

Abstract
Interdisciplinary scientific evaluation of the human microbiota has identified three enteric microbial biotransformations of particular relevance for human health and well-being, especially cancer. Two biotransformations are counterproductive; one is productive. First, selective bacteria can reverse beneficial hepatic hydroxylation to produce toxic secondary bile acids, especially deoxycholic acid. Second, numerous bacterial species can reverse hepatic detoxification—in a sense, retoxify hormones and xeonobiotics—by deglucuronidation. Third, numerous enteric bacteria can effect a very positive biotransformation through the production of butyrate, a small chain fatty acid with anti-cancer activity. Each biotransformation is addressed in sequence for its relevance in representative gastrointestinal and extra-intestinal cancers. This is not a complete review of their connection with every type of cancer. The intent is to introduce the reader to clinically relevant microbial biochemistry plus the emerging evidence that links these to both carcinogenesis and treatment. Included is the evidence base to guide counseling for potentially helpful dietary adjustments.
Key Words: Microbiota, bile acids, deoxycholic acid, beta-glucuronidase, small chain fatty acids, butyrate
INTRODUCTION
As far back as the 1890s, a connection between cancer and bacteria was noted with the successful treatment of inoperable sarcomas by injections of bacteria.1 Since then, the most widely understood link between cancer and bacteria has been the induction of gastric MALT (mucosa-associated lymphoid tissue) lymphoma by the bacterial pathogen Helicobacter pylori. This microbe has been officially recognized as a carcinogen. And, surprisingly, successfully treating the gastric lymphoma means first successfully treating the bacteria.
But the relationship between gastric cancer and H pylori is much more complex than the easily understood 1:1 causality of a given carcinogen with a given illness or a given pathogen with a given illness. For example, we now know that H pylori alone is not enough to induce stomach cancer. Promotion requires the presence of a complex microbiota. Mice with just H pylori develop fewer tumors than regular mice.2 Moreover, the presence of H pylori infection lowers the risk of esophageal cancer.3,4 These observations emphasize the need to move from a specific pathogen/infection model to an ecological model of the microbiota as a system.
The importance of an ecological approach has certainly been suggested by studies on germ-free mice that document the microbiota have tumor-promoting capacity in multiple carcinogenesis models both directly (eg, colon)5 and indirectly (eg, liver).6 And, likewise, in regular mice, treatment with antibiotics to eliminate bacteria can reduce the development of colon7 and liver cancers.8
In late 2013, the microbiota-cancer link was firmly established by several rigorous studies that addressed both prevention and treatment. First came documentation of a causal link between dysbiosis of the intestinal microbiota and colon tumorigenesis.9 Next came two reports that documented how the microbiota can alter a patient's response to chemotherapeutic agents.10,11
For clinicians, translating these basic science insights and breakthroughs to everyday practice may seem impractical. After all, few have the technology to document the complexity of a given patient's intestinal microbiota. However, all clinicians and researchers do have access to three accessible, measurable, and modifiable products of the microbiota.
For this reason, this review focuses on the three microbial biotransformations readily measurable in stool samples: deoxycholic acid (DCA), beta-glucuronidase, and butyrate. Each is addressed in sequence for its relevance in selected gastrointestinal and extra-intestinal cancers. This is not a complete review of their connection with every type of cancer. The intent is to introduce the reader to clinically relevant microbial biochemistry plus the emerging evidence that links these to both carcinogenesis and treatment. Included is the evidence base to guide counseling for potentially helpful dietary adjustments.
1. FIRST MICROBIAL BIOTRANSFORMATION
Microbial Enzyme: 7-α-dehydroxylase
Microbial Biotransformation: Dehydroxylation
Functional Result: Production of toxic secondary bile acids
Background
Bile acids and bile salts are best known as the highly effective detergents necessary for the fat solubilization and emulsification of dietary lipid and lipid-soluble vitamin absorption throughout the small intestine.
