

Abstract
microRNAs (miRNA) are regulators of cellular pathways and alterations of normal miRNA expression levels have been shown to increase tumorigenesis. miR-24 has been demonstrated as having both tumor suppressive and oncogenic properties depending on cell context. Here we demonstrate a possible role for pre-miR-24-2 as a tumor suppressor in the MCF-7 breast cancer cell line through the preferential processing of mature miR-24-2* over miR-24. Specifically, we show that the ectopic expression of miR-24-2* in MCF-7 breast cancer cells results in a suppression of cellular survival both in vivo and in vitro. Notably, the overexpression of miR-24-2* results in a dampening of cell survival through the targeted suppression of PKCα. Additionally, a similar biological change is observed in vivo where MCF-7 cells overexpressing pre-miR-24-2 have decreased tumorigenicity and tumor incidence. Taken together our data demonstrate that when overexpressed biogenesis of the pre-miR-24-2 favors miR-24-2* in the MCF-7 breast cancer cell line and suggests a tumor suppressive role for miR-24-2* observed through the inhibition of PKCα-mediated cellular survival.
Keywords: miRNA maturation, PKCα cellular survival, breast cancer, strand preference, miR-24-2*
Introduction
microRNAs (miRNAs) are important regulators of all facets of cellular function, regulating gene expression through translational inhibition by binding to the 3′untranslated region (UTR) of target mRNA. Interestingly, miRNAs are dysregulated in many human cancers and are emerging as administrators of cellular transformation and cancer progression. Due to the role of miRNAs in cancer development and progression, many miRNAs are being classified by their oncogenic or tumor suppressor activity [1-4].
miR-24 governs cellular development and proliferation, acting as a tumor suppressor or oncogene in a cell type-specific manner [5]. In myeloid cells miR-24 was shown to be an oncogene, acting to increase cellular proliferation through the repression of a MAPK phosphatase, mitogen-activated protein kinase phosphatase 7 (MKP-7), a negative regulator of MAPK signaling [6]. miR-24 has also been implicated as an oncogene in prostate cancer cells where it was shown that miR-24 inhibits apoptosis through inhibition of Fas associated protein 1 (FAF1) expression, a regulator of programmed cell death [7]. In 2009, Walker et al revealed that miR-24 represses apoptosis in the neural retina through negative regulation of caspase-9 and apoptotic peptidase activating factor 1 (APAF-1), further demonstrating a role for miR-24 as an oncogene [8]. In contrast, miR-24 has been described as a tumor suppressor in colon cancer cell lines by targeting and repressing dihydrofolate reductase (DHFR), a protein associated with enhanced proliferation [9]. Additionally, multiple studies have demonstrated that miR-24 regulates the cell cycle both positively and negatively [5, 10]. Many studies have reported miRNA profiles correlating microRNA expression levels to breast cancer tumor grade and receptor status [11]. Breast cancer profiling demonstrated that miR-24 is negatively regulated by estrogen and is expressed at lower levels in primary breast samples versus metastatic solid tumors [12, 13]. The passenger strand in the pre-miR-24-2 stem loop, miR-24-2*, is increased in MCF-7 breast cancer cells when compared to non-malignant mammary epithelia [14]. This suggests that opposing oncogenic and tumor suppressive roles may be performed by the pre-miR-24-2 hairpin loop in breast carcinomas.
Protein Kinase C alpha (PKCα) is a conventional PKC isoform which phosphorylates serine/threonine residues activating pathways involved in normal and neoplastic cellular functions such as apoptosis, proliferation, and differentiation [15]. Once activated, PKCα functions through activation of downstream signaling such as the mitogen-activated protein kinase (MAPK) cascade [16]. PKCα has been demonstrated to directly phosphorylate Raf-1, an upstream activator of the MAPK pathway, which ultimately results in increased phosphorylation of the extracellular signal regulated kinases 1 and 2 (ERK1/2). Through direct activation of the Raf-MEK1/2-Erk1/2 pathway, PKCα is able to induce survival genes which aid in the transformation and progression of neoplasms [17, 18]. Increased expression of PKCα in breast cancers is commonly associated with a more malignant phenotype [19]. In addition to effects on survival and proliferation, PKCα has been demonstrated to promote the metastatic capacity and aggressiveness of neoplasms through the Erk pathway [20-22].
In this study we demonstrate that stable overexpression of pre-miR-24-2 in the MCF-7 human breast cancer cell line promotes tumor suppressive signaling and biological changes. Specifically, we demonstrate that the overexpression of pre-miR-24-2 leads to increased levels of miR-24-2* and the decreased expression of its mRNA target, PKCα. Additionally, overexpression of pre-miR-24-2 is correlated with decreased PMA-mediated cellular survival in vitro. Finally, we show that pre-miR-24-2 alters tumor cell survival in vivo evident through both decreased tumor incidence and tumorigenicity. Taken together, our data demonstrate the possible role for miR-24-2* as a tumor suppressor in the ER-positive MCF-7 breast cancer cell line.
Materials and Methods
Cells and reagents
MCF-7 human breast cancer cell line was acquired from American Type Culture Collection (Manassas, VA). The MCF-7 and MCF-7-TN cell lines were cultured as previously described [23]. MCF-7-TN-R cells were generated by exposing MCF-7 cells to increasing concentration of TNFα until resistance was established [24]. Cells were maintained in Dulbecco's modified Eagle's medium (DMEM; pH 7.4; Invitrogen Corp., Carlsbad, CA) supplemented with 10% fetal bovine serum (Hyclone, Salt Lake City, UT), 1% non-essential amino acids, minimal essential amino acids, sodium pyruvate, penicillin/ streptomycin, and insulin under mycoplasma-free conditions at 37°C in humidified 5% CO2 and 95% as previously described [25].
Animals
SCID/beige immuno-compromised female ovariectomized mice (4-6 weeks old) were obtained from Charles River Laboratories (Wilmington, MA). The animals were allowed a period of adaptation in a sterile and pathogen-free environment with food and water ad libitum. Mice were divided into 2 treatment groups of five mice each: MCF-7-vector and MCF-7- miR-24-2. Cells were harvested in the exponential growth phase using a PBS/EDTA solution and washed. Viable cells (5 × 106) in 50 μl of sterile PBS suspension were mixed with 100 μl Reduced Growth Factor Matrigel (BD Biosciences, Bedford, MA). Bilateral injections were administered into the mammary fat pad using 27 ½ gauge sterile syringes. All procedures in animals were carried out under anesthesia using a mix of isofluorane and oxygen. Tumor size was measured every 2-3 days using digital calipers. The volume of the tumor was calculated using the formula: 4/3π LS2 (L= larger radius; S = shorter radius). At necroscopy, animals were euthanized by cervical dislocation after exposure to CO2. Tumors were removed and frozen in liquid nitrogen or fixed in 10% formalin for further analysis. All procedures involving these animals were conducted in compliance with State and Federal laws, standards of the U.S. Department of Health and Human Services, and guidelines established by Tulane University Animal Care and Use Committee. The facilities and laboratory animals program of Tulane University are accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care.
