

Abstract
High-resolution oligonucleotide array comparative genomic hybridization (aCGH) and spectral karyotyping (SKY) were applied to a panel of malignant mesothelioma (MMt) cell lines. SKY has not been applied to MMt before, and complete karyotypes are reported based on the integration of SKY and aCGH results. A whole genome search for homozygous deletions (HDs) produced the largest set of recurrent and non-recurrent HDs for MMt (52 recurrent HDs in 10 genomic regions; 36 non-recurrent HDs). For the first time, LINGO2, RBFOX1/A2BP1, RPL29, DUSP7, and CCSER1/FAM190A were found to be homozygously deleted in MMt, and some of these genes could be new tumor suppressor genes for MMt. Integration of SKY and aCGH data allowed reconstruction of chromosomal rearrangements that led to the formation of HDs. Our data imply that only with acquisition of structural and/or numerical karyotypic instability can MMt cells attain a complete loss of tumor suppressor genes located in 9p21.3, which is the most frequently homozygously deleted region. Tetraploidization is a late event in the karyotypic progression of MMt cells, after HDs in the 9p21.3 region have already been acquired.
Keywords: Malignant mesothelioma, oligonucleotide array CGH, spectral karyotyping, homozygous deletions, tumor suppressors
Malignant mesothelioma (MMt) is a rare and very aggressive tumor of the mesothelium, and the most prevalent subtype is malignant pleural mesothelioma where 80% of cases are associated with asbestos exposure (1). The peritoneum and the pericardium are additional MMt tumor sites. MMt incidence is 0.9 new cases per 100,000 persons per year. The 5-year relative survival is 7–11% overall, which positions MMt immediately above pancreatic cancer (5–6%), which is at the bottom of the survival list by cancer site (2). The mean latency from exposure to diagnosis varies from 12.3–51 years, depending on the type of asbestos. Despite decades of long latency for most of the cases, there is no identified stage of a slow growing or indolent tumor, which might be reflected by the aggressive behavior of MMt (3).
Early karyotypic studies, which used chromosome banding analyses of MMt biopsy specimens, effusions, and cell lines, first revealed the complexity of the genetic changes involved in this malignancy (4–7). Most MMts display multiple clonal chromosomal abnormalities; all chromosomes contributed to numerical changes, and losses were more common than gains. Furthermore, all chromosomes except the Y were found to participate in structural changes (7). A number of recurrent gains and losses have been found, including monosomy of chromosomes 4 and 22; polysomy of chromosomes 5, 7, and 20; and losses of 1p21–p22, 3p21, 6q15–q21, 9p21–p22, 11p11–p13, 13q, 14q, and 22q12 (7). Nevertheless, G-banding studies have not resulted in the identification of any chromosomal aberrations specific to MMt, which are necessary for the purposes of diagnosis, differential evaluation, and subcategorization of these tumors.
Additional studies of chromosomal gains and losses by comparative genomic hybridization (CGH) revealed even more recurrent chromosomal imbalances present in MMt. Chromosomal-CGH (cCGH) studies confirmed many findings of the G-banding and loss of heterozygosity (LOH) studies, and unveiled new focal areas of copy number alterations (CNAs). The most frequent whole chromosome or arm losses were 4q, 5q, 6q, 8p, 9p, 12p, 14q, and 22q; the most frequent focal losses were 1p11–p22, 3p21, 4q31.1–qter, 6q14, 6q22, 6q24, 6q25–qter, 8p12–p21, 8p21–pter, 9p21, 13q12–q14, 14q24–qter, 15q11.1–q21, and 22q13. The most frequent whole chromosome or arm gains were 5p and 20q, and the most frequent focal gains were 5q14–q23, 7p21–pter, 7q31–q35, 8q24–qter, 15q22–qter, and 17q22–q24 (8–11).
cCGH has limited resolution, between 3–5 Mb for detecting deletions and at least 2 Mb for amplifications (12), which precludes the detection of small deletions and amplifications. Array CGH (aCGH) overcomes this problem by increasing the resolution limit to the gene or nucleotide level, depending on the array platform coverage. aCGH studies have identified many changes similar to the ones previously reported by cCGH, such as most whole chromosome or chromosome arm gains and losses. In addition, many new focal copy number losses and gains have been found by aCGH in MMt cancer cells (13–19). Losses are more frequent than gains, suggesting a prevalent role for tumor suppressor genes. Many studies utilizing LOH, G-banding, cCGH, and aCGH have suggested that areas in 9p21.3, 22q12.2, and 13q12.11, which contain the CDKN2A/p16INK4A, CDKN2A/p14ARF, CDKN2B/p15, NF2, and LATS2 tumor suppressor genes and possibly the MTAP gene, were causally involved in MMt tumorigenesis as well as in other tumor types. The CDKN2A/CDKN2B genes (9p21.3) were found to be homozygously deleted in a high percentage of MMt cell lines and tumors (20–22), and this is the most frequently homozygously deleted region in many other tumor types (23–26). The NF2 gene (22q12.2) undergoes frequent biallelic inactivation by HDs in MMt tumors and cell lines (19,27–29). Finally, LATS2(13q12.11) has been found to be homozygously deleted in approximately 20% of MMt tumors and cell lines (30), and behaves as a tumor suppressor gene in some other cancer types (31). Recently, BAP1, which is considered to be a tumor suppressor gene, has been described as the main target of deletion in the 3p21.1 region in sporadic MMts (32–34) as well as germline mutations in MMt families (35). In addition to tumor suppressor genes, involvement of other genes, such as EGFR, VEGF, JUN, osteopontin, mesothelin, and members of the PI3K/AKT pathway among others, have also been proposed for MMt (14,36–38).
In this study, spectral karyotyping (SKY) and high-resolution oligonucleotide aCGH were utilized for dissecting genomic changes present in a panel of MMt cell lines. SKY has not been applied to MMt before, and aCGH analysis includes a particular search for HDs. Cell lines are a much better system than primary tumors to detect deletions that could be masked by normal cell contamination in tumors (39). Here, we report the largest set of recurrent and nonrecurrent HDs for MMt (88 HDs in 17 cell lines: 52 recurrent HDs in 10 genomic regions and 36 non-recurrent HDs).
Integration of SKY and aCGH data allowed the reconstruction of multistep events that lead to the formation of HDs. The advantages of combining SKY and aCGH datasets are numerous, including: 1) aCGH data confirms band location and refines locations of breakpoints of structural rearrangements detected by SKY; 2) aCGH confirms the balanced or unbalanced nature of structural rearrangements; 3) SKY contributes to the understanding of the etiology of genomic copy number changes detected by aCGH.
Materials and methods
Cell lines
MMt cell lines (H28, H290, H513, H2052, H2369, H2373, H2461, H2591, H2595, H2596, H2691, H2722, H2731, H2795, H2804, H2810, H2818, and H2869) were grown in RPMI 1640 supplemented with 10% fetal bovine serum and 5 mM L-glutamine. The samples comprised seven epithelioid (H513, H2461, H2591, H2595, H2795, H2810, and H2818), four sarcomatoid (H2373, H2596, H2691, and H2731), one biphasic (H2869), and six unknown cell lines (H28, H290, H2052, H2369, H2722, and H2804).
Oligonucleotide aCGH
A high-resolution aCGH was performed on 18 MMt cell lines; however, one cell line, H2804, was excluded from the analysis due to the quality of the hybridization. Agilent Human Genome Microarray Kit 105k (Design 014698, with median distance between probes 22 kb; Agilent Technologies Inc., Santa Clara, CA) was used in the aCGH analysis. Array probes were mapped from the Human Genome Assembly NCBI35/hg17 to GRCh37/hg19 with the LiftOver program (http://genome.ucsc.edu/cgibin/hgLiftOver): 99,951 probes were mapped successfully, whereas conversion failed for 125 probes that were omitted. DNA labeling with Cy5- and Cy3-dUTP, array hybridization, and washing were done according to the Agilent Oligonucleotide Array-Based CGH for Genomic DNA Analysis Protocol” (Version 5.0, Publication Number: G4410-90011 V.5.1, November 2007), starting with 3 μg of AluI- and RsaI-digested cell line and human female DNA. Arrays were scanned and data were extracted from array images with Agilent Feature Extraction Software (version 9.0) with default settings. A comprehensive study of HDs was performed by analyzing the data with the R-2.10.0 Language and statistical computing environment (40), and cghMCR package (41) from Bioconductor (42). Briefly, background intensity was subtracted using the “minimum” method; data were transformed to a log2 ratio of Cy5- and Cy3-signals and median-normalized within arrays. After segmentation with the Circular Binary Segmentation (CBS) algorithm (DNAcopy package) (43,44), minimal common deleted (mcd) regions with log2 ratios less than −2 (losses) were selected with the cghMCR package. aCGH data were also analyzed by Nexus CN 5.0 and 6.0 (BioDiscovery Inc., Hawthorne, CA) and by Agilent Genomic Workbench Lite Edition 6.5.0.18.exe. Nexus CN uses BioDiscovery's Fast Adaptive States Segmentation Technique (FASST2) algorithm to make copy number calls and the STAC (Significant Testing for Aberrant Copy number) method to determine the smallest significant regions with a statistically significant high frequency of aberrations over the background level of aberration (45). This latter method is performed after the FASST2 segmentation is applied. STAC excludes large copy number changes, such as whole-chromosome or whole-arm gains and losses, and concentrates instead on smaller focal copy number changes. X and Y chromosomes were excluded from all analyses. A gene annotation enrichment analysis and clustering were performed, with DAVID (http://david.abcc.ncifcrf.gov/) (46), with all the genes from significant CNAs.
Polymerase chain reaction (PCR)
PCR performed on genomic DNA from the H2869 and H28 MMt cell lines and Promega male genomic DNA used Applied Biosystems AmpliTaq Gold with GeneAmp 10 × PCR buffer, 10 mM dNTPs, and AmpliTaq Gold polymerase 5 U/μL stock solutions. Primers were synthesized by Invitrogen and resuspended in water to 100 μM, from which a 10-μM working solution was prepared. Final concentrations were 1 × PCR buffer, 0.2 mM dNTPs, 1 μM of each of the primers, and 0.05 U/μL of the polymerase. PCR conditions were 10 minutes at 95°C followed by 35 cycles of 30 seconds at 94°C, 30 seconds at 55°C, 1 minute at 72°C, and a final step of 10 minutes at 72°C.