Each day, approximately 500 mg of cholesterol undergoes hydroxylation as well as oxidation of the sterol side chain to become a bile acid. The two primary bile acids produced in the liver are cholic acid (CA) and chenodeoxycholic acid (CDCA). These are conjugated to glycine or taurine, ionized into amphipathic salts, secreted actively, and carried in the bile to the gallbladder for concentration and storage (Figure 1).12
Figure 1.
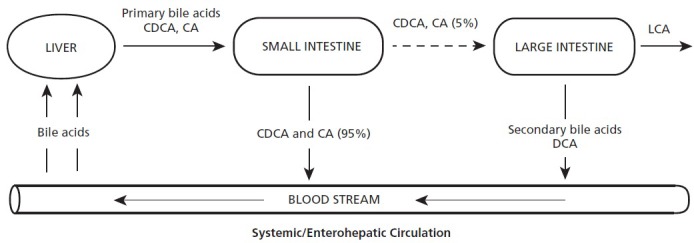
The primary bile acids chenodeoxycholic acid (CDCA) and cholic acid (CA) are produced in the liver, stored in the gallbladder, and, when prompted, discharged into the small intestine. These support the digestion of fats, and 95% are reabsorbed in the distal ileum and returned to the liver via the enterohepatic circulation. These can also circulate to the entire body. Approximately 5% pass on to the large intestine, where they may be transformed into the potential toxins deoxycholic acid (DCA) and lithocholic acid (LCA). DCA is subject to uptake, systemic circulation, and return to the liver where it can be concentrated and stored in the gall bladder.
With meals, bile acids are released from the gallbladder into the duodenum and flow to the terminal ileum, where they are absorbed by passive diffusion and transported back to the liver via the portal vein. They are then taken up by the liver and re-exported into the bile. This enterohepatic circulation from liver to intestine and back occurs in 4 to 12 cycles per day for each bile acid molecule.13
A very small percentage of bile salts are not absorbed from the small intestine and instead enter the large intestine where approximately 0.0001% of all colonic bacteria have the capacity to reverse the hepatocyte synthesis that produced the bile acids.14 Enteric bacteria first deconjugate the bile salts and then, if the correct species and strains are present, dehydroxylate them. The result is not cholesterol but what are termed secondary bile acids, deoxycholic acid (DCA) and lithocholic acid (LCA). DCA, but not LCA, can be reabsorbed through passive nonionic diffusion across the colonic epithelium. DCA, like CA and CDCA, participates in the enterohepatic circulation from intestines back to the liver. This accounts for the approximately 25-fold difference in concentration of DCA and LCA in the gallbladder.15
Surprisingly, bile acids are also more than detergents. High concentrations of the secondary bile acid DCA have been linked to several cancers. DCA does not appear to be directly toxic but instead is a promoter of carcinogenesis.16,17 This is likely due to its role as a signaling molecule related to the control of lipid, bile acid, and carbohydrate metabolism.18
DCA can activate multiple cell signaling pathways related to carcinogenesis including protein kinase C, ERK1/2 via the epidermal growth factor receptor (EGFR), beta-catenin, Jun-N-terminal kinase 1 and 2 (JNK 1/2), and p38 MAPK.15,19 DCA is closely linked to inflammatory pathways because NF-kappa B DNA binding activity and subsequent pro-inflammatory cytokine transcription, occurs only in the presence of a dissociating agent such as DCA.20
Bile acid activated receptors are found not only in epithelial cells in the enterohepatic system but also in multiple extra-intestinal sites including the breast, the adrenal glands, and immune cells. These receptors are comprised of the G-protein–coupled receptor TGR5 (GP-BAR1, G-protein-coupled bile acid receptor) as well as the superfamily of nuclear receptors including the farnesoid-X-receptor (FXR), the constitutive androstane receptor (CAR), the pregnane-x-receptor (PXR) and the vitamin D receptor (VDR). These nuclear receptors regulate the cell cycle, mitosis, proliferation, and apoptosis.