RNA Extraction and Quantitative Real Time RT-PCR
MCF-7-vector and MCF-7-miR-24-2 cells were grown in 10% DMEM media and treated with 20ng/ml PMA or DMSO for 24 hours. Cells were harvested and total RNA extraction was performed using Qiagen RNeasy RNA purification system (Valencia, CA) according to the manufacturer's protocol. microRNA was extracted using Qiagen miRNeasy purification system (Valencia, CA) according to the manufacturer's protocol. The quantity and quality of the RNA and miRNA were determined by measuring the absorbance at 260 and 280 nm using the NanoDrop ND-1000 (NanoDrop, Wilmington, DE). 2ug of total RNA was reverse-transcribed using the BioRad iScript kit and qPCR was performed using BioRad SYBR green (BioRad, Hercules, CA). Actin, MMP-1, E2F2, PKCα, and E-cadherin genes were amplified, n ≥ 3 independent biological repeats. For microRNA expression, total RNA was extracted using Qiagen miRNeasy purification system (Qiagen, Valencia, CA) according to the manufacturer's protocol. For miRNA, 1.5 ug of total RNA was reverse–transcribed using the RT2 transcription kit (SA Bioscience, Frederick, MD) and qPCR was performed using SYBR green (SA Biosciene, Frederick, MD), pre-miR-24, (Qiagen, Valencia, CA), miR-24-2* (SA Bioscience, Frederick, MD), miR-24 (SABiosciences, Frederick, MD), and U6 (SABiosciences, Frederick, MD) primers. Data was analyzed by comparing relative target gene expression to β-actin control for mRNA and U6 for miRNA and raw U6 CT values are displayed as Supplemental Figure S1. Relative gene expression was analyzed using 2-ΔΔCt method [26].
Cell Viability Assay
Viability assays were performed as previously described [27, 28]. Briefly, cells were plated at a density of 7.5 × 105 cells per well in a 96-well plate and allowed to attach overnight. Cells were then treated with DMSO, TNF-α (1ng/mL) (PeproTech, Inc; Rocky Hill, NJ) PMA (20ng/mL) (Sigma Aldrich; St. Louis, MO), TNF-α + PMA for 24 h. Following treatment, 20 μL of 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT, 5 mg/ml; Fisher Scientific) reagent was incubated in each well for 4 h. Cells were lysed with 20% SDS in 50% dimethylformamide. Absorbances were read on an ELx808 Microtek plate reader (Bio-Tek Instruments, Winooski, VT) at 550 nm, with a reference wavelength of 630 nm.
Transfection of MCF-7 Cell Line
The protocol for the generation of stable cells lines was modified from previously published transfection methods from our lab [29]. 1 × 106 MCF-7 cells were plated in 10cm2 plates and allowed to adhere at 37°C. After 24 hours, plates were transfected with 5ug of pMSCV-puro-pre-miR-24-2-GFP or pMSCV-puro-vector-GFP plasmid in 100ul Opti-MEM. Transfection was accomplished using 15 ul Lipofectamine (3:1 ratio of Lipofectamine to DNA) per manufacturer's instructions (Invitrogen Corp, Carslbad CA). Media was changed the following day and cells were grown in 10% DMEM supplemented as described above. Three days post transfection cells were treated with1ug/ul puromycin every two days for 2 weeks selecting for only cells resistant to puromycin and expressing GFP. Once stable cells were obtained, cells were then pooled and maintained in 10% DMEM as described above. Pools were assessed for stable expression of pre-miR-24-2, miR-24, and miR-24-2* by qRT-PCR. Transient transfections of miR-24 mimic, miR-24-2* mimic or vector were carried out as follows. Parental MCF-7 cells were plated at 1 × 106 cells per dish in a 10cm2 dish in 5% phenol free DMEM, cells were allowed to adhere at 37°C. After 24 hours plates were transfected with 5ug of miR-24 mimic, miR-24-2* mimic, or vector (Thermo Scientific). Transfection was accomplished using Lipofectamine as per manufacturers protocol (Invitrogen Corp, Carslbad Ca). After 24 hours cells were collected for total RNA extraction and qPCR was performed for PKCα mRNA levels.
Western Blot Analysis
MCF-7 cells were grown in DMEM supplemented with 10% FBS as described above. Cells were washed with PBS and lysed with M-Per lysis buffer supplemented with 1% protease inhibitor and 1% phosphatase inhibitors I and II (Invitrogen Corp, Carlsbad CA). Supernatant containing whole protein extracts was obtained through centrifugation at 12,000 RPM for ten minutes at 4 °C. Amount of protein extracted per sample was determined by measuring the absorbance at 260 and 280 nm using the NanoDrop ND-1000. Proteins were heat denatured and 40ug of protein were loaded per lane on Bis-Tris-nuPAGE gel (Invitrogen Corp, Carlsbad CA). Protein was transferred to nitrocellulose using iBlot and iBlot transfer stacks per manufacturer's protocol (Invitrogen Corp, Carlsbad CA). To block nonspecific binding, nitrocellulose membrane was incubated in 3% milk in 1% TBS-T for 1 hour. Overnight incubation of membrane with primary antibody for PKCα (1:1,000; Cell Signaling Technology, Davers MA) at 4 °C was followed by three fifteen minute washes in 1% TBS-T. Next membrane was incubated for 1 hour in secondary antibody (1:10,000; Licor Biotechnology, Lincon NE) followed by three ten minute washes in 1% TBS-T. Protein band density was determined using a Licor gel imager (Licor Biotechnology, Lincon, NE).
PKC-alpha 3′UTR-Luciferase Assay
Cells were plated in 24-well plates at a density of 5 × 105 cells/well and allowed to attach overnight. After 18 hours, cells were transfected for 24 hours in serum-free DMEM with 300ng of PKC-alpha 3′UTR-luciferase plasmid, and 20ng of miR-24 mimic, miR-24-2* mimic, or vector (Thermo Scientific) by using 6 : l of Effectene transfection reagent (QIAGEN, Valencia, CA ) per microgram of DNA. Cells were incubated at 37 °C. After 24 hours, the medium was removed, and 100ul of lysis buffer was added per well and incubated for 15 min at room temperature. Luciferase activity for the cell extracts was determined using luciferase substrate (Promega, Madison, WI) in an Auto Lumat Plus luminometer (Berthold, Oak Ridge, TN). 3′UTR PKCα plasmid was generated through amplification of the PKCα 3′UTR using primers: forward PRKCA 4929 (GACTACGCGTTGTGTGCACATCACACCATT) and reverse PRKCA 5589 (GACTAAGCTTAACATTCCCTGGATCTGCTG). Primer number represents the segment of DNA sequence obtained from Ensembl. Primers were designed with Primer3. JY genomic DNA was used to amplify PKCα 3′UTR segments and PCR product was ligated into the vector pMIR-REP-dCMV plasmid with MluI and HindIII.
Statistical Analysis
Statistical Analyses were performed using Graph Pad Prism 5. Student's t-test was used to determine p values with statistically significant values of p <0.05.
Results
Increased expression of pre-miR-24-2 in MCF-7 breast cancer cells leads to decreased expression of PKCα
As products of the pre-miR-24-2 hair pin loop have been demonstrated as having differences in expression levels among breast cancer samples or cell lines, where miR-24 expression was shown to be lower in more metastatic solid tumors compared to non-metastatic and miR-24-2* is increased in MCF-7 breast cancer cell line versus non malignant mammary epithelia [13, 14], we set out to determine if miR-24 had an oncogenic or tumor suppressive function in a classical breast carcinoma cell line. We chose to overexpress the pre-miR-24-2 miRNA in the ER-positive MCF-7 cell line to determine the effects of miR-24 on breast cancer progression. MCF-7 cells were transduced using 5ug of either a pMSCV-puro-vector-GFP plasmid or a pMSCV-puro-pre-miR-24-2-GFP plasmid (Supplemental Figure S2). Both plasmids contained sequences encoding antibiotic resistance to puromycin and a GFP tag. Following transduction, stable MCF-7-vector and −pre-miR-24-2 pools were selected using puromycin. Once established no distinct change in cellular morphology was observed between the two cell lines (data not show).