The primers utilized for CDKN2A exon 2a were: forward primer 5′ GTCCCCTTGCCTGGAAAGAT 3′ and reverse primer 5′ CAGCCCCTCCTCTTTCTTCC 3′. GAPDH primers were used as a control of the integrity of the templates.
SKY
For the SKY analysis, cells were harvested by mitotic shake-off after Colcemid treatment (0.025 μg/mL: 1–2 hours). They were processed by standard cytogenetic methods using 0.075M potassium chlorideand a methanol-acetic acid (3:1) fixative. Metaphase spreads were prepared under optimized humidity conditions with a Thermotron Cytogenetic Drying Chamber (Thermotron, Holland, MI).
We used chromosome-specific painting probes (Spectral Imaging, Migdal Ha'Emek, Israel), and hybridizations were performed according to the company's protocol. Image acquisition was performed using an SD200 Spectral (Applied Spectral Imaging, Carlsbad, CA) mounted on a Leica DMRXA microscope (Leica, Wetzlar, Germany) through a custom-designed optical filter (SKY v.3; Chroma Technology, Brattleboro, VT). Applied Spectral Imaging software (Spectral Imaging and SKY View) was used for image acquisition and analysis.
Complete karyotypes were defined for 10 MMt cell lines (H28, H513, H2461, H2722, H2731, H2795, H2804, H2810, H2818, and H2869). Characterization of rearrangements for chromosome 9 was done in three additional cell lines (H2369, H2373, and H290). We report SKY results by using the short form of the 2009 International System for Human Cytogenetic Nomenclature (ISCN).
Results
Karyotypic abnormalities detected by SKY
Complete spectral karyotypes were determined for 10 MMt cell lines (Table 1). All cell lines had abnormal aneuploid karyotypes, with structural and numerical chromosomal alterations (Figure 1A). Structural alterations included translocations, deletions, duplications, insertions, and inversions.
Table 1. Karyotypes of MMt cell lines.
| H2461 |
| 34∼38,48,57,<2n->,XY,der(1)t(1;5)(q11,q11.2)[10],+der(1)t(1;5)(q11,q11.2)[7],der(1;12)(q10;q10)[10], +der(1;12)(q10;q10)[2],der(3)t(3;17)(p14.2;q12[9],-4[8],+i(5)(p10)[9],+6[3],+7[2],+i(7)(p10)[10], +8[2],+8[2],-9[9],+10[3],-13[7],-14[9],der(15)t(9;15)(q12;p13)[9],-16[4],-17[10],-18[3],-19[9], +20[2],-21[9],-22[10],der(22)t(1;22)(q12;p13)[9][cp10] |
| H2818 |
| 38∼42,52,73,<2n->,XY,+X[2],+Y[2],+1[2],+2[3],del(2)(p13.2)[7],+3[2],-4[8],+5[2],der(5)del(5)(q14q23)t(5;9)(p14;q21)[7],-6[5],del(6)(q21)[5],der(6)t(6;17)(p?16;p12)[2],+der(6)t(6;17)(p?16;p12)[2],+t(6;17)(q?16;p12)[2],der(6)t(6;21)(q11;q11)[4],+del(7)(p12)[2],-8[5],-9[3],+10[2],+11[2],del(11)(?p12)[4],+12[4],-13[6],-14[8],+15[2],+16[2],-16[3],-17[3],der(17)t(6;17)(q16;p12)[2],-18[4],-21[6],-22[5][cp8] |
| H2869 |
| 36∼40,67,<2n->,XY,der(1)t(1;7)(q11;p11)t(5;7)(?;p21)[4],der(1;8)(q10;q10)[3],der(1;8)(q10; q10)del(1)(q12)[4],-3[4],-4[7],+5[2],der(7)t(5;7)(?;p21)[7],der(8)t(1;8)(q21.2;p23)[7],+der(8)t(1;8)(q21.2;p23)[2],-11[3],del(11)(p11)[6],r(12)[7],+13[2],-14[7],-16[3],der(16)t(12;16)(?;p11.2)[4],-18[7],+20[2],der(20)t(1;20)(q12p12;q13)[8],-21[3],-22[5][cp8] |
| H2795 |
| 38∼49,<2n+/->,X,der(Y)t(Y;1)(p11;q12)[10],der(Y)t(Y;1)(p11;q12)[3],der(1)t(Y;1)(q12;q12)t(1;17)(p35.3;p12)[10],+der(1)t(Y;1)(q12;q12)t(1;17)(p35.3;p12)[5],+2[3],+der(2)t(2;18)(q32;?q11.2)[3],t(2;18)(q32;?q11.2)[10],+t(2;18)(q32;?q11.2)[2],der(3)t(3;7)(p25;?)[8],der(3)t(3;7)(p25;?)[3],+4[2],+der(6;8)(p10;q10)[8],del(7)(q31)[4],-9[3],der(11)t(7;11)(?;q23)[7],+12[2],+12[2],-12[3],+der(13;20)(q10;?10)?amp(13;20)[6],+14[3],-15[3],+16[3],der(18)t(2;18)(q32;?q11.2)[2],-20[4],+21[4],+22[2],-22[4][cp10] |
| H2731 |
| 42∼56,<2n+>,X,-X[3],-Y,+1[6],+2[6],+3[4],+3[2],-3[3],+4[3],der(4;10)(p10;p10)[7], +5[3],+6[5],+6[3],del(7)(?q11)[7],+der(7)t(7;13)(q31.2;q12.3)[8],+der(7)t(7;13)(q31.2;q12.3)[5],+der(7;13)t(7;13)t(7;13)[2],del(9)(p11)[4],der(9;11)(q10;q10)[6],+der(9;11)(q10;q10)[4],der(9;22)(q10;q10)[2],+i(9)(p10)[4],-10[3],der(10)t(?1or6or16;10)(?;p12)[5],+der(10)t(?1or6or16;10)(?;p12)[4],+der(11;13)(?q11;q12)[2],+12[3],13[9],+der(14)t(13;14)(q11;q13)[6],der(14)t(5;14)(?;p11)[2],+16[2],der(16)t(16;17)(q10;q10)[7],+17[5],der(17)t(17;19)(?p13;?)[2],-18[7],+der(19;20)t(17;19)(?p13;?q13)t(19;20)(?q10;q10)[4],der(19;22)(?;q10)del(22)[2],+20[5],+21[3],-21[3],i(21)(q10)[3],-22[4],del(22)[2],der(22)t(12;22)(?;p11)[5][cp9] |
| H513 |
| 59∼68,<3n->,XX,del(1)(p32)[5],der(1)t(1;16)(?p11;q22)[6],der(2)t(2;6)(p11;p24)[6],-3[7],-4[2], del(5)(?q14q31)[5],+i(5)(p10)[7],+i(5)(p10)[6],+i(5)(p10)[2],del(6)(?p11)[2],der(6)t(6;20)(p21;?)x2[7], +der(6)t(6;6)(p24;p11)t(2;6)(p11;p24)[6],+7[4],del(7)(q31)[6],-8[7],del(8)(p12)[3], der(9)inv(q11q22)t(9;13)(q34;q11)[3],der(9)t(9;21)(q13;q11)[5],inv(9)(q11q22)[4], -10[5],der(10)t(8;10)(q12;q25)[6]x2,der(11;19)t(19;20)(q13;?)t(11;19)(p11;q11)[7], +der(11;19)t(19;20)(q13;?)t(11;19)(p11;q11)[6],+min(11)[3],+der(12)t(3;12)(?;q13)[6],+13[3],-14[4], +15[2],der(15)t(15;17)(q12;q21)[7],+der(15)t(15;17)(q12;q21)[2],-17[3],der(17)t(5;17)(p10;q10)[3], der(17)t(17;18)(q21;q21)[3],-18[7],-19[4],-20[4],-21[7],-21[5],-22[4][cp7] |
| H2810 |
| 51∼93,<3n+>,XX,-X[2],-Y,del(1)(q32)[8],del(1)(q32)[5],der(2;4)(q10;q10)[5],der(2;4)(q10;q10)t(4;21)(?q21;q11)[2],del(3)(p21)[6],der(3;22)(q10;q10)[2],der(3)t(3;12)(p14;q15)[8],der(3)t(3;12)(p14;q15)[7],+der(3)t(3;12)(p14;q15)[3],+der(3)t(3;15)(p10;p10)[2],-4[4],del(4)(p15)[2],der(4)t(1;4)(?;q27)[7],der(4)t(1;4)(?;q27)[2],+5[3],+6[4],-6[2],-7[4],-7[3],ace(7)[3],ace(7)[2],del(7)(q11.2)[4],+der(7)t(3;7)(?;q31)[3],der(8)t(Y;8)(?;p12)[6],der(8)t(Y;8)(?;p12)[2],der(8)t(Y;16)t(Y;8)(?;p12)[2],-9[3],+der(9)t(7;9)(?;p24.1)[2],+der(9)t(9;22)(p13;?)[2],der(9)t(9;15)(p24.1;?)[7],-10[4],+11[3],der(11)t(11;15)(p15;q21)[7],+der(11)t(11;15)(p15;q21)[7],del(12)(q11)[6],del(12)(q11)[2],der(12)t(12;12)(q24;?)[3],der(12)t(12;4;14)[3],-13[8],-14[6],der(14)ins(12)[7],der(14)ins(12)[4],-15[8],del(15)[6],der(15)t(15;22)[2],+16[3],der(16)t(11;16)(?;q23)t(11;13)(?;?)[7],der(16)t(11;16)(?;q23)t(11;13)(?;?)[3],+17[2],der(17)t(17;21)(p11;q11)t(2;17)(?;q25)[8],+der(17)t(17;21)(p11;q11)t(2;17)(?;q25)[7],+18[3],+19[4],-19[4],+20[4],-21[8],-22[5],+der(22)t(4;22)t(4;14)[2][cp8] |
| H2804 |
| 49∼79,{104},<3n+/->,XXY,-X[2],+Y[2],der(1)t(1;6)(q11;?)[8],der(1)t(1;6)(q11;?)[7],+der(1;14)(q10;q10)[6],+der(1;14)(q10;q10)[4],i(1)(q10)[7],-2[3],del(3)(p11)[8],+del(3)(p11)[4],t(3;18)(p11;q21)[8],t(3;18)(p11;q21)[7],+t(3;18)(p11;q21)[3],+t(3;18)(p11;q21)[2],-4[4],der(4)t(4;9)(q13;q21)[5],+der(4)t(4;9)(q13;q21)[2],t(4;9)(q13;q21)[7],+t(4;9)(q13;q21)[4],+5[2],+5[2],-5[3],del(6)(q12)[6],+del(6)(q12)[2],der(7)t(?1;7)(q?;q11)[4],der(7)t(?1;7)(q?;q11)[2],der(7)t(7;13)(q31;q11)[7],+der(7)t(7;13)(q31;q11)[5],+8[5],del(8)(p11.2),+9[3],-10[4]psudc(10)t(10;11)(p12:?p14)del(10)(q25)[8],psudc(10)t(10;11)(p12:?p14)del(10)(q25)[4],-11[8],-11[3],+12[4],der(13)t(13;19)(q21;?)[6],der(13)t(13;19)(q21;?)[5],-14[7],+15[2],der(15)t(5:15)(p11;p11)[7],15[2],der(15)t(5:15)(p11;p11)[5],-16[6],+17[3],+17[2],-18[4],-19[5],der(19)t(3;19;13)[6],der(19)t(3;19;13)[2],+20[3],-21[6],-22[7][cp8] |
