

Abstract
Excluding 53BP1 from chromatin is required to attenuate the DNA damage response during mitosis, yet the functional relevance and regulation of this exclusion is unclear. Here we show that 53BP1 is phosphorylated during mitosis on two residues, T1609 and S1618, located in its well-conserved ubiquitination-dependent recruitment (UDR) motif. Phosphorylating these sites blocks the interaction of the UDR motif with mononuclesomes containing ubiquitinated histone H2A and impedes binding of 53BP1 to mitotic chromatin. Ectopic recruitment of 53BP1- T1609A/S1618A to mitotic DNA lesions was associated with significant mitotic defects that could be reversed by inhibiting non-homologous end joining. We also reveal that protein phosphatase complex, PP4C/R3β dephosphorylates T1609 and S1618 to allow the recruitment of 53BP1 to chromatin in G1 phase. Our results identify key sites of 53BP1 phosphorylation during mitosis, identify the counteracting phosphatase complex that restores the potential for DDR during interphase, and establish the physiological importance of this regulation.
INTRODUCTION
53BP1 (p53 binding protein 1) is a multi-domain protein with a complex and unique role in the repair of double-strand DNA breaks (DSBs). Recruitment of 53BP1 to DSB sites is essential for its function in the DNA damage response (DDR), and the minimal region (residues 1220 to 1711) required for its recruitment includes the oligomerization domain, tudor domain and a carboxy terminal extension termed the ubiquitination dependent recruitment (UDR) motif (Fradet-Turcotte et al., 2013; Huyen et al., 2004; Iwabuchi et al., 2003; Zgheib et al., 2009). At the chromatin end, dimethylated lysine 20 of histone H4 (Botuyan et al., 2006; Pei et al., 2011), and ubiquitinated lysine 15 of histone H2A (Fradet-Turcotte et al., 2013)are necessary for the recruitment of 53BP1 to chromatin. Recent studies suggest that 53BP1 plays a critical role in choice of DSB repair pathway by promoting non-homologous end joining (NHEJ) mediated repair of a DSB and specifically countering the function of the homologous-recombination (HR) repair protein BRCA1 at a DSB(Bouwman et al., 2010; Bunting et al., 2010; Chapman et al., 2012). This is evident as loss of 53BP1 in a BRCA1-deficient cell restores HR-mediated DSB repair. Function of 53BP1 in DDR is regulated in the course of the cell cycle(Giunta and Jackson, 2011). 53BP1 is hyperphosphorylated during mitosis and this correlates with its exclusion from chromatin and DNA lesions (Giunta et al., 2010; Nelson et al., 2009; van Vugt et al., 2010). The phosphorylation of 53BP1 dissipates as cells move into the G1-phase and participation of 53BP1 in DSB repair is completely restored. We hypothesized that dephosphorylation of 53BP1 is necessary for its role in DSB repair in G1 cells.
We and others have shown that protein phosphatase, PP4C, a PP2A-like phosphatase, regulates the activity of critical DNA repair factors, H2AX, RPA2 and KAP-1(Chowdhury et al., 2008; Lee et al., 2012; Lee et al., 2010a; Liu et al., 2012; Nakada et al., 2008). To systematically identify proteins dephosphorylated by, PP4C, we recently conducted a quantitative phosphoproteomic screen based on the rationale that sites hyperphosphorylated in the absence of PP4C are putative substrates (Lee et al., 2012). We identified two phosphoresidues of 53BP1, threonine 1609 (T1609) and serine 1618 (S1618) that were hyperphosphorylated in the absence of PP4C. Here we demonstrate that the residues T1609 and S1618 are phosphorylated during mitosis to prevent the recruitment of 53BP1 to DNA lesions. These residues get dephosphorylated by a PP4C/R3β complex as cells transit to the G1 phase, and this dephosphorylation event is necessary for the participation of 53BP1 in the DDR. Furthermore, allowing the recruitment of 53BP1 to DNA breaks in mitosis via mutations of T1609 and S1618 leads to defective chromosome segregation.
RESULTS
53BP1 is a bonafide substrate of PP4C/R3β
The two residues T1609 and S1618 are located in the UDR motif which is essential for 53BP1 recruitment to DSB sites (Fig. 1A)(Fradet-Turcotte et al., 2013) and has been shown to be phosphorylated during mitosis (Dephoure et al., 2008), (Grosstessner-Hain et al., 2011).We used isotopically-encoded synthetic phosphopeptide analogs to verify the phosphorylation state and site assignment of our data for pT1609 and pS1618 (Supplementary Fig. 1A). Hyperphosphorylation of 53BP1 during mitosis is detected by a mobility shift during gel electrophoresis (Giunta et al., 2010; van Vugt et al., 2010) therefore we silenced the subunits of PP4 (Gingras et al., 2005) and evaluated the mobility of 53BP1 during the transition from mitosis to G1. Silencing PP4C (the catalytic subunit) and PP4R3β, a regulatory subunit, leads to a shift in 53BP1 mobility in G1 cells (Supplementary Fig. 1B). Silencing other PP4 regulatory subunits (R2, R1 and R3α) did not impact the mobility of 53BP1 (Supplementary Fig. 1B). Consistent with these results, only PP4C and PP4R3β interact with 53BP1 and this association is significantly enhanced in cells synchronized in mitosis (Fig. 1B). Next we sought to confirm that the residues T1609 and S1618 are indeed phosphorylated in mitosis, and that their phosphorylation status is influenced by a PP4C/PP4R3β complex. We generated a phospho-specific antibody that recognizes the combination of phospho-T1609 and phospho-S1618, and observed that mutating either of these residues significantly reduces the antibody signal (Supplementary Fig. 1C). Using the phospho-T1609/pS1618 antibody and an antibody against phospho-S1618 we observe that these two residues are phosphorylated in a population of mitotic cells, but T1609 and S1618 are not phosphorylated as these cells progress to the G1 phase. We observe a detectable increase in phospho-T1609 and phospho-S1618 in the G1 phase of PP4C or PP4R3β silenced cells (Fig. 1C). To further validate these results we monitored the kinetics of dephosphorylation as cells transit from mitosis to G1 (Supplementary Fig. 1D) and observed that depletion of PP4C or PP4R3β blocks the dephosphorylation of T1609 and S1618 (Fig. 1D).
Figure 1. 53BP1 is a bonafide substrate of PP4C/PP4R3β.

A. Schematic representation of human 53BP1 and alignment of the region flanking T1609 and S1618 showing evolutionary conservation of this region. Sequence alignment was performed with ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2).
B. Interaction of 53BP1 with PP4C and PP4R3β. U2OS cells, stably expressing FH-Empty vector (FH-c) or FH-53BP1 were harvested after 8h synchronization in mitosis (M) using 100 ng/ml Nocodazole or left in an asynchronous state (Asyn). Whole cell extracts were immunoprecipitated with anti-FLAG agarose beads and analyzed by immunoblotting using indicated antibodies.
C. Impact of PP4C and PP4R3β silencing on 53BP1 phosphorylation. HeLa cells were transfected with indicated siRNAs against PP4 subunits for 60h and synchronized in mitosis with 100 ng/ml Nocodazole for 8h. Cells were released by mitotic shake-off into media without drug and harvested after 5h (G1 phase). Whole-cell lysates were probed with 53BP1 phospho-1618 (p1618) and 53BP1 phospho-1609/1618 (p1609–1618) antibodies. Total 53BP1 and tubulin were used as loading controls. Phospho-Ser10-histone H3 (pS10-H3) was used to indicate mitotic (M) cells. Cells were probed in parallel with antibodies against PP4R1, PP4R2, PP4R3α, PP4R3β, and PP4C to determine knockdown efficiency and specificity.
D. Kinetics of 53BP1 hyperphosphorylation in PP4C/PP4R3β silenced cells during transition from mitosis to G1 phase. Upper panel: Schematic to study kinetics of 53BP1 hyperphosphorylation. Lower panel: HeLa cells transfected with scrambled control, PP4C or PP4R3β siRNAs, synchronized in mitosis with 100 ng/ml Nocodazole, released by mitotic shake-off, and harvested at indicated time points for western blot analysis. Cell lysates were probed with indicated antibodies.
