor secondary antibodies and an Odyssey imaging system. (D) Schematic showing the complementation of ROP18-deficient parasites by insertion at the UPRT locus (uprt::ROP18). (E) Diagnostic PCR demonstrating complementation of ROP18 at the UPRT locus (rop18::DHFR*/uprt::ROP18). (F) Survival curves of parasites in CD1 mice. Wild-type RH and GT1 strains were compared to their respective knockouts (∆rop18). The GT1 knockout was complemented to re-express ROP18 (∆rop18/ROP18) at wild-type levels (see panel C). Mice were injected i.p. with the number of parasites indicated in parentheses, and survival was monitored for 30 days.
To determine the role of ROP18 in the type I GT1 strain, we injected the ∆rop18 parasites into CD1 mice at different dosages and monitored the survival of mice for 30 days. For comparison, we also injected RH and a ∆rop18 mutant in this background. As described previously (9), RHΔrop18 parasites were only slightly attenuated, and mice lived only 2 to 3 days longer when challenged with 300 tachyzoites (Fig. 4F). In contrast, most (75%) mice survived infection at a dose of 300 tachyzoites of GT1Δrop18 parasites, while injection of 3,000 parasites resulted in approximately 60% mortality (Fig. 4F). The reduced virulence of GT1Δrop18 parasites was fully restored in the complemented line (Fig. 4F). These results suggest that ROP18 plays a more important role in mediating pathogenesis in the type I natural isolate GT1 than in the laboratory strain RH.
DISCUSSION
Although existing genetic tools work well in selected laboratory strains of T. gondii, natural isolates, in particular those that complete the entire life cycle, are much more refractory to manipulation. To overcome this limitation, we developed a CRISPR/CAS9 system for targeted gene disruption in T. gondii. CRISPR/CAS9 was able to target genes for disruption with high efficiency and also facilitated gene disruption and/or deletion by insertion of a selectable marker following site-specific targeting using an sgRNA. Remarkably, gene disruption by insertion of a selectable marker did not require homology arms for efficient integration, suggesting that this process occurs by NHEJ. In contrast, when relatively long regions of homology were used, CRISPR/CAS9 promoted efficient homologous recombination, resulting in complete deletion of a coding region that spanned a 4-kb region using a single site-specific break introduced by an sgRNA. Application of CRISPR/CAS9 to the type I strain GT1, which has previously been refractory to genetic manipulation, allowed efficient disruption of an important virulence factor ROP18, as well as complementation by reintroduction of the gene into a neutral locus. These results demonstrate the utility of CRISPR/CAS9 for genetic manipulations in diverse genetic backgrounds, without the need to generate specific lines that have enhanced levels of homologous recombination.
Although reverse genetic technologies for T. gondii are well developed, the targeting of specific loci is limited by the fact that recombination tends to occur by nonhomologous integration of foreign DNA into the chromosome (21, 24, 25). Typically, homologous recombination requires long homology arms surrounding a selectable marker of interest to favor double-crossover replacement or single crossover followed by resolution of a pseudodiploid to generate knockouts (21, 24). The high frequency of nonhomologous recombination in T. gondii suggests that integration occurs by chromosome breaks that are repaired by NHEJ. Double-strand chromosomal breaks are normally repaired by NHEJ, which relies on a complex of proteins, including the heterodimer KU70/KU80, which is conserved in a wide range of organisms from bacteria to humans (26). Disruption of this system affects nonhomologous repair but does not affect the process of homologous recombination. Hence, using ∆ku80 T. gondii lines, it is possible to achieve homologous recombination rates of nearly ~95% and to target genes for deletion by double crossover (5, 7) or tag the endogenous locus by single crossover to replace the 3′ end of the gene with an epitope-tagged copy (6). The utility of this system was further enhanced by importing the positive-negative selectable marker hypoxanthine-xanthine-guanosine phosphoribosyl transferase (HXGPRT), described previously (27), thus allowing markers to be recycled (5). Although this system provides a very efficient means of gene disruption in T. gondii, it is thus far limited to two laboratory-adapted strains that represent the type I (RH strain) (5, 6) and type II (Pru) (7) clonal lineages. However, it is difficult to extrapolate from these results to infer the possible roles of genes in other clonal strains or in diverse lineages. There are also some intrinsic limitations to using ∆ku80 mutants, as this defect results in genome instability, thus limiting the use of certain selectable markers that may induce mutation (5) and precluding their use in meiotic crosses. Additionally, studies have implicated NHEJ in telomere stability (26), such that long-term maintenance of ∆ku80 lines may result in genome rearrangements. All of these limitations make it desirable to find a means of enhancing gene targeting in T. gondii without the use of modified strains.
