

Abstract
Tauroursodeoxycholic acid (TUDCA) is the taurine conjugate of ursodeoxycholic acid (UDCA), a US Food and Drug Administration–approved hydrophilic bile acid for the treatment of certain cholestatic liver diseases. There is a growing body of research on the mechanism(s) of TUDCA and its potential therapeutic effect on a wide variety of non-liver diseases. Both UDCA and TUDCA are potent inhibitors of apoptosis, in part by interfering with the upstream mitochondrial pathway of cell death, inhibiting oxygen-radical production, reducing endoplasmic reticulum (ER) stress, and stabilizing the unfolded protein response (UPR). Several studies have demonstrated that TUDCA serves as an anti-apoptotic agent for a number of neurodegenerative diseases, including amyotrophic lateral sclerosis, Alzheimer's disease, Parkinson's disease, and Huntington's disease. In addition, TUDCA plays an important role in protecting against cell death in certain retinal disorders, such as retinitis pigmentosa. It has been shown to reduce ER stress associated with elevated glucose levels in diabetes by inhibiting caspase activation, up-regulating the UPR, and inhibiting reactive oxygen species. Obesity, stroke, acute myocardial infarction, spinal cord injury, and a long list of acute and chronic non-liver diseases associated with apoptosis are all potential therapeutic targets for T/UDCA. A growing number of pre-clinical and clinical studies underscore the potential benefit of this simple, naturally occurring bile acid, which has been used in Chinese medicine for more than 3000 years.
Key Words: Apoptosis, bile acids, chaperone, cytoprotection, endoplasmic reticulum stress, misfolded proteins, neurodegenerative diseases, unfolded protein response, tauroursodeoxy-cholic acid (TUDCA), ursodeoxycholic acid (UDCA)
INTRODUCTION
Tauroursodeoxycholic acid (TUDCA) is the taurine conjugate of ursodeoxycholic acid (UDCA), a secondary bile acid produced only by intestinal bacteria. T/UDCA has been studied for its ameliorating effects on inflammatory metabolic diseases, including atherosclerosis, diabetes, and renal disease.1 UDCA was originally US Food and Drug Administration (FDA)–approved for the treatment of certain cholestatic liver diseases based on its choleretic effects and ability to protect hepatocytes from hydrophobic bile acids. Studies examining the mechanisms of TUDCA and UDCA action have shown that these compounds act as potent inhibitors of apoptosis by interfering with the mitochondrial pathway of cell death, inhibiting oxygen-radical production, and reducing endoplasmic reticulum stress and caspase activation (Figure 1).2 Normally, the endoplasmic reticulum synthesizes proteins and folds them properly with the help of endoplasmic reticulum (ER) chaperones. Too many unfolded proteins signal an ER stress response associated with reduced protein synthesis and increased expression of chaperones, malfunction of the unfolded protein response (UPR) and ultimately cell death.3
Figure 1.
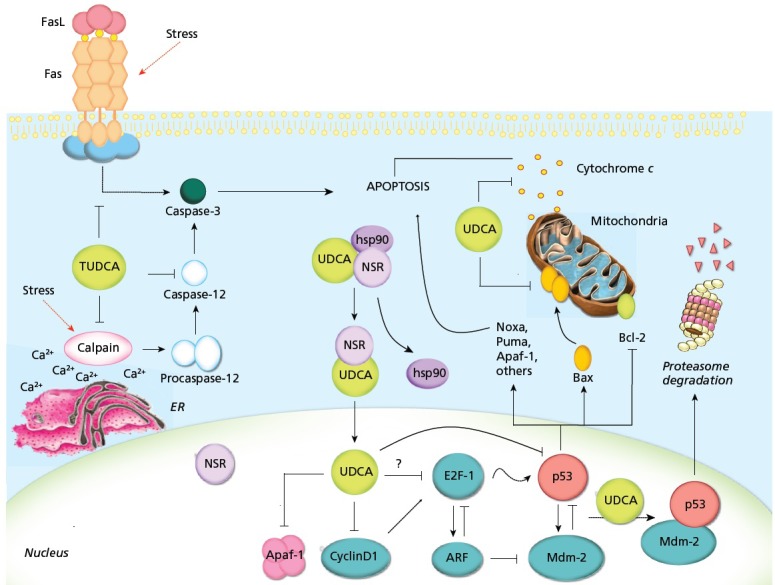
Proposed mechanisms of T/UDCA inhibition of apoptosis. T/UDCA negatively modulates the mitochondrial pathway by inhibiting Bax translocation, ROS formation, cytochrome c release, and caspase-3 activation. T/UDCA protects against mitochondrial membrane permeabilization and decreased ΔΨm, which reduces ROS production and apoptosis. T/UDCA can also interfere with the death receptor pathway, inhibiting caspase-3 activation. Moreover, T/UDCA inhibits apoptosis associated with ER stress by modulating intracellular calcium levels and inhibiting calpain and caspase-12 activation. Importantly, T/UDCA interacts with NSR, leading to NSR/hsp90 dissociation and nuclear translocation of the T/UDCA/NSR complex. Once in the nucleus, T/UDCA reduces apoptosis by modulating the E2F-1/p53/Bax pathway, inhibiting MDM2/p53 association, decreasing BAX, PUMA and NOXA expression, reducing p53 transactivation and DNA binding activity, and increasing p53 degradation. Finally, T/UDCA downregulates cyclin D1 and Apaf-1, further inhibiting the mitochondrial apoptotic cascade.
Abbreviations: Cyt c, cytochrome c; Hsp90, heat shock protein 90; T/UDCA, tauroursodeoxycholic/ursodeoxycholic acid.
Reprinted with permission from Amaral et al, 2009.2
Recent studies have shown that T/UDCA can prevent UPR dysfunction and ameliorate ER stress. It does so in part by improving protein folding capacity via the activation of transcription factor 6 and by assisting in the transfer of mutant proteins.4 TUDCA is a co-transporter of Na+/taurocholate and activates α5β1 protein into its active conformation by transferring the β1 unit.5 It also inhibits the phosphorylation of initiation factor 2α(eIF2α), which is normally activated by protein kinase RNA-like ER kinase (PERK).4
Cisplatin is an effective anti-cancer chemotherapeutic drug that is known to cause sensory neuropathy in patients who receive the drug. N18D3 hybrid neurons exposed to cisplatin treatment showed morphological changes such as cell shrinkage and cytoplasmic blebbing, as well as condensed and fragmented nuclear morphology, which are characteristics of apoptotic cell death. A clear 180- to 200-base pair internucleosomal DNA cleavage was induced by cisplatin treatment. Cisplatin treatment not only induced cytotoxicity in N18D3 hybrid neurons but also induced a substantial accumulation of p53 protein. P53 has been reported to play a major role in cisplatin-induced apoptotic cell death. Pre-incubation with UDCA was found to be effective in reducing cisplatin neurotoxicity and completely blocked cell death in N18D3 neurons, in part by suppressing pro-apoptotic p53 protein.6 P53 is also a known tumor suppressor protein as it promotes growth arrest and apoptosis and is dysfunctional in certain cancers. TUDCA has recently been seen to modulate the control of p53 via Mdm-2. Mdm-2 is the direct repressor of p53 and the modulation of Mdm-2 and p53 appears to be a key target of TUDCA.7 The inhibition of p53 action is also a potential treatment for diseases that are associated with increased activation of p53 activity.8
TUDCA improves apolipoprotein E4 (APOE4) macrophage survival and function. APOE4 and APOE3 have important functions in binding to LDL receptors. APOE is a protein associated with several classes of plasma lipoproteins expressed in the liver and other tissues, including those of the central nervous system, vascular smooth muscle cells, adrenals, macrophages, and adipocytes. It contributes to cholesterol transport and modulates metabolic disease progression via lipid transport-independent mechanisms. APOE3 and APOE4 are isoforms of the polymorphic APOE that bind to LDL receptors and other LDL receptor family proteins with similar affinity. However, APOE3 appears to protect against metabolic disorders while APOE4 is a major genetic risk factor of inflammatory metabolic diseases, including atherosclerosis, diabetes, and Alzheimer's disease (AD). Increased cell death was observed in APOE4 macrophages when stimulated with LPS or oxidized LDL and was due mainly to potentiation of ER stress signaling and JNK phosphorylation.1 TUDCA attenuated LPS- and oxLDL-induced apoptosis of APOE4 macrophages to levels observed in APOE3 macrophages.
TUDCA acts as a mitochondrial stabilizer and anti-apoptotic agent in several models of neurodegenerative diseases, including AD, Parkinson's diseases (PD), and Huntington's diseases (HD). Based on mechanistic studies conducted primarily in rodent models, TUDCA may provide a novel and effective treatment in neurological disorders with its neuroprotective activities. TUDCA shows cytoprotective properties through the inhibition of apoptosis,2,9 and it has been convincingly demonstrated that T/UDCA crosses the blood brain barrier in humans.10 That said, not everyone is convinced, and there is at least one report recommending that the use of T/UDCA beyond PBC is unjustified.11
ALZHEIMER'S DISEASE
Alzheimer's disease is a progressive and untreatable neurodegenerative disease that affects specific areas of the brain, including the hippocampus and frontal cortex. Deficits in memory and other cognitive skills compromise independent living in AD patients. The mechanisms of neuronal dysfunction and cell death in AD are not entirely understood, but there is growing evidence to suggest that apoptosis plays a key role in the loss of cell number. AD is characterized by extracellular accumulation of amyloid β-peptide (Aβ) plaques and intracellular neurofibrillary tangles (NFT) containing hyperphosphorylated tau-proteins. Aβ, a protein derived from the cleavage of the amyloid-precursor protein (APP) in high quantities aggregates to toxic amyloid plaques, affecting physiological mechanisms of the cell and induces neuronal death.