E. Inhibition of kinases PLK-1 and p38-MAPK in PP4C/PP4R3β silenced cells does not affect hyperphosphorylation of 53BP1 at T1609 and S1618. Upper panel: Schematic to study PLK-1 and p38-MAPK inhibition in PP4C/PP4R3β silenced cells. Lower panel: HeLa cells were transfected with indicated siRNAs and treated with Nocodazole as in Fig 1D. Kinase inhibitors against PLK-1 (BI2536, 20nM) and p38-MAPK (SB202190, 10µM) were added to the cells1h following release from mitosis. Cells were harvested at various time-points after mitotic release and lysates were probed with indicated antibodies.
F. Recombinant PP4C incubated with mitotic extracts shows a dose-dependent decrease in phosphorylation at T1609 and S1618 whereas the catalytically inactive PP4C D82A mutant protein fails to dephosphorylate even at high concentrations. Extract was also incubated with λ-protein phosphatase (PP) as a positive control.
A recent phosphoproteomic study (Grosstessner-Hain et al., 2011) reported that S1618 is phosphorylated by PLK1. We confirmed these results using the pS1618 specific phospho-antibody and the PLK-1 inhibitor, BI2536 (Supplementary Fig. 1E). T1609 gets phosphorylated during mitosis (Dephoure et al., 2008), but the kinase phosphorylating T1609 has not been identified. Based on the prediction algorithm of the Group-based Prediction System (GPS 2.1.2) (Xue et al., 2008) several isoforms of the p38 mitogen-activated protein kinase (MAPK) were predicted to phosphorylate T1609 with the highest prediction scores (Supplementary Fig. 1F). p38 MAPK is active during mitosis and has been implicated in mitotic checkpoints (Lee et al., 2010b). Two different inhibitors of the p38 MAPK, SB203580 and SB202190 along with the pT1609/pS1618 antibody was used to demonstrate that phosphorylation of T1609 is likely to be mediated by p38 MAPK (Supplementary Fig. 1F). The phosphorylation of S1618 is not affected by the inhibitors of p38 MAPK. However sequence based predictions have limited accuracy, and chemical inhibitors may have ‘off-target’ effects, therefore we cannot formally rule out that other kinases, such as CDK1 or NEK2, are also involved in the phosphorylation of T1609.
The elevated levels of phospho-T1609 and phospho-S1618 in PP4C/PP4R3β silenced G1 cells could be due to ectopic activation of PLK1 and p38 MAPK. In that scenario continued PLK1 and p38 MAPK activity would be necessary for maintaining the pT1609/pS1618 signal in the G1 phase of PP4-deficient cells. To evaluate this possibility, we inhibited PLK1 and the p38 MAPK immediately after mitotic exit. Addition of the inhibitors for PLK1 and the p38 MAPK leads to a complete loss of pT1609/pS1618 signal within 3 hours in mitotic cells (Supplementary Fig. 1E and 1F). Depletion of PP4C, or PP4R3β, causes persistence of phospho-T1609 and phospho-S1618 in G1 cells even in the presence of inhibitors for PLK1 and the p38 MAPK (Fig. 1E) for up to 6 hours. This result suggests that elevated level of phospho-T1609 and phospho-S1618 in PP4C/PP4R3β depleted cells is due to the lack of dephosphorylation, and not ectopic phosphorylation of these residues.
To determine whether phospho-T1609 and phospho-S1618 are directly targeted by PP4C, we immunopurified endogenous phospho-53BP1 from mitotic cells and performed dephosphorylation assays. PP4C dephosphorylates both phospho-T1609 and phospho-S1618 in a dose-dependent manner (Fig. 1F). Catalytically inactive PP4C mutant and λ phosphatase served as controls. Together these results strongly suggest that PP4C directly dephosphorylates phosphoresidues, T1609 and S1618 of 53BP1.
Dephosphorylation of 53BP1 is necessary for recruitment to DSBs
We hypothesized that PP4C/PP4R3β mediated dephosphorylation of 53BP1 is necessary for its localization at DSB repair foci in G1 cells. To test this idea we evaluated the localization of 53BP1 in the G1 phase of synchronized HeLa cells at various time points after ionizing radiation (IR), in the presence and absence of PP4C and PP4R3β. There is a striking reduction of 53BP1 foci in PP4C and PP4R3β silenced cells (Fig. 2A). To further confirm the cell cycle phase specificity of this phenotype we utilized the Fucci system (Sakaue-Sawano et al., 2008) to visualize the G1 phase (mKO2-CDT1) in RPE-1 cells. As anticipated, absence of PP4C and PP4R3β causes a significant reduction in 53BP1 foci in G1 cells (Fig. 2B). Localization of upstream factors of 53BP1, such as RNF-168, is not affected by silencing PP4C and PP4R3β (Supplementary Fig. 2).
Figure 2. PP4C/PP4R3β silencing abrogates 53BP1 foci in G1 cells.

A, B: Schematic shows protocol for assessing 53BP1 activity in G1 cells irradiated immediately after mitotic shake-off. PP4C or PP4R3β was depleted by siRNA transfection of HeLa cells (A) or Fucci-RPE1 cells (B). Mitotic cells were collected after 8h Nocodazole treatment, irradiated immediately after release (10 Gy), fixed and co-stained with antibodies for 53BP1 (A, B) and γ-H2AX (A) at indicated times in G1 phase. CDT1/RFP is an internal Fucci cell marker that illuminates cells in G1 phase. Quantification shows percent 53BP1 positive cells in G1. Cells displaying ≥ 10 foci were counted as positive. The data are expressed as mean ± S.D; n = 3. More than 100 cells were quantified per condition.
To evaluate the precise impact of dephosphorylating 53BP1, the two residues T1609 and S1618 that are hyperphosphorylated in the absence of PP4C were altered to phosphonull (T1609A and S1618A) and phosphomimetic mutants (T1609E and S1618D). These mutations were introduced individually and in combination (53BP1-AA and 53BP1-ED) in FLAG and HA (FH)-tagged full-length 53BP1. Using the FLAG antibody the recruitment of these mutants to DSB sites was monitored in HeLa cells. None of the individual mutants have a significant impact on localization of 53BP1 to DSB sites (Supplementary Figure 3A). The 53BP1-AA mutant is also recruited to DNA repair foci at levels compared to wild-type 53BP1 (Fig. 3A and Supplementary Fig. 3A). However, there is almost a complete block in the localization of the 53BP1-ED mutant to DSBs marked by γ-H2AX. Since these experiments were conducted in cells expressing endogenous 53BP1 we further confirmed our results by expressing the wildtype 53BP1, and the 53BP1 phosphomutants (AA and ED) in 53BP1-deficient mouse embryonic fibroblasts (Supplementary Fig. 3B).
Figure 3. Constitutive phosphorylation of 53BP1 at T1609 and S1618 impedes foci formation at DSBs.

A. Phosphomimetic 53BP1 mutant is not recruited to DSBs. Upper panel: Schematic to study 53BP1 focal recruitment. Lower left panel: HeLa cells stably expressing full-length FH-53BP1 (WT, AA, or ED) were prepared for immunofluorescence after mitotic shake-off and 10Gy IR as described in Fig 2A,B. 53BP1 foci were visualized using anti-FLAG antibody. γ-H2AX was stained to mark sites of DNA damage. Lower right panel: Quantification shows percent 53BP1 positive cells in G1 for WT and phospho-mutants. Cells displaying ≥ 10 foci were counted as positive. The data is expressed as mean ± S.D; n = 3; > 100 cells quantified per mutant. Lower right panel inset: Immunoblot showing equal expression levels of different FH-53BP1 constructs (WT, AA, and ED). Irradiated cells released into G1 were analyzed in parallel for expression using anti-FLAG antibody. Tubulin was used as a loading control.
B. The failure of 53BP1 to form foci in PP4R3β silenced cells is rescued by expressing the 53BP1 AA mutant. Left panel: HeLa cells expressing FH-53BP1 WT or indicated phosphomutants were transfected with control siRNA or siRNA against PP4R3β and prepared for immunofluorescence as described in Figure 2A,B. Anti-FLAG antibody was used to visualize 53BP1. γ-H2AX was stained to mark sites of DNA damage. Right panel: Quantification shows percent 53BP1 positive cells in G1 for WT and phospho-mutants. Cells displaying ≥ 10 foci were counted as positive. The data is expressed as mean ± S.D; n = 3; > 100 cells quantified per mutant.