| H28 |
| 61∼85,<3n+/4n->,XXYY,-Y[7],-Y[3],+1[6],der(1)del(1)(p21)del(1)(p32)[2],der(1;3)(q10;?q10)del(3)(?q24)[2],+der(1;16)(q10;?q10)[8],+2[6],+3[3],+der(3)t(3;5)(p21;?)x2[10],der(4)t(2;4)(p11;p11)[8],der(4)t(4;11)(p15;q12.1)[7],+5[5],+6[2],der(6)t(X;6)(?;p25)[9],+der(6)t(X;6)(?;p25)[7],+7[4],del(7)(q31)[10],+8[4],der(9)t(9;17)(p13;q21)[10],+der(9)t(9;17)(p13;q21)[7],+10[5],-12[4],-14[8],der(14)t(9;14)(q13;p11)x2[10],der(14)t(9;14)(q13;p11)[2],+15[5],+16[2],-18[10],-18[3],+19[2],+der(19)t(13;19)(q21;q13)[8],+20[2],-20[3],+21[6],-22[5][cp10] |
| H2722 |
| 54∼76,<3n+>,XXY,-X[3],-Y[3],der(1)t(1;3)(p35.2;?)[11],+der(1)t(1;3)(p35.2;?)[10],+del(2)(p22)[10],der(3)t(3;8)(?p14;?p22)x2[10],der(3)t(3;15)(p11;q11)[9],-4[10],-4[4],+5[7],-6[4],+7[7],3pter->3p24::8p11->8p22::3p14->3p24::8p11->8pter[11],+3pter->3p24::8p11->8p22::3p14->3p24::8p11->8pter[9],-9[11],-9[5],der(10)t(10;12)(?q21;q13)[11],+der(10)t(10;12)(?q21;q13)[8],der(12)t(10;12)(p11;p12)ins(12;10)(q13;??)[11],+der(12)t(10;12)(p11;p12)ins(12;10)(q13;??)[8],-13[10],der(15)t(9;15)(q11;q14)[11],+der(15)t(9;15)(q11;q14)[11],del(16)(q12)[4],der(16)t(6;16)(?;q11)[9],-17[4],-18[3],+19[9],+20[6],+21[8],-22[9][cp11] |
Figure 1.
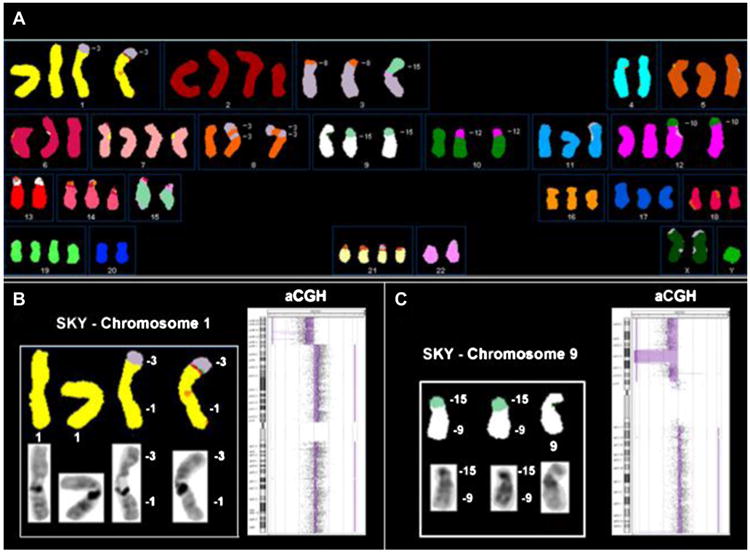
A total of 139 unique structurally rearranged chromosomes were found in the 10 cell lines. The lowest number of structural rearrangements was seven (cell line H2461), and the highest number of structural rearrangements was 27 (cell line H2810) (Table 1). The number of numerical chromosomal alterations (calculated as a sum of deviations of each chromosome from the ploidy level) varied between eight in cell line H2869 and up to 25 in cell line H2804.
Cell lines were separated into three groups based on their ploidy level: hypo-diploid (H2461, H2818, H2869, H2795), hypo-triploid (H2731, H513, H2810), and hyper-triploid (H28, H2804, H2722). The number of structural rearrangements was lowest in the hypo-diploid group (7–9 rearranged chromosomes per cell line), intermediate in the hyper-triploid group (11–15 per cell line), and highest among the hypo-triploid cell lines (21–27 per cell line). The number of numerical alterations was also lower in the hypo-diploid group (8–15 gains and losses per cell line) compared with the other two groups (18–25 numerical alterations per cell line).
Chromosomes 1 and 9 were the most frequently involved in structural rearrangements (in 19 rearrangements each), chromosomes 3 and 7 were involved in 13 rearrangements each, and chromosome 17 was involved in 12 rearrangements. The most frequent structural abnormalities detected by SKY were non-reciprocal translocations (103 rearrangements) and deletions (32 rearrangements). Three reciprocal translocations were detected: t(2;6)(p11;p24) in cell line H513, t(2;18)(q32;q11.2) in H2795, and t(17;19)(?p13;?q13) in cell line H2804. Additional complex rearrangements, which contained possible reciprocal translocations, were detected in cell line H2795 (Table 1). We were able to delineate 130 breakpoints (BPs) of translocations and deletions. Most of the BPs (75 BPs, 57.7%) were located in the centromeric/pericentromeric regions.
Whole-arm rearrangements included six isochromosomes found in four cell lines: i(1)(q10) in cell line H2804, i(5)(p10) in two cell lines (H513 and H2461), i(7)(p10) in cell line H2461, i(9)(p10), and i(21)(p10) in cell line H2731. All autosomes were involved in the whole-arm translocations, with chromosomes 1 and 9 among the most frequently involved. Six different whole-arm deletions were found in five cell lines.
Forty-two percent of BPs resided on the chromosomal arms. Chromosomal band 1q12 contained six BPs, 7q31 and 17q21 – four BPs, 13q21 and 19q13 – three BPs, 1p32, 1p36.1, 3q24, 4p15, 7p21, 9p13, 9p23, 9q12, 12q13 – two BPs each. Breakpoint location, when possible, was confirmed based on aCGH data. Figures 1B and C illustrate the advantage of combining SKY and aCGH data to examine genomic alterations, which include confirmation and refinement of BP of chromosomal rearrangements as well as understanding the precise nature of particular gains and losses through structural unbalanced rearrangements. No recurrent non-reciprocal or reciprocal translocations were found, besides i(5)(p10) in cell lines H513 and H2461.
Recurrent deletions, which were detected based on G-banding-like converted DAPI images, included del(1)(p32) in cell lines H513 and H28, and del(7)(q31) in cell lines H2795, H513, and H28.
Structural chromosomal abnormalities resulted in recurrent losses of whole chromosomal arms or fragments of 1p, 3p, 8p, 9p, 11p, 12p, 13q, 15q, and 17p, whereas recurrent numerical alterations included losses of whole chromosomes 4, 13, 14, 18, 19, 20, 21, and 22. Similarly, structural chromosomal abnormalities resulted in recurrent gains of whole chromosomal arms or fragments 1q, 5p, 7p, and 9q. Recurrent numerical alterations included gains of whole chromosomes 5, 7, and 20.
The most frequent finding was a partial or complete loss of chromosome 22 detected in the 10 studied cell lines (100%), sometimes only in a fraction of cells (present in <50% of cells in H2795, H2810, H2731, and H28), and partial or complete loss of 9p as well as loss of the whole chromosomes 4 and 14 in 70% of cell lines each. Partial loss of 1p was present in 60% of studied MMt cell lines as well as partial or complete loss of 11p and chromosomes 13 and 18. Losses of 3p, 8p, and chromosome 21 were present in 50% of cell lines each; loss of 17p was found in 40% of cell lines; and partial or complete loss of chromosome 19 in 30% of MMt cell lines. Loss of chromosome 20 was detected in 60% of cell lines, mainly as a gain present in a minor fraction of cells in each cell line (20–50% of cells), and as a loss in 30% of cell lines. Gain of 7p or of the whole chromosome 7 was found in 80% of cell lines and a gain of 1q in 50% of cell lines. Gain of 5p and of 9q was found in 40% and 30% of MMt cell lines, respectively.
Karyotypic heterogeneity (i.e., differences among metaphase spreads for the same cell line) was detected in cell lines as ploidy variability (in 9 of 10 MMt cell lines) as well as structural and numerical chromosomal heterogeneity in every MMt cell line studied by SKY (data not shown).