In AD brains, the primary modification of tau has been proposed as the abnormal phosphorylation. Caspase-3 cleavage of tau in the C-terminal region has also been detected in AD brains and promotes tau assembly.12 The rTg4510 mouse model is a taupathy model with massive neurodegeneration in specific cortical and limbic structures. Results from a study of the rTg4510 mouse model suggested that apoptosis is an early event associated with tau cleavage in the hippocampus and the frontal cortex, occurring prior to NFT formation and massive neuronal death. Caspase-3–cleaved intermediate tau species appear to represent a toxic form of the molecule in rTg4510 brains, resulting in protein aggregation in NFT and neuronal dysfunction. Apoptosis and caspase-3 cleavage of tau induced fibrillar Aβ were inhibited significantly by TUDCA.13
Apoptosis, a type of programmed cell death, is an energy-dependent process. There are a series of biochemical and morphological modifications, which include condensation of chromatin, shrinkage of cytoplasm, and the formation of apoptotic bodies. Apoptosis occurs primarily via extrinsic death-receptor and/or intrinsic mitochondrial pathways. Oxidative stress, DNA damage, or protein misfolding leads to mitochondrial membrane permeability, release of apoptogenic factors into the cytoplasm, and disruption of the mitochondrial membrane potential, ultimately resulting in cell death. Currently, there is no effective treatment for patients with AD. Approved drugs can enhance transmitter levels but do not slow disease progression. TUDCA has established itself as a potent inhibitor of apoptosis and may be a possible therapeutic intervention for neurodegenerative diseases such as AD.
Phosphatidylinositide 3'-OH kinase (PI3K) promotes survival downstream of apoptosis-inducing stimuli. A growing number of cellular intermediates are activated by PI3K, including the serine/threonine protein kinase Akt, which are capable of suppressing apoptosis. Aβ peptide is a strong inducer of the Bax pro-apoptotic mitochondrial pathway and a weak activator of Akt phosphorylation.14 There is significant dysregulation of anti-apoptotic Bcl-2 and proapoptotic Bax proteins in human AD tissues.12,13 TUDCA modulates Aβ-induced apoptosis by activating a PI3K survival pathway and thereby suppressing Bax translocation. Rat cortical neuron response to incubation with Aβ show that cytochrome c is significantly depleted from mitochondria. Release of cytochrome c was accompanied by caspase-3 activation, DNA degradation, and nuclear fragmentation. Bax protein levels increased in mitochondria during Aβ-induced apoptosis, and this was associated with increased release of cytochrome c. Rat cortical neurons exposed to Aβ peptide with TUDCA treatment showed significant reduction of Bax translocation, thus inhibited cytochrome c release, caspase activation, and DNA and nuclear fragmentation. In addition, TUDCA activated the PI3K-dependent survival pathway. Most notably, PI3K/Akt activation by TUDCA was sufficient to retain Bax in the cytoplasm after Aβ treatment.14
TUDCA also modulates phosphorylation and translocation of Bad via PI3K in glutamate-induced apoptosis of rat cortical neurons. Glutamate is an excitatory neurotransmitter in the CNS that regulates neuronal plasticity and induction of cell death. Cell death induced by glutamate may be involved in chronic neurodegenerative disorders, such as AD. Rat cortical neurons exposed to glutamate induced cytochrome c release, caspase activation, and morphologic changes of apoptosis. Significant reduction of glutamate-induced apoptosis of rat cortical neurons was observed in pretreatment with TUDCA. The Bcl-2 family of proteins controls the regulation of mitochondrial membrane function. Glutamate modulates the expression of Bcl-2 family proteins and induces cytochrome c release, caspase activation, and nuclear fragmentation. Incubation with TUDCA promoted phosphorylation and translocation of pro-apoptotic Bad from mitochondria to the cytosol, thereby inhibiting apoptosis and suggesting an important target for the anti-apoptotic function of TUDCA. The phosphorylation of Bad by TUDCA was also found to occur through a PI3K-dependent mechanism.15
One of the earliest sites of AD pathology is associated with reduced synapse density, and synaptic loss is highly correlated with cognitive impairment. TUDCA modulates synaptic deficits induced by amyloid and reduced the down-regulation of the postsynaptic density-95 protein (PSD-95) in an AD mouse model. TUDCA also prevented the reduction in dendritic spine number and decrease spontaneous miniature excitatory synaptic activity.16
The mitochondrial membrane is an important target in Aβ-induced cytotoxicity. An electron paramagnetic resonance (EPR) spectroscopy analysis showed that Aβ disrupted the mitochondrial membrane lipid and protein structure, inducing oxidative injury, which increases membrane permeability and release of caspase-activating factors. Lipid polarity and protein mobility were disrupted by Aβ and increased cytochrome c release. Aβ induced distress of mitochondrial function and structure were diminished by pretreatment of TUDCA.17 TUDCA was evaluated in several studies and has been shown to reduce Aβ toxicity by interfering with its production and accumulation. It inhibited Aβ-induced apoptosis by promoting mitochondrial membrane stability and reducing the release of cytochrome c and downstream activation of caspases.
In addition to mitochondria playing a central role in the apoptotic process, ER is also a critical organelle in AD. ER stress as discussed earlier leads to accumulation of unfolded or misfolded proteins like Aβ peptide. UPR is triggered by ER stress, and severe or prolonged activation of UPR results in apoptotic cell death. ER stress also leads to activation of several kinases that have functional effects on neuronal homeostasis, including apoptosis signal–regulating kinase 1 (ASK1), which triggers c-Jun N-terminal kinase (JNK) signaling. ASK1-mediated JNK activation has the potential to stimulate AD pathogenesis. Caspase-2 activation is a requirement of Aβ-induced cell death, and TUDCA prevented its activation. TUDCA also revoked Aβ-induced JNK/caspase-2 signaling and modulated Aβ-induced caspase-12-mediated apoptosis triggered by ER subcellular compartment. ER stress markers down-regulated by Aβ were partially restored by TUDCA.18
Accumulation of Aβ in the brain is associated with mutations in amyloid precursor protein (APP) and pre-senilin 1 (PS1) genes. TUDCA treatment in APP/PS1 mice decreased Aβ production and inhibited accumulation of Aβ deposits in the brain. Decreased Aβ levels were observed in both hippocampus and frontal cortex of TUDCA-treated APP/PSI mice. These findings suggested that TUDCA interferes with Aβ production, possibly by regulation of lipid metabolism mediators. Activation of astrocytes and microglia in areas of Aβ plaques contributes to an inflammatory process that develops around the injury in the brain. Activated astrocytes and microglia were visualized by GFAP immunoreactivity and Iba-I immunoreactivity, respectively, and observed in APP/PSI mice. Significantly less GFAP immunoreactivity and Iba-I immunoreactivity were observed in TUDCA-treated APP/PSI mice, suggesting that TUDCA inhibits activation of astrocytes and microglia. Marked improvement in the integrity of MAP2-positive neuronal fibers was observed around the amyloid plaques in the brains of TUDCA-treated APP/PSI. It was suggested that the rates of neuronal degeneration in APP/PSI mice treated with TUDCA were significantly decreased.19
Memory loss and cognitive decline are major hallmarks observed in AD patients. Learning and working-memory tasks rely predominantly on hippocampal function. TUDCA prevented downstream abnormal conformations of tau, which may have beneficial consequences in slowing cognitive decline.18 Based on the contextual fear-conditioning test, it was suggested that TUDCA may prevent memory deficits in APP/PS1 mice via attenuation of Aβ-associated neurodegeneration.19 Findings from another study in APP/PSI mice suggested that dietary TUDCA supplementation improved the use of spatial search strategies during a maze in APP/PS1 mice. It also showed improvement in social memory and passive avoidance learning in APP/PSI mice.20
Cerebral amyloid angiopathy (CAA) is common feature of AD. This age-associated condition is characterized by the deposition of amyloid peptides in cortical and leptomeningeal vessels that cause capillary disruption and endothelial dysfunction. CAA plays a significant role in intracerebral hemorrhage. There is limited information regarding the effect of amyloid peptides on endothelial vessel wall cells that are in contact with vascular amyloid deposits in CAA. E22Q is a glutamine to glutamic acid substitution at residue 22 and is associated with hereditary cerebral hemorrhage with amyloidosis Dutch type. TUDCA protected brain microvascular endothelial cells from apoptotic insult triggered by the potent vasculotropic E22Q peptide. TUDCA also prevented AβE22Q-induced mitochondrial Bax translocation, cytochrome c release and subsequent cell death.21 Such studies provide new perspectives in modulating amyloid-induced toxicity in CAA and suggest TUDCA as a potential therapeutic intervention for AD.