TargetScan is a program developed to predict mRNA targets for miRNAs based on complementary matches in seed sequences. Based on these predictions, miR-24 is predicted to target many genes involved in cancer progression through cell cycle regulation and cellular proliferation. Potential TargetScan targets PKCα, PKCη, and E-cadherin, along with the previously defined miR-24 target E2F2 [30], were chosen for validation in our cell line due to their roles in cell cycle progression, epithelial-to-mesenchymal transition (EMT), and proliferation [30]. Validation of changes in target mRNA expression was determined by qPCR in MCF-7-pre-miR-24-2 cells compared to MCF-7-vector cells. The MCF-7-pre-miR-24-2 cell line demonstrated significantly decreased mRNA levels of PKCα (Figure 1A), E-cadherin, and E2F2 compared to vector cells (Supplemental Figure S3). There was no significant change in mRNA levels for PKCη (data not shown). Of the three targets downregulated by miR-24, PKCα was chosen for further study based on the significant role PKCα plays in tumor progression via enhanced proliferation and cellular survival. MCF-7-vector and MCF-7-pre-miR-24-2 cell lines were collected and total protein extracted for western blot analysis for PKCα. The MCF-7-pre-miR-24-2 cell line revealed a decrease in total PKCα protein levels compared to vector cells (Figure 1B), confirming that the observed decrease in PKCα mRNA levels translates to decreased protein levels.
Figure 1. Pre-miR-24-2 decreases PKCα and PKCα-mediated gene expression in vitro.
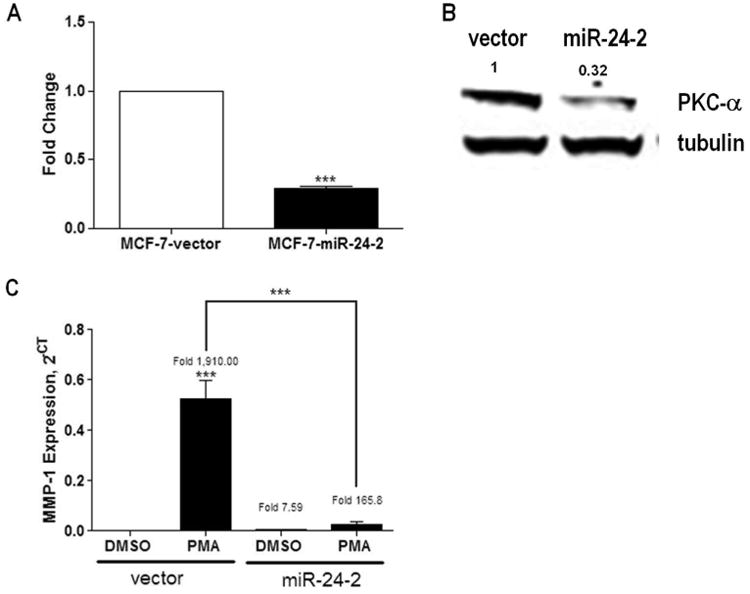
(A) MCF-7-vector and −pre-miR-24-2 cell lines were harvested for total RNA. qPCR was performed for PKCα. Cycle number was normalized to beta-actin and vector control designated as 1. (B) Cells were harvested for total protein extraction and western blot analysis was performed for total PKCα expression. Loading control was alpha tubulin, n=1. (C) qPCR was performed for MMP-1 following 24 hours of treatment with 20ng/ml PMA or vehicle control. Cycle number was normalized to beta-actin and vector designated as 1, results represent relative expression value. *** Significantly different from DMSO treated vector, p < 0.001. Error bars represent SEM, n > 3 independent biological repeats.
Loss of PKCα in MCF-7 cells overexpressing pre-miR-24-2 decreases downstream PKCα mediated gene expression
To determine if decreased expression of PKCα altered the MCF-7 phenotype, MCF-7-pre-miR-24-2 and -vector cells were treated with phorbol 12-myristate 13-acetate (PMA) followed by qPCR. PMA, a phorbol ester, directly activates PKCα in MCF-7 cells mediating increased MAPK signaling and resulting in increased cell survival signals [1, 31]. PMA-induced PKCα activation is evident by increased expression of matrix metalloproteinase-1 (MMP-1) [32]. Following treatment with PMA, both MCF-7-vector and −pre-miR-24-2 cells demonstrated a significant fold increase in MMP-1 expression compared to vehicle treated vector cells (Figure 1C). While there was an observed significant increase in MMP-1 in both the MCF-7-vector and-pre-miR-24-2 cell lines following PMA treatment, the MCF-7-vector cell line demonstrated a significantly higher induction compared to −pre-miR-24-2 cells (p<0.01). The diminished effect of PMA on MMP-1 expression in the MCF-7-pre-miR-24-2 cell line suggests miR-24 may be inhibiting activation of the MAPK pathway through abrogation of PKCα expression.
Overexpression of pre-miR-24-2 in MCF-7 breast cancer cells decreases PMA-mediated cell survival
Elevated levels of PKCα have been associated with increased cell survival and drug resistance in breast carcinomas through the increased activation of ERK signaling. Therefore, we performed MTT viability assays to determine the effects of pre-miR-24-2 overexpression on cell survival in the MCF-7 cell line. Previously, we have demonstrated that treatment with TNFα induces apoptosis while PMA treatment protects from TNFα-induced apoptosis [33, 34]. MCF-7-pre-miR-24-2 cells and -vector cells were treated with PMA, TNFα, TNFα + PMA, or vehicle control for 24 hours. Each cell line was normalized to its own respective DMSO control and, following treatment with TNFα, both cell lines showed a significant decrease in cell viability compared to respective vehicle treated control. Interestingly, while MCF-7-vector cells demonstrated the expected cell survival advantage following treatment with TNFα and PMA, this was not observed in MCF-7-pre-miR-24-2 cells. With average changes in viability of 84.99% +/- 8.9, there was no significant change in MCF-7-vector cells treated with a combination of TNFα + PMA compared to vehicle treated vector cells. However, MCF-7-pre-miR-24-2 cells treated with both TNFα + PMA demonstrated a significant decrease in cell viability with only 60.30% +/- 8.5, p<0.01 of cells being viable compared to vehicle treated MCF-7-pre-miR-24-2 cells (Figure 2A). These results suggest a role for pre-miR-24-2 in the inhibition of PKCα-mediated cell survival.
Figure 2. Pre-miR-24-2 decreases PKCα-mediated cellular survival in vitro and in vivo.
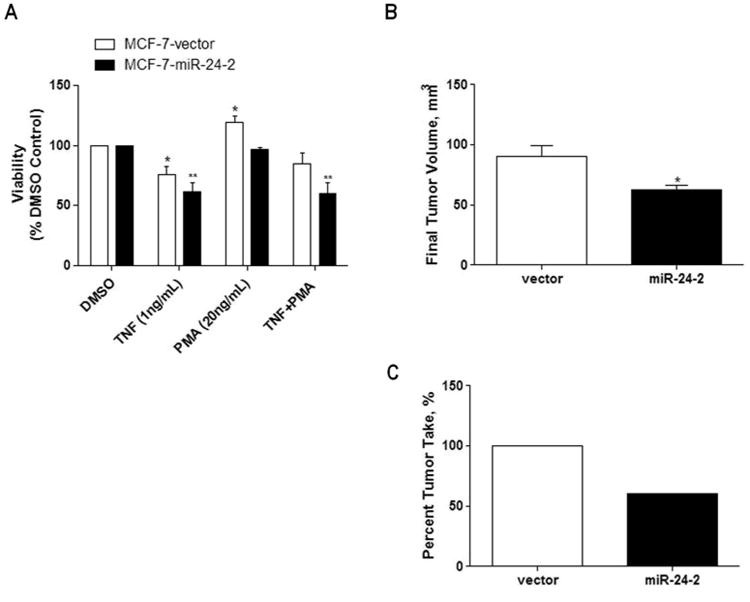
(A) MCF-7-vector and -miR-24-2 cells were grown in 5% DMEM for 48 hours prior to treatment with TNF (1ng/mL), PMA (29ng/mL), or TNF + PMA for 24 hours. Cells were then treated with MTT for 4 hours and lysed. Treatment groups were normalized as % vehicle treated cell line. * Significantly different from vehicle treated vector, p < 0.05. ** Significantly different from vehicle treated vector, p < 0.001. Error bars represent SEM, n = 5 independent biological repeats. (B) 5 × 106 MCF-7-vector or −miR-24-2 cells were injected into the mammary fat pad of SCID/beige female mice. Final tumor volume and tumor formation was obtained at day 52. (A) Final tumor volume. n=10 for MCF-7-vector cells and n=6 MCF-7-miR-24-2 cells. * Significantly different from vector, p < 0.05. Error bars represent SEM. (C) Final tumor formation of n=10 injections for MCF-7-vector and-miR-24-2 injected mice.