Although a relatively recent development, CRISPR/CAS9 has proven useful for a number of types of genome modification in model organisms. Site-specific double-strand DNA breaks (DSB) induced by CRISPR/CAS9 in cultured mammalian cells can be repaired by either the mutagenic NHEJ, leading to insertion and deletion mutations in targeted genes, or homologous recombination in the presence of a donor template to knock in specific gene alleles or selection markers (18). CRISPR/CAS9 has also been used to target multiple genes in one step and to perform genomic editing of specific alleles by homologous recombination (20). In zebra fish, CRISPR/CAS9 has been used for gene disruption and for epitope tagging by homologous recombination (although the efficiency is very low) (16). In Caenorhabditis elegans, CRISPR/CAS9 was used to insert selectable markers into specific genes using homology templates containing long flanking (2-kb) arms (28). CRISPR/CAS9 has also been used to delete large chromosomal regions of haploid human cells using two guide RNAs targeting two different regions in the same cell (29). CRISPR/CAS9 stimulated homologous recombination has been used to correct genetic disorders in a few disease models by replacing the mutated genes with wild-type copies through homology-directed repair (30, 31).
Here, we imported the CRISPR/CAS9 system into T. gondii to perform targeted gene disruption and gene deletion. We took advantage of the fact that CAS9 works efficiently using a single fused guide RNA consisting of a scaffold RNA and a site-specific RNA that matches the target gene of interest (14, 32). We engineered sgRNA that contained 20 bp of direct homology to the gene of interest positioned upstream of the PAM sequence for CAS9 from S. pyogenes. CRISPR/CAS9 led to efficient double-strand breaks at a site corresponding to 17 to 18 bp of the sgRNA, the repair of which led to small deletions and insertions in the UPRT target. Although we have not extensively examined the genome of T. gondii for potential off-target effects due to CRISPR-mediated mutation, this possibility is less of a concern than in mammalian systems, simply due to the smaller genome and hence decreased probability of off-target matches for 20-bp guide RNAs. Nonetheless, caution should be exercised in the design of gRNAs to ensure they are unique in the genome and terminate at the NGG PAM sequence. To address the requirement for sequence specificity, we tested two sgRNAs that had 2-bp mismatches, engineered close to the site of the double-strand break that is initiated by CRISPR/CAS9. These changes decreased the efficiency of disruption of the target UPRT 200- to 3,000-fold, suggesting that for targeted gene disruption, the process is highly efficient.
The targeting of UPRT for disruption by CRISPR/CAS9 was extremely efficient and easily assayed using FUDR resistance. However, applying this technology to other loci that may lack a selectable phenotype, or for which reagents might not be available to assess disruption, will be less straightforward. In this regard, the ability to introduce a selectable marker at the site of double-strand break induced by the sgRNA should provide a highly efficient means to target any nonessential gene. An added advantage of CRISPR/CAS9 is that insertion appears not to require homology arms, as the insertion of DHFR* into the UPRT locus that had been targeted by an sgRNA was as efficient regardless of the length or even presence of homology arms that match the targeted region. These results strongly suggest that short homology arms do not drive homologous recombination but instead are inserted by NHEJ where the fragment provided as a selectable marker is used to repair the break in the chromosomes introduced by CRISPR/CAS9. Although we did not verify this here, it is likely that under these circumstances (i.e., short or no homology regions) the DHFR* cassette can insert in either orientation, although if the goal is gene disruption, this does not affect the outcome. In terms of targeting genes for disruption, CRISPR/CAS9 is much more efficient for targeted disruption than ∆ku80 lines, which require homology arms of ≥500 bp in length for efficient targeting. Hence, CRISPR/CAS9 should be highly effective for rapid disruption of genes and for introducing transgenes to specific sites. For the loci examined here (UPRT and ROP18), we achieved high frequencies of gene disruption. However, the efficiency may vary for different genes depending on the specific guide RNA used, the selection cassette, and the transformation efficiency. The UPRT locus provides a convenient nonessential locus for targeting that also provides a means of negative selection. We used this strategy here to complement the ∆rop18 mutant, but in principle it could be used to introduce any transgene of interest.