Of all these bile acid receptors, FXR is the ligand-activated transcription factor responsible for bile acid and triglyceride synthesis, bile acid uptake and export, plus bile acid conjugation and detoxification.21 Additionally, FXR appears to play a significant role in many cancers. It has high affinity for the primary bile acids CA and CDCA as well as the major secondary bile acids DCA and LCA.22 Four isoforms exist so tissue-to-tissue variability in function may exist for primary and secondary bile acids. FXR appears to be activated by unconjugated bile acids.23
Pertinent Microbial Biotransformation
Bile salts are synthesized from cholesterol in hepatocytes via cholesterol-7α-hydroxylase (CYP 7A1). The primary bile acids produced, CA and CDCA, after deconjugation are then 7-α-dehydroxylated in the large intestine by enteric bacteria to form the secondary bile acids DCA and LCA. This biotransformation occurs only with intestinal bacteria. This is inhibited at low colonic pH associated with the fermentation of resistant starches.24–26
Responsible Microbiota Bacteria
Secondary bile acids are produced by large intestine anaerobic bacteria from the genus Clostridium, specifically clostridial cluster XIVa. These are gram-positive, spore-forming anerobes that are members of the phylum Firmicutes.27 Only members of this cluster with the bai operon can produce these secondary bile acids.15
Laboratory Measurements
Fecal dexoycholic acid (DCA).
Representative Consequences for Cancer
Esophageal and Gastroesophageal Cancers
Despite the widespread use of proton pump inhibitors (PPIs), and despite the rapid decline in the prevalence of Helicobacter pylori, the incidence of both esophageal and gastroesophageal cancers has increased at a dramatically greater rate than for any other cancer.
One under-recognized factor in these cancers is the role of unconjugated bile acids including DCA from gastroduodenal reflux. Bile acids are not expected to be found at the gastro-esophageal junction, but their presence has been clearly documented and correlates with the degree of pathology seen in the progression from Barrett's esophagus (BE) to esophageal adenocarcinoma.28 In patients with both GERD and BE, refluxed fluids show high concentrations of DCA.29 Use of acid-suppressing medications, such as PPIs, ironically can result in bacterial overgrowth in the stomach and small intestine with increased production of unconjugated secondary bile acids, particularly DCA.30,31 This is important because in biopsies or cell lines derived from such patients, ex vivo and in vitro bile acid exposure induces expression of multiple inflammatory mediators, oxidative stress, and DNA damage.32 This is confirmed in in vivo animal models where mice fed a zinc-deficient diet supplemented with DCA demonstrate increased oxidative stress and development of BE-like pathologic changes.33
The bile acid receptor FXR is over expressed in Barrett's esophagus and esophageal adenocarcinoma. FXR mediates multiple bile acid–induced alterations in gene expression relevant to cancer cell growth.34 Overexpression is associated with higher tumor grade, larger tumor size, and lymph node metastasis. Inhibition of FXR (by guggulsterone) induced apoptosis in vitro and reduced tumor formation and growth in nude mouse xenografts. This reduced viability of esophageal and cancer cells occurred in a time-dependent and dose-dependent manner.35
Breast Cancer
Although breast tissue is not considered to be a bile acid target, the intestinal microbiota and secondary bile acids were recognized as potential agents in breast cancer as far back as 1971.36 The reasoning is as follows. Extra-intestinal effects are possible when secondary bile acids produced in the large intestine are passively absorbed and circulate via the blood stream to other tissues. Most surprisingly, intestinal bile acids are found in breast cyst fluids in concentrations up to 50 times that of the serum.37 Human studies using labeled chenodeoxylate administration prior to breast cyst aspiration demonstrated rapid uptake and concentration of intestinal bile acids into benign breast cysts.38 However, with an unsupplemented diet, day-to-day variation is minimal.39