Overexpression of pre-miR-24-2 decreases tumorigenesis in vivo
To determine if the decreased cellular survival observed in MCF-7-pre-miR-24-2 cells in vitro translated to a decrease in tumorigenicity in vivo, SCID/beige female ovariectomized mice were inoculated bilaterally with either MCF-7-vector or MCF-7-pre-miR-24-2 cells in the mammary fat pad. At necroscopy (day 52 post cell injection), MCF-7-pre-miR-24-2 tumors had an average final tumor volume of 62.53 +/- 3.74 mm3, significantly smaller than the final tumor volume for –vector tumors of 90.61 +/- 9.12 mm3, p < 0.05 (Figure 2B). Additionally, MCF-7-pre-miR-24-2 injected mice demonstrated a decrease in tumor incidence, having only 60% tumor formation compared to 100% tumor formation displayed by the MCF-7-vector injected animals (Figure 2C). Taken together these results demonstrate over expression of pre-miR-24-2 decreases tumor cell survival, evident through both decreased tumor growth and tumor formation.
Cleavage of pre-miR-24-2 results in increased levels on miR-24-2* but not miR-24
qPCR was performed for pre-miR-24-2 stem loop, miR-24, and miR-24-2* in the MCF-7-vector and -pre-miR-24-2 cell lines. MCF-7-pre-miR-24-2 cells demonstrated significantly increased levels of both the pre-miR-24-2 stem loop and mature miR-24-2* compared to vector cells (Figure 3A). Surprisingly, mature miR-24 expression was not significantly increased in MCF-7-pre-miR-24-2 cells compared to vector cells. To determine if the observed alteration in pre-miR-24-2 maturation was an artifact of plasmid construct or due to alterations in miRNA biogenesis, we next sought to overexpress the pMSCV-pre-miR-24-2 construct in the MCF-7-TN cell line, an apoptosis resistant MCF-7 variant. Once stabile cell lines were established, qPCR for miR-24 and miR-24-2* revealed significantly increased expression of both miRNAs (Figure 3B). This suggests a variation in the selection of guide strand over passenger strand in stem loop processing exists in MCF-7 breast carcinoma cells. Supplemental Figure S4 demonstrates the biogenesis of pre-miR-24-2 to miR-24 and miR-24-2*.
Figure 3. MCF-7 breast cancer cells preferentially increase miR-24-2* expression.
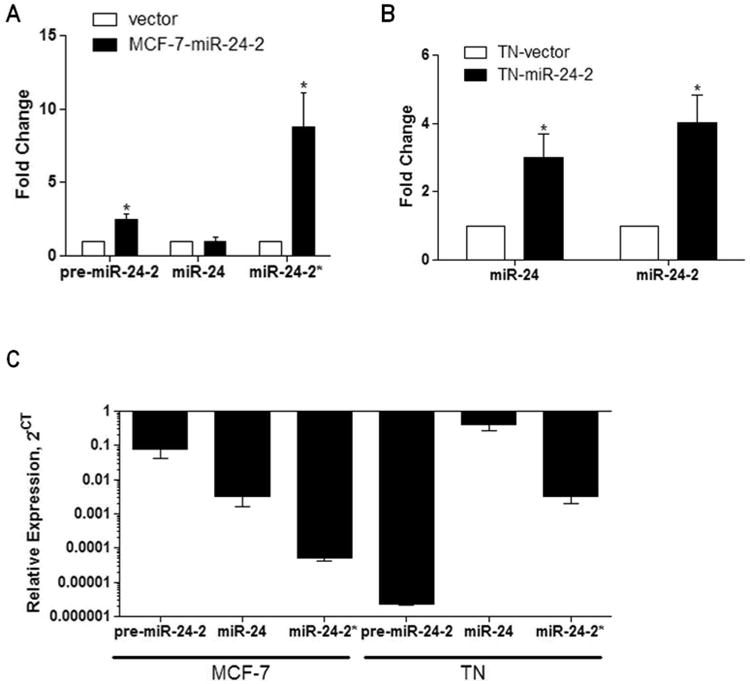
(A) MCF-7-vector and −pre-miR-24-2 cell lines (n=4) and (B) MCF-7-TN-vector and -pre-miR-24-2 cell lines (n=3) were harvested for total RNA. qPCR was performed for pre-miR-24-2, mature miR-24, and miR-24-2*. Cycle number was normalized to U6 RNA and vector. Vector cells were designated as 1. * Significantly different from vector, p < 0.05. Error bars represent SEM, of independent biological repeats. (C) qPCR was performed for miR-24, miR-24-2*, and pre-miR-24-2* in MCF-7 and MCF-7-TN cell lines. Results represent relative expression levels, n = 3 independent biological repeats.
Expression of extended PKCα isoform favors miR-24-2* binding and subsequent repression of PKCα in the MCF-7 cell line
As passenger strand miRNAs are generally considered to be quickly degraded and have little to no expression, we next sought to determine relative expression levels of pre-miR-24-2, miR-24, and miR-24-2* in both the MCF-7 and MCF-7-TN cell lines. Following analysis it was observed that miR-24-2* is expressed in both cell lines; however, its expression levels are lower than that of miR-24 (Figure 3C). To further determine if miR-24-2* can be expressed at levels high enough to elicit a phenotypic response, we used miRBase, an online data base with published miRNA sequences for mature and star miRNAs [35]. Based on deep sequencing analysis of previously published data, miR-24-2* is observed to have expression levels that can be equivalent to the generally favored miR-24, and differences in miRNA sequence may dictate alterations in expression levels (Table 1) [36-39]. Based on this data we believe it is possible for miR-24-2* to be expressed at levels high enough to elicit a phenotypic change. To investigate a possible mechanism through which miR-24-2* may influence PKCα expression, we used an in house algorithm called “Seedfinder” to identify all 7-mer and 8-mer miR-24-2* seeds throughout the human genome. The resulting .bed file containing miR-24-2* target loci were loaded onto the University of California Santa Cruz Genome Browser to visualize possible miR-24-2* seeds within the PKCα locus [40]. As shown in Figure 4, a miR-24-2* 8-mer seed is present within a PKCα isoform that contains an extended 3′ UTR. Alignment data derived from previously published MCF-7 RNA-seq data [41] shows that this extended isoform is dominant (Figure 4A) and that transcription termination in MCF-7 cells occurs downstream from the miR-24-2* 8-mer seed sequence (Figure 4B). Similar studies with miR-155 have previously demonstrated loss of miRNA target recognition through differential isoform expression [42, 43]. SeedFinder was also used to generate targets for miR-24 seed sites and analysis of both miR-24 and miR-24-2* targets demonstrates miR-24-2* as having overall less targets than miR-24, and few of these targets overlap between the two miRNAs (Supplemental Figure S5). All miR-24 and miR-24-2* targets containing at least one 8-mer site or multiple 7-mer sites are listed as Supplemental File 1. We next used Pathway Interaction Database (PID) [42], a free online database, to derive network maps for genes predicted to be targeted by either miR-24 or miR-24-2*. Of the top twenty pathways generated from predicted targets for each miRNA, we saw some correlation between the two miRNAs where both miRNAs governed aspects of IL-6 signaling events in addition to RHOA activity (Supplemental Table 1).