Generating clean knockouts of an entire genetic locus is often desirable to remove any concern that a truncated version of the RNA or protein may still be generated from a partial disruption. CRISPR/CAS9 also proved extremely efficient for gene deletion by homologous recombination at a site that was targeted for a double-strand break using a site-specific sgRNA. In this case, it was important to provide relatively long homology arms that lay outside the gene of interest. Although nearly all of the clones isolated from such experiments showed evidence of disruption of the target UPRT locus, only about 50% showed clear evidence of double crossovers by homologous recombination. Using simple diagnostic PCR screening, it should be possible to isolate clones where the entire locus has been deleted from experiments pairing an sgRNA CRISPR plasmid with a template containing long homology regions flanking a selectable marker. Generation of such homology-targeting plasmids by Gateway makes this feasible on a moderately high throughput scale, since the design and construction of vectors are modular. The selectable marker could also easily be recycled, by flanking it with loxP sites and introducing Cre recombinase to remove the marker once the targeted gene disruption has been achieved (33) (the HXGPRT marker would provide the added utility of being both positive and negative selection). Finally, given the efficiency of CRISPR/CAS9 for targeted gene disruption using insertion of the DHFR* cassette, this system might be exploited for genome-wide screens of gene disruptions, as has recently been done for several systems (34, 35).
A second major advance provided by CRISPR/CAS9 is the efficient genetic manipulation of diverse T. gondii strains that are otherwise refractory to transformation. In previous studies, we identified ROP18 as an important virulence determinant in a genetic cross between the type I strain GT1 and the type III strain CTG (8). Because of difficulties in generating gene knockouts in the GT1 line, we instead used a ∆ku80 mutant of RH to test the role of ROP18 in the type I background (9). Loss of this gene in the RH strain resulted in only a modest decrease in acute virulence (9), in contrast to the large difference seen in the acute virulence of the parental strains (8). Although this discrepancy could be accounted for by other genes, the original mapping studies estimated that ~65% of the acute virulence phenotype was attributed to ROP18 (8). Using CRISPR/CAS9, we were able to efficiently disrupt ROP18 in the GT1 strain, revealing that it plays a much larger role in this background. Although the reasons for this difference are presently unclear, previous studies have shown that the RH strain differs from other type I strains in gene expression (11) and in various growth-related phenotypes in vitro (22), which might mask the role of specific genes in virulence. In addition, previous studies have predicted that alleles of ROP18 play a wider role in virulence of diverse strains, such as those found in South America (36). However, these studies were based on transgenic expression of ROP18 alleles in a type III strain, which is normally hypomorphic for ROP18. CRISPR/CAS9 now offers the possibility of deleting ROP18 in these lineages, thus providing a better assessment of its role in pathogenesis.
In addition to working in a wider range of backgrounds, CRISPR/CAS9 also has obvious advantages in terms of efficiency of transformation. In the example of ROP18, all 6 randomly selected pyrimethamine-resistant clones proved to be clean knockouts. This contrasts with numerous failed efforts to disrupt ROP18 or KU80 in this background using conventional technology (our unpublished results, consisting of more than three failed attempts each by several members of the lab). Moreover, CRISPR/CAS9 provided a rapid means of complementing the gene by reinsertion into the UPRT locus. Random integration is widely used to introduce transgenes in T. gondii, yet it has the disadvantage of not knowing the precise genetic location of the insertion, thus potentially complicating the interpretation of the phenotype. Efficient targeting into the UPRT locus, which is a nonessential gene in T. gondii, provides a neutral location for complementing genes, and this process was aided by using CRISPR/CAS9. The efficiency of CRISPR/CAS9 for performing genetic manipulations in natural genetic backgrounds extends the power of reverse genetics beyond a few laboratory strains of T. gondii. As well as encompassing other members of the clonal lineages, it quite likely will also be applicable to the diverse members of other haplogroups that have recently been described for T. gondii (4). Thus, CRISPR/CAS9 adds another powerful tool to the repertoire of genetic techniques available in T. gondii.
MATERIALS AND METHODS
Parasites strains and growth conditions.