PARKINSON'S DISEASE
Parkinson's disease is a progressive neurodegenerative disease characterized by cardinal symptoms such as resting tremor, rigidity, postural instability, and brady-kinesia. It is caused by relentless loss of nigrostriatal dopaminergic neurons in the substantia nigra of the brain. There are many pathogenic mechanisms in PD, but one of the most common is a vicious cycle of oxidative damage and decrease in anti-oxidative glutathione in PD brain tissue.22 Signs of apoptosis include mitochondrial dysfunction, chromatin condensation, and caspase activation in dying cells. Mitochondrial dysfunction leads to downstream increase in reactive oxygen species (ROS), inflammatory responses, and activation of cell death pathways. Syndromes related to PD are caused by ingestion of complex I inhibitors. The complex I inhibitor 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) causes degeneration of nigrostriatal dopaminergic neurons and characterizes a toxin paradigm for PD.23 Rotenone is another complex I inhibitor that causes multiple changes in the mitochondria relevant to PD. It is known to induce free radical generation, inhibition of complex I, and apoptosis.24 In the pathogenesis of PD, the JNK pathway constitutes a central stress activated response, and increased levels are found in PD brains.23 UDCA and TUDCA have been studied and found to play a role as a neuroprotectant in PD by protecting against apoptosis and regulating JNK activity and cellular redox thresholds.
A number of genes have been associated with familial cases of PD. Genetic modulation of α-synuclein, parkin, and DJ-1 all disrupt mitochondrial function in Caenorhabditis elegans. TUDCA treatment yielded protection against rotenone-induced toxicity. Non-tg lines were fully protected by TUDCA, and K08E3.7 and α-synuclein-expressing strains showed increased survival with TUDCA treatment. A gene associated with cell death that takes place during the development of the nematode C elegans is ced-3. TUDCA does not protect C elegans lacking ced-3, suggesting that TUDCA acts in part by inhibiting apoptosis.24
UDCA and its highly water-soluble formula, Yoo's solution (YS) were shown to protect the human dopaminergic neuronal cell line SH-SY5Y against sodium nitroprusside (SNP)–induced cytotoxicity.23 SNP was used as a neurotoxicant due to its strong NO-generating characteristics and cytotoxicity in dopaminergic cells. UDCA significantly attenuated programmed cell death processes such as nuclear fragmentation, caspase activation, MMP decrease, and cytochrome c release. ROS such as superoxide, hydrogen peroxide, hydroxyl radical, and RNS including NO directly contribute to oxidative damage of dopaminergic cells. UDCA decreased the generation of total ROS and NO. Glutathione (GSH), an endogenous tripeptide anti-oxidant, exerts critical protective effects as a representative intracellular anti-oxidant. Under normal conditions, GSH is properly maintained in most cells and suppresses the spontaneous oxidative damage induced by PD-related toxicants. UDCA maintained the GSH level under oxidative stress caused by SNP.23
The neuroprotective role of TUDCA against MPTP toxicity was observed in a study with mice lacking glutathione S-transferase pi (GSTP), an endogenous JNK inhibitor. Pre-treatment with TUDCA significantly reduced the depletion of DA neurons and dopaminergic fiber loss caused by MPTP. TUDCA protected against MPTP-induced neurodegeneration by delaying and decreasing dopaminergic cell loss. Pre-treatment with TUDCA modified the cellular environment and attenuated the deleterious events of MPTP by blocking ROS production and JNK activation in GSTP null mice.23
TUDCA administration is also neuroprotective for nigral dopamine neurons transplanted into rodent models of PD. Numerous studies have demonstrated that the transplantation of fetal nigral dopamine neurons results in a 90% to 95% loss of transplanted neurons because of apoptosis. Parkinsonian rats were transplanted with nigral dopamine neurons from fetal rats incubated in 50 uM TUDCA or saline. Rats treated with dopamine neurons incubated in TUDCA exhibited a significant reduction in rotational asymmetry compared to rats transplanted with dopamine neurons alone.25 Histological analysis of the transplanted cells revealed a significantly greater number of tyrosine-positive cells that survived transplantation in the TUDCA-treated cells vs the saline-treated cells. Incubation of isolated dopamine neurons with TUDCA resulted in significantly fewer TUNEL-positive neurons in comparison with vehicle controls. Together, these results suggested that TUDCA can enhance survival of transplanted dopamine neurons via reduction of apoptosis. Interestingly, a recent study that screened more than 2000 compounds identified ursocholanic acid and its chemically-related compound UDCA as highly promising drug therapies for future neuroprotective trials in PD.26
HUNTINGTON'S DISEASE
Huntington's disease is a progressive, fatal, autosomal dominant neurodegenerative disorder caused by abnormal expansion of the trinucleotide (CAG) repeat sequence in exon 1 of the gene encoding the huntingtin protein. HD is characterized by selective neuronal loss and dysfunction in the striatum and cortex with corresponding motor and cognitive impairment. The expansion results in selective death of neurons by interfering with mitochondrial homeostasis. Mitochondria compromise in HD is demonstrated by abnormal energy metabolite concentrations and utilizations, stress-induced mitochondrial depolarization, free radical production, and associated oxidative damage.27 Mitochondrial perturbation produces reactive oxygen species and opens the mitochondrial permeability transition (MPT) pore, releasing cytochrome c that initiates a caspase-mediated apoptotic cascade. 3-Nitropropionic acid (3-NP), an irreversible inhibitor of succine dehydrogenase, appears to be associated with metabolic compromise and oxidative stress causing apoptotic neuronal cell death in the striatum and hippocampus of HD patients.28 There is currently no effective treatment for HD, but TUDCA may offer potential therapeutic benefit by stabilizing the mitochondrial membrane, inhibiting the mitochondrial permeability transition, decreasing free radical formation, and derailing the apoptotic process.27
The effects of TUDCA were studied in both 3-NP and transgenic mouse models of HD. Rat neuronal RN33B cells incubated with 3-NP showed the characteristic signs of apoptosis including condensed chromatin and nuclear fragmentation with formation of apoptotic bodies. 3-NP triggers mitochondrial cytochrome c release, which is independent of MPT pore opening, and this results in the activation of cytosolic caspase-3 and cleavage of the nuclear enzyme PARP.28 The R6/2 mouse model of HD reflects more accurately the true pathophysiology of HD associated with the trinucleotide CAG expansion.29 Mice in the study exhibited severe neuropathophysiology and associated neurodegeneration with sensorimotor deficits. Striatal atrophy and formation of neuronal intranuclear inclusions were also prominent.
3-NP cultures treated with TUDCA significantly increased neuronal survival by inhibiting swelling and release of cytochrome c in isolated mitochondria, DNA fragmentation, caspase activation, and apoptosis in RN33B cells.27,28 In vivo, TUDCA preserved striatal mitochondria structure and significantly prevented apoptosis in the brain (Figure 2). 3-NP treatment resulted in swollen striatal mitochondria that exhibited abnormal membrane structure and terminal deoxynucelotidyltransferase-mediated dUTP nick end labeling (TUNEL)-labeling of striatal cells, and these were significantly reduced with administration of TUDCA.27 TUDCA inhibited apoptotic events, in part by preventing Bax translocation, depolarizing the mitochondrial membrane, and producing ROS.28 Striatal sections from TUDCA-treated Tg mice contained fewer TUNEL-positive cells compared to mice that received vehicle alone.30 Both studies of 3-NP and R6/2 Tg mouse model of HD showed reduced striatal lesion volumes when treated with TUDCA. A common pathological hallmark of HD is the formation of intracellular inclusions composed primarily of misfolded huntingtin protein and ubiquitin. Striatal inclusions in TUDCA-treated mice were both smaller in size and reduced in number compared to untreated Tg HD mice, improving the hallmark of HD pathology.
Figure 2.
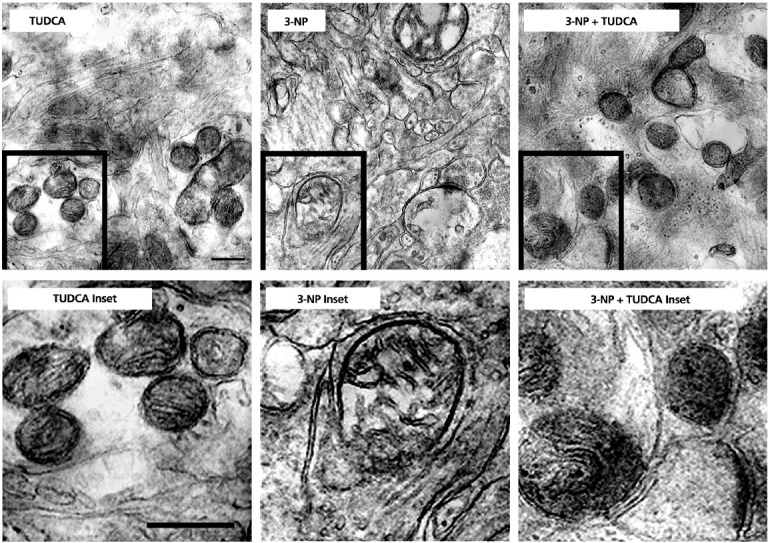
TUDCA protects striatal mitochondria exposed to 3-NP in vivo. Top left: TUDCA rats exhibit normal striatal mitochondrial size and shape, with intact membrane structure. Bottom left: Enlarged view of TUDCA inset. Top center: Chronic (28 day) 3-NP administration results in abnormal mitochondria. Framed mitochondrion at the same magnification shows dramatic swelling, structural alterations, and loss of membrane integrity. Bottom center: Enlarged view of 3-NP inset. Top right: TUDCA protects mitochondria chronically exposed to 3-NP, as evidenced by normal mitochondrial size, shape, and structure. Bottom right: Enlarged view of 3-NP + TUDCA inset. Magnification is the same for all images. Scale bar, 1 μm.