Global high-resolution array copy number analysis
To determine areas of CNAs, a FASST2 segmentation (see Materials and methods) was applied. The cell lines showed multiple recurrent genomic CNAs. Individual and combined genome view profiles are shown in Figure 2. Loss of a whole chromosome or chromosome arm were observed in 3p, 4, 6q, 8p, 9p, 10p, 11p, 12p, 13q, 14q, 15q, 16q, 17p, 18q, 18, 19p, and 22q, ranging from approximately 60% for 22q to 30% for 6q. Gain of a whole arm was observed in 1q, 5p, 7p, 8q, 9q, 17q, and 20q, ranging from approximately 17% for 1q and 17q to 41% for 7p. The most frequent focal gain and losses are listed in Supplementary Table 1. The percentage of cell lines that displayed losses was higher than the percentage of cell lines that showed gains (Figure 2A).
Figure 2.
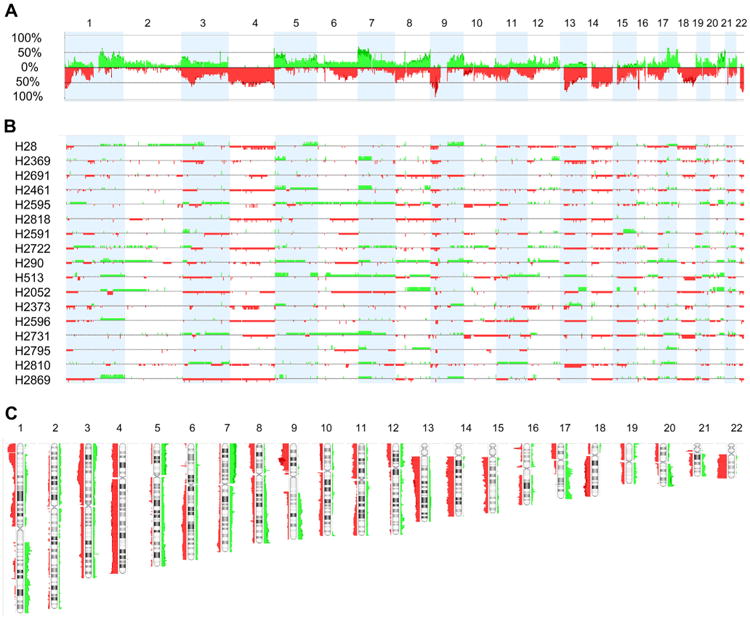
To determine the smallest significant regions harboring CNAs, a STAC number method was applied (see Materials and methods). STAC excludes whole-chromosome or chromosome arms copy number changes from its analysis and concentrates on smaller, focal CNAs instead. The length of the smallest significant regions varied from approximately 22 kb to 7 Mb and covered 277 genes (Supplemental Tables 1 and 2). All together, the regions of gain covered 150 genes and the regions of loss 127 genes. By this method, the region most frequently deleted was also in chromosome 9p21 (see above); all 17 cell lines were homozygously deleted. Some of the genes in these areas are known to be mutated or involved in cancer, such as CDKN2A/p16INK4A and CDKN2A/p14ARF in 9p21.3, RPL22 and MTOR in 1p36, and RBFOX1/A2BP1 in 16p13.3. Conversely, the most frequently gained regions were present in 65% of the 17 cell lines and involved three different areas located in chromosomes 1q23.1, 5p15.33, and 17q12–q21.31. As in the case of the deleted genes mentioned previously, some of the genes in the regions of copy number gains, such as ERBB2 in 17q21.1, FCRL4/IRTA1 in 1q21.1, and ZDHHC11 in 5p15.33, are known to play a role in cancer (Supplemental Tables 1 and 2).
Both regions of gains and losses had miRNAs: five miRNA genes were involved in one region of loss and seven miRNA genes were in different regions of gain (Supplemental Tables 1 and 2).
The percentage of known germinal copy number variations (CNVs) that overlapped with the smallest significant regions of gains and losses ranged from 0–100% (Supplemental Table 1). Approximately one fourth of these significant regions were located in areas completely covered by known CNVs.
HD analysis
The R-2.10.0 Language and the bioconductor package were used to identify HDs. The minimal deleted region with a log2 ratio less than −2, was determined with the cghMCR package (see Materials and methods; Supplemental Table 3). The HDs were considered recurrent when they were present in at least two cell lines or non-recurrent when present in single cell lines. A total of 88 HDs were found in the entire set of 17 MMt cell lines, varying from 1 to 9 per cell line (Supplemental Table 4). The total number of recurrent HDs was 52. Their lengths ranged from 11 kb to >10 Mb and contained 66 genes (Table 2 and Supplemental Table 4). These recurrent HDs corresponded to 10 genomic regions: 9p21.3, 9p21.2, 16p13.3, 22q11.23, 22q12.2, 3q26, 8p11.22, 3p21.2, 4q22.1, and 13q12.11 (Table 3).
Table 2. Genes present in homozygously deleted regions in MMt cell lines.
| Recurrent homozygous HD genes | Non-recurrent HD genes | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
||||||||||
| Chromosome | No. of cell lines | Chromosome | No. of cell lines | Chromosome | Cell line | No. of cell lines | |||||
|
|
|
|
|
|
|
|
|||||
| Gene | Location | HD | Allelic loss | Gene | Location | HD | Allelic loss | Gene | Location | With HD | With allelic loss |
| CDKN2A/p14ARF | 9p21.3 | 17 | 0 | IFNA7 | 9p21.3 | 6 | 8 | RERE | 1p36.23 | H2869 | 11 |
| CDKN2A/p16INK4A | 9p21.3 | 16 | 1 | IFNA10 | 9p21.3 | 6 | 8 | UBR4 | 1p36.13 | H28 | 9 |
| CDKN2B | 9p21.3 | 16 | 1 | IFNA16 | 9p21.3 | 6 | 8 | AGBL4 | 1p33 | H2461 | 4 |
| CDKN2B-AS | 9p21.3 | 16 | 1 | IFNA17 | 9p21.3 | 6 | 8 | RFWD2 | 1q25.2 | H2052 | 5 |
| UBA52P6 | 9p21.3 | 16 | 1 | IFNA14 | 9p21.3 | 6 | 8 | SCARNA3 | 1q25.1 | H2052 | 4 |
| C9orf53 | 9p21.3 | 16 | 1 | C9orf11/EQTN | 9p21.2 | 6 | 8 | PTPN14 | 1q41 | H2591 | 1 |
| MTAP | 9p21.3 | 14 | 2 | MOBKL2B | 9p21.2 | 6 | 8 | CABC1, CDC42BPA | 1q42.13 | H2731 | 1 |
| IFNA13 | 9p21.3 | 10 | 5 | C9orf35/MOB3B | 9p21.2 | 6 | 8 | OR2T10 | 1q44 | H28 | 3 |
| IFNA2 | 9p21.3 | 10 | 5 | IFNK | 9p21.2 | 6 | 8 | C2orf50 | 2p25.1 | H2373 | 1 |
| IFNP11 | 9p21.3 | 10 | 5 | C9orf72 | 9p21.2 | 6 | 8 | PQLC3 | 2p25.1 | H2373 | 3 |
| IFNP12 | 9p21.3 | 10 | 5 | KIAA1797 | 9p21.3 | 5 | 9 | ROCK2 | 2p25.1 | H2373 | 2 |
| IFNA8 | 9p21.3 | 10 | 5 | PTPLAD2 | 9p21.3 | 5 | 9 | LRRC3B, NEK10 | 3p24.1 | H290 | 6 |
| IFNWP2 | 9p21.3 | 10 | 5 | IFNB1 | 9p21.3 | 5 | 9 | CTNNB1, ULK4 | 3p22.1 | H28 | 10 |
| IFNA1 | 9p21.3 | 10 | 5 | IFNW1 | 9p21.3 | 5 | 9 | WDR51A | 3p21.2 | H2595 | 9 |
| IFNWP19 | 9p21.3 | 10 | 5 | MIR491 | 9p21.3 | 4 | 10 | BAP1, PHF7, SEMA3G, TNNC1 | 3p21.1 | H2722 | 9 |
| IFNE | 9p21.3 | 10 | 5 | NF2 | 22q12.2 | 4 | 9 | UGT2B17 | 4q13.2 | H290 | 10 |
| MIR31 | 9p21.3 | 10 | 5 | GSTT1 | 22q11.23 | 4 | 6 | UGT2B29P | 4q13.2 | H290 | 11 |
| DMRTA1 | 9p21.3 | 9 | 6 | MIR876 | 9p21.1 | 3 | 11 | UGT2B28 | 4q13.2 | H2373 | 10 |
| ELAVL2 | 9p21.3 | 9 | 6 | MIR873 | 9p21.1 | 3 | 10 | HLA-DRB5, HLA-DRB6 | 6p21.32 | H2373 | 14 |
| C9orf134/IZUMO3 | 9p21.3 | 8 | 7 | ADAM5P | 8p11.22 | 3 | 6 | CUX1 | 7q22.1 | H2691 | 1 |
| TUSC1 | 9p21.2 | 8 | 7 | ADAM3A | 8p11.22 | 3 | 2 | CSMD3 | 8q23.3 | H2461 | 5 |
| IFNWP15 | 9p21.3 | 6 | 8 | CCSER1/FAM190A | 4q22.1 | 2 | 10 | MIR2053 | 8q23.3 | H2461 | 6 |
| IFNWP9 | 9p21.3 | 6 | 8 | MLLT3 | 9p21.3 | 2 | 10 | PTPRD | 9p24.1 | H2691 | 11 |
| IFNWP18 | 9p21.3 | 6 | 8 | LATS2 | 13q12.11 | 2 | 9 | KRT18P36, RPS26P2 | 9p21.1 | H2595 | 12 |
| IFNWP5 | 9p21.3 | 6 | 8 | RPL29 | 3p21.2 | 2 | 8 | C9orf30, TMEFF1 | 9q31.1 | H2818 | 1 |
| IFNA22P | 9p21.3 | 6 | 8 | DUSP7 | 3p21.2 | 2 | 8 | SAPS3 | 11q13.2 | H2369 | 0 |
| KLHL9 | 9p21.3 | 8 | 7 | CNTN5 | 11q22.1 | H2596 | 3 | ||||
| IFNA6 | 9p21.3 | 8 | 7 | KLRC3, KLRC2 | 12p13.2 | H2373 | 4 | ||||
| IFNA5 | 9p21.3 | 8 | 6 | PLBD1, GUCY2C | 12p13.1-12p12.3 | H290 | 5 | ||||
| IFNP20 | 9p21.3 | 8 | 6 | GPC6 | 13q31.3 | H2461 | 6 | ||||
| IFT74 | 9p21.2 | 7 | 9 | ACOT6 | 14q24.3 | H2818 | 6 | ||||
| LRRC19 | 9p21.2 | 7 | 9 | NRXN3 | 14q31.1 | H2369 | 10 | ||||
| TEK | 9p21.2 | 7 | 9 | IGHVII-40-1, IGHV3-41 | 14q32.33 | H2052 | 10 | ||||
| NCRNA00032/LINC00032 | 9p21.2 | 7 | 9 | STK11 | 19p13.3 | H2369 | 6 | ||||
| LINGO2 | 9p21.2 | 7 | 8 | ZNF573, WDR87, SIPA1L3 | 19q13.1 | H2369 | 3 | ||||
| C9orf82/CAAP1 | 9p21.2 | 7 | 7 | MACROD2 | 20p12.1 | H2691 | 4 | ||||
| PLAA | 9p21.2 | 7 | 7 | NCRNA00186 | 20p12.1 | H2691 | 6 | ||||
| RBFOX1/A2BP1 | 16p13.3 | 7 | 4 | NIPSNAP1 | 22q12.2 | H2591 | 11 | ||||
| IFNA21 | 9p21.3 | 6 | 8 | C22orf30, DEPDC5 | 22q12 | H2369 | 12 | ||||
| IFNA4 | 9p21.3 | 6 | 8 | APOBEC3B, APOBEC3C, APOBEC3D | 22q13.1 | H2596 | 11 | ||||
Table 3. Smallest recurrent homozygously deleted regions, codeleted in the same MMt cell lines, and their main target genes.