C. Kinetics of 53BP1 recruitment to DNA lesions. To quantify the kinetics of 53BP1 recruitment to DSBs, a multi-photon laser (MPL) system was used. U2OS cells stably expressing full-length GFP-53BP1 (WT, AA, or ED) were treated with Nocodazole for 6h and released into G1. Left panels: Representative freeze-frame images from live-cell movies for each sample. A total of 10 cells were monitored per condition for 53BP1 recruitment to DSBs. Right panel: Quantification of 53BP1 recruitment was done by comparison of average signal intensity at 8 min in individual cells represented by a single dot; mean ± S.D; n = 3.
Depletion of PP4C/PP4R3β has an impact on 53BP1 foci formation in G1 cells that was comparable to the phenotype induced by the expression of phosphomimetic 53BP1-ED mutant (Fig. 2A and Fig. 3A). Theoretically if indeed the impact of PP4R3β on 53BP1 recruitment was due to the dephosphorylation of S1618 and T1609, then the effect of silencing PP4R3β would be reversed by expressing the 53BP1 phosphonull (53BP1-AA) mutant. Consistent with this notion, localization of 53BP1 to DSB sites in PP4R3β-silenced cells was restored by the 53BP1-AA mutant, but not wildtype 53BP1 or the 53BP1-ED mutant (Fig. 3B).This result strongly suggests that PP4C/PP4R3β-mediated dephosphorylation of 53BP1 at T1609 and S1618 is necessary for its localization to DSBs in G1 cells.
The phosphorylation of 53BP1 at T1609 and S1618 correlates with the loss of 53BP1 foci, and this can be due to defective recruitment of 53BP1 to DSB sites or impaired retention of 53BP1 at DSB sites. To address this issue and rigorously quantify the kinetics of 53BP1 recruitment to DSBs we utilized a multi-photon laser (MPL) system that induces DSBs in defined subfemtoliter volumes of the nucleus (Botvinick and Shah, 2007; Hartlerode et al., 2012). Full length 53BP1 fused with GFP was expressed in U2OS cell, and kinetics assessed by live cell imaging. As before, cells synchronized in the G1-phase were utilized for these assays. Wildtype 53BP1 and the 53BP1-AA mutant are recruited rapidly, within a few minutes, to the laser induced DSBs, but consistent with our previous results there is virtually no recruitment of the 53BP1-ED mutants (Fig.3C and Supplementary Videos 1–3). This result indicates that constitutive phosphorylation of 53BP1 at T1609 and S1618 blocks its recruitment to DNA lesions.
Functional relevance of dephosphorylating 53BP1 at T1609 and S1618
Distinct nuclear bodies visible in G1 cells represent endogenous DNA damage that occurs during DNA replication and are carried through mitosis (Harrigan et al., 2011; Lukas et al., 2011). Low doses of aphidicolin increases the frequency of nuclear bodies in cells without triggering the cellular checkpoints. 53BP1 is recruited to nuclear bodies in G1 cells and plays a role in the resolution of these DNA lesions (Lukas et al., 2011). We transfected U2OS cells with GFP-tagged wild-type 53BP1 and the 53BP1 phospho-mutants (AA and ED), treated the cells with aphidocolin and stained them with cyclin A to distinguish G2/M cells (Fig.4A). Consistent with their exclusion from DNA lesions, 53BP1-ED phosphomimetic mutants are not recruited to nuclear bodies whereas the 53BP1-AA phosphonull mutants are localized at nuclear bodies at levels comparable to wild-type 53BP1. Together these results suggest that recruitment of 53BP1 to endogenous and exogenous DNA lesions is compromised by constitutive phosphorylation of the T1609 and the S1618 residue.
Figure 4. Constitutive phosphorylation of 53BP1 at T1609 and S1618 alters 53BP1 recruitment to nuclear bodies and induces resistance to PARP inhibitor.
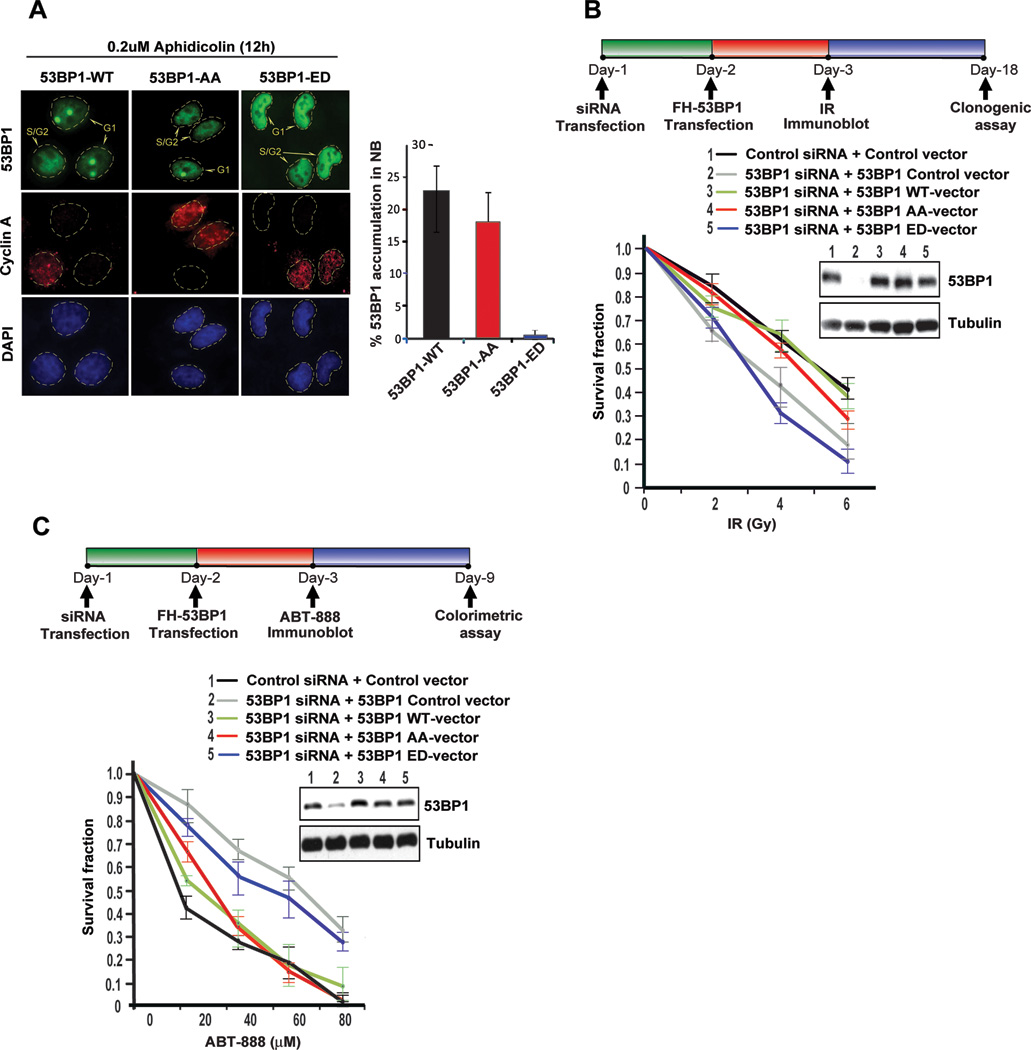
A. Recruitment of 53BP1 to replication stress induced nuclear bodies in G1. U2OS cells, stably expressing full-length GFP-53BP1 (WT, AA or ED) were exposed to low dose of Aphidicolin (0.2 µM) for 12 h and stained with anti-Cyclin A, which illuminates S/G2 cells. Representative images and quantification of 53BP1 accumulation in nuclear bodies are shown; >100 Cyclin A negative cells were quantified; mean ± S.D; n = 3.