Reprinted with permission from Keene et al, 2001.27
In addition to these results, TUDCA also ameliorated locomotor and sensorimotor deficits in both HD systems. Open field task was used to measure R6/2 mouse hypoactivity, and TUDCA-treated R6/2 mice were significantly more active compared with vehicle controls.30 3-NP rats treated with TUDCA resulted in little or no loss of ability to habituate to a stimulus, meaning there was little observation of reduced overall activity. Rota-Rod tests were used to evaluate sensorimotor abilities longitudinally. Four rotational velocities were used to identify differences in performance on simple, intermediate, and difficult tasks. TUDCA-treated R6/2 mice performed markedly better than control Tg mice, suggesting that TUDCA plays a role in improving sensorimotor deficits in HD mice.
OCULAR DISORDERS
TUDCA has been shown to ameliorate a number of retinal disorders. Retinitis pigmentosa (RP) is a genetic disease in which a single point mutation in the rhodopsin-encoding gene (RHO) leads to loss of rod photoreceptor cells via protein misfolding and endoplasmic reticulum stress. The majority of cells in the outer retina are made up of rods, which are metabolically active cells that consume the majority of oxygen delivered to that region of the retina. Once rods die, oxygen consumption is greatly reduced, resulting in a large increase in oxygen in the outer retina. Progressive oxidative damage and death of cones result primarily from the increase of NADPH oxidase and generation of ROS.31 Other causes of blindness include bright light exposure, retinal detachment, age-related macular degeneration and diabetic retinopathy. Many studies have shown that TUDCA exhibit anti-apoptotic properties in neurodegenerative diseases affecting the retina by reducing photoreceptor apoptosis.
TUDCA prevented apoptosis and preserved function and morphology of photoreceptor cells in both genetic and environmental mouse models of human retinal degeneration. These included the Pde6brd10 (rd1o) mouse, a genetic model of retinitis pigmentosa, and the environmental model of blindness in the light-induced retinal degeneration (LIRD) mouse. Electroretinograms (ERGs) were recorded as an outcome measure of retinal function. The results showed that ERG a-wave and b-wave amplitudes were greater in mice treated with TUDCA compared to those treated with vehicle.32 Retinas of TUDCA-treated mice possessed thicker outer nuclear layers, more photoreceptor cells, and more fully developed photoreceptor outer segments in both mouse models (Figure 3). Overall, signs of apoptosis in rd10 and LIRD mouse models of retinal degeneration were dramatically suppressed with TUDCA treatment.31–33
Figure 3.
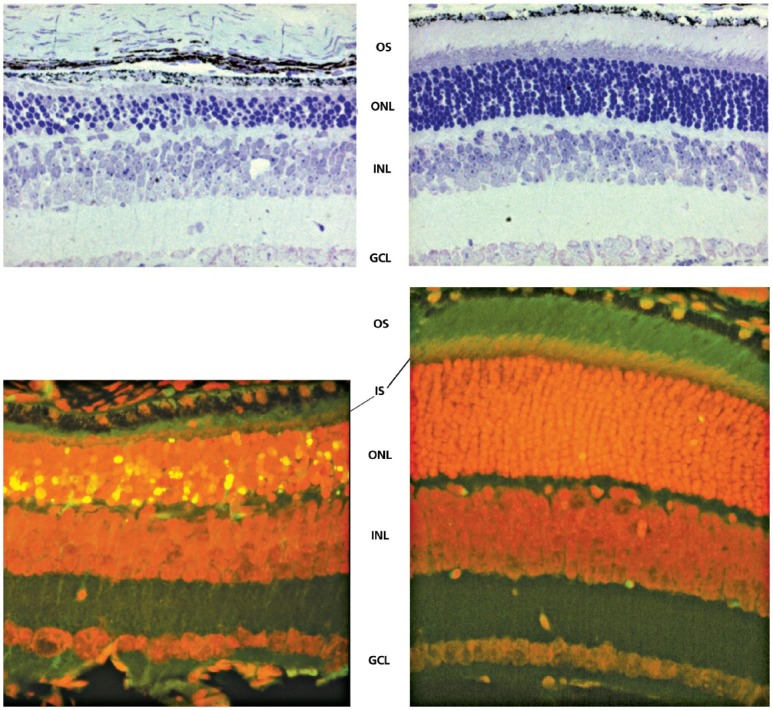
Effect of TUDCA on rd10 mouse retinal morphology. Representative micrographs stained with toluidine blue showing significant preservation of photoreceptor nuclei in the outer nuclear layer (ONL) in TUDCA-treated eyes (top panel, right) compared with vehicle-treated eyes (top panel, left). Counts of photoreceptor nuclei were significantly greater in eyes with TUDCA treatment compared to vehicle only. Combined inner segment (IS) and outer segment (OS) length was similarly preserved, with no change in thickness of inner nuclear layer (INL) or ganglion cell layer (GCL). Fluorescence microscopy using a B-2A long pass emission fluorescence filter allows further observation of the preservation of photoreceptor IS and OS present in TUDCA-treated (bottom panel, right) vs vehicle-treated (bottom panel, left) retinal sections. TUNEL-positive nuclei (green/yellow signal) are seen to be abundant in vehicle-treated sections, but are rare in TUDCA-treated sections. TUDCA treatment provided significant preservation of photoreceptor nuclei number in the ONL. Treatment had no discernable effect on the INL or GCL.
Reprinted with permission from Boatright et al, 2006.32
The action of TUDCA in a separate P23H mouse model of RP was shown to preserve cone and rod structure and function, in addition to contacts with their postsynaptic neurons. The number of photoreceptor rows present in the outer nuclear layer (ONL) was observed postnatally to assess the protective action of TUDCA. TUNEL labeling was performed to quantify the degree of photoreceptor cell apoptosis, and the number of TUNEL-positive cells per retinal section was found to be significantly higher in control P23H mice compared to TUDCA-treated mice. TUDCA also prevented the loss of cell dendrites of the retina that establish connections with both rod and cone photoreceptors, including horizontal and bipolar cells. Results from the study also indicated that TUDCA slowed the loss of mitochondria in photoreceptors.34 In addition to these and other models of retinal disease,35,36 TUDCA administration also preserved photoreceptor function after retinal detachment by reducing oxidative stress and caspase activity.37
A leading cause of vision loss in people over the age of 50 years is age-related macular degeneration (AMD). AMD can be characterized by the development of chorodial neovascularization (CNV), resulting in inflammation, angiogenesis, and apoptosis. A recent study used a laser-induced CNV model to demonstrate the suppressing effects of TUDCA on CNV and vascular endothelial growth factor (VEGF) expression. VEGF is known to be a mediator of CNV in exudative AMD in humans, and anti-VEGF agents suppress CNV and improve vision. Rats treated with systemic TUDCA before and after CNV induction showed less fluorescein leakage from CNV and reduced CNV lesion sizes. VEGF levels of retina in the TUDCA group were significantly lower than in the control group, suggesting that systemic administration of TUDCA was associated with suppression of early VEGF elevation in the retina after laser injury. It also suggested that the inhibition of VEGF up-regulation was associated with the observed reduction in CNV size and vascularity.38
The development of retinal problems resulting from diabetes is a consequence of endothelial cell (EC) injury due to toxic levels of glucose. In the working-age population, diabetic retinopathy is the most common cause of blindness. Damaged endothelial tight junctions or impaired inner blood-retinal barrier (BRB) result in vascular leakage and retinal barrier. As the disease progresses, ECs undergo apoptosis leading to consequent neovascularization. Activating transcription factor 4 (ATF4), an ER stress-inducible transcription factor, is a key regulator of endothelial inflammation in diabetic retinopathy. High glucose levels, demonstrated through activation of signal transducer and activator of transcription 3 (STAT3) induce ER stress and activate ETF4 in retinal ECs. This results in the increased production of inflammatory factors and retinal vascular leakage commonly seen in diabetic retinopathy.39 C/EBP homologous protein (CHOP) is a target gene of ATF4, and intermittent exposure to high glucose causes an increase in ATF4 and CHOP expression and inflammatory cytokine expression. Human retinal pericytes (HRP) pretreated with TUDCA followed by incubation with intermittent high glucose showed that VEGF expression was largely abolished. TUDCA also effectively suppressed the induction of CHOP expression by intermittent high glucose.40 In cultured retinal neural cells induced by exposure to elevated glucose concentration, TUDCA markedly reduced cell death.41 The release of apoptosis-inducing factor (AIF) from the mitochondria and the accumulation of AIF in the nucleus were partially prevented by TUDCA. After cells exposed to elevated glucose concentration were exposed to TUDCA treatment, decreased levels of biomarkers of oxidative stress such as protein carbonyl groups and ROS production were observed. Retinal neural cell cultures were protected from cell death induced by elevated glucose concentration with TUDCA treatment through decreasing mito-nuclear translocation of AIF. Together, these studies demonstrated that TUDCA can ameliorate cellular damage in a variety of retinal disorders.42
STROKE
TUDCA has been shown to exert neuroprotective effects in a rodent model of ischemic brain injury.43 Rats were subjected to transient cerebral ischemia by a temporary occlusion of the middle cerebral artery for 60 minutes by the insertion of a filament. TUDCA was administered one hour after the removal of the occluding filament. Assessment of infarct volume 7 days after ischemic injury revealed that animals treated with TUDCA had a 50% reduction in infarct volume and an 80% reduction in TUNEL-positive apoptotic cells. Reductions in mitochondrial swelling and in apoptotic caspase-3 protein levels were also observed. These results suggest that TUDCA was cytoprotective in ischemic brain injury and exerted its actions, in part by inhibiting apoptosis.