| Cytoband and gene | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| 9p21.3 | 9p21.2 | 16p13.3 | 22q11.23 | 22q12.2 | 3q26 | 8p11.22 | 3p21.2 | 4q22.1 | 13q12.11 | |
|
|
|
|
|
|
|
|
|
|
|
|
| Cell line | CDKN2A | LINGO2 | RBFOX1/A2BP1 | GSTT1a | NF2 | No known genesa | ADAM5P and ADAM3Aa | RPL29 and DUSP7 | CCSER1/FAM190A | LATS2 |
| H2052 | X | X | X | X | X | X | ||||
| H2722 | X | X | X | X | ||||||
| H2595 | X | X | X | |||||||
| H2596 | X | X | X | |||||||
| H2795 | X | X | ||||||||
| H28 | X | X | X | |||||||
| H290 | X | X | ||||||||
| H2691 | X | X | X | X | X | |||||
| H2869 | X | X | X | |||||||
| H2591 | X | X | X | X | X | |||||
| H2369 | X | X | X | |||||||
| H2461 | X | X | X | X | ||||||
| H2373 | X | X | X | X | ||||||
| H2810 | X | X | ||||||||
| H513 | X | X | ||||||||
| H2731 | X | |||||||||
| H2818 | X | |||||||||
| No. of cell lines | 17 | 7 | 7 | 5 | 4 | 4 | 3 | 2 | 2 | 2 |
Area completely included in a copy number variation region.
To find the main targets of these deletions, the minimal common deleted (mcd) regions were defined as the minimal areas where most of the HDs overlapped. The HD regions and the genes deleted in these regions were: two regions in 9p21, one where the main targets seemed to be CDKN2A/p14ARF, CDKN2A/p16INK4A, and CDKN2B/p15 and the other where the target seemed to be LINGO2; 16p13.3 and RBFOX1/A2BP1; 22q11.23 and GSTT1; 22q12.2 and NF2; 8p11.22 and pseudogenes ADAM5P and ADAM3A; 13q12.11 and LATS2; 3p21.2 and RPL29 and DUSP7, and 4q22.1 and CCSER1/FAM190A (Table 3).
CCSER1/FAM190A was deleted in two cell lines where the HDs did not overlap; however, since the HDs seemed to be targeting the same gene, it was considered as a recurrent HD. GSTT1, ADAM5, and ADAM3A as well as the 3q26 region (without known genes) were completely included within a CNV region (most stringent Database of Genomic Variants track in Nexus CN 6.0). The frequency of all the recurrent HDs varied from 100%, homozygously deleted in all 17 MMt cell lines, for the 9p21.3 region, to 12%, homozygously deleted in only two cell lines each, for the 13q12.11 and LATS2; 3p21.2 and RPL29 and DUSP7; and 4q22.1 and CCSER1/FAM190A. In addition, 36 non-recurrent HDs contained 57 genes; their HD lengths varied from 13 kb to approximately 688 kb (Table 2, Supplemental Tables 3 and 4). Among the genes located in the areas of non-recurrent HDs, BAP1 located in 3p21.1 was homozygously deleted in H2722 and showed allelic loss in nine cell lines from the 17-MMt cell line panel. In addition, both recurrent and nonrecurrent homozygously deleted genomic sites usually displayed allelic loss in other cell lines without HD (Table 2)
Delineation of chromosomal rearrangements leading to 9p21.3 HDs
The combination of SKY and aCGH data allowed reconstruction of chromosomal rearrangements that led to copy number changes (Figure 1C). Areas of HDs on 9p21.3 exhibited two distinct profile patterns: 1) a simple peak going from a log2 ratio 0 down to a log2 ratio <−2, which is an indicator of the presence of the same deletion on all homologous chromosomes 9 (in cell lines H2869, H2052, H2373, H2795); 2) a peak going to a log2 ratio <−2 embedded in a larger area of log2 <0, which is an indicator of allelic loss of genetic material on the p arm of a homologous chromosome 9 (in cell lines H28, H2691, H2369, H513, H2461, H2818, H2731, H2591, H2810, H2596, H2722, H290, and H2595) (Supplemental Figure 1).
Delineation of karyotypic changes by SKY revealed that cell lines from the first group (H2869, H2373, H2795; all with chromosome counts in the hypo/near-diploid range) contained two copies of chromosome 9 with cytogenetically visible (H2373, H2795) or sub-microscopic (H2869) deletions in p21.3. These findings indicate that chromosome 9 with a p21.3 deletion was duplicated and the normal chromosome 9 was lost (Table 4).
Table 4. Integration of SKY and aCGH data for chromosome 9 rearrangements leading to HD in 9p21.3 region in malignant mesothelioma cell lines.
| Cell line | Ploidy | 9p losses by CGH | 9p rearrangements by SKY | Events leading to HD | Cartoon |
|---|---|---|---|---|---|
| H2869 | 2n− | Homozygous microdeletion 9p21.3 | 9×2 | Microdeletion on one copy of chr9, loss of normal chr9, and gain of second copy of chr9 with microdeletion | A |
| H2373 | 2n− | Homozygous deletion 9p21.2-21.3 | Del(9)(p21.2-p21.3)×2 | Deletion on one copy of chr9, loss of normal chr9, and gain of second copy of chr9 with deletion | A |
| H2795 | 2n± | Homozygous deletion 9p21.1-p21.3 | del(9)(p21.1-p21.3)×2 | Deletion on one copy of chr9, loss of normal chr9, and gain of second copy of chr9 with deletion | A |
| H2818 | 2n− | Homozygous deletion on 9p21.3, loss of 9p | 9, der(5)del(5)(q14q23)t(5;9)(p14;q21) | Microdeletion on one copy of chr9, loss of 9q21-pter due to unbalanced translocation | B |
| H2461 | 2n− | Homozygous microdeletion on 9p21.3, loss of 9p | 9, der(15)t(9;15)(q12;p13) | Microdeletion on one copy of chr9, loss of 9q12-pter due to unbalanced translocation | B |
| H513 | 3n− | homozygous microdeletion on 9p21.3, loss of 9p21.2-pter | 9, der(9)t(9;13)×2 | Microdeletion on one copy of chr9, loss of 9p21.2-pter due to unbalanced translocation, and gain of the second copy of der(9) | C (or E) |
| H2731 | 2n+ | Homozygous deletion on 9p21.3 | i(9)(p10), del(9)(p11), der(9;11) (q10;q10)×2 | Microdeletion on one copy of chr9, loss of second copy of 9p due to unbalanced translocation,isochromosome i(9)(p10) formation, and gain of the second copy der(9;11) | C (or E) |
| H28 | 3n±/4n− | Homozygous microdeletion on 9p21.3, loss of 9p13.3-pter | 9x2, der(9)t(9;17)(p13;q21)×2 | Microdeletion on one copy of chr9, loss of 9p13.3 due to unbalanced translocation, and tetraploidization | D |
| H2369 | 3n+/4n− | Homozygous microdeletion on 9p21.3, loss of 9p11-pter | 9x2, der(5;9)(p10;q10)×2 | Microdeletion on one copy of chr9, loss of 9p due to unbalanced translocation, and tetraploidization | D |
| H2722 | 3n+ | Homozygous deletion 9p21.1-p21.3, loss of 9p11-pter | del(9)(p21.1-p21.3), der(15)t(9;15) (q11;q14)×2 | Deletion on one copy of chr9, loss of second 9p due to unbalanced translocation, tetraploidization, and loss one copy of del(9) | E |
| H290 | 3n+/5n− | homozygous deletion 9p21.1-p21.3, loss of 9p21.1-pter, gain of 9p13.3-p21.1 | 9x(2-5 copies), der(5)t(5;9)(?; p21.1) ×(0-3copies) | Deletion 9p21.1-p21.3, loss of 9p13-pter due to unbalanced translocation, tetraploidization, further gains and losses, and structural rearrangements | E,F |
| H2810 | 3n±/4n± | Homozygous deletion 9p21.3, loss of 9p24.1-pter | 9×2, der(9)t(9;22)(p13;?), der(9)t(9;15) (p24.1;?), der(9)t(7;9)(?;p24.1) | Deletion 9p21.1-p21.3, loss of 9p13-pter due to unbalanced translocation, tetraploidization, further gains and losses, and structural rearrangements | E,F |
In the second group of cell lines, in addition to the p21.3 deletion on one homologue of chromosome 9, allelic loss occurred on the second homologous chromosome due to an unbalanced translocation that led to the loss of a whole 9p arm or part of a 9p arm (cell lines H2818 and H2461) (Table 4). After the first two events involving chromosome 9 occurred, some of cell lines in the second group gained additional copies of rearranged chromosomes (cell lines H513 and H2731), whereas others went through tetraploidization (cell lines H28 and H2369) and further numerical or structural rearrangements seen as losses or gains of primary derivatives 9, as well as formation of secondary derivatives due to additional translocations (cell lines H2722, H290, and H2810) (Table 4).