The T. gondii strains used in this study were maintained by growth in human foreskin fibroblasts (HFF) cultured in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 10 µg/ml gentamicin, and 10 mM glutamine (Thermo, Fisher Scientific, Waltham, MA). Wild-type RH and GT1 and the parasite mutant lines RH Δku80 and RH Δku80Δrop18 were described previously (9). Transgenic parasites were obtained by electroporation of constructs into cells and selection with 3 µM pyrimethamine (Sigma-Aldrich, St. Louis, MO) or 10 µM FUDR (Sigma-Aldrich, St. Louis, MO).
Plasmid construction.
All the plasmids and primers used in this study are listed in Tables S1 and S2 in the supplemental material, and further details on plasmid construction can be found in the supplemental methods in the supplemental material. A map and sequence file for the plasmid expressing CAS9 and a single guide RNA (sgRNA) targeting the UPRT gene in T. gondii can be found in Fig. S1 in the supplemental material. The UPRT-targeting CRISPR plasmid (sgUPRT) was constructed in two steps: first, the U6 promoter from T. gondii driving expression of the UPRT-specific sgRNA was cloned into the backbone plasmid pSAG1-Ble-SAG1, as described in the supplemental methods. This plasmid was further modified to express the CAS9-NLS-GFP cassette under the SAG1 promoter. All other CRISPR/CAS9 plasmids were obtained through Q5 mutagenesis (New England Biolabs, Ipswich, MA) of this plasmid to change the UPRT targeting gRNA to other specific sgRNAs using primers listed in Table S2. To generate a plasmid for inserting DHFR* into the UPRT gene, upstream (750-bp) and downstream (900-bp) regions directly adjacent to the sgUPRT target sequence were used to flank DHFR* (24). To generate a construct for deleting the entire coding sequence of UPRT, flanking regions 5′ (~1 kb) and 3′ (~1 kb) outside the coding region were used to surround the DHFR* cassette. To test the efficiency of short homology regions for site-specific integration, the DHFR* cassette was amplified using 20-bp flanking regions from the UPRT locus (surrounding the sgUPRT site) (Table S2) or the DHFR* cassette was used as an amplicon generated without homology regions. To generate a construct for disrupting the ROP18 locus, 5′ (820-bp) and 3′ (850-bp) regions flanking the sgROP18 target region were used to surround the DHFR* cassette (Table S1). The ROP18 gene was complemented using a previously described plasmid for targeting the type I ROP18 allele to the UPRT locus (23).
Site-specific mutagenesis of the UPRT gene.
To test the efficiency of targeted mutation at the UPRT locus, RH strain parasites (107 cells) were transfected with indicated CRISPR plasmids (Table S1) by electroporation. Transfected cells were allowed to grow for 48 h in the absence of drug selection and egress naturally. To estimate viability, 200 parasites were subject to plaque assays in HFF monolayers grown for 7 days without FUDR. In parallel, aliquots containing 6,000 or 600,000 parasites were tested for growth in HFF cells in 10 µM FUDR. Monolayers were stained with 0.1% crystal violet, and the number of plaques produced under each condition was recorded. To confirm the mutations in the UPRT gene, we obtained FUDR-resistant clones from transfected parasites by subcloning after 5 to 6 days of selection. DNA from clones was PCR amplified to obtain the 1.2-kb region flanking the target region (i.e., PCR3) and sequenced using the Sanger dideoxy method (Genewiz Inc., South Plainfield, NJ) (Table S2).
Targeted disruption and deletion of the UPRT gene.