The loss of cells in the brain from intracerebral hemorrhage (ICH) involves both necrotic and apoptotic cell death. TUDCA was investigated as a treatment regimen following ICH in rats for reducing the loss of brain cells from apoptosis.44 ICH was induced by injection of collagenase into the striatum, causing vascular leakage into the parenchyma. Animals treated with TUDCA exhibited a 50% reduction in infarct volume, and apoptosis was also reduced by 50% in the areas adjacent to the site of hemorrhage as revealed by TUNEL staining. Animals treated with TUDCA exhibited elevated levels of anti-apoptotic Bcl-2 in the ischemic hemisphere. Moreover, downstream caspase activity was reduced 40% to 60% in the ischemic hemisphere in animals treated with TUDCA. In addition to reducing levels of apoptosis, TUDCA treatment was associated with significant improvements in limb placement, stepping ability and amelioration of locomotor asymmetry.
DIABETES
Type I diabetes is associated with the loss of insulin-producing islet cells of the pancreas, primarily via targeting and destruction by the immune system. Type II diabetes is typically characterized by normal insulin production by pancreatic islet cells but an increase in insulin resistance resulting in elevated blood glucose levels. Several reports confirm the ability of TUDCA to improve the hyperglycemia associated with both types of diabetes. It has been shown that high glucose levels cause ER stress and defective autophagy, which facilitate podocyte injury in the glomerulus.45 TUDCA inhibited the defective autophagy and prevented podocyte injury.46 Adipocyte autophagy was suppressed in an insulin-resistant mouse model, and this resulted in both increased inflammation and ER stress. Treatment with TUDCA reduced the ER stress and inflammation, but the suppression of autophagy was not entirely reversed.47
Acinar cells are another important factor in diabetes and pancreatitis. Acinar cells in the pancreas are exocrine glands that are signaled to secrete their hormones and substances by cholecystokinin (CCK) signaling. When rat acinar cells were stimulated with CCK-8 and TUDCA, there was increased amylase secretion and 50% reduction in activation of trypsin, a molecule that hydrolyzes proteins.48 TUDCA treatment resulted in increased chaperone binding, reduced ER stress, and stabilization of the UPR in a model of acute pancreatitis.49
The incubation of free fatty acids with β cells has been shown to initiate type II diabetes. Reduction of ER stress by administration of TUDCA significantly reduced the effects of the free fatty acids on the β cells.50,51 Type I diabetes is characterized by the death and dysfunction of pancreatic β-cells, and deficits in the expression of UPR mediators activating transcription factor 6 (ATF6) and X-box binding protein 1 (XBP1).52 Mice with type I diabetes and exhibiting deficits in ATF6 and XBP1 expression were treated with TUDCA in the pre-diabetic stage. With treatment, they exhibited restored expression of ATF6 and XBP1, reduced infiltration of immune cells in the pancreas, decreased β-cell apoptosis, and preserved insulin secretion.52
ER stress also contributes to insulin insensitivity.53,54 In vivo tissues were tested with TUDCA and insulin sensitivity increased 30%.55 TUDCA also restored impaired islets and insulin secretion by reducing ER stress. TUDCA in pig islets also increased ATP content, which is beneficial for islet protection after transplantation.56 ER stress also activates protein tyrosine phosphatase 1B (PTP1B), which lowers insulin signaling, and TUDCA administration was found to lower expression of PTP1B.57 Together, these studies demonstrate the pleiotropic effects of TUDCA on correcting deficits associated with type I and II diabetes.58,59
OBESITY
Adipose tissue is important in maintaining metabolic homeostasis, and its dysfunction can lead to obesity and type II diabetes. Increased adipose tissue can result in elevated ER stress induced by free fatty acid (FFA)-mediated reactive oxygen species (ROS) generation and up-regulation of inflammatory cytokines.60 Several studies now report the improved function of adipose tissue with exposure to TUDCA, in part by reducing ER stress, maintaining adipocyte autophagy, stabilizing the UPR and inhibiting adipogenesis.61 Obesity can also lead to deficits in myocardial function. In one study, ob/ob obese mice were found to exhibit cardiac hypertrophy, compromised contraction of cardiomyocytes, and hypertension in comparison to normal controls. Treatment of ob/ob mice with TUDCA led to a decrease in cardiac hypertrophy, decreased hypertension, and a normalization of cardiac contractility. Moreover, TUDCA-treated ob/ob mice exhibited normalization of sarco(endo)plasmic reticulum Ca(2+)-ATPase (SERCA) expression and activity.62
CARDIOVASCULAR DISEASE
Hypertension is a major risk factor for cardiovascular disease. ER stress has been shown to increase production of endothelium-derived contractile factors (EDCFs), such as vasoconstrictor prostanoids that increase resistance, resulting in hypertension.63,64 TUDCA treatment in rats for 1 week showed decreased signs of ER stress such as expression of COX-1 and kinase phosphorylation and decreased levels of prostanoids and blood pressure. TUDCA also decreased activation of transcription factor 6 and ameliorated the decrease in mitochondrial calcium in rats with hypertension. It also suppressed proliferation in pulmonary artery smooth muscle cells and reduced the elevated blood pressure.65 TUDCA injections into the lateral cerebroventricles of mice reduced ER stress in the brain and its associated Ang II-dependent hypertension.66
Contractile function is critical to the function of the heart.67 Up-regulation of the SERCA2a protein increased LV diastolic dysfunction in rats, and this was almost entirely reversed with TUDCA treatment in diabetic rats.68 ER stress induces cardiomyoctye contractile dysfunction and is associated with intracellular Ca2+ defects, which improved upon treatment with TUDCA.69 One study also showed TUDCA can promote cardioprotection through the prevention of ER stress, which inhibits erythropoietin-induced suppression of mitochondrial permeability transition pore opening.70 In additional cardiac studies, TUDCA was able to oxidize low-density lipoprotein deposition and reduce AV calcification71, inhibit neointimal hyperplasia and the proliferation of vascular smooth muscle cells (VSMCs)72, and significantly reduce the injury from myocardial infarction.73,74 These improvements result from the myriad of mechanisms described above, including mitochondrial protection and reduction in ER stress.
GASTROINTESTINAL DISORDERS
TUDCA has been used to treat experimental gastrointestinal disorders.75 Inflammatory bowel diseases (IBDs) are immune-mediated disorders of the gastrointestinal tract that are chronically relapsing. ER stress mechanisms have been implicated in the pathogenesis of both acute and chronic conditions of intestinal inflammation. Chemical chaperones exhibit beneficial effects associated with ER stress mechanisms in the GI system by inhibiting the UPR. ER chaperone glucose regulated protein (GRP)78 releases three ER transmembrane proteins when unfolded proteins accumulate. This initiates the three branches of the UPR signaling cascade. Research focusing on the regulation of classical UPR dependent genes like GRP78 and CHOP suggested that TUDCA could reduce ER stress and stabilize the UPR in intestinal epithelial cells. TUDCA was able to inhibit the activation of the early signaling steps in the three branches of UPR, preventing the formation and binding of activated transcription factors to the ERSE and UPRE elements of the GRP78 promoter for transcriptional activation. In this particular case, TUDCA acted as a potent chemical chaperone in the treatment of ER stress-associated diseases like IBD.76
In another study, researchers found that TUDCA reduced the damaging effects of taurodeoxycholic acid (TDCA) on fundus gastric mucosa. Bile acids, particularly deoxycholic acid, have been indicated to directly cause injury to the gastric mucosa. TDCA was studied in an amphibian model of gastric mucosa to assess the effects it has on inducing tissue damage, effects on electrical transepithelial parameters, acid secretion, and histology in absence or presence of TUDCA. TDCA caused a reduction in transepithelial potential difference and transepithelial resistance and a decrease in acid secretion from mucosal exposure. Neck cells were also affected by TDCA. TUDCA did not cause a significant change in electrical parameters and affected only oxyntic cells. A reduction in short circuit current and transepithelial resistance was observed with mucosal exposure to the combination of TUDCA and TDCA. The results suggested that TUDCA protected the gastric mucosa from the damaging effects of TDCA and that TUDCA may play a role in treating gastritis associated with bile reflux.77
RENAL INJURY
Acute kidney injury (AKI) is associated with cell death by apoptosis.78 In one study that assessed the efficacy of TUDCA in AKI, it was found that GRP78 and CHOP expression was blocked, which reduced caspase 12 activation and apoptosis in nephrons of the kidney.79 Treated rats were found to exhibit nephroprotection after ischemia/reperfusion induced AKI. TUDCA also inhibited the mitochondrial pathway of apoptosis and up-regulated survival pathways.80 Dysfunction of the UPR in the proximal tubules can cause proteinuria-induced tubulointerstitial lesions that result in tubular damage and dysfunction.81 In these conditions, TUDCA has been shown to stabilize the UPR and its associated pathways and prevent apoptosis.