B. Radiosensitivity of 53BP1 phosphomutants. Endogenous 53BP1 was silenced in HeLa cells with siRNA and replaced with siRNA-resistant 53BP1 constructs (WT, AA, or ED). At 72 h after siRNA transfection, cells were irradiated at the indicated doses and viability was evaluated by clonogenic survival. Immunoblots performed to confirm siRNA efficiency and expression of siRNA-resistant constructs are shown. Data are expressed as mean ± S.D. n =3.
C. Phosphomimetic 53BP1 mutant rescues PARP inhibitor sensitivity in BRCA1-deficient cells. As in Figure 3B, endogenous 53BP1 was replaced with siRNA-resistant 53BP1 constructs (WT, AA, or ED) in the BRCA1-deficient ovarian cancer line UWB1.289, and sensitivity to clinical-grade PARP inhibitor ABT888 was assessed by CellTiter-Glo® colorimetric viability assay. Immunoblots performed to confirm siRNA efficiency and expression of siRNA-resistant constructs are shown. Data are expressed as mean ± S.D. n =3.
Based on these results we hypothesized that 53BP1 constitutively phosphorylated at T1609 and S1618 is functionally equivalent to the loss of 53BP1. 53BP1-deficient cells have increased radiosensitivity and loss of 53BP1 in BRCA1-deficent tumors induces resistance to PARP inhibitors. Therefore we assessed the radiosensitivity of HeLa cells where endogenous 53BP1 was replaced with the phosphomutants (AA and ED). Cells expressing the 53BP1-ED mutant are significantly more radiosensitive than cells expressing the wild-type 53BP1 and the 53BP1-AA mutant (Fig 4B). Similarly, the BRCA1-mutant ovarian line UWB1.289 was manipulated to replace endogenous 53BP1 with the phosphomutants, (AA and ED) and used to assess sensitivity to clinical grade PARP inhibitor, ABT-888. As anticipated the expression of the 53BP1-ED mutant confers resistance to ABT-888 whereas cells expressing the wildtype 53BP1 or the 53BP1-AA mutant remain sensitive to ABT-888 (Fig 4C). Together these results highlight the importance of T1609 and S1618 in regulating 53BP1 activity and suggest that the phosphorylation status of the UDR motif in 53BP1 may have relevance in cancer.
Phosphorylation of T1609 and S1618 directly blocks chromatin association of 53BP1
Next we sought to investigate the mechanism by which the phosphorylation of 53BP1 at T1609 and S1618 impedes its recruitment to DNA breaks. Even in the presence of DSBs, 53BP1 is excluded from chromatin in mitotic cells (Giunta et al., 2010; Giunta and Jackson, 2011; Nelson et al., 2009; van Vugt et al., 2010). Therefore it is feasible that phosphorylation of 53BP1 at T1609 and S1618 influences its sub-nuclear localization. To investigate this possibility we fractionated U2OS cells expressing FH-tagged wild-type 53BP1, 53BP1-AA and 53BP1-ED mutants and evaluated the amount of 53BP1 in chromatin-enriched fractions and nuclear-soluble fractions in G1 cells exposed to IR after synchronization in mitosis by nocodazole. In cells expressing wild-type and the phosphonull 53BP1-AA mutant there was a detectable increase in 53BP1 in the chromatin-enriched fraction after IR. However, for the cells expressing the phosphomimetic 53BP1-ED mutant, 53BP1 is largely retained in the nuclear soluble fraction with relatively low levels in chromatin (Fig. 5A). Association of 53BP1 with histone H4 dimethylated at Lys20 (H4K20me2)is critical for localization to chromatin(Botuyan et al., 2006; Pei et al., 2011). We focused on the G1 phase and assessed the interaction of 53BP1 with H4K20me2 after cells enter the G1 stage. Relative to wildtype 53BP1 and the phosphonull 53BP1-AA mutant there is a significant decrease in the interaction of H4K20me2 with phosphomimetic 53BP1-ED mutant in G1 cells (Fig. 5B). Another 53BP1 interacting protein, RPA2 (Yoo et al., 2005) interacts with wildtype 53BP1, and the 53BP1-AA and 53BP1-ED mutants at comparable levels. These results indicate phosphorylation of the two residues T1609 and S1618 impedes the recruitment of 53BP1 to DSB sites by blocking the DNA damage induced localization of 53BP1 to chromatin.
Figure 5. Constitutive phosphorylation of 53BP1 alters sub-nuclear localization and prevents its direct interaction with RNF168-ubiquitinated mononucleosomes.

A. Sub-nuclear localization of 53BP1 is altered by hyperphosphorylation of T1609/S1618. U2OS cells, expressing full length FH-tagged 53BP1 (WT, AA or ED) were synchronized in mitosis using Nocodazole and biochemical fractionation was conducted at indicated time points after 10 Gy IR. Nuclear soluble (NS) and chromatin-enriched (Chr) fractions were probed using anti-FLAG antibody. KU70 and histone H3 (H3) was probed for loading and fractionation controls, respectively.
B. Interaction of 53BP1 with H4K20Me2. U2OS cells expressing full-length FH-tagged 53BP1 (WT, AA, or ED) or empty vector (FH-c) were synchronized in mitosis, exposed to 10Gy IR immediately after release, and harvested at indicated time points. 53BP1 was immunopurified using anti-FLAG agarose beads. The immunoprecipitate was analyzed by immunoblotting using antibodies against H4K20Me2 and RPA2, which is a known interacting partner of 53BP1. Total 53BP1 in the input and pull-down fraction is also shown.
C. Pull-down assays of RNF168-ubiquitinated H4KC20me2 nucleosomes with GST-fusion proteins. Left panel: Installation of a dimethyl-lysine analogue at the mutated cysteine of H4K20C and ubiquitination of mononucleosomes was confirmed by immunoblotting using specific antibodies. Right panel: Pull-down assays of non-ubiquitinated and RNF168-ubiquitinated H4KC20me2 nucleosomes with the indicated GST-fusion proteins. Input lanes represent 33% of the nucleosome amount used in the pull-down assays; n = 3.
D. Phosphorylation of 53BP1 foci-forming region (FFR) by incubation with mitotic extract prevents its interaction with RNF168-ubiquitinated H4KC20me2 nucleosomes. GST-fusion proteins were pre-incubated with a HeLa mitotic extract in presence of 1mM ATP and a combination of PLK-1 inhibitor (BI2536, 100nM) and p38-MAPK inhibitor (SB202190, 10µM) (Kinase-i) when indicated. Following pre-incubation, GST proteins were further incubated with RNF168-ubiquitinated H4KC20me2 nucleosomes. Input represents 33% of the nucleosomes amount used in the pull-down assays; n = 3.
The direct association of 53BP1 with chromatin is mediated via the interaction of its Tudor domain with H4K20me2(Botuyan et al., 2006; Pei et al., 2011)and its UDR motif with the mono-ubiquitinated Lys15 residue of histone H2A (H2A-K15-ub) (Fradet-Turcotte et al., 2013). To determine the mechanism by which the phosphorylation of T1609 and S1618 influences the interaction of 53BP1 with chromatin, we expressed recombinant focus forming region (FFR) of 53BP1 (1220–1711aa) which includes the Tudor domains and the UDR motif. First we confirmed that similar to the full-length 53BP1, the ED mutant of the 53BP1-FFR is also excluded from DSB sites in cells (Supplemental Figure 4A). Next, we assessed binding of wildtype 53BP1-FFR and 53BP1- FFR (ED) to the H4K20me2-derived peptide, and observed that both proteins interacted efficiently with the methylated peptide but not the unmethylated peptide (Supplemental Fig. 4B). This result suggests that phosphorylation of T1609 and S1618 does not impact the interaction of the Tudor domain of 53BP1 with H4K20me2. Considering that T1609 and S1618 are in the UDR motif, it is feasible that their phosphorylation status influences the binding of the UDR motif with ubiquitinated H2A. Recombinant monomeric nucleosome core particles (NCP) were recently used by Durocher and colleagues to demonstrate the direct interaction of the UDR motif of 53BP1 with H2A-K15-ub (Fradet-Turcotte et al., 2013). For pull down assays with recombinant GST-53BP1-FFRs (wildtype and ED mutant) we used NCPs that contained H4K20me2 and were ubiquitinated at H2A by RNF168 (Fig. 5C, left panel). As anticipated, wild-type GST-53BP1-FFR only interacts with nucleosomes containing ubiquitinated H2A. On the contrary, the GST-53BP1-FFR (ED) mutant does not interact with NCPs irrespective of H2A ubiquitination (Fig. 5C, right panel). To confirm that the GST-53BP1-FFR (ED) mutant truly reflects mitotic phosphorylation of 53BP1, we used mitotic extracts supplemented with ATP to phosphorylate wild-type GST-53BP1-FFR prior to pull-down with NCPs (Fig. 5D). As observed with the GST-53BP1-FFR (ED) mutant, the phosphorylation impedes interaction of wildtype GST-53BP1-FFR with NCPs, and the interaction is restored by incubation of mitotic extracts with inhibitors for PLK1 and the p38 MAPK. Together these results suggest that the phosphorylation of the two residues T1609 and S1618 in the UDR motif of 53BP1 directly impedes its interaction with ubiquitinated H2A, and prevents the interaction of 53BP1 with chromatin in mitotic cells.