To study the efficiency of CRISPR/CAS9-mediated gene disruption by integration of a selectable marker, sgUPRT or sgROP18 targeting CRISPR/CAS9 plasmids (see Table S1 in the supplemental material) was combined with various amplicons containing DHFR* expression cassettes for transfection. Amplicons were PCR amplified using primers listed in Table S2 in the supplemental material, purified by agarose gel electrophoresis, and extracted using the Qiaquick gel extraction kit (Qiagen Inc., Valencia, CA). Recovered DNAs were quantified using a Nanodrop 2000 instrument (Nanodrop Instruments, Wilmington, DE). Mixtures of CRISPR/CAS9 plasmids with the purified DHFR* amplicons (5:1 mass ratio) were cotransfected into RH parasites by electroporation. Pyrimethamine-resistant parasites were obtained by selection with 3 µM pyrimethamine, and resistant cells were then used for determination of viability and FUDR resistance by plaque assay as described above. In parallel, single-cell clones were obtained by limiting dilution, and lysates were generated as described previously (4). The specificity of DHFR* integration and loss of genes by deletion were tested by PCR using primers listed in Table S2. PCRs were performed using Taq DNA polymerase (New England Biolabs, Ipswich, MA) in a 25-µl reaction mixture containing 2 µl of lysate as the template according to the manufacturer’s directions. In general, reactions were performed for 35 cycles with denaturation at 95°C for 30 s, annealing at 57°C for 45 s, and extension for 90 s at 68°C. Products were analyzed by electrophoresis in agarose gels with ethidium bromide. In some cases, PCR3 was conducted using Q5 DNA polymerase (New England Biolabs, Ipswich, MA) to efficiently amplify the inserted DHFR* fragment, giving a band of 4.4 kb, which was inefficiently amplified using Taq polymerase under the conditions described above. When PCR3 was performed with Q5 polymerase, 25-µl reaction mixtures containing 2 µl template were amplified for 35 cycles with denaturation at 95°C for 30 s, annealing at 60°C for 30 s, and extension for 3 min at 68°C.
Generation of ROP18 knockout and complemented lines.
To disrupt ROP18 in GT1, we cotransfected the sgROP18 CRISPR plasmid along with an amplicon containing ROP18 homology regions surrounding a pyrimethamine-resistant DHFR* cassette. Selection by growth for 7 to 8 days in pyrimethamine (3 µM) was used to get stably resistant clones that were subsequently screened by PCR for correct integration of DHFR* into the ROP18 locus (see Table S2 in the supplemental material). PCR-positive clones were further analyzed by Western blotting to confirm the loss of ROP18 expression. To complement the ROP18-deficient parasites, we transfected the GT1 rop18::DHFR* parasites with the sgUPRT CRISPR plasmid (mass ratio, 1:5 [CRISPR/CAS9 to ROP18 complement]) to target integration to the UPRT locus, along with a previously described ROP18-complementing plasmid (Ty-tagged ROP18 driven by the IMC1 promoter) (23). Parasites were selected with 10 µM FUDR for 5 to 6 days, and single clones were screened by immunofluorescence for Ty expression using monoclonal antibody (MAb) BB2 (37). Positive clones were further analyzed by PCR for correct integration at the UPRT locus (see Table S2) and by Western blotting for ROP18 expression.
Immunofluorescence microscopy.
To estimate the frequency of CRISPR/CAS9-mediated gene disruptions, RH parasites (~107) were transfected with 7.5 µg of indicated CRISPR plasmids (Table S1) and analyzed by immunofluorescence staining for GFP 24 h posttransfection. HFF monolayers were fixed with 4% formaldehyde, permeabilized with 0.1% Triton X-100, and incubated with MAb 3E6 (Life Technologies, Carlsbad, CA) to detect GFP. Parasites were detected with rabbit anti-TgALD (38). Alexa Fluor 594-conjugated goat anti-rabbit IgG and Alexa Fluor 488-conjugated goat anti-mouse IgG secondary antibodies (Life Technologies, Carlsbad, CA) were used to detect primary antibodies. Images were acquired using a Zeiss Axioskop 2 MOT Plus microscope equipped with a 63×, NA (numerical aperture) 1.6 oil immersion lens and an AxioCam MRm monochrome camera (Carl Zeiss, Inc., Thornwood, NJ).
Western blotting.
To assess ROP18 expression levels, parasite lysates were subjected to Western blot analysis as previously described (39). Rabbit anti-ROP18 (9) was used to stain ROP18, and mouse anti-GRA7 (40) was used as a loading control. IRDye800CW-conjugated goat anti-mouse IgG and IRDye680RD-conjugated goat anti-rabbit IgG were used as secondary antibodies, and the blot was scanned using the Li-Cor Odyssey imaging system (Li-Cor Biosciences, Lincoln, NE).
Virulence in laboratory mice.