CONCLUSIONS
With the exception of greater solubility, and somewhat different pharmacokinetics, TUDCA and UDCA, are essentially identical molecules that both have the unique ability to increase cell survival and inhibit programmed cell death, or apoptosis. The protective mechanisms of T/UDCA have been studied in a wide range of cell types and animal models of human disease. T/ UDCA is a strong inhibitor of apoptosis by interfering early on with the mitochondrial pathway of cell death and associated processes such as ROS and reducing endoplasmic reticulum stress. T/UDCA serves as a neuroprotectant against acute injuries such as stroke, as well as chronic neurodegenerative diseases including AD, PD, and HD. It protects against cell death in transgenic mouse models of retinitis pigmentosa, ameliorates the insulin resistance associated with obesity, inhibits ER stress associated with type II diabetes, and is approved in South Korea for the treatment of patients with amyotrophic lateral sclerosis.82 It has been reported to be effective in a phase II clinical study for the treatment of transthyretin amyloidosis in combination with doxycycline.83 T/UDCA has been shown to be effective in animal models of spinal cord injury,84 head trauma, glaucoma, acute renal failure, pancreatitis, metabolic syndrome, psoriasis,85,86 myocardial infarction, neuropathic pain,87 ischemia reperfusion syndromes, GI disorders, and diabetic retinopathy in addition to the obvious liver-related disorders. Obese patients seem to benefit from T/UDCA via the inhibition of ER stress and UPR dysfunction. Patients with macular degeneration, hair loss and early onset of aging may benefit from the remarkable properties of T/UDCA.
The mechanism(s) by which TUDCA and UDCA exhibit anti-apoptotic properties involve numerous targets including reduced Bax translocation to mitochondrial membrane, cytochrome c release, caspase activation, DNA and nuclear fragmentation, reduced ROS, stabilization of the UPR, inhibition of p53 transactivation—and the list goes on and on. Simply stated, T/ UDCA is a pleiotropic agent with multiple cellular targets that not only inhibit apoptosis but also upregulate survival pathways. It is well established that the UPR, endoplasmic reticulum stress, and apoptosis are all important factors in underlying causes of many diseases and that the inhibition of these events can dramatically change the course or onset of disease. It is not surprising, therefore, that the Chinese have used bear bile for 3000 years for the treatment of numerous health problems.88–90 After all, T/UDCA makes up more than 50% of the bile acid pool in bear bile compared to 2% in humans! Nature, through this circulating factor, may have given black bears the ability to hibernate for long periods of time without untoward effects. Perhaps the next millennium will witness the use of this ancient remedy for a wide-ranging number of clinical disorders; and while humans do not hibernate, they do plan on taking long trips through space in route to Mars. Already, novel and more active analogues of these molecules are being synthesized.91
Disclosures The authors completed the ICMJE Form for Disclosure of Potential Conflicts of Interest and disclosed that Dr Low is a consultant for Saneron, Genomix, Metselex, Regenevida, and Insera Therapeutics and Dr Steer is a consultant for Metselex and Regenevida. None of these organizations provided funding for the writing of this review.
Authors' Contributions Sheila Vang and Katie Longley participated in outlining and writing the review article and contributed equally as co-first authors. Clifford Steer and Walter Low participated in outlining, updating and writing the review article.
Contributor Information
Sheila Vang, Department of Integrative Biology and Physiology, University of Minnesota, Minneapolis (Ms Vang), United States..
Katie Longley, Department of Integrative Biology and Physiology, University of Minnesota, Minneapolis (Ms Longley), United States..
Clifford J. Steer, Department of Medicine, University of Minnesota Medical School, Minneapolis, and Department of Genetics, Cell Biology and Development, University of Minnesota (Dr Steer), United States..
Walter C. Low, Department of Neurosurgery, University of Minnesota Medical School and Department of Integrative Biology and Physiology, University of Minnesota (Dr Low), United States..
REFERENCES
- 1.Cash JG, Kuhel DG, Basford JE, et al. Apolipoprotein E4 impairs macrophage efferocytosis and potentiates apoptosis by accelerating endoplasmic reticulum stress. J Biol Chem. 2012;287(33):27876–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amaral JD, Viana RJ, Ramalho RM, Steer CJ, Rodrigues CMP. Bile acids: regulation of apoptosis by ursodeoxycholic acid. J Lipid Res. 2009;50(9):1721–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chakrabarti A, Chen AW, Varner JD. A review of the mammalian unfolded protein response. Biotechnol Bioeng. 2011;108(12):2777–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Omura T, Asari M, Yamamoto J, et al. Sodium tauroursodeoxycholate prevents paraquat-induced cell death by suppressing endoplasmic reticulum stress responses in human lung epithelial A549 cells. Biochem Biophys Res Commun. 2013;432(4):689–94 [DOI] [PubMed] [Google Scholar]
- 5.Beuers U. β1 integrin is a long-sought sensor for tauroursodeoxycholic acid. Hepatology. 2013;57(3):867–9 [DOI] [PubMed] [Google Scholar]
- 6.Park IH, Kim MK, Kim SU. Ursodeoxycholic acid prevents apoptosis of mouse sensory neurons induced by cisplatin by reducing P53 accumulation. Biochem Biophys Res Commun. 2008;377(4):1025–30 [DOI] [PubMed] [Google Scholar]
- 7.Amaral JD, Castro RE, Solá S, Steer CJ, Rodrigues CMP. p53 is a key molecular target of ursodeoxycholic acid in regulating apoptosis. J Biol Chem. 2007;282(47):34250–9 [DOI] [PubMed] [Google Scholar]
- 8.Amaral JD, Xavier JM, Steer CJ, Rodrigues CMP. Targeting the p53 pathway of apoptosis. Curr Pharm Des. 2010;16(22):2493–503 [DOI] [PubMed] [Google Scholar]
- 9.Amaral JD, Solá S, Steer CJ, Rodrigues CMP. Role of nuclear steroid receptors in apoptosis. Curr Med Chem. 2009;16(29):3886–902 [DOI] [PubMed] [Google Scholar]
- 10.Parry GJ, Rodrigues CMP, Aranha MM, et al. Safety, tolerability and cerebro-spinal fluid penetration of ursodeoxycholic acid in patients with amyotrophic lateral sclerosis. Clin Neuropharm. 2010;33:17–21 [DOI] [PubMed] [Google Scholar]
- 11.Kotb MA. Molecular mechanisms of ursodeoxycholic acid toxicity & side effects: ursodeoxycholic acid freezes regeneration & induces hibernation mode. Int J Mol Sci. 2012;13:8882–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ramalho RM, Viana RJ, Castro RE, Steer CJ, Low WC, Rodrigues CMP. Apoptosis in transgenic mice expressing the P301L mutated form of human tau. Mol Med. 2008;14(5-6):309–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramalho RM, Viana RJ, Low WC, Steer CJ, Rodrigues CMP. Bile acids and apoptosis modulation: an emerging role in experimental Alzheimer's disease. Trends Mol Med. 2008;14(2):54–62 [DOI] [PubMed] [Google Scholar]
- 14.Solá S, Castro RE, Laires PA, Steer CJ, Rodrigues CMP. Tauroursodeoxycholic acid prevents amyloid-beta peptide-induced neuronal death via a phosphatidylinositol 3-kinase-dependent signaling pathway. Mol Med. 2003;9(9-12):226–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Castro RE, Solá S, Ramalho RM, Steer CJ, Rodrigues CMP. The bile acid tauroursodeoxycholic acid modulates phosphorylation and translocation of Bad via phosphatidylinositol 3-kinase in glutamate-induced apoptosis of rat cortical neurons. J Pharmacol Exp Ther. 2004;311(2):845–52 [DOI] [PubMed] [Google Scholar]
- 16.Ramalho RM, Nunes AF, Dias RB, et al. Tauroursodeoxycholic acid suppresses amyloid β-induced synaptic toxicity in vitro and in APP/PS1 mice. Neurobiol Aging. 2013;34(2):551–61 [DOI] [PubMed] [Google Scholar]
- 17.Rodrigues CMP, Solá S, Brito MA, Brondino CD, Brites D, Moura JJ. Amyloid beta-peptide disrupts mitochondrial membrane lipid and protein structure: protective role of tauroursodeoxycholate. Biochem Biophys Res Commun. 2001;281(2):468–74 [DOI] [PubMed] [Google Scholar]
- 18.Viana RJ, Nũnes AF, Rodrigues CM. Endoplasmic reticulum enrollment in Alzheimer's disease. Mol Neurobiol. 2012;46(2):522–34 [DOI] [PubMed] [Google Scholar]