Ectopic recruitment of 53BP1 to chromatin in mitosis causes mitotic defects
Dephosphorylation of T1609 and S1618 in G1 is necessary for the recruitment of 53BP1 to chromatin and specifically to DSBs. A key question is whether the loss of phosphorylation at these two residues is sufficient to allow the recruitment of 53BP1 to DNA lesions in mitosis. 53BP1 and upstream factors (RNF8 and RNF168) are excluded from DSBs during early and mid-mitosis. In late-mitosis (anaphase/telophase) RNF8 and RNF168 are recruited to DSBs while 53BP1 continues to be excluded (Giunta et al., 2010; Giunta and Jackson, 2011). A possible interpretation of this result is that RNF168-mediated ubiquitination of H2A at DNA lesions in late mitosis is not sufficient to recruit 53BP1. We hypothesized that the phosphorylation of 53BP1 at T1609 and S1618 prevents its recruitment to DNA lesions in late mitosis. To test our hypothesis we utilized U2OS cells expressing RFP-H2B (to visualize chromatin) and GFP-tagged wild-type 53BP1 or 53BP1 phospho-mutants (AA and ED). Cells were treated with low doses of aphidicolin to induce replication-induced DNA lesions and time-lapse imaging was conducted to monitor the sub-cellular localization of 53BP1 during mitosis. Consistent with previous studies wildtype 53BP1 is not recruited to chromatin during mitosis, and the 53BP1 foci become visible in G1 cells. As anticipated the 53BP1-ED mutant remains excluded from chromatin both in mitosis and G1 cells. However, the 53BP1-AA mutant formed distinct foci during late mitosis, (transition of anaphase/telophase), (Fig. 6A and Supplementary Video 4–5). Quantification of the time required for 53BP1 to form foci after anaphase onset showed that the 53BP1-AA mutant was forming foci significantly faster than wildtype 53BP1 (Fig. 6A). Interestingly, in some cells the 53BP1-AA mutant is recruited to chromatin as early as 10–20 minutes after anaphase onset when the chromosomes remain condensed (Fig. 6A, 20’ time point). To examine the functional consequences of 53BP1 recruitment to DNA lesions in mitosis we synchronized cells in metaphase and exposed them to low dose of IR (0.5 Gy) prior to release. Formation of γ-H2AX foci in mitotic cells confirmed the presence of DSBs (Supplemental Fig. 5). As anticipated unlike wild type 53BP1 the 53BP1-AA mutant is recruited to chromatin during the transition from anaphase to telophase (Fig. 6B and supplementary videos 6–8). Surprisingly cells expressing 53BP1-AA mutant exposed to IR had a significant increase in mitotic errors manifested as increases in lagging chromosomes and micronuclei (Fig. 6B and C). Micronuclei (MN) formed by whole chromosome mis-segregation contain kinetochores (Fenech, 2010), therefore we evaluated the presence of kinetochores in MN generated by IR in cells expressing 53BP1-AA mutant. In cells expressing the wildtype 53BP1 the proportion of kinetochore-positive and kinetochore-negative MN were comparable. In contrast cells expressing the 53BP1-AA mutant have a striking increase in kinetochore-positive MN (Fig. 6D). 53BP1 promotes aberrant NHEJ at uncapped telomeres leading to genomic instability (Dimitrova et al., 2008). Interestingly, blocking NHEJ by incubation with the DNA-PK inhibitor ‘rescues’ the mitotic defects induced by the 53BP1-AA mutant (Fig. 6D).These results suggest that phosphorylation of 53BP1 at T1609 and S1618 during mitosis is necessary and sufficient to prevent its recruitment to DNA lesions in late mitosis. Furthermore, inappropriate recruitment of 53BP1 to mitotic DNA lesions impairs chromosome segregation.
Figure 6. Loss of phosphorylation at T1609 and S1618 of 53BP1 allows its recruitment to DNA lesions in mitotic cells and promotes genomic instability.

A. Premature recruitment of 53BP1-AA mutant to DNA lesions during mitosis. To examine the kinetics of 53BP1 foci formation, U2OS cells expressing RFP-H2B were transfected with different GFP-53BP1 constructs (WT, AA, ED), treated with 0.2 uM Aphidicolin for 12 hrs and released prior to live cell imaging. Left panels: representative time-lapse still images of cells expressing RFP-H2B and GFP tagged 53BP1-WT (top panels) or 53BP1-AA (middle panels) or 53BP1-ED (bottom panels) during G2, mitosis (early and late) and the following G1 phase. Time is shown in minutes (t=0, anaphase onset). Upper right panels: late mitotic cells (red box region) with premature 53BP1-AA foci formation are illustrated in overlaid images along with high magnification images (boxes 1 and 2). GFP-53BP1 and RFP-H2B are shown in green and red, respectively. Bottom right panels: Corresponding quantification of the timing of 53BP1 foci formation (p <0.0001, non-parametric t-test). Averages are shown in red bars.
B, C. Premature mitotic 53BP1-AA foci formation is accompanied by increased mitotic defects. B. U2OS cells expressing RFP-H2B were transfected with different GFP-53BP1 constructs (WT, AA) and exposed to 0.5Gy IR during mitosis prior to live cell imaging. Overlaid images with GFP-53BP1 (green) and RFP-H2B (red, also shown at the bottom with RFP-H2B only). Cells containing premature mitotic 53BP1-AA foci progress through cell cycle with increased mitotic defects, such as lagging chromosomes (dashed boxes) and micronuclei (MN, boxes).
C. Left panels: Corresponding mitotic defects that persist as MN in G1. Note that some MN contains 53BP1-AA (Box, 130 min, GFP in primary nuclei is overexposed to accentuate 53BP1 signal in MN). Upper right panel: Corresponding quantification for the timing of 53BP1 foci formation in indicated conditions (p <0.0001, non-parametric t-test). Lower right panel: percentage of cells that display lagging chromosomes and MN in indicated conditions. Only newly arising defects in cells exiting mitosis were scored (p <0.0001, non-parametric t-test). Errors bars indicate S.E.M.
D. U2OS cells expressing different GFP-53BP1 constructs (WT or AA) were treated with Nocodazole for 6h. Resulting mitotic cells were irradiated at low-dose (0.5 Gy) and released into normal media. At 6h following release, cells were fixed and co-stained with antibodies for γ-H2AX and CREST (kinetochore marker) to visualize kinetochore-positive (CREST+) MN. Where indicated, transfected cells were pretreated with DNA-PK inhibitor (DNA-PKi) (NU7441, 10µM) 1h prior to irradiation. Quantification of CREST− and CREST+ MN in cells expressing WT or AA GFP-53BP1 with or without DNA-PKi is shown. The data are expressed as mean ± S.D; n = 3 (> 100 cells were quantified). Refer to Supplementary Figure 5B for representative CREST staining.