Mice were obtained from The Jackson Laboratory (Bar Harbor, ME) and maintained in an Association for Assessment and Accreditation of Laboratory Animal Care International-approved facility at Washington University School of Medicine. All animal experiments were conducted according to the U.S. Public Health Service Policy on Humane Care and Use of Laboratory Animals, and protocols were approved by the Institutional Care Committee at the School of Medicine, Washington University in St. Louis. To determine the virulence in mice, parasites were injected intraperitoneally (i.p.) into 8- to 10-week-old female CD-1 mice (5 mice/condition). The survival of mice was monitored for 30 days, and blood was drawn from surviving mice at day 30 and tested by enzyme-linked immunosorbent assay (ELISA) to confirm infection. Mice that were seronegative by ELISA were not included in the analysis. Cumulative mortality was plotted as a Kaplan-Meier survival plot and analyzed in Prism (GraphPad Software, Inc., La Jolla, CA).
SUPPLEMENTAL MATERIAL
Supplemental methods. Download
Plasmids used in this study.
Primers used in this study.
A map and sequence file for the plasmid expressing CAS9 and a single guide RNA (sgRNA) targeting the UPRT gene in T. gondii. Download
ACKNOWLEDGMENTS
We are grateful to Josh Beck and Michael Behnke for plasmids used here and to members of the Sibley laboratory for useful advice.
This work was supported in part by grants from the National Institutes of Health (AI034036 and AI036629).
Footnotes
Citation Shen B, Brown KM, Lee TD, Sibley LD. 2014. Efficient gene disruption in diverse strains of toxoplasma gondii using CRISPR/CAS9. mBio 5(3):e01114-14. doi:10.1128/mBio.01114-14.
REFERENCES
- 1. Dubey JP. 2010. Toxoplasmosis of animals and humans. CRC Press, Boca Raton, FL [Google Scholar]
- 2. Sibley LD, Ajioka JW. 2008. Population structure of Toxoplasma gondii: clonal expansion driven by infrequent recombination and selective sweeps. Annu. Rev. Microbiol. 62:329–351. 10.1146/annurev.micro.62.081307.162925 [DOI] [PubMed] [Google Scholar]
- 3. Hunter CA, Sibley LD. 2012. Modulation of innate immunity by Toxoplasma gondii virulence effectors. Nat. Rev. Microbiol. 10:766–778. 10.1038/nrmicro2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Su C, Khan A, Zhou P, Majumdar D, Ajzenberg D, Dardé ML, Zhu XQ, Ajioka JW, Rosenthal BM, Dubey JP, Sibley LD. 2012. Globally diverse Toxoplasma gondii isolates comprise six major clades originating from a small number of distinct ancestral lineages. Proc. Natl. Acad. Sci. U. S. A. 109:5844–5849. 10.1073/pnas.1203190109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fox BA, Ristuccia JG, Gigley JP, Bzik DJ. 2009. Efficient gene replacements in Toxoplasma gondii strains deficient for nonhomologous end joining. Eukaryot. Cell 8:520–529. 10.1128/EC.00357-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huynh MH, Carruthers VB. 2009. Tagging of endogenous genes in a Toxoplasma gondii strain lacking Ku80. Eukaryot. Cell 8:530–539. 10.1128/EC.00358-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fox BA, Falla A, Rommereim LM, Tomita T, Gigley JP, Mercier C, Cesbron-Delauw MF, Weiss LM, Bzik DJ. 2011. Type II Toxoplasma gondii KU80 knockout strains enable functional analysis of genes required for cyst development and latent infection. Eukaryot. Cell 10:1193–1206. 10.1128/EC.00297-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Taylor S, Barragan A, Su C, Fux B, Fentress SJ, Tang K, Beatty WL, Hajj HE, Jerome M, Behnke MS, White M, Wootton JC, Sibley LD. 2006. A secreted serine-threonine kinase determines virulence in the eukaryotic pathogen Toxoplasma gondii. Science 314:1776–1780. 10.1126/science.1133643 [DOI] [PubMed] [Google Scholar]