Molecular characterization of the 9p21.3 HD
A more detailed analysis of the most frequently homozygously deleted region, situated in 9p21.3, was performed. This was also the most frequent CNA observed, since all MMt cell lines had it. The mcd region was located inside the CDKN2A gene, and it was limited by Agilent probes A_14_P112983 (positioned in the human genome at 21,990,522-21,990,581 bp) and A_14_P129522 (positioned at 21,968,042-21,968,099 bp, build GRCh37/hg19) and covered an approximately 22-kb area. The CDKN2A locus is >26 kb long, and it is transcribed into various transcripts. The most common transcripts correspond to tumor suppressor p14ARF and tumor suppressor p16INK4A and are shown in Figure 3. These two transcripts have different promoters, differ in their first exons (exon 1 for p14ARF and exon 2a for p16INK4A), and have different reading frames. As a result, they are two different proteins. The genomic DNA coding for exon 1 of p14ARF was clearly missing in all 17 MMt cell lines, whereas exon 2a corresponding to p16INK4A was missing in 16 cell lines according to aCGH. In one cell line, H2869, because of the probe spacing in the array, it was not possible to define more precisely the limits of the mcd region (Figure 3). We then performed a PCR for CDKN2A exon 2a and observed that it was homozygously deleted (data not shown); therefore, both p14ARF and p16INK4A were disrupted in the H2869 MMt cell line.
Figure 3.
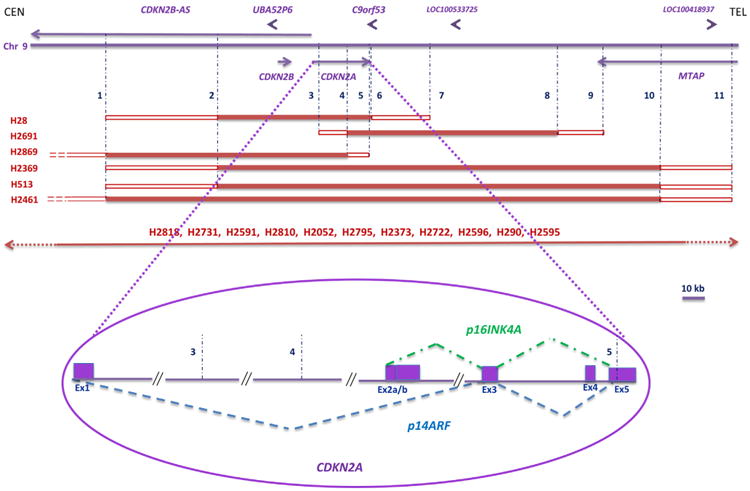
Other genes were also co-deleted with CDKN2A (Table 2; Supplementary Table 3). The closest neighboring genes included tumor suppressor genes CDKN2B/p15 and CDKN2B-AS, UBA52P6, and C9orf53 in 16 cell lines and MTAP in 14 cell lines.
The size of the HDs in the 9p21.3 region varied from approximately 68 kb for cell line H28 to >10 Mb for cell line H2595. It is noteworthy that 6 of 17 MMt cell lines had HDs between 68–248 kb and the remaining 11 cell lines had HDs >1 Mb. These HDs were also much larger than the HDs in other chromosome regions, the largest of which was a 688-kb HD (Supplementary Table 4). The larger recurrent HD region in 9p21, which extended to 9p22 in some MMt cell lines, had 58 genes including 27 members of the type I interferon genes, 4 microRNA genes, and 22 other known genes; remaining were open reading frames, a pseudogene, and an uncharacterized protein gene.
In six of the MMt cell lines, the LINGO2 gene located in the proximal region of 9p21.2 was co-deleted together with the p21.3 region. LINGO2 seemed to be a separate target, as shown by an intragenic HD in one of the cell lines, H28; this HD was clearly separated from the HD that involved CDKN2A.
Discussion
Here, we report an integrated aCGH/SKY genomic analysis in a panel of MMt cell lines.
For the first-time, SKY was applied to MMt cells. Numerous karyotypic abnormalities have been delineated, including ploidy changes, as well as structural and numerical chromosomal rearrangements. As in previous studies of MMt tumors and cell lines by conventional cytogenetic methods, no recurrent translocations have been found besides i(5)(p10) in two cell lines, H513 and H2461. The majority of translocations (96%) detected by SKY were unbalanced and, together with cytogenetically visible deletions, produced a variety of chromosomal aberrations that resulted in recurrent gains and losses of chromosomal segments or whole arms of certain chromosomes. Among unbalanced translocations, whole-arm aberrations were the most frequent, in agreement with our previous finding on a panel of 60 cancer cell lines of different types (47). Integration of SKY and high-resolution genome-wide scans of CNAs using oligonucleotide aCGH in 17 MMt cell lines allowed identification of large and focal CNAs as well as BPs with higher precision.
The frequency of clonal balanced (reciprocal) translocations found by SKY was estimated as 0.40 translocations per cell line. This frequency of balanced translocations is the same as in epithelial cancer cell lines (0.40 per cell line) and leukemia cell lines (0.44 per cell line), where balanced translocations are considered to be initiation events (47). This demonstrates the possible significance of reciprocal translocations in MMt tumorigenesis.
Gains and losses of the whole chromosomes are also frequently present, with recurrent losses of chromosome 4, 13, 14, and 22, which is in agreement with previously reported data for MMt tumors and cell lines (4–7,10,48,49).
Genomic profiling of a panel of established MMt cell lines revealed that recurrent whole-arm CNAs harbored the majority of recurrent focal gains and losses. Many whole-arm and focal CNAs overlapped with previous findings in other MMt cell lines and, most important, in MMt tumors (8–11,13–19). Of note is a recent second-generation sequencing of one MMt tumor (50). The genome profile of this one tumor was in good agreement with the results of the 17 MMt cell lines presented here.
Areas of recurrent focal losses were enriched with genes that, when deleted or down-regulated, could decrease apoptosis (MFN2, TNFRSF1B, TNFRSF9, TNFRSF25, DFFA, NPPA, and PIK3CD), increase cell proliferation (NCF1, CSF3), and promote G1–S transition (CDKN2A, MTOR). Genes in regions of gains included several oncogenes (ERBB2, ELN, RPL22, and IRTA1), as well as genes, up-regulation of which could decrease apoptosis (mir-1, CSF3, mir-133, and BECN1), increase cell proliferation (ERBB2, ELN), and promote cell cycle progression (ERBB2).
Areas of significant focal copy number changes were enriched with genes involved in cell migration (NCF1, GRB7, LIMK1, RAMP2, ELN, ERBB2, CDKN2A, UTS2), tight junction (WNK4, CLDN3, CLDN4, TJP2), autophagy (GTF2IRD2, BECN1, NCR3, MTOR, CDKN2A, ICMT), sugar transport (SLC2A7, SLC2A5, SLC45A1), oxidation reduction (MTHFR, AOC2, AOC3, CYP2E1, DHRS3, H6PD, PGD, KCNAB2, PLOD1), and regulation of ubiquitination (FBXO2, CDKN2A, PSMD3, PSME3, VPS28).
For the first time, a comprehensive whole-genome search for homozygous deletions (HDs) was undertaken for the panel of MMt cell lines utilizing the R-2.10.0 Language and the cghMCR package of Bioconductor. Here, we report the largest set of recurrent and non-recurrent HDs in MMt described to date. HDs significantly facilitate narrowing down the region where there is a suspected presence of tumor suppressor genes. A total of 88 HDs were found, many of which were embedded in regions of confirmed focal loss; 52 of the 88 HDs were recurrent HDs located within 10 genomic regions; the remaining 36 HDs were non-recurrent.
The most frequently homozygously deleted region was located in 9p21.3 that targeted CDKN2A/p14ARF and CDKN2A/p16INK4A (deleted in 100% of the MMt cell lines) and CDKN2B/p15 (deleted in 94% of the MMt cell lines), followed by 9p21.2 that targeted LINGO2 and 16p13.3 that targeted RBFOX1/A2BP1 in 41% each; 22q11.23 and GSTT1 in 29%; 22q12.2 and NF2 and 3q26 with no known genes in 23% each; 8p11.22 and ADAM5P and ADAM3A in 18% each; 3p21.2 and RPL29 and DUSP7, 4q22.1 and CCSER1/FAM190A, and 13q12.11 and LATS2 in 12% each of the 17 MMt cell lines (Table 3).
Only three of the 10 recurrent HD areas have been previously reported in MMt tumors and cell lines (9p21.3, 22q12.2, and 13q12.11), and all three harbor known tumor suppressor genes: CDKN2A/p16INK4A, CDKN2A/p14ARF, CDKN2B/p15, NF2, and LATS2 (20–30,51).