- 19.Nũnes AF, Amaral JD, Lo AC, et al. TUDCA, a bile acid, attenuates amyloid precursor protein processing and amyloid-β deposition in APP/PS1 mice. Mol Neurobiol. 2012;45(3):440–54 [DOI] [PubMed] [Google Scholar]
- 20.Lo AC, Callaerts-Vegh Z, Nũnes AF, Rodrigues CM, D'Hooge R. Tauroursodeoxycholic acid (TUDCA) supplementation prevents cognitive impairment and amyloid deposition in APP/PS1 mice. Neurobiol Dis. 2013;50:21–9 [DOI] [PubMed] [Google Scholar]
- 21.Viana RJ, Nũnes AF, Castro RE, et al. Tauroursodeoxycholic acid prevents E22Q Alzheimer's Abeta toxicity in human cerebral endothelial cells. Cell Mol Life Sci. 2009;66(6):1094–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chun HS, Low WC, Ursodeoxycholic acid suppresses mitochondria-dependent programmed cell death induced by sodium nitroprusside in SH-SY5Y cells. Toxicology. 2012;292(2-3):105–12 [DOI] [PubMed] [Google Scholar]
- 23.Castro-Caldas M, Carvalho AN, Rodrigues E, et al. Tauroursodeoxycholic acid prevents MPTP-induced dopaminergic cell death in a mouse model of Parkinson's disease. Mol Neurobiol. 2012;46(2):475–86 [DOI] [PubMed] [Google Scholar]
- 24.Ved R, Saha S, Westlund B, et al. Similar patterns of mitochondrial vulnerability and rescue induced by genetic modification of alpha-synuclein, parkin, and DJ-1 in Caenorhabditis elegans. J Biol Chem. 2005;280(52):42655–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duan WM, Rodrigues CMP, Zhao LR, Steer CJ, Low WC. Tauroursodeoxycholic acid improves the survival and function of nigral transplants in a rat model of Parkinson's disease. Cell Transplant. 2002;11(3):195–205 Erratum in: Cell Transplant.2002;11(6): 619 [PubMed] [Google Scholar]
- 26.Mortiboys H, Aasly J, Bandmann O. Ursocholanic acid rescues mitochondrial function in common forms of familial Parkinson's disease. Brain. 2013;136(10):3038–50 [DOI] [PubMed] [Google Scholar]
- 27.Keene CD, Rodrigues CMP, Eich T, et al. A bile acid protects against motor and cognitive deficits and reduces striatal degeneration in the 3-nitropropionic acid model of Huntington's disease. Exp Neurol. 2001;171(2):351–60 [DOI] [PubMed] [Google Scholar]
- 28.Rodrigues CMP, Stieers CL, Keene CD, et al. Tauroursodeoxycholic acid partially prevents apoptosis induced by 3-nitropropionic acid: evidence for a mitochondrial pathway independent of the permeability transition. J Neurochem. 2000;75(6):2368–79 [DOI] [PubMed] [Google Scholar]
- 29.Tkac I, Dubinsky JM, Keene CD, Gruetter R, Low WC. Neurochemical changes in Huntington R6/2 mouse striatum detected by in vivo 1H NMR spectroscopy. J Neurochem. 2007;100(5):1397–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Keene CD, Rodrigues CM, Eich T, Chhabra MS, Steer CJ, Low WC. Tauroursodeoxycholic acid, a bile acid, is neuroprotective in a transgenic animal model of Huntington's disease. Proc Natl Acad Sci U S A. 2002;99(16):10671–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oveson BC, Iwase T, Hackett SF, et al. Contituents of bile, bilirubin and TUDCA, protect against oxidative stress-induced retinal degeneration. J Neurochem. 2011;116:144–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boatright JH, Moring AG, McElroy C, et al. Tool from ancient pharmacopoeia prevents vision loss. Mol Vision. 2006;12:1706–14 [PubMed] [Google Scholar]
- 33.Phillips MJ, Walker TA, Choi HY, et al. Tauroursodeoxycholic acid preservation of photoreceptor structure and function in the rd10 mouse through postnatal day 30. Invest Ophthalmol Vis Sci. 2008;49(5):2148–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fernández-Sánchez L, Lax P, Pinilla I, Martín-Nieto J, Cuenca N. Tauroursodeoxycholic acid prevents retinal degeneration in transgenic P23H rats. Invest Ophthalmol Vis Sci. 2011;52(8):4998–5008 [DOI] [PubMed] [Google Scholar]
- 35.Boatright JH, Nickerson JM, Moring AG, Pardue MT. Bile acids in treatment of ocular disease. J Ocul Biol Dis Infor. 2009;2(3):149–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drack AV, Dumitrescu AV, Bhattarai S, et al. TUDCA slows retinal degeneration in two different mouse models of retinitis pigmentosa and prevents obesity in Bardet-Biedl Syndrome type 1 mice. Invest Ophthalmol Vis Sci. 2012;53:100–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mantopoulos D, Murakami Y, Comander J, et al. Tauroursodeoxycholic acid (TUDCA) protects photoreceptors from cell death after experimental retinal detachment. PLoS ONE. 2011;6(9):e24245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Woo SJ, Kim JH, Yu HG. Ursodeoxycholic acid and tauroursodeoxycholic acid suppress choroidal neovascularization in a laser-treated rat model. J Ocul Pharmacol Ther. 2010;26(3):223–9 [DOI] [PubMed] [Google Scholar]
- 39.Chen Y, Wang JJ, Li J, et al. Activating transcription factor 4 mediates hyper-glycaemia-induced endothelial inflammation and retinal vascular leakage through activation of STAT3 in a mouse model of type 1 diabetes. Diabetologia. 2012;55(9):2533–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhong Y, Li J, Chen Y, Wang JJ, Ratan R, Zhang SX. Activation of endoplasmic reticulum stress by hyperglycemia is essential for Müller cell-derived inflammatory cytokine production in diabetes. Diabetes. 2012;61(2):492–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gaspar JM, Martins A, Cruz R, Rodrigues CMP, Ambrósio AF, Santiago AR. Tauroursodeoxycholic acid protects retinal neural cells from cell death induced by prolonged exposure to elevated glucose. Neuroscience. 2013;253:380–8 [DOI] [PubMed] [Google Scholar]
- 42.Zhang T, Baehr W, Fu Y. Chemical chaperone TUDCA preserves cone photo-receptors in a mouse model of Leber congenital amaurosis. Invest Ophthalmol Vis Sci. 2012;53(7):3349–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rodrigues CMP, Spellman SR, Solá S, et al. Neuroprotection by a bile acid in an acute stroke model in the rat. J Cereb Blood Flow Metab. 2002;22(4):463–71 [DOI] [PubMed] [Google Scholar]
- 44.Rodrigues CMP, Solá S, Nan Z, et al. Tauroursodeoxycholic acid reduces apoptosis and protects against neurological injury after acute hemorrhagic stroke in rats. Proc Natl Acad Sci U S A. 2003;100(10):6087–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou L, Zhang J, Fang Q, et al. Autophagy-mediated insulin receptor down-regulationcontributes to endoplasmic reticulum stress-induced insulin resistance. Mol Pharmacol. 2009;76:596–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fang L, Zhou Y, Cao H, Wen P, Jiang L, He W, Dai C, Yang J. Autophagy attenuates diabetic glomerular damage through protection of hyperglycemia-induced podocyte injury. PLoS ONE. 2013;8(4):e60546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshizaki T, Kusunoki C, Kondo M, et al. Autophagy regulates inflammation in adipocytes. Biochem Biophys Res Commun. 2012;417(1):352–7 [DOI] [PubMed] [Google Scholar]
- 48.Malo A, Krüger B, Seyhun E, et al. Tauroursodeoxycholic acid reduces endoplasmic reticulum stress, trypsin activation, and acinar cell apoptosis while increasing secretion in rat pancreatic acini. Am J Physiol Gastrointest Liver Physiol. 2010;299(4):G877–86 [DOI] [PubMed] [Google Scholar]
- 49.Seyhun E, Malo A, Schäfer C, et al. Tauroursodeoxycholic acid reduces endoplasmic reticulum stress, acinar cell damage, and systemic inflammation in acute pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2011;301(5):G773–82 [DOI] [PubMed] [Google Scholar]
- 50.Tang C, Koulajian K, Schuiki I, et al. Glucose-induced beta cell dysfunction in vivo in rats: link between oxidative stress and endoplasmic reticulum stress. Diabetologia. 2012;55(5):1366–79 [DOI] [PubMed] [Google Scholar]
- 51.Zhu Q, Zhong JJ, Jin JF, Yin XM, Miao H. Tauroursodeoxycholate, a chemical chaperone, prevents palmitate-induced apoptosis in pancreatic β-cells by reducing ER stress. Exp Clin Endocrinol Diabetes. 2013;121(1):43–7 [DOI] [PubMed] [Google Scholar]