DISCUSSION
Phosphorylation of 53BP1 has been comprehensively investigated (Jowsey et al., 2007), and recent studies demonstrate that generating phosphonull mutants of a combination of 28 phosphosites (PI3-like kinase; S/TPQ motifs) in the N-terminus of 53BP1 disrupts interaction with critical factors such as Rif1 (Callen et al., 2013; Chapman et al., 2013; Di Virgilio et al., 2013; Escribano-Diaz et al., 2013; Zimmermann et al., 2013) and abrogates its function in class-switch recombination and DNA repair (Bothmer et al., 2011; Di Virgilio et al., 2013; Ward et al., 2006). However there is no understanding of how dephosphorylation may directly regulate 53BP1 activity.
Large scale phosphoproteomic studies have revealed that a significant number of the DDR factors are constitutively phosphorylated in the course of the cell cycle (Dephoure et al., 2008; Grosstessner-Hain et al., 2011; Hegemann et al., 2011) and over one-third of observed phosphorylation sites are down-regulated within minutes of DNA damage (Bennetzen et al., 2010; Bensimon et al., 2010). How the constitutive phosphorylation and potential dephosphorylation of the DDR proteins impact their function remains unclear. Studies on the role of phosphatases in the DDR, including ones from our group, have been limited to demonstrating their significance in later stages of the DDR, largely in relieving the cell cycle checkpoint and restoring the cell to its ‘pre-DNA damage’ state (Lee and Chowdhury, 2011; Peng and Maller, 2010). Our results on the PP4C/PP4R3β complex mediated dephosphorylation of 53BP1 counter the existing paradigm regarding the role of phosphatases in DSB repair. Here we provide an example where a phosphatase by regulating the critical step of 53BP1 recruitment to DSB sites, influences the process of DNA repair, and the choice of DSB repair pathway.
Mitotic cells are distinct from inter-phase cells in their ability to tolerate the presence of DSBs as cells progress through the mitotic phase without triggering a checkpoint response (Heijink et al., 2013). More recent studies have demonstrated that the ‘sensors’ of the DSBs such as the MRN complex and the Ku complex (Gomez-Godinez et al., 2010; Peterson et al., 2011) are recruited to DSBs in mitosis and γ-H2AX is normally phosphorylated (Giunta et al., 2010; Nelson et al., 2009; van Vugt et al., 2010). A fundamental and important question is why do cells block the ‘secondary’ steps of the DSB repair process in mitosis, specifically the recruitment of 53BP1 to DNA lesions. Our results with the 53BP1-AA mutant suggest that allowing 53BP1 to localize to DSBs and chromatin in late mitosis counter-intuitively promotes genomic instability, and leads to mitotic defects that are manifested in increased number of kinetochore positive micronuclei and lagging chromosomes. The underlying mechanism for this observation remains unclear but it is feasible that stress induced telomere de-protection in mitosis (Hayashi et al., 2012) coupled with the ability of 53BP1 to promote end-joining of telomeres by increasing chromatin mobility (Dimitrova et al., 2008) may contribute to this striking phenotype. Blocking NHEJ in the 53BP1-AA expressing cells does resolve the mitotic defects giving some credence to this idea. Another important question is whether other DDR factors have a similar regulatory mechanism where dephosphorylation is a pre-requisite for their participation in the DDR. The evolutionary conservation of the phosphoresidues (T1609 and S1618) and the flanking amino acids give some credence to the idea that similar ‘dephosphorylation-dependent chromatin recruitment motifs’, maybe present in other DNA repair proteins. Preliminary computational predictions support this notion (unpublished results, SD and DC) but significant experimental work is necessary to confirm the functional relevance of these predictions. Future studies will elucidate the molecular details of how protein phosphatases regulate the activity of 53BP1 and DNA repair proteins in the early steps of the DDR.
EXPERIMENTAL PROCEDURES
In vitro dephosphorylation assay
The in vitro dephosphorylation assay was performed as described (Lee et al., 2012; Lee et al., 2010a). PP4C WT and D82A mutant proteins were purified using the Bac-to-Bac Baculovirus Expression System (Invitrogen) according to the manufacturer's manual. For the dephosphorylation assay, phosphorylated FH-53BP1 was prepared by immunoprecipitation of 53BP1 from nocodazole-arrested mitotic cells with anti-FLAG antibody conjugated agarose. Phosphatase reactions with phosphorylated FH-53BP1 were performed in 20 mM Tris-HCl, pH 7.4, 50 mM NaCl, 0.2 mM EDTA, 0.2% β-mercaptoethanol for 30 min at 30°C. Reactions were resolved on 12% (v/v) SDS-PAGE and relative phosphatase activity was determined by loss of phospho-FH-53BP1 immunoreactivity as determined by p1618 and p(1609–1618) antibody staining.
Co-immunoprecipitation, Immunoblotting and Chromatin Fractionation
These techniques were performed as previously described in (Lee et al., 2012; Lee et al., 2010a). Briefly, cell lysates from U2OS cells, expressing 53BP1 WT or phosphomutants, were incubated with anti–Flag agarose (Sigma) at 4 °C for 16 h and immunoprecipitated proteins were resolved by SDS-PAGE and analyzed by immunoblot.
Immunoblots were visualized using the Odyssey Infrared Imaging System. After primary antibody incubation, blots were incubated with goat anti–mouse IR Dye 800CW or goat anti–rabbit IR Dye 680 (LI-COR) and scanned using the LI-COR scanner. Images were visualized and quantified by Odyssey V3.0 software (http://biosupport.licor.com). Chromatin fractionation was performed as described previously (Lee et al, 2010). Quantification of fractions for equal loading was done using NanoDrop 1000 (Thermo Scientific).
Immunofluorescence
Immunofluorescence was performed as previously described (Lee et al., 2010a). Briefly, U2OS, RPE1-Fucci, 53BP1−/− MEF cells were plated on glass slides, fixed, permeablized and incubated with primary and secondary antibodies prior to being mounted using DapiFuoromount-G (SouthernBiotech). Secondary Alexa Fluor IgG antibodies used were: 488 goat anti-rabbit, 488 goat anti-mouse, 594 goat anti-rabbit, 594 goat anti-mouse, 647 donkey anti-goat, goat anti-rabbit 568, and goat anti-human 660 (Invitrogen). A stack of images for each cell were collected with 100× objective (1.35 N.A.) using Olympus FV1000 confocal microscope and presented as maximum intensity projection.
Live-cell Imaging
MPL-induced DNA Damage and Fluorescence Image acquisition. A Mai Tai@ Ultrafast Ti:Sapphire Laser (Spectra-Physics, Santa Clara, CA, USA, 100 fs pulse, 80 MHz repetition rate) was aligned into a Nikon Ti microscope (Nikon, Melville, NY, USA) via a custom-built open beam optical path. The laser was expanded to overfill the back aperture of a 60×1.4 numerical aperture objective (Nikon). A 675 nm low pass dichroic mirror (Chroma Technology Corp., Bellows Falls, VT, USA) was mounted in the microscope to reflect the laser into the objective. An average power level of 25 mW at 780 nm was used for all DNA damage experiments. The power was measured in the optical path outside of the microscope and is approximately three times higher than the power at the sample. Focus formation was monitored by accumulation of a GFP-tagged full-length 53BP1 wild type or AA and ED mutant via epifluorescence excitation. Cells were incubated in a HEPES Leibovitz-15 based live cell media to permit a pH buffered environment for imaging on the CCD camera (The PhotometricsCoolSNAPHQ Monochrome camera). Each set of data collection began with the capture of four fluorescence images prior to DNA damage followed by exposure to the laser for five seconds. A time-lapse movie was collected for 10 min after DNA damage induction with a frame interval of 10 seconds for the first 5 minutes and 30 seconds for the second 5 minutes. Data movies were analyzed in MATLAB@ (The MathWorks, Natick, MA, USA) with a custom algorithm to follow focus movement, quantify intensity and generate a plot of fluorescence accumulation over time.
Viability Assays
Clonogenic and Colorimetric Assays were performed as previously described described (Lee et al., 2012; Lee et al., 2010a). Briefly, for clonogenic assays plated cells were irradiated at indicated doses on the following day and allowed to form colonies for 2 weeks before being stained by 0.1% crystal violet solution. Surviving colonies of greater than1mm diameter were counted. For colorimetric assays cells were allowed to form colonies for 5 days before being quantified by CellTiter-Glo® Luminescent Cell Viability Assay (Promega).