Table 1. Deep Sequencing Reads for miR-24-2 Stem Loop.
| hsa-miR–24–2–5p | hsa–miR–24–3p | Count | RPM | Ref |
|---|---|---|---|---|
| ........CCCGUGCCUACUGAGCUGAAACACAGU...................................... | 1 | 1.08e+03 | 36 | |
| ........CCCGUGCCUACUGAGCUGAAACA.......................................... | 1 | 1.08e+03 | 36 | |
| ........CCCGUGCCUACUGAGCUGAAACACA........................................ | 1 | 1.08e+03 | 36 | |
| ........CCCGUGCCUACUGAGCUGAAACACAG....................................... | 1 | 1.08e+03 | 36 | |
| ...........GUGCCUACUGAGCUGAAACACAGU...................................... | 147 | 26.5 | 37, 38 | |
| ...........GUGCCUACUGAGCUGAAACACAG....................................... | 89 | 4.95 | 37 | |
| ...........GUGCCUACUGAGCUGAAACACA........................................ | 51 | 3.75 | 37 | |
| ...........GUGCCUACUGAGCUGAAACAC......................................... | 7 | 1.88 | 37 | |
| ...........GUGCCUACUGAGCUGAAACACAGUU..................................... | 4 | 2.2 | 37 | |
| ...........GUGCCUACUGAGCUGAAACA.......................................... | 2 | 2.83 | 37 | |
| ............UGCCUACUGAGCUGAAACACAGU...................................... | 7 | 1.57 | 37 | |
| ...............CUACUGAGCUGAAACAC......................................... | 1 | 625 | 36 | |
| ...............CUACUGAGCUGAAACA.......................................... | 1 | 625 | 36 | |
| ................UACUGAGCUGAAACACA........................................ | 1 | 625 | 36 | |
| ................UACUGAGCUGAAACAC......................................... | 1 | 625 | 36 | |
| .................ACUGAGCUGAAACACA........................................ | 2 | 1.31e+03 | 36 | |
| .................ACUGAGCUGAAACAC......................................... | 1 | 625 | 36 | |
| ..................CUGAGCUGAAACACA........................................ | 1 | 625 | 36 | |
| ...............................................ACUGGCUCAGUUCAGCAGGAACA... | 7 | 2.26 | 37 | |
| ...............................................ACUGGCUCAGUUCAGCAGGAACAG.. | 3 | 1.65 | 37 | |
| ...............................................ACUGGCUCAGUUCAGCAGGA...... | 2 | 1.1 | 37 | |
| ...............................................ACUGGCUCAGUUCAGCAGGAAC.... | 1 | 8.85 | 39 | |
| ................................................CUGGCUCAGUUCAGCAGGAACAG.. | 64 | 4.42 | 37 | |
| ................................................CUGGCUCAGUUCAGCAGGAACA... | 43 | 2.98 | 37 | |
| ................................................CUGGCUCAGUUCAGCAGGAAC.... | 2 | 1.1 | 37 | |
| .................................................UGGCUCAGUUCAGCAGGAACAG.. | 27857 | 1.64e+03 | 37, 38, 39 | |
| .................................................UGGCUCAGUUCAGCAGGAACA... | 4009 | 376 | 37, 39 | |
| .................................................UGGCUCAGUUCAGCAGGAAC.... | 1837 | 640 | 37, 38, 39 | |
| .................................................UGGCUCAGUUCAGCAGGAA..... | 304 | 70.4 | 36, 37, 39 | |
| .................................................UGGCUCAGUUCAGCAGGA...... | 81 | 58.6 | 36, 37, 39 | |
| .................................................UGGCUCAGUUCAGCAGG....... | 59 | 1.73e+03 | 36, 37, 39 | |
| .................................................UGGCUCAGUUCAGCAGGAACAGG. | 48 | 7.83 | 36, 37, 38 | |
| ..................................................GGCUCAGUUCAGCAGGAACAG.. | 64 | 6.6 | 37, 39 | |
| ..................................................GGCUCAGUUCAGCAGGAACA... | 7 | 6.96 | 37, 39 | |
| ...................................................GCUCAGUUCAGCAGGAACAG.. | 3 | 35.4 | 37, 38 | |
| ...................................................GCUCAGUUCAGCAGGAACAGG. | 2 | 0.72 | 37 | |
| ......................................................CAGUUCAGCAGGAAC.... | 3 | 1e+03 | 36, 39 | |
| ......................................................CAGUUCAGCAGGAACA... | 2 | 1.31e+03 | 36 | |
| ......................................................CAGUUCAGCAGGAACAG.. | 1 | 625 | 36 | |
Results obtained from miRBase [35]
Figure 4. MCF-7 Breast Cancer Cells Express Multiple PKCα Isoforms.
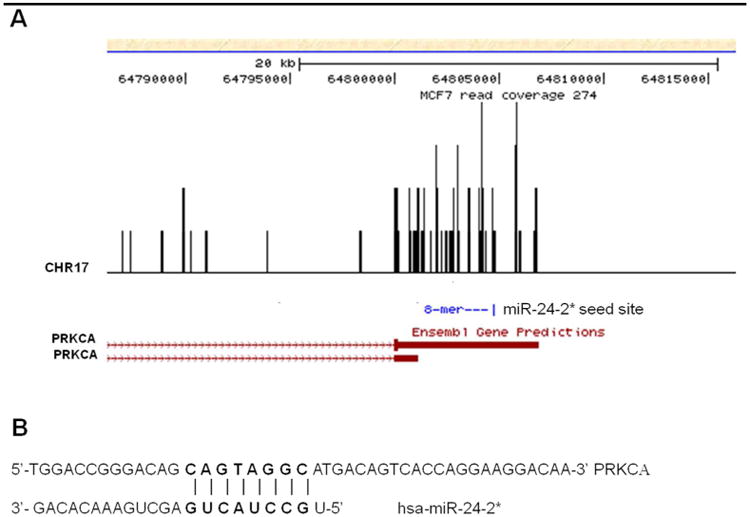
(A) UCSC Genome Browser was used to determine PKCα isorofms for the MCF-7 breast cancer cell line. Analysis miR-24-2* target sites along PKCα transcripts was determined through an in house algorithm “Seedfinder”. (B) Depiction of miR-24-2* seed sequence targeting of PKCα 3′UTR.
Since a pre-miR-24-2 hairpin was stably expressed in the MCF-7 breast cancer cell line, we next sought to determine the individual effects of each mature miRNA strand on PKCα expression. The parental MCF-7 cell line was transiently transfected with a mimic for either miR-24, miR-24-2*, or vector. After 24 hours cells, were collected for total RNA isolation and qPCR was performed for PKCα expression. The miR-24-2* mimic significantly repressed PKCα mRNA levels while there was no observed effect seen following transfection of the miR-24 mimic (Figure 5A). To determine if PKCα is a direct target of miR-24-2*, a 3′UTR luciferase was performed following 24 hours of transient transfection of miR-24 mimic, miR-24-2* mimic, or vector in the MCF-7 cell line. The miR-24-2* mimic significantly repressed the 3′UTR of PKCα compared to vector (normalized to 100). There was no significant change observed following transient transfection of the miR-24 mimic compared to vector control (Figure 5B).
Figure 5. miR-24-2* Directly Targets the 3′UTR of PKCα.