Here, we report for the first time that LINGO2, RBFOX1/A2BP1, RPL29, DUSP7, and CCSER1/FAM190A were recurrently homozygously deleted in the MMt cell line panel. Some of these genes could be new candidate tumor suppressor genes. Of note, LINGO2 and RBFOX1/A2BP1 seemed to be homozygously deleted in a mutually exclusive way, with the exception of one cell line in which both genes were homozygously deleted (Table 3). Both of these genes have already been implicated in other types of cancer. RBFOX1/A2BP1 has been found to be deleted in gastric cancer, and deletions were associated with poor prognosis in colon cancer (52,53). LINGO2 is possibly involved in gliomas (54). Regarding the RPL29, DUSP7, and CCSER1/FAM190A genes, information about their roles in tumorigenesis is limited. DUSP7 was found to be deleted in more than 20% of a set of 53 MMt tumors; however, no mutations were found (32). The CCSER1/FAM190A gene had internal rearrangements in 19 of 48 non-selected human cancers (55).
Other recurrent HDs that targeted GSTT1, pseudogenes ADAM5P and ADAM3A, and the 3q26 region with no known genes were completely located inside CNV regions and may not have been related to the tumorigenesis and progression of MMt.
In addition to recurrent HDs, a large set of non-recurrent HDs, which included 36 HDs, were also found in the 17 MMt cell lines described here (Supplemental Table 4). To determine the possible relevance of non-recurrent HDs to MMt, the frequency of allelic loss was established and we found that regions where these HDs were located had frequently recurrent allelic loss in other MMt cell lines. For example, 1p36.23, where the RERE gene is located, had an HD in one MMt cell line and showed allelic loss for that region in 11 of the remaining MMt cell lines, whereas CTNNB1/β1-catenin was homozygously deleted in one cell line and had allelic loss in 10 additional MMt cell lines (Table 2). This suggests that HD may not be the main mechanism of inactivation of genes in the area; however, since HDs facilitate the search for tumor suppressor genes by narrowing the potential regions in the genome where they may be located, these non-recurrent HDs could be a useful tool to find genes with a putative role in MMt tumorigenesis. The presence among these genes of BAP1, which is a new tumor suppressor gene in MMts and is mainly inactivated by point mutations and, in some cases, by HDs (32,34,35), highlights the importance of non-recurrent HD regions found in this study.
In the well-known 9p21.3 region, we found that both CDKN2A/p14ARF and CDKN2A/p16INK4A were homozygously deleted in 100% of the MMt cell lines. Although initially, CDKN2A/p16INK4A was considered the main target of HD in the 9p21.3 region, we found that both tumor suppressors are affected. Research in mice supports the independent and complementary roles of CDKN2A/p16INK4A and CDKN2A/p14ARF. Mice with deletions of both p16INF4A and p19ARF (homologous to human p14ARF gene) had accelerated asbestos-induced MMt development compared with mice that had deletions in either region (56). CDKN2A/p16INK4A affects the RB pathway, whereas CDKN2A/p14ARF is involved in G1 and G2 arrest through the p53 pathway (25,57). The p53 gene is rarely mutated in MMt (58), supporting inactivation of the p53 pathway by different mechanisms. CDKN2A/p14ARF, CDKN2A/p16INK4A, and CDKN2B/p15 are very frequently simultaneously homozygously deleted in MMt; this provides a way of disrupting the p53, RB, and TGF-β pathways, which are three important cancer pathways, with one HD.
The 9p21.3 region of recurrent HDs is often very large (in 65% of the MMt cell lines analyzed in this study, it was >1 Mb) and may include additional targets. Outside the minimal common homozygously deleted region in 9p21.3 were other genes, such as MTAP, the interferon (IFN) family of genes, and other genes (Table 2). The HD of some of these genes might confer an additional growth selective advantage, and yet, other genes, such as the IFN genes, could be just passengers. LINGO2 seems to be a separate target. In many cases, it was deleted at the centromeric end of a large HD that spanned the 9p21.3–p21.2 region. In one cell line, it was targeted by separate small HDs.
Integration of SKY and aCGH data was used for the reconstruction of chromosomal rearrangements that led to the formation of HDs. Delineation of chromosomal rearrangements and ploidy changes by SKY combined with detection of CNAs and their breakpoints by aCGH in MMt cells revealed that HDs in the 9p21.3 region were acquired in at least two different ways. In one group of cell lines, chromosome 9 with a p21.3 microdeletion/small deletion was duplicated and the normal chromosome 9 was lost (possibly due to a non-disjunction or gene conversion event that led to the formation of HDs) (Figure 4A). In the second group of cell lines, in addition to a 9p21.3 microdeletion/small deletion, allelic loss occurred on the 9p of the second homologous chromosome due to unbalanced translocations (Figure 4B). Moreover, some of the cell lines in the second group went through tetraploidization after the first two events (microdeletion/small deletion and unbalanced translocations) occurred (Figure 4C,D), and some of them also experienced additional numerical or structural rearrangements (losses or gains of primary derivatives of 9 as well as formation of secondary derivatives due to additional translocations) (Figure 4E,F). Chromosomal heterogeneity detected in MMt cell lines points to a presence of structural and numerical chromosomal instability in these cells. It is reasonable to suggest that only with the acquisition of structural or numerical chromosomal instability could MMt cells attain complete loss of the 9p21.3 genomic region and loss of the tumor suppressor genes located there. Our data also suggest that tetraploidization is a late event in the karyotypic progression of MMt cells, after HDs in the 9p21.3 region have already been acquired.
Figure 4.

Aneuploidy of MMt karyotypes as well as the presence of structural and numerical chromosomal heterogeneity makes MMt tumors possible candidates for treatment by anti-cancer drugs that target cells with aneuploid, karyotypically unstable phenotypes (59).
Supplementary Material
Acknowledgments
The authors are grateful to W. Michael Kuehl for critical reading of the manuscript.
Footnotes
Supplementary data: Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.cancergen.2013.04.006.
References
- 1.Carbone M, Kratzke RA, Testa JR. The pathogenesis of mesothelioma. Semin Oncol. 2002;29:2–17. doi: 10.1053/sonc.2002.30227. [DOI] [PubMed] [Google Scholar]
- 2.Howlader N, Noone AM, Krapcho M, et al. SEER Cancer Statistics Review, 1975-2009 (Vintage 2009 Populations) National Cancer Institute; Bethesda, MD: [Accessed on XXX, XX, 2012]. Available at http://seer.cancer.gov/csr/1975_2009_pops09/ [Google Scholar]
- 3.Carbone M, Ly BH, Dodson RF, et al. Malignant mesothelioma: facts, myths, and hypotheses. J Cell Physiol. 2012;227:44–58. doi: 10.1002/jcp.22724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gibas Z, Li FP, Antman KH, et al. Chromosome changes in malignant mesothelioma. Cancer Genet Cytogenet. 1986;20:191–201. doi: 10.1016/0165-4608(86)90074-9. [DOI] [PubMed] [Google Scholar]
- 5.Popescu NC, Chahinian AP, DiPaolo JA. Nonrandom chromosome alterations in human malignant mesothelioma. Cancer Res. 1988;48:142–147. [PubMed] [Google Scholar]
- 6.Tiainen M, Tammilehto L, Mattson K, et al. Nonrandom chromosomal abnormalities in malignant pleural mesothelioma. Cancer Genet Cytogenet. 1988;33:251–274. doi: 10.1016/0165-4608(88)90035-0. [DOI] [PubMed] [Google Scholar]
- 7.Sandberg AA, Bridge JA. Updates on the cytogenetics and molecular genetics of bone and soft tissue tumors. Mesothelioma Cancer Genet Cytogenet. 2001;127:93–110. doi: 10.1007/3-540-30792-3_7. [DOI] [PubMed] [Google Scholar]
- 8.Kivipensas P, Bjorkqvist AM, Karhu R, et al. Gains and losses of DNA sequences in malignant mesothelioma by comparative genomic hybridization. Cancer Genet Cytogenet. 1996;89:7–13. doi: 10.1016/0165-4608(95)00358-4. [DOI] [PubMed] [Google Scholar]
- 9.Balsara BR, Bell DW, Sonoda G, et al. Comparative genomic hybridization and loss of heterozygosity analyses identify a common region of deletion at 15q11.1-15 in human malignant mesothelioma. Cancer Res. 1999;59:450–454. [PubMed] [Google Scholar]
- 10.Murthy SS, Testa JR. Asbestos, chromosomal deletions, and tumor suppressor gene alterations in human malignant mesothelioma. J Cell Physiol. 1999;180:150–157. doi: 10.1002/(SICI)1097-4652(199908)180:2<150::AID-JCP2>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 11.Jensen RH, Tiirikainen M, You L, et al. Genomic alterations in human mesothelioma including high resolution mapping of common regions of DNA loss in chromosome arm 6q. Anti-cancer Res. 2003;23:2281–2289. [PubMed] [Google Scholar]
- 12.Kallioniemi OP, Kallioniemi A, Piper J, et al. Optimizing comparative genomic hybridization for analysis of DNA sequence copy number changes in solid tumors. Genes Chromosomes Cancer. 1994;10:231–243. doi: 10.1002/gcc.2870100403. [DOI] [PubMed] [Google Scholar]
- 13.Zanazzi C, Hersmus R, Veltman IM, et al. Gene expression profiling and gene copy-number changes in malignant mesothelioma cell lines. Genes Chromosomes Cancer. 2007;46:895–908. doi: 10.1002/gcc.20475. [DOI] [PubMed] [Google Scholar]
- 14.Taniguchi T, Karnan S, Fukui T, et al. Genomic profiling of malignant pleural mesothelioma with array-based comparative genomic hybridization shows frequent non-random chromosomal alteration regions including JUN amplification on 1p32. Cancer Sci. 2007;98:438–446. doi: 10.1111/j.1349-7006.2006.00386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lindholm PM, Salmenkivi K, Vauhkonen H, et al. Gene copy number analysis in malignant pleural mesothelioma using oligonucleotide array CGH. Cytogenet Genome Res. 2007;119:46–52. doi: 10.1159/000109618. [DOI] [PubMed] [Google Scholar]