Nucleosomes and Recombinant Proteins
Assembled histone H4K20me20-containing mononucleosomes were a kind gift of Daniel Durocher (University of Toronto). RNF168 ubiquitination of mononucleosomes was done as described previously (Fradet-Turcotte et al., 2013). using 2.5µg of nucleosomes in a final volume of 50µl. The installation of ubiquitin was confirmed by immunoblotting. The details of the purification of the 53BP1 fragments have been provided in the supplementary section.
Pull-down Assays
Nucleosome pull-downs were done by incubating 15µl of ubiquitination reaction with 3µg of GST proteins immobilized on glutathione sepharose 4B (GE Healthcare). The buffer composition and other details have been provided in the supplementary section. For some assays, GST proteins were first phosphorylated in vitro using a mitotic extract prepared from Nocodazole-treated HeLa cells. In indicated reactions a combination of 100nM BI2536 and 10µM SB202190 kinase inhibitors was also added in the phosphorylation reaction.
Mitotic 53BP1 Imaging
U2OS cells expressing RFP-H2B were transfected with wild-type or mutant GFP-53BP1 constructs. For Aphidicolin experiments, transfected cells were treated with 0.2µM Aphidicolin for 12 hrs and released to regular medium prior to imaging. For irradiation experiments, transfected cells were arrested in mitosis with 100 ng/ml of Nocodazole (Noc) for 6 hrs and released to regular medium prior to imaging. Live-cell imaging was performed using a TE2000-E2 inverted Nikon microscope equipped with the Nikon Perfect Focus system enclosed within a temperature- and CO2-controlled environment. In parallel, Noc-arrested irradiated cells were fixed and stained for γ-H2AX (1:300). For consistency, only cells with comparable and moderate level 53BP1 were subjected for the quantitation by gauging fluorescent intensity of 53BP1. Time from anaphase onset to the first appearance of discrete 53BP1 foci was quantified.
Supplementary Material
Highlights.
53BP1 is phosphorylated in mitosis to block recruitment to chromatin and DNA breaks.
Ectopic recruitment of 53BP1 to DNA lesions in mitosis promotes genomic instability.
PP4C/R3β phosphatase complex dephosphorylates 53BP1 during mitosis to G1 transition.
Dephosphorylation of 53BP1 is necessary for its function in the DNA damage response.
ACKNOWLEDGEMENTS
The 53BP1−/− MEFs and the 53BP1-GFP siRNA-resistant construct were gifts from P.Jeggo (Sussex). The GFP-RNF168 expressing U2OS cells and modified nuclesomes core particles (NCPs) were a gift from D Durocher (Toronto). Human CREST serum was a gift from Dr. Arno Kromminga. Xiao-Feng Zheng for making the graphical abstract. DC is supported by R01CA142698 (NCI), R01 AI101897-01 (NIAID), Basic Scholar Grant (American Cancer Society), Ann-Fuller Foundation, Mary Kay Foundation and start-up funds from DFCI. DL is supported by Basic Science Research Program through the National Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planing (NRF-2013R1A1A1061207) and Chonnam National University, 2013
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Bennetzen MV, Larsen DH, Bunkenborg J, Bartek J, Lukas J, Andersen JS. Site-specific phosphorylation dynamics of the nuclear proteome during the DNA damage response. Molecular & cellular proteomics : MCP. 2010;9:1314–1323. doi: 10.1074/mcp.M900616-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensimon A, Schmidt A, Ziv Y, Elkon R, Wang SY, Chen DJ, Aebersold R, Shiloh Y. ATM-dependent and -independent dynamics of the nuclear phosphoproteome after DNA damage. Sci Signal. 2010;3:rs3. doi: 10.1126/scisignal.2001034. [DOI] [PubMed] [Google Scholar]
- Bothmer A, Robbiani DF, Di Virgilio M, Bunting SF, Klein IA, Feldhahn N, Barlow J, Chen HT, Bosque D, Callen E, et al. Regulation of DNA end joining, resection, and immunoglobulin class switch recombination by 53BP1. Mol Cell. 2011;42:319–329. doi: 10.1016/j.molcel.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botvinick EL, Shah JV. Laser-based measurements in cell biology. Methods in cell biology. 2007;82:81–109. doi: 10.1016/S0091-679X(06)82003-0. [DOI] [PubMed] [Google Scholar]
- Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol. 2010;17:688–695. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callen E, Di Virgilio M, Kruhlak MJ, Nieto-Soler M, Wong N, Chen HT, Faryabi RB, Polato F, Santos M, Starnes LM, et al. 53BP1 mediates productive and mutagenic DNA repair through distinct phosphoprotein interactions. Cell. 2013;153:1266–1280. doi: 10.1016/j.cell.2013.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman JR, Barral P, Vannier JB, Borel V, Steger M, Tomas-Loba A, Sartori AA, Adams IR, Batista FD, Boulton SJ. RIF1 is essential for 53BP1-dependent nonhomologous end joining and suppression of DNA double-strand break resection. Mol Cell. 2013;49:858–871. doi: 10.1016/j.molcel.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman JR, Sossick AJ, Boulton SJ, Jackson SP. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. Journal of cell science. 2012 doi: 10.1242/jcs.105353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury D, Xu X, Zhong X, Ahmed F, Zhong J, Liao J, Dykxhoorn DM, Weinstock DM, Pfeifer GP, Lieberman J. A PP4-phosphatase complex dephosphorylates gamma-H2AX generated during DNA replication. Mol Cell. 2008;31:33–46. doi: 10.1016/j.molcel.2008.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dephoure N, Zhou C, Villen J, Beausoleil SA, Bakalarski CE, Elledge SJ, Gygi SP. A quantitative atlas of mitotic phosphorylation. Proc Natl Acad Sci U S A. 2008;105:10762–10767. doi: 10.1073/pnas.0805139105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Virgilio M, Callen E, Yamane A, Zhang W, Jankovic M, Gitlin AD, Feldhahn N, Resch W, Oliveira TY, Chait BT, et al. Rif1 prevents resection of DNA breaks and promotes immunoglobulin class switching. Science. 2013;339:711–715. doi: 10.1126/science.1230624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimitrova N, Chen YC, Spector DL, de Lange T. 53BP1 promotes nonhomologous end joining of telomeres by increasing chromatin mobility. Nature. 2008;456:524–528. doi: 10.1038/nature07433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escribano-Diaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkac J, Cook MA, Rosebrock AP, Munro M, Canny MD, et al. A Cell Cycle-Dependent Regulatory Circuit Composed of 53BP1-RIF1 and BRCA1-CtIP Controls DNA Repair Pathway Choice. Mol Cell. 2013 doi: 10.1016/j.molcel.2013.01.001. [DOI] [PubMed] [Google Scholar]
- Fenech M. The lymphocyte cytokinesis-block micronucleus cytome assay and its application in radiation biodosimetry. Health physics. 2010;98:234–243. doi: 10.1097/HP.0b013e3181b85044. [DOI] [PubMed] [Google Scholar]
- Fradet-Turcotte A, Canny MD, Escribano-Diaz C, Orthwein A, Leung CC, Huang H, Landry MC, Kitevski-LeBlanc J, Noordermeer SM, Sicheri F, et al. 53BP1 is a reader of the DNA-damage-induced H2A Lys 15 ubiquitin mark. Nature. 2013;499:50–54. doi: 10.1038/nature12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras AC, Caballero M, Zarske M, Sanchez A, Hazbun TR, Fields S, Sonenberg N, Hafen E, Raught B, Aebersold R. A novel, evolutionarily conserved protein phosphatase complex involved in cisplatin sensitivity. Molecular & cellular proteomics : MCP. 2005;4:1725–1740. doi: 10.1074/mcp.M500231-MCP200. [DOI] [PubMed] [Google Scholar]