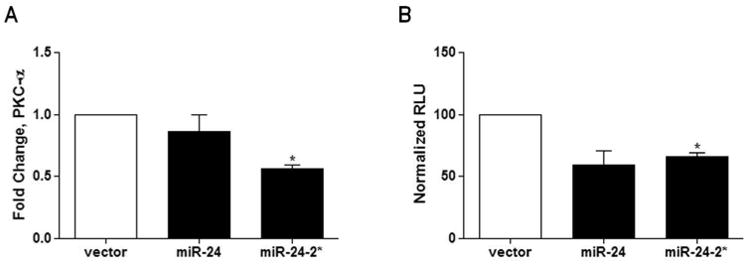
(A) MCF-7 cells were transiently transfected with 5ug of miR-24 mimic, miR-24-2* mimic or vector. After 24 hours cells were collected for total RNA extraction and qPCR was performed for PKCα expression. Normalization was to actin and vector cells designated as 1. (B) PKCα 3′UTR luciferase was performed in MCF-7 cells transiently transfected with miR-24 mimic, miR-24-2* mimic, or vector. MCF-7 cells were transfected with 300ng of PKCα 3′UTR luciferase reporter plasmid and 20ng of either miR-24 mimic, miR-24-2* mimic, or vector for 24 hours prior to cell lysis. Cells were then read on a luminometer. * Significantly different from vector, p ≤ 0.05. Error bars represent SEM, n= 3 of independent biological repeats.
Discussion
Identifying miRNAs acting as possible tumor suppressors or oncomirs allows for a deeper understanding of the mechanisms that regulate signaling pathways which fuel cancer progression. miR-24 has previously been described as a cell type specific oncogene and tumor suppressor. Here, we have provided evidence for the first time suggesting that the over-expression of the pre-miR-24-2 transcript acts to suppress cell survival in the MCF-7 breast cancer cell line both in vivo and in vitro consistent with a tumor suppressive activity. In vitro we demonstrate that the passenger strand to the pre-miR-24-2 hair pin, miR-24-2*, suppresses cellular survival through the negative regulation of PKCα expression. Additionally, our observed decrease in cellular survival correlated with decreased tumorigenicity and tumor incidence in vivo.
Surprisingly, we observed no change in the expression of mature miR-24 in MCF-7-pre-miR-24-2 transfected cells. Recent reports by Srivastava et al demonstrated transient transfection of the pre-miR-24-2 hairpin in MCF-7 cells resulted in the increase of miR-24 and suppression of the miR-24 target BCL2 [45]. Differences in observed miRNA preference may be due to alterations in cellular processing mechanisms that may have occurred following stable expression versus transient expression. Additionally, as precursor structure can dictate alterations in the biogenesis of miRNAs, and Dicer has been demonstrated to account for the majority of alterations in miRNA heterogeneity [46], perhaps some differences exist between the transient pre-miR-24-2 structure used by Srivastava et al and the one used in our experiments, as their structure was a pre-miR-24-2 mimic and our pre-miR-24-2 hairpin was derived from Drosha processing [46, 47]. Following cleavage by Dicer, the miR*, or passenger strand, is frequently degraded and the guide strand is moved into the RISC complex for mRNA binding and gene expression suppression. While mechanisms for preferential selection of the guide strand over the passenger strand have previously been evaluated and are generally recognized as a result of instability of the 5′ ends following Dicer cleavage, a separate mechanism may exist to account for the increased miR-24-2* over miR-24 expression observed in our cell system [48, 49]. Recent studies by Paris et al demonstrate that ER-β is capable of altering miRNA biogenesis and increasing star strand miRNAs associated with the miR-24-1 cluster through ERβ association with the Drosha complex. Additional studies have shown that other signaling molecules, such as ERα and Smad proteins, can alter the biogenesis of some miRNAs [50, 51, 52]. Deep sequencing data of miRNA expression profiles obtained from miRBase demonstrates that some star miRNAs, including miR-24-2*, can be expressed at high levels that may be equivalent to their conventional guide strand counterparts [35]. It has been speculated by Hu et al that these changes in miRNA expression patterns may arise from small changes in the miRNA sequence at the 5′ end [53].
Our previous work has demonstrated that the PKCα isoform is critical for MCF-7 cell survival, implicating it as a key mediator in maintaining cell survival in breast cancer [34]. Through the overexpression of miR-24-2* in MCF-7 cells, we see a decrease of both the RNA and protein levels of PKCα leading to a decrease in PMA-induced cell survival. Previously, it has been shown that PMA increases expression of miR-24 [54]; taken together with our data, this suggests the possibility of a negative feedback loop between pre-miR-24-2 and PKCα. Targeting of PKCα and its downstream effectors, such as MAPK signaling proteins, has been an ongoing process in the development of novel cancer therapeutics. The discovery of novel inhibitors of PKCα, such as miRNA, holds the possibility for development of innovative treatment options in breast cancer therapy. Pre-miR-24-2 maturation in the MCF-7 breast cancer cell line demonstrates preference of the star strand over the guide strand. Here we demonstrate that a star miRNA, miR-24-2*, is capable of eliciting phenotypic changes in theMCF-7 cell line, through the direct inhibition of the 3′UTR of PKCα. Overexpression of miR-24-2* functionally exhibits tumor suppressive activity through the suppression of cellular survival.
Supplementary Material
Supplemental Figure 1 U6 Remains Constant between MCF-7-vector and MCF-7-pre-miR-24-2 Cell Lines. Raw CT values for U6 derived from qPCR when normalizing for miR-24, miR-24-2* expression levels in MCF-7-vector and MCF-7-pre-miR-24-2 cell lines.
Acknowledgments
Grant Support: This research was supported by the National Institutes of Health, National Cancer Institute R01CA125806-01A2 (ME Burow), the Office of Naval Research N00014-16-1-1136 (ME Burow) and the National Center for Research Resources P20RR020152 (BM Collins-Burow).