- 52.Engin F, Yermalovich A, Ngyuen T, et al. Restoration of the unfolded protein response in pancreatic β cells protects mice against type 1 diabetes. Sci Transl Med. 2013;5(211)ra156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Galán M, Kassan M, Choi SK, et al. A novel role for epidermal growth factor receptor tyrosine kinase and its downstream endoplasmic reticulum stress in cardiac damage and microvascular dysfunction in type 1 diabetes mellitus. Hypertension. 2012; 60(1):71–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Purkayastha S, Zhang H, Zhang G, Ahmed Z, Wang Y, Cai D. Neural dysregulation of peripheral insulin action and blood pressure by brain endoplasmic reticulum stress. Proc Natl Acad Sci U S A. 2011;108(7):2939–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kars M, Yang L, Gregor MF, et al. Tauroursodeoxycholic acid may improve liver and muscle but not adipose tissue insulin sensitivity in obese men and women. Diabetes. 2010;59(8):1899–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee YY, Hong SH, Lee YJ, Chung SS, Jung HS, Park SG, Park KS. Tauroursodeoxycholate (TUDCA), chemical chaperone, enhances function of islets by reducing ER stress. Biochem Biophys Res Commun. 2010;397(4):735–9 [DOI] [PubMed] [Google Scholar]
- 57.Panzhinskiy E, Hua Y, Culver B, Ren J, Nair S. Endoplasmic reticulum stress upregulates protein tyrosine phosphatase 1B and impairs glucose uptake in cultured myotubes. Diabetologia. 2013;56(3):598–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Özcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–61 [DOI] [PubMed] [Google Scholar]
- 59.Özcan U, Yilmaz E, Özcan L, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kawasaki N, Asada R, Saito A, Kanemoto S, Imaizumi K. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci Rep. 2012;2:799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Younce C, Kolattukudy P. MCP-1 induced protein promotes adipogenesis via oxidative stress, endoplasmic reticulum stress and autophagy. Cell Physiol Biochem. 2012;30(2):307–20 [DOI] [PubMed] [Google Scholar]
- 62.Ceylan-Isik AF, Sreejayan N, Ren J. Endoplasmic reticulum chaperon tauroursodeoxycholic acid alleviates obesity-induced myocardial contractile dysfunction. J Mol Cell Cardiol. 2011;50(1):107–16 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 63.Liang B, Wang S, Wang Q, Zhang W, Viollet B, Zhu Y, Zou MH. Aberrant endoplasmic reticulum stress in vascular smooth muscle increases vascular contractility and blood pressure in mice deficient of AMP-activated protein kinase-α2 in vivo. Arterioscler Thromb Vasc Biol. 2013;33(3):595–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Spitler KM, Matsumoto T, Webb RC. Suppression of endoplasmic reticulum stress improves endothelium-dependent contractile responses in aorta of the spontaneously hypertensive rat. Am J Physiol Heart Circ Physiol. 2013;305(3):H344–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dromparis P, Paulin R, Stenson TH, Haromy A, Sutendra G, Michelakis ED. Attenuating endoplasmic reticulum stress as a novel therapeutic strategy in pulmonary hypertension. Circulation. 2013;127(1):115–25 [DOI] [PubMed] [Google Scholar]
- 66.Young CN, Cao X, Guruju MR, et al. ER stress in the brain subfornical organ mediates angiotensin-dependent hypertension. J Clin Invest. 2012;122(11):3960–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang Y, Xia Z, La Cour KH, Ren J. Activation of Akt rescues endoplasmic reticulum stress-impaired murine cardiac contractile function via glycogen synthase kinase-3β-mediated suppression of mitochondrial permeation pore opening. Antioxid Redox Signal. 2011November1;15(9):2407–24 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 68.Takada A, Miki T, Kuno A, et al. Role of ER stress in ventricular contractile dysfunction in type 2 diabetes. PLoS ONE. 2012;7(6):e39893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Turdi S, Hu N, Ren J. Tauroursodeoxycholic acid mitigates high fat diet-induced cardiomyocyte contractile and intracellular Ca2+ anomalies. PLoS ONE. 2013;8(5):e63615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Miki T, Miura T, Hotta H, et al. Endoplasmic reticulum stress in diabetic hearts abolishes erythropoietin-induced myocardial protection by impairment of phospho-glycogen synthase kinase-3beta-mediated suppression of mitochondrial permeability transition. Diabetes. 2009;58(12):2863–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cai Z, Li F, Gong W, et al. Endoplasmic reticulum stress participates in aortic valve calcification in hypercholesterolemic animals. Arterioscler Thromb Vasc Biol. 2013;33(10):2345–54 [DOI] [PubMed] [Google Scholar]
- 72.Kim SY, Kwon YW, Jung IL, Sung JH, Park SG. Tauroursodeoxycholate (TUDCA) inhibits neointimal hyperplasia by suppression of ERK via PKCα-mediated MKP-1 induction. Cardiovasc Res. 2011;92(2):307–16 [DOI] [PubMed] [Google Scholar]
- 73.Rivard AL, Steer CJ, Kren BT, et al. Administration of tauroursodeoxycholic acid (TUDCA) reduces apoptosis following myocardial infarction in rat. Am J Chin Med. 2007;35(2):279–95 [DOI] [PubMed] [Google Scholar]
- 74.Mitra A, Basak T, Datta K, Naskar S, Sengupta S, Sarkar S. Role of α-crystallin B as a regulatory switch in modulating cardiomyocyte apoptosis by mitochondria or endoplasmic reticulum during cardiac hypertrophy and myocardial infarction. Cell Death Dis. 2013;4:e582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Calmus Y, Weill B, Ozier Y, Chéreau C, Houssin D, Poupon R. Immunosuppressive properties of chenodeoxycholic and ursodeoxycholic acids in the mouse. Gastroenterology. 1992;103(2):617–21 [DOI] [PubMed] [Google Scholar]
- 76.Berger E, Haller D. Structure-function analysis of the tertiary bile acid TUDCA for the resolution of endoplasmic reticulum stress in intestinal epithelial cells. Biochem Biophys Res Commun. 2011;409(4):610–5 [DOI] [PubMed] [Google Scholar]
- 77.Piepoli AL, Caroppo R, Armentano R, Caruso ML, Guerra V, Maselli MA. Tauroursodeoxycholic acid reduces damaging effects of taurodeoxycholic acid on fundus gastric mucosa. Arch Physiol Biochem. 2002;110(3):197–202 [DOI] [PubMed] [Google Scholar]
- 78.Havasi A, Borkan SC. Apoptosis and acute kidney injury. Kidney Int. 2011;80(1):29–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gao X, Fu L, Xiao M, et al. The nephroprotective effect of tauroursodeoxycholic acid on ischaemia/reperfusion-induced acute kidney injury by inhibiting endoplasmic reticulum stress. Basic Clin Pharmacol Toxicol. 2012;111(1):14–23 [DOI] [PubMed] [Google Scholar]
- 80.Gupta S, Li S, Abedin MJ, et al. Prevention of acute kidney injury by tauroursodeoxycholic acid in rat and cell culture models. PLoS ONE. 2012;7(11):e48950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Takeda N, Kume S, Tanaka Y, et al. Altered unfolded protein response is implicated in the age-related exacerbation of proteinuria-induced proximal tubular cell damage. Am J Pathol. 2013;183(3):774–85 [DOI] [PubMed] [Google Scholar]
- 82.Min J-H, Hong Y-H, Sung J-J, et al. Oral solubilized ursodeoxycholic acid therapy in amyotrophic lateral sclerosis: A randomized cross-over trial. Korean Med Sci. 2012;27:200–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Obici L, Cortese A, Lozza A, et al. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: a phase II study. Amyloid. 2012;19 (Suppl 1):34–6 [DOI] [PubMed] [Google Scholar]
- 84.Colak A, Kelten B, Sağmanligil A, et al. Tauroursodeoxycholic acid and secondary damage after spinal cord injury in rats. J Clin Neurosci. 2008;15(6):665–71 [DOI] [PubMed] [Google Scholar]
- 85.Yamaguchi Y, Itami S, Nishida K, et al. Taurin-conjugated ursodeoxycholic acid has a reversible inhibitory effect on human keratinocyte growth. J Dermatol Sci. 1998;18(1):35–42 [DOI] [PubMed] [Google Scholar]
- 86.Itoh S, Kono M, Akimoto T. Psoriasis treated with ursodeoxycholic acid: three case reports. Clin Exp Derm. 2007;32(4):398–400 [DOI] [PubMed] [Google Scholar]
- 87.Leong ML, Gu M, Speltz-Paiz R, et al. Neuronal loss in the rostral ventromedial medulla in a rat model of neuropathic pain. J Neurosci. 2011;31(47):17028–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Smith J. A bear market. J Life Sci. May2007; pp 20–3
- 89.Wang N, Feng Y, Cheung F, et al. A comparative study on the hepatoprotective action of bear bile and coptidis rhizoma aqueous extract on experimental liver fibrosis in rats. BMC Compl Alt Med. 2012;12:239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhou C-S, Yin Z-Y, Zhao W-X, et al. Bear bile inhibits the immunosuppression activity of hepatic stellate cells in vivo. Hepato-Gastroenterology. 2012;59:1529–36 [DOI] [PubMed] [Google Scholar]
- 91.Dosa PI, Ward T, Castro RE, Rodrigues CMP, Steer CJ. Synthesis and evaluation of water-soluble prodrugs of ursodeoxycholic acid (UDCA), an anti-apoptotic bile acid. Chem Med Chem. 2013;8:1002–11 [DOI] [PubMed] [Google Scholar]