- 9. Fentress SJ, Behnke MS, Dunay IR, Mashayekhi M, Rommereim LM, Fox BA, Bzik DJ, Taylor GA, Turk BE, Lichti CF, Townsend RR, Qiu W, Hui R, Beatty WL, Sibley LD. 2010. Phosphorylation of immunity-related GTPases by a Toxoplasma gondii-secreted kinase promotes macrophage survival and virulence. Cell Host Microbe 8:484–495. 10.1016/j.chom.2010.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Khan A, Fux B, Su C, Dubey JP, Darde ML, Ajioka JW, Rosenthal BM, Sibley LD. 2007. Recent transcontinental sweep of Toxoplasma gondii driven by a single monomorphic chromosome. Proc. Natl. Acad. Sci. U. S. A. 104:14872–14877. 10.1073/pnas.0702356104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Yang N, Farrell A, Niedelman W, Melo M, Lu D, Julien L, Marth GT, Gubbels MJ, Saeij JP. 2013. Genetic basis for phenotypic differences between different Toxoplasma gondii type I strains. BMC Genomics 14:467. 10.1186/1471-2164-14-467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, Horvath P. 2007. CRISPR provides acquired resistance against viruses in prokaryotes. Science 315:1709–1712. 10.1126/science.1138140 [DOI] [PubMed] [Google Scholar]
- 13. Mali P, Esvelt KM, Church GM. 2013. Cas9 as a versatile tool for engineering biology. Nat. Methods 10:957–963. 10.1038/nmeth.2649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. 2012. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337:816–821. 10.1126/science.1225829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Friedland AE, Tzur YB, Esvelt KM, Colaiácovo MP, Church GM, Calarco JA. 2013. Heritable genome editing in C. elegans via a CRISPR-Cas9 system. Nat. Methods 10:741–743. 10.1038/nmeth.2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hruscha A, Krawitz P, Rechenberg A, Heinrich V, Hecht J, Haass C, Schmid B. 2013. Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish. Development 140:4982–4987. 10.1242/dev.099085 [DOI] [PubMed] [Google Scholar]
- 17. Jiang W, Zhou H, Bi H, Fromm M, Yang B, Weeks DP. 2013. Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice. Nucleic Acids Res. 41:e188. 10.1093/nar/gks1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mali P, Yang L, Esvelt KM, Aach J, Guell M, DiCarlo JE, Norville JE, Church GM. 2013. RNA-guided human genome engineering via Cas9. Science 339:823–826. 10.1126/science.1232033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ren X, Sun J, Housden BE, Hu Y, Roesel C, Lin S, Liu LP, Yang Z, Mao D, Sun L, Wu Q, Ji JY, Xi J, Mohr SE, Xu J, Perrimon N, Ni JQ. 2013. Optimized gene editing technology for Drosophila melanogaster using germ line-specific Cas9. Proc. Natl. Acad. Sci. U. S. A. 110:19012–19017. 10.1073/pnas.1318481110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, Jaenisch R. 2013. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153:910–918. 10.1016/j.cell.2013.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Donald RG, Roos DS. 1995. Insertional mutagenesis and marker rescue in a protozoan parasite: cloning of the uracil phosphoribosyltransferase locus from Toxoplasma gondii. Proc. Natl. Acad. Sci. U. S. A. 92:5749–5753. 10.1073/pnas.92.12.5749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Khan A, Behnke MS, Dunay IR, White MW, Sibley LD. 2009. Phenotypic and gene expression changes among clonal type I strains of Toxoplasma gondii. Eukaryot. Cell 8:1828–1836. 10.1128/EC.00150-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Behnke MS, Fentress SJ, Mashayekhi M, Li LX, Taylor GA, Sibley LD. 2012. The polymorphic pseudokinase ROP5 controls virulence in Toxoplasma gondii by regulating the active kinase ROP18. PLoS Pathog. 8:e1002992. 10.1371/journal.ppat.1002992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Donald RG, Roos DS. 1993. Stable molecular transformation of Toxoplasma gondii: a selectable dihydrofolate reductase-thymidylate synthase marker based on drug-resistance mutations in malaria. Proc. Natl. Acad. Sci. U. S. A. 90:11703–11707. 10.1073/pnas.90.24.11703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Roos DS, Sullivan WJ, Striepen B, Bohne W, Donald RG. 1997. Tagging genes and trapping promoters in Toxoplasma gondii by insertional mutagenesis. Methods 13:112–122. 10.1006/meth.1997.0504 [DOI] [PubMed] [Google Scholar]