Abbreviations
- miRNA
microRNA
- PKCα
protein kinase C alpha
- UTR
untranslated region
- MKP-7
mitogen-activated protein kinase phosphatase 7
- FAF-1
fas associated protein 1
- APAF-1
apoptotic peptidase activity factor 1
- DHFR
dihydrofolate reductase
- MAPK
mitogen-activated protein kinase
- ERK
extracellular signal regulated kinase
- ER
estrogen receptor
- PMA
phorbol 12-myristate 13-acetate
- EMT
epithelial to mesenchymal transition
- MMP1
matrix metalloproteinase
- TNFα
tumor necrosis factor alpha
References
- 1.Visone R, Croce CM. miRNAs and Cancer. The American Journal of Pathology. 2009;174(4):1131–1138. doi: 10.2353/ajpath.2009.080794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nature Reviews. 2009;10:704–714. doi: 10.1038/nrg2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garzon R, Calin GA, Croce CM. microRNAs in Cancer. Annual Rev Medicine. 2009;60:167–179. doi: 10.1146/annurev.med.59.053006.104707. [DOI] [PubMed] [Google Scholar]
- 4.Alvarez-Garcia I, Miska EA. microRNA functions in animal development and human disease. Development. 2005;132(21):4653–4662. doi: 10.1242/dev.02073. [DOI] [PubMed] [Google Scholar]
- 5.Chhabra R, Dubey R, Saini N. Cooperative and individualistic functions of the microRNAs in the miR-23a∼27a∼24-2 cluster and its implication in human disease. Molecular Cancer. 2010;9:232. doi: 10.1186/1476-4598-9-232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zaidi SK, Dowdy CR, Winjnen AJ, Lian JB, Azra R, Stein JL, Croce CM, Stein GS. Altered Runx1 Subnuclear Targeting Enhances Myeloid Cell Proliferation and Blocks Differentiation by Activating a miR-24/MKP-7/ MAPK Network. Cancer Research. 2009;69(21):8249–8255. doi: 10.1158/0008-5472.CAN-09-1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qin W, Shi Y, Zhao B, Yao Chengguo, Jin L, Jiexian M, Jin Y. miR-24 Regulates Apoptosis by Targeting the Open Reading Frame (ORF) Region of FAF1 in Cancer Cells. Plos One. 2010;2(5):e9429. doi: 10.1371/journal.pone.0009429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Walker JC, Harland RM. microRNA-24a is required to repress apoptosis in the developing neural retina. Genes and Development. 2009;23:1046–1051. doi: 10.1101/gad.1777709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mishra PJ, Song B, Mishra PJ, Wang Y, Humeniuk R, Banerjee D, Merlino G, Ju J, Bertino JR. mir-24 tumor suppressor activity is regulated independent of p53 and through a target site polymorphism. PLOS ONE. 2009;12(e8445):1–10. doi: 10.1371/journal.pone.0008445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lal A, Kim HH, Abdelmohsen K, Kuwano Y, Pullmann R, Srikantan S, Subrahmanyam R, Martindale JL, Yang X, Ahmed F, Navarro F, Dykxhoorn D, Lieberman J, Gorospe M. p16ink4a translation suppressed by miR-24. PLoS ONE. 2008;3(3):e1864. doi: 10.1371/journal.pone.0001864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blenkiron C, Goldstein LD, Thorne NP, Spiteri I, Chin S, Dunning MJ, Barbosa-Morais NL, Teschendorff AE, Green AR, Ellis IO, Tavare S, Caldas C, Miska EA. MicroRNA expression profiling of human breast cancer identifies new markers of tumor subtype. Genome Biology. 2007;8(10):R214.2. doi: 10.1186/gb-2007-8-10-r214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mailot G, Lacroix-Triki M, Pierredon S, Gratadou L, Schmidt S, Benes V, Roche H, Dalenc F, Auboeuf D, Millevoi S, Vagner S. Widespread estrogen-dependent repression of microRNAs involved in breast tumor cell growth. Cancer Research. 2009;69(21):8332–8340. doi: 10.1158/0008-5472.CAN-09-2206. [DOI] [PubMed] [Google Scholar]
- 13.Baffa R, Fassan M, Volinia S, O'Hara B, Liu CG, Palazzo JP, Gardiman M, Rugge M, Gomella LG, Croce CM, Rosenberg A. MicroRNA expression profiling of human metastatic cancers identifies cancer gene targets. Journal of Pathology. 2009;219:214–221. doi: 10.1002/path.2586. [DOI] [PubMed] [Google Scholar]
- 14.Zhang H, Su S, Zhou Q, Lu Y. Differential expression profiles of microRNAs between breast cancer cells and mammary epithelial cells. Chinese Journal of Cancer. 2009;28(5):1–9. [PubMed] [Google Scholar]
- 15.Newton AC. Protein Kinase C: structure, function, and regulation. Journal of Biological Chemistry. 1995;270(48):28495–28498. doi: 10.1074/jbc.270.48.28495. [DOI] [PubMed] [Google Scholar]
- 16.Nakashima S. Protein Kinase C alpha (PKCalpha): regulation and Biological function. Journal of Biochemistry. 2002;132(5):669–675. doi: 10.1093/oxfordjournals.jbchem.a003272. [DOI] [PubMed] [Google Scholar]
- 17.Kolch W, Heidecker G, Kochs G, Hummel R, Vahidi H, Mischak H, Finkenzeller G, Marme D, Rapp UR. Protein kinase C-alpha activates RAF-1 by direct phosphorylation. Nature. 1993;364:249–252. doi: 10.1038/364249a0. [DOI] [PubMed] [Google Scholar]
- 18.Schonwasser DC, Marais RM, Marshall CJ, Parker PJ. Activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by conventional, novel, and atypical protein kinase C isotypes. Molecular and Cellular Biology. 1998;18(2):790–798. doi: 10.1128/mcb.18.2.790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lahn M, Kohler G, Sundell K, Su C, Paterson BM, Bumol TF. Protein Kinase C alpha expression in breast and ovarian cancer. Oncology. 2004;67(1):1–10. doi: 10.1159/000080279. [DOI] [PubMed] [Google Scholar]
- 20.Ways DK, Kukoly CA, deVente J, Hooker JL, Bryant WO, Posekany KJ, Fletcher DJ, Cook PP, Parker PJ. MCF-7 breast cancer cells transfected with protein kinase C-alpha exhibit altered expression of other protein kinase C isoforms and display a more aggressive neoplastic phenotype. Journal Clinical Investigation. 1995;95(4):1906–1915. doi: 10.1172/JCI117872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gupta AK, Galoforo SS, Berns CM, Martinez AA, Corry PM, Guan KL, Lee YJ. Elevated levels of ERK2 in human breast carcinoma MCF-7 cells transfected with protein kinase C alpha. Cell Prolif. 1996;29(12):655–663. doi: 10.1111/j.1365-2184.1996.tb00979.x. [DOI] [PubMed] [Google Scholar]
- 22.Blobe GC, Sachs CW, Khan WA, Fabbro D, Stabel S, Wetsel WC, Obeid LM, Fine RL, Hannun YA. Selective regulation of expression of protein kinase C (PKC) isoenzymes in multidrug-resistant MCF-7 cells. Functional significance of enhanced expression of PKC alpha Journal Biol Chem. 1993;268(1):658–664. [PubMed] [Google Scholar]
- 23.Struckhoff AP, Bittman R, Burow ME, et al. Novel ceramide analogs as potential chemotherapeutic agents in breast cancer. The Journal of pharmacology and experimental therapeutics. 2004;309:523–532. doi: 10.1124/jpet.103.062760. [DOI] [PubMed] [Google Scholar]
- 24.Antoon JW, White MD, Slaughter EM, et al. Targeting NF-kB mediated breast cancer chemoresistance through selective inhibition of sphingosine kinase-2. Cancer biology & therapy. 2011;11 doi: 10.4161/cbt.11.7.14903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rhodes L, Antoon J, Muir SE, Elliot S, Beckman B, et al. Effects of human mesenchymal stem cells on ER-positive human breast carcinoma cells mediated through ER-SDF-1/CXCR4 crosstalk. Molecular Cancer. 2010;9:295. doi: 10.1186/1476-4598-9-295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schmittgen TD, Zakrajsek BA, Mills AG, Gorn V, Singer MJ, Reed MW. Quantitative reverse transcription-polymerase chain reaction to study mRNA decay: comparison of endpoint and real-time methods. Analytical Biochemistry. 2000;285:194–204. doi: 10.1006/abio.2000.4753. [DOI] [PubMed] [Google Scholar]