- Giunta S, Belotserkovskaya R, Jackson SP. DNA damage signaling in response to double-strand breaks during mitosis. J Cell Biol. 2010;190:197–207. doi: 10.1083/jcb.200911156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giunta S, Jackson SP. Give me a break, but not in mitosis: the mitotic DNA damage response marks DNA double-strand breaks with early signaling events. Cell Cycle. 2011;10:1215–1221. doi: 10.4161/cc.10.8.15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Godinez V, Wu T, Sherman AJ, Lee CS, Liaw LH, Zhongsheng Y, Yokomori K, Berns MW. Analysis of DNA double-strand break response and chromatin structure in mitosis using laser microirradiation. Nucleic Acids Res. 2010;38:e202. doi: 10.1093/nar/gkq836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosstessner-Hain K, Hegemann B, Novatchkova M, Rameseder J, Joughin BA, Hudecz O, Roitinger E, Pichler P, Kraut N, Yaffe MB, et al. Quantitative phospho-proteomics to investigate the polo-like kinase 1-dependent phospho-proteome. Molecular & cellular proteomics : MCP. 2011;10:M111 008540. doi: 10.1074/mcp.M111.008540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrigan JA, Belotserkovskaya R, Coates J, Dimitrova DS, Polo SE, Bradshaw CR, Fraser P, Jackson SP. Replication stress induces 53BP1-containing OPT domains in G1 cells. J Cell Biol. 2011;193:97–108. doi: 10.1083/jcb.201011083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartlerode AJ, Guan Y, Rajendran A, Ura K, Schotta G, Xie A, Shah JV, Scully R. Impact of histone H4 lysine 20 methylation on 53BP1 responses to chromosomal double strand breaks. PloS one. 2012;7:e49211. doi: 10.1371/journal.pone.0049211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi MT, Cesare AJ, Fitzpatrick JA, Lazzerini-Denchi E, Karlseder J. A telomere-dependent DNA damage checkpoint induced by prolonged mitotic arrest. Nat Struct Mol Biol. 2012;19:387–394. doi: 10.1038/nsmb.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegemann B, Hutchins JR, Hudecz O, Novatchkova M, Rameseder J, Sykora MM, Liu S, Mazanek M, Lenart P, Heriche JK, et al. Systematic phosphorylation analysis of human mitotic protein complexes. Sci Signal. 2011;4:rs12. doi: 10.1126/scisignal.2001993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heijink AM, Krajewska M, van Vugt MA. The DNA damage response during mitosis. Mutation research. 2013 doi: 10.1016/j.mrfmmm.2013.07.003. [DOI] [PubMed] [Google Scholar]
- Huyen Y, Zgheib O, Ditullio RA, Jr, Gorgoulis VG, Zacharatos P, Petty TJ, Sheston EA, Mellert HS, Stavridi ES, Halazonetis TD. Methylated lysine 79 of histone H3 targets 53BP1 to DNA double-strand breaks. Nature. 2004;432:406–411. doi: 10.1038/nature03114. [DOI] [PubMed] [Google Scholar]
- Iwabuchi K, Basu BP, Kysela B, Kurihara T, Shibata M, Guan D, Cao Y, Hamada T, Imamura K, Jeggo PA, et al. Potential role for 53BP1 in DNA end-joining repair through direct interaction with DNA. J Biol Chem. 2003;278:36487–36495. doi: 10.1074/jbc.M304066200. [DOI] [PubMed] [Google Scholar]
- Jowsey P, Morrice NA, Hastie CJ, McLauchlan H, Toth R, Rouse J. Characterisation of the sites of DNA damage-induced 53BP1 phosphorylation catalysed by ATM and ATR. DNA Repair (Amst) 2007;6:1536–1544. doi: 10.1016/j.dnarep.2007.04.011. [DOI] [PubMed] [Google Scholar]
- Lee DH, Chowdhury D. What goes on must come off: phosphatases gatecrash the DNA damage response. Trends Biochem Sci. 2011;36:569–577. doi: 10.1016/j.tibs.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Goodarzi AA, Adelmant GO, Pan Y, Jeggo PA, Marto JA, Chowdhury D. Phosphoproteomic analysis reveals that PP4 dephosphorylates KAP-1 impacting the DNA damage response. Embo J. 2012;31:2403–2415. doi: 10.1038/emboj.2012.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Pan Y, Kanner S, Sung P, Borowiec JA, Chowdhury D. A PP4 phosphatase complex dephosphorylates RPA2 to facilitate DNA repair via homologous recombination. Nat Struct Mol Biol. 2010a;17:365–372. doi: 10.1038/nsmb.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Kenny AE, Rieder CL. P38 mitogen-activated protein kinase activity is required during mitosis for timely satisfaction of the mitotic checkpoint but not for the fidelity of chromosome segregation. Molecular biology of the cell. 2010b;21:2150–2160. doi: 10.1091/mbc.E10-02-0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Xu L, Zhong J, Liao J, Li J, Xu X. Protein phosphatase PP4 is involved in NHEJ-mediated repair of DNA double-strand breaks. Cell Cycle. 2012;11:2643–2649. doi: 10.4161/cc.20957. [DOI] [PubMed] [Google Scholar]
- Lukas C, Savic V, Bekker-Jensen S, Doil C, Neumann B, Pedersen RS, Grofte M, Chan KL, Hickson ID, Bartek J, et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat Cell Biol. 2011;13:243–253. doi: 10.1038/ncb2201. [DOI] [PubMed] [Google Scholar]
- Nakada S, Chen GI, Gingras AC, Durocher D. PP4 is a gamma H2AX phosphatase required for recovery from the DNA damage checkpoint. EMBO reports. 2008;9:1019–1026. doi: 10.1038/embor.2008.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson G, Buhmann M, von Zglinicki T. DNA damage foci in mitosis are devoid of 53BP1. Cell Cycle. 2009;8:3379–3383. doi: 10.4161/cc.8.20.9857. [DOI] [PubMed] [Google Scholar]
- Pei H, Zhang L, Luo K, Qin Y, Chesi M, Fei F, Bergsagel PL, Wang L, You Z, Lou Z. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature. 2011;470:124–128. doi: 10.1038/nature09658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng A, Maller JL. Serine/threonine phosphatases in the DNA damage response and cancer. Oncogene. 2010;29:5977–5988. doi: 10.1038/onc.2010.371. [DOI] [PubMed] [Google Scholar]
- Peterson SE, Li Y, Chait BT, Gottesman ME, Baer R, Gautier J. Cdk1 uncouples CtIP-dependent resection and Rad51 filament formation during M-phase double-strand break repair. J Cell Biol. 2011;194:705–720. doi: 10.1083/jcb.201103103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell. 2008;132:487–498. doi: 10.1016/j.cell.2007.12.033. [DOI] [PubMed] [Google Scholar]
- van Vugt MA, Gardino AK, Linding R, Ostheimer GJ, Reinhardt HC, Ong SE, Tan CS, Miao H, Keezer SM, Li J, et al. A mitotic phosphorylation feedback network connects Cdk1, Plk1, 53BP1, and Chk2 to inactivate the G(2)/M DNA damage checkpoint. PLoS Biol. 2010;8:e1000287. doi: 10.1371/journal.pbio.1000287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward I, Kim JE, Minn K, Chini CC, Mer G, Chen J. The tandem BRCT domain of 53BP1 is not required for its repair function. J Biol Chem. 2006;281:38472–38477. doi: 10.1074/jbc.M607577200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue Y, Ren J, Gao X, Jin C, Wen L, Yao X. GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Molecular & cellular proteomics : MCP. 2008;7:1598–1608. doi: 10.1074/mcp.M700574-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo E, Kim BU, Lee SY, Cho CH, Chung JH, Lee CH. 53BP1 is associated with replication protein A and is required for RPA2 hyperphosphorylation following DNA damage. Oncogene. 2005;24:5423–5430. doi: 10.1038/sj.onc.1208710. [DOI] [PubMed] [Google Scholar]
- Zgheib O, Pataky K, Brugger J, Halazonetis TD. An oligomerized 53BP1 tudor domain suffices for recognition of DNA double-strand breaks. Mol Cell Biol. 2009;29:1050–1058. doi: 10.1128/MCB.01011-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M, Lottersberger F, Buonomo SB, Sfeir A, de Lange T. 53BP1 regulates DSB repair using Rif1 to control 5' end resection. Science. 2013;339:700–704. doi: 10.1126/science.1231573. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.