- 16.Ivanov SV, Miller J, Lucito R, et al. Genomic events associated with progression of pleural malignant mesothelioma. Int J Cancer. 2009;124:589–599. doi: 10.1002/ijc.23949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Serio G, Gentile M, Pennella A, et al. Characterization of a complex chromosome aberration in two cases of peritoneal mesothelioma arising primarily in the hernial sac. Pathol Int. 2009;59:415–421. doi: 10.1111/j.1440-1827.2009.02387.x. [DOI] [PubMed] [Google Scholar]
- 18.Cheung M, Pei J, Pei Y, et al. The promyelocytic leukemia zincfinger gene, PLZF, is frequently downregulated in malignant mesothelioma cells and contributes to cell survival. Oncogene. 2010;29:1633–1640. doi: 10.1038/onc.2009.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jean D, Thomas E, Manie E, et al. Syntenic relationships between genomic profiles of fiber-induced murine and human malignant mesothelioma. Am J Pathol. 2011;178:881–894. doi: 10.1016/j.ajpath.2010.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng JQ, Jhanwar SC, Klein WM, et al. p16 alterations and deletion mapping of 9p21-p22 in malignant mesothelioma. Cancer Res. 1994;54:5547–5551. [PubMed] [Google Scholar]
- 21.Xiao S, Li D, Vijg J, et al. Codeletion of p15 and p16 in primary malignant mesothelioma. Oncogene. 1995;11:511–515. [PubMed] [Google Scholar]
- 22.Kratzke RA, Otterson GA, Lincoln CE, et al. Immunohistochemical analysis of the p16INK4 cyclin-dependent kinase inhibitor in malignant mesothelioma. J Natl Cancer Inst. 1995;87:1870–1875. doi: 10.1093/jnci/87.24.1870. [DOI] [PubMed] [Google Scholar]
- 23.Beroukhim R, Mermel CH, Porter D, et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bignell GR, Greenman CD, Davies H, et al. Signatures of mutation and selection in the cancer genome. Nature. 2010;463:893–898. doi: 10.1038/nature08768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ruas M, Peters G. The p16INK4a/CDKN2A tumor suppressor and its relatives. Biochim Biophys Acta. 1998;1378:F115–F177. doi: 10.1016/s0304-419x(98)00017-1. [DOI] [PubMed] [Google Scholar]
- 26.Kamb A, Gruis NA, Weaver-Feldhaus J, et al. A cell cycle regulator potentially involved in genesis of many tumor types. Science. 1994;264:436–440. doi: 10.1126/science.8153634. [DOI] [PubMed] [Google Scholar]
- 27.Bianchi AB, Mitsunaga SI, Cheng JQ, et al. High frequency of inactivating mutations in the neurofibromatosis type 2 gene (NF2) in primary malignant mesotheliomas. Proc Natl Acad Sci U S A. 1995;92:10854–10858. doi: 10.1073/pnas.92.24.10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sekido Y. Inactivation of Merlin in malignant mesothelioma cells and the Hippo signaling cascade dysregulation. Pathol Int. 2011;61:331–344. doi: 10.1111/j.1440-1827.2011.02666.x. [DOI] [PubMed] [Google Scholar]
- 29.Cheng JQ, Lee WC, Klein MA, et al. Frequent mutations of NF2 and allelic loss from chromosome band 22q12 in malignant mesothelioma: evidence for a two-hit mechanism of NF2 inactivation. Genes Chromosomes Cancer. 1999;24:238–242. [PubMed] [Google Scholar]
- 30.Murakami H, Mizuno T, Taniguchi T, et al. LATS2 is a tumor suppressor gene of malignant mesothelioma. Cancer Res. 2011;71:873–883. doi: 10.1158/0008-5472.CAN-10-2164. [DOI] [PubMed] [Google Scholar]
- 31.Visser S, Yang X. LATS tumor suppressor: a new governor of cellular homeostasis. Cell Cycle. 2010;9:3892–3903. doi: 10.4161/cc.9.19.13386. [DOI] [PubMed] [Google Scholar]
- 32.Bott M, Brevet M, Taylor BS, et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat Genet. 2011;43:668–672. doi: 10.1038/ng.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshikawa Y, Sato A, Tsujimura T, et al. Frequent deletion of 3p21.1 region carrying semaphorin 3G and aberrant expression of the genes participating in semaphorin signaling in the epithelioid type of malignant mesothelioma cells. Int J Oncol. 2011;39:1365–1374. doi: 10.3892/ijo.2011.1158. [DOI] [PubMed] [Google Scholar]
- 34.Yoshikawa Y, Sato A, Tsujimura T, et al. Frequent inactivation of the BAP1 gene in epithelioid-type malignant mesothelioma. Cancer Sci. 2012;103:868–874. doi: 10.1111/j.1349-7006.2012.02223.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Testa JR, Cheung M, Pei J, et al. Germline BAP1 mutations predispose to malignant mesothelioma. Nat Genet. 2011;43:1022–1025. doi: 10.1038/ng.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ramos-Nino ME, Testa JR, Altomare DA, et al. Cellular and molecular parameters of mesothelioma. J Cell Biochem. 2006;98:723–734. doi: 10.1002/jcb.20828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tomasetti M, Santarelli L. Biomarkers for early detection of malignant mesothelioma: diagnostic and therapeutic application. Cancers. 2010;2:523–548. doi: 10.3390/cancers2020523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sekido Y. Genomic abnormalities and signal transduction dysregulation in malignant mesothelioma cells. Cancer Sci. 2010;101:1–6. doi: 10.1111/j.1349-7006.2009.01336.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Solomon DA, Kim JS, Ressom HW, et al. Sample type bias in the analysis of cancer genomes. Cancer Res. 2009;69:5630–5633. doi: 10.1158/0008-5472.CAN-09-1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ihaka R, Gentleman R. R: a language for data analysis and graphics. J Comput Graph Stat. 1996;5:299–314. [Google Scholar]
- 41.Aguirre AJ, Brennan C, Bailey G, et al. High-resolution characterization of the pancreatic adenocarcinoma genome. Proc Natl Acad Sci U S A. 2004;101:9067–9072. doi: 10.1073/pnas.0402932101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gentleman R, Carey VJ, Huber W, et al. Bioinformatics, and computational biology solutions using R and Bioconductor. New York: Springer; 2005. [Google Scholar]
- 43.Olshen AB, Venkatraman ES, Lucito R, et al. Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics. 2004;5:557–572. doi: 10.1093/biostatistics/kxh008. [DOI] [PubMed] [Google Scholar]
- 44.Venkatraman ES, Olshen AB. A faster circular binary segmentation algorithm for the analysis of array CGH data. Bioinformatics. 2007;23:657–663. doi: 10.1093/bioinformatics/btl646. [DOI] [PubMed] [Google Scholar]
- 45.Diskin SJ, Eck T, Greshock J, et al. STAC: A method for testing the significance of DNA copy number aberrations across multiple array-CGH experiments. Genome Res. 2006;16:1149–1158. doi: 10.1101/gr.5076506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 47.Roschke AV, Tonon G, Gehlhaus KS, et al. Karyotypic complexity of the NCI-60 drug-screening panel. Cancer Res. 2003;63:8634–8647. [PubMed] [Google Scholar]
- 48.Flejter WL, Li FP, Antman KH, et al. Recurring loss involving chromosomes 1, 3, and 22 in malignant mesothelioma: possible sites of tumor suppressor genes. Genes Chromosomes Cancer. 1989;1:148–154. doi: 10.1002/gcc.2870010207. [DOI] [PubMed] [Google Scholar]
- 49.Hagemeijer A, Versnel MA, Van Drunen E, et al. Cytogenetic analysis of malignant mesothelioma. Cancer Genet Cytogenet. 1990;47:1–28. doi: 10.1016/0165-4608(90)90258-c. [DOI] [PubMed] [Google Scholar]
- 50.Bueno R, De Rienzo A, Dong L, et al. Second generation sequencing of the mesothelioma tumor genome. PLoS One. 2010;5:e10612. doi: 10.1371/journal.pone.0010612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Illei PB, Rusch VW, Zakowski MF, et al. Homozygous deletion of CDKN2A and codeletion of the methylthioadenosine phosphorylase gene in the majority of pleural mesotheliomas. Clin Cancer Res. 2003;9:2108–2113. [PubMed] [Google Scholar]
- 52.Andersen CL, Lamy P, Thorsen K, et al. Frequent genomic loss at chr16p13.2 is associated with poor prognosis in colorectal cancer. Int J Cancer. 2011;129:1848–1858. doi: 10.1002/ijc.25841. [DOI] [PubMed] [Google Scholar]
- 53.Tada M, Kanai F, Tanaka Y, et al. Prognostic significance of genetic alterations detected by high-density single nucleotide polymorphism array in gastric cancer. Cancer Sci. 2010;101:1261–1269. doi: 10.1111/j.1349-7006.2010.01500.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kotliarov Y, Kotliarova S, Charong N, et al. Correlation analysis between single-nucleotide polymorphism and expression arrays in gliomas identifies potentially relevant target genes. Cancer Res. 2009;69:1596–1603. doi: 10.1158/0008-5472.CAN-08-2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Scrimieri F, Calhoun ES, Patel K, et al. FAM190A rearrangements provide a multitude of individualized tumor signatures and neo-antigens in cancer. Oncotarget. 2011;2:69–75. doi: 10.18632/oncotarget.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Altomare DA, Menges CW, Xu J, et al. Losses of both products of the Cdkn2a/Arf locus contribute to asbestos-induced mesothelioma development and cooperate to accelerate tumorigenesis. PLoS One. 2011;6:e18828. doi: 10.1371/journal.pone.0018828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sherr CJ, Weber JD. The ARF/p53 pathway. Curr Opin Genet Dev. 2000;10:94–99. doi: 10.1016/s0959-437x(99)00038-6. [DOI] [PubMed] [Google Scholar]
- 58.Toyooka S, Kishimoto T, Date H. Advances in the molecular biology of malignant mesothelioma. Acta Med Okayama. 2008;62:1–7. doi: 10.18926/AMO/30990. [DOI] [PubMed] [Google Scholar]
- 59.Roschke AV, Kirsch IR. Targeting karyotypic complexity and chromosomal instability of cancer cells. Curr Drug Targets. 2010;11:1341–1350. doi: 10.2174/1389450111007011341. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.