- 27.Antoon JW, Liu J, Gestaut MM, Burow ME, Beckman BS, Foroozesh M. Design, synthesis, and biological activity of a family of novel ceramide analogues in chemoresistant breast cancer cells. J Med Chem. 2009;52(18):5748–52. doi: 10.1021/jm9009668. [DOI] [PubMed] [Google Scholar]
- 28.Antoon JW, White MD, Meacham WD, Slaughter EM, Muir SE, Elliott S, Rhodes LV, Ashe HB, Weise TE, Smith CD, Burow ME, Beckman BS. Antiestrogenic effects of the novel sphingosine kinase-2 inhibitor ABC294640. Endocrinology. 2010;151(11):5124–35. doi: 10.1210/en.2010-0420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou C, Nitschke AM, Xiong W, Zhang Q, Tang Y, Bloch M, Elliott S, Zhu Y, Bazzone L, Yu D, Weldon CB, Schiff R, McLachlan JA, Beckman BS, Wiese TE, Nephew KP, Shan B, Burow ME, Wang G. Proteomic analysis of tumor necrosis factor-alpha resistant human breast cancer cells reveals a MEK5/Erk5-mediated epithelial-mesenchymal transition phenotype. Breast Cancer Res. 2008;10:R105. doi: 10.1186/bcr2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lal A, Navarro F, Maher C, Maliszewski LE, Yan N, O'Day E, Chowdhury D, Dykxhoorn DM, Tsai P, Hofmann O, Becker KG, Gorospe M, Lieberman J. miR-24 inhibits cell proliferation by targeting E2F2, MYC, and other cell-cycle genes via binding to “seedless” 3′UTR microRNA recognition elements. Molecular Cell. 2009;35(5):610–625. doi: 10.1016/j.molcel.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nishizuka Y. The role of protein kinase C in cell surface signal transduction and tumour promotion. Nature. 1984;308(5961):693–698. doi: 10.1038/308693a0. [DOI] [PubMed] [Google Scholar]
- 32.Vetter M, Blumenthal SG, Lindemann RK, Manns J, Wesselborg S, Thomssen C, Dittmer J. Ets 1 is an Effector of Protein Kinase C alpha in Cancer Cells. Oncogene. 2005;24(4):650–661. doi: 10.1038/sj.onc.1208234. [DOI] [PubMed] [Google Scholar]
- 33.Burow Me, Weldon CB, Collins-Burow BM, Ramsey N, Mckee A, Klippel A, McLachlan JA, Vlejan S, Beckman BS. Cross-talk between phosphatidylinositol 3-kinase and sphingomyelinase pathways as a mechanism for cell survival/death decisions. J Biol Chem. 2000;275(13):9628–9635. doi: 10.1074/jbc.275.13.9628. [DOI] [PubMed] [Google Scholar]
- 34.Weldon CB, McKee A, Collins-Burow BM, Melnik LI, Scandurro AB, McLachlan JA, Burow ME, Beckman BS. PKC-mediated survival signaling in breast carcinoma cells: A role for MEK1-AP1 signaling. International Journal of Oncology. 2005;26:763–768. [PubMed] [Google Scholar]
- 35.Kozomara A, Griffiths-Jones S. miRBase: integrating microRNA annotation and deep-sequencing data. NAR. 2011;39(Database Issue):D152–D157. doi: 10.1093/nar/gkq1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Witten D, Tibshirani R, Gu SG, Fire A, Lui WO. Ultra-high throughput sequencing-based small RNA discovery and discrete statistical biomarker analysis in a collection of cervical tumours and matched controls. BMC Biol. 2010;8:58. doi: 10.1186/1741-7007-8-58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stark MS, Tyagi S, Nancarrow DJ, Boyle GM, Cook AL, Whiteman DC, et al. Characterization of the Melanoma miRNAome by Deep Sequencing. PLoS One. 2010;5:e9685. doi: 10.1371/journal.pone.0009685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhu JY, Pfuhl T, Motsch N, Barth S, Nicholls J, Grasser F, et al. Identification of novel Epstein-Barr virus microRNA genes from nasopharyngeal carcinomas. J Virol. 2009;83:3333–3341. doi: 10.1128/JVI.01689-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bar M, Wyman SK, Fritz BR, Qi J, Garg KS, Parkin RK, et al. MicroRNA discovery and profiling in human embryonic stem cells by deep sequencing of small RNA libraries. Stem Cells. 2008;26:2496–2505. doi: 10.1634/stemcells.2008-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rhead B, Karolchik D, Kuhn RM, Hinrichs AS, Zweig AS, Fujita PA, Diekhans M, Smith KE, Rosenbloom KR, Raney BJ, Pohl A, Pheasant M, Meyer LR, Learned K, Hsu F, Hillman-Jackson J, Harte RA, Giardine B, Dreszer TR, Clawson H, Barber GP, Haussler D, Kent WJ. The UCSC Genome Browser database: update 2010. Nucleic Acids Research. 2010;38(database issue):D613–9. doi: 10.1093/nar/gkp939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–476. doi: 10.1038/nature07509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu G, Fewell C, Taylor C, Deng N, Hedges D, Zhang K, Lacey M, Zhang H, Yin Q, Cameron J, Lin Z, Zhu D, Flemigton EK. Transcriptome and targetome analysis in MIR155 expressing cells using RNA-seq. RNA. 2010;16:1610–1622. doi: 10.1261/rna.2194910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deng N, Puetter A, Zhang K, Johnson K, Zhao Z, Taylor C, Flemington EK, Zhu D. Isoform-level microRNA-155 target prediction using RNA-seq. Nucleic Acids Research. 2011;39(9):e61. doi: 10.1093/nar/gkr042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.The website of the National Cancer Institute. http://www.cancer.gov.
- 45.Srivastava N, Manvati S, Srivastava A, Pal R, Kalaiarasan P, Shilpi C, et al. miR-24-2 controls H2AFX expression regardless of gene copy number alteration and induces apoptosis by targeting antiapoptotic gene BCL2: a potential for therapeutic intervention. Breast Cancer Research. 2011;13:R39. doi: 10.1186/bcr2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park J, Tian Y, Simanshu D, Chang H, Jee D, Patel D, et al. Dicer recognizes the 5′ end of RNA for efficient and accurate processing. Nature. 2011;475:201–207. doi: 10.1038/nature10198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Starega-Roslan J, Koscianska E, Kozlowski P, Krzyzosiak W. The role of the precursor structure in the biogenesis of microRNA. Cell Mol. 2011;68:2859–2871. doi: 10.1007/s00018-011-0726-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Winter J, Jung S, Keller S, Gregory RI, Diederichs S. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nature Cell Biology. 2009;11:228–234. doi: 10.1038/ncb0309-228. [DOI] [PubMed] [Google Scholar]
- 49.Khvorova A, Reynolds A, Jayasena SD. Functional siRNA and miRNA exhibit strand bias. Cell. 2003;115(2):209–216. doi: 10.1016/s0092-8674(03)00801-8. [DOI] [PubMed] [Google Scholar]
- 50.Yamagata K, Fujiyama S, Ito S, Ueda T, Murata T, Naitou M, et al. Maturation of microRNA is hormonally regulated by a nuclear receptor. Molecular Cell. 2009;36:340–347. doi: 10.1016/j.molcel.2009.08.017. [DOI] [PubMed] [Google Scholar]
- 51.Paris O, Ferraro L, Grober O, Ravo M, Filippo M, Giurato G, et al. Direct regulation of microRNA biogenesis and expression by estrogen receptor beta in hormone-responsive breast cancer. Oncogene. 2012:1–11. doi: 10.1038/onc.2011.583. [DOI] [PubMed] [Google Scholar]
- 52.Hu H, Yan Z, Xu Y, Hu H, Menzel C, Zhou Y. Sequence features associated with microRNA strand selection in humans and flies. BMC Genomics. 2009;10:413. doi: 10.1186/1471-2164-10-413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Siomi H, Siomi M. Posttranscriptional regulation of microRNA biogenesis in animals. Molecular Cell. 2010;38:323–332. doi: 10.1016/j.molcel.2010.03.013. [DOI] [PubMed] [Google Scholar]
- 54.Takagi S, Nakajima M, Kida K, Yamaura Y, Fukami T, Yokoi T. MicroRNAs regulate human hepaotcyte nuclear factor 4alpha, modulating the expression of metabolic enzymes and cell cycle. Journal of Biological Chemistry. 2010;285(7):4415–4422. doi: 10.1074/jbc.M109.085431. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1 U6 Remains Constant between MCF-7-vector and MCF-7-pre-miR-24-2 Cell Lines. Raw CT values for U6 derived from qPCR when normalizing for miR-24, miR-24-2* expression levels in MCF-7-vector and MCF-7-pre-miR-24-2 cell lines.