- 26. Critchlow SE, Jackson SP. 1998. DNA end-joining: from yeast to man. Trends Biochem. Sci. 23:394–398. 10.1016/S0968-0004(98)01284-5 [DOI] [PubMed] [Google Scholar]
- 27. Donald RG, Carter D, Ullman B, Roos DS. 1996. Insertional tagging, cloning, and expression of the Toxoplasma gondii hypoxanthine-xanthine-guanine phosphoribosyltransferase gene. Use as a selectable marker for stable transformation. J. Biol. Chem. 271:14010–14019. 10.1074/jbc.271.24.14010 [DOI] [PubMed] [Google Scholar]
- 28. Chen C, Fenk LA, de Bono M. 2013. Efficient genome editing in Caenorhabditis elegans by CRISPR-targeted homologous recombination. Nucleic Acids Res. 41:e193. 10.1093/nar/gkt805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ohshima J, Lee Y, Sasai M, Saitoh T, Su Ma J, Kamiyama N, Matsuura Y, Pann-Ghill S, Hayashi M, Ebisu S, Takeda K, Akira S, Yamamoto M. 2014. Role of mouse and human autophagy proteins in IFN-gamma-induced cell-autonomous responses against Toxoplasma gondii. J. Immunol. 192:3328–3335. 10.4049/jimmunol.1302822 [DOI] [PubMed] [Google Scholar]
- 30. Wu Y, Liang D, Wang Y, Bai M, Tang W, Bao S, Yan Z, Li D, Li J. 2013. Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell 13:659–662. 10.1016/j.stem.2013.10.016 [DOI] [PubMed] [Google Scholar]
- 31. Schwank G, Koo BK, Sasselli V, Dekkers JF, Heo I, Demircan T, Sasaki N, Boymans S, Cuppen E, van der Ent CK, Nieuwenhuis EE, Beekman JM, Clevers H. 2013. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell 13:653–658. 10.1016/j.stem.2013.11.002 [DOI] [PubMed] [Google Scholar]
- 32. Jinek M, East A, Cheng A, Lin S, Ma E, Doudna J. 2013. RNA-programmed genome editing in human cells. Elife 2:e00471. 10.7554/eLife.00471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Heaslip AT, Nishi M, Stein B, Hu K. 2011. The motility of a human parasite, Toxoplasma gondii, is regulated by a novel lysine methyltransferase. PLoS Pathog. 7:e1002201. 10.1371/journal.ppat.1002201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelsen TS, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. 2014. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343:84–87. 10.1126/science.1247005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang T, Wei JJ, Sabatini DM, Lander ES. 2014. Genetic screens in human cells using the CRISPR-Cas9 system. Science 343:80–84. 10.1126/science.1246981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Khan A, Taylor S, Ajioka JW, Rosenthal BM, Sibley LD. 2009. Selection at a single locus leads to widespread expansion of Toxoplasma gondii lineages that are virulence in mice. PLoS Genet. 5:e1000404. 10.1371/journal.pgen.1000404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bastin P, Bagherzadeh Z, Matthews KR, Gull K. 1996. A novel epitope tag system to study protein targeting and organelle biogenesis in Trypanosoma brucei. Mol. Biochem. Parasitol. 77:235–239. 10.1016/0166-6851(96)02598-4 [DOI] [PubMed] [Google Scholar]
- 38. Starnes GL, Coincon M, Sygusch J, Sibley LD. 2009. Aldolase is essential for energy production and bridging adhesin-actin cytoskeletal interactions during parasite invasion of host cells. Cell Host Microbe 5:353–364. 10.1016/j.chom.2009.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Parussini F, Tang Q, Moin SM, Mital J, Urban S, Ward GE. 2012. Intramembrane proteolysis of Toxoplasma apical membrane antigen 1 facilitates host-cell invasion but is dispensable for replication. Proc. Natl. Acad. Sci. U. S. A. 109:7463–7468. 10.1073/pnas.1114661109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Alaganan A, Fentress SJ, Tang K, Wang Q, Sibley LD. 2014. Toxoplasma GRA7 effector increases turnover of immunity-related GTPases and contributes to acute virulence in the mouse. Proc. Natl. Acad. Sci. U. S. A. 111:1126–1131. 10.1073/pnas.1313501111 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental methods. Download
Plasmids used in this study.
Primers used in this study.
A map and sequence file for the plasmid expressing CAS9 and a single guide RNA (sgRNA) targeting the UPRT gene in T. gondii. Download