Additionally, the bile acid receptor FXR is also found in normal breast ductal epithelial cells as well as breast cancer cell lines and tissue specimens.40 The FXR plays several roles in breast cancer. The primary bile acid CDCA activates FXR for beneficial purposes including growth inhibition of MCF-7, MDA-MB-468 and tamoxifen-resistant breast cancer cells (MCF-7 TR1). Specifically, CDCA in vitro treatment significantly reduced epidermal growth factor (EGF)-induced growth and blocked HER2/MAPK signaling.41 Additionally, in breast cancer cell lines MCF-7 and MDA-MB-468, the FXR CDCA-like ligand GW4064 induced SHP, the atypical nuclear receptor that down-regulates genes by interacting with other nuclear receptors including the estrogen receptor to prevent gene transcription. In this case, the FXR bile acid receptor activation results in inhibited induction of aromatase.40
In contrast to CDCA, the secondary bile acids DCA and LCA activate FXR in non-beneficial ways. In this case, FXR activation results in multiple pro-cancer effects relevant to breast health including: (1) estrogen-receptor activation,42,43 (2) promotion of cancer cell survival,44 (3) induced migration of metastatic human breast cancer MDA-MB-231 cells45 and (4) expression of drug resistance proteins.46 This appears to be quite important. Post-menopausal women with newly diagnosed breast cancers have demonstrated mean serum levels of DCA that were 52% higher (P=.012) than those of controls.47
Possible Therapeutic Interventions: 7-α-dehydroxylase
Low-animal fat, low meat, low processed food diet. Western or standard American diets are associated with elevated serum levels of bile acids39 and elevated fecal levels of the potentially toxic secondary bile acids DCA and LCA.48,49 Persons on a low animal fat or vegetarian diet require less primary bile acid production and demonstrate reduced concentrations of 7-α-dehydroxylating bacteria.7,8,50 Omnivorous diets are associated with increased, and vegetarian diets are associated with reduced, concentrations of clostridial cluster XIVa bacteria.51,52
Resistant starch diet. Natural sources of resistant starches include cereal starches, legume starches, green banana and potatoes, raw, cooked and cooled, or as unmodified potato starch. Resistant starches have multiple health benefits including reduced colonic pH and decreased DCA and LCA production.
Discontinuation of acid suppressing medications. Use of acid suppressing medications are associated with a high prevalence of bacterial overgrowth and markedly increased amounts of unconjugated secondary bile acids.12
Curcumin/Turmeric in diet or by supplementation 500 mg per day. In one randomized study of 33 patients with Barrett's esophagitis, esophageal biopsies demonstrated in vivo increased apoptosis and reduced NF-kappa B activation. In vitro studies of tissues from these patients demonstrated that curcumin abrogated bile-driven effects.53
Z-guggulsterone (gugulipid). This is a plant sterol from the resin of Cammiphora mukul that is an effective antagonist of FXR that lowers cholesterol54 and may have anti-esophageal cancer effects.13,14 and multiple anti-breast cancer effects including migration prevention and induced apoptosis55,56 The gugu plant has been used in the Ayurvedic healing tradition for states similar to metabolic syndrome as well as cancer.57
Ursodeoxycholic acid (UCDA) This secondary bile acid, sold as Ursodiol, may serve as an anti-dote to the toxic secondary bile acids DCA and LCA. UCDA prevents indomethacin-induced intestinal barrier dysfunction,58 protects mitochondria against DCA-induced oxidative stress,59 and attenuates chemically-induced colitis and colitis-associated adenocarcinoma and squamous cell carcinoma.60
Probiotic supplementation: Secondary bile acid production may be reduced by administration of lactobacilli and bifidobactera probiotics. Specifically, several species can assimilate or accumulate the primary bile acid cholic acid.61,62 Less cholic acid can mean less deoxycholic acid. Likewise, increased cholic acid substrate supports increased population of clostridial cluster XlVa species and increased DCA production.63,64
Precaution
Taurine supplementation: The amino acid taurine, after deconjugation from bile acids, is transformed in the colonic bacteria into hydrogen sulfide. This itself is a risk factor for both inflammatory bowel disease and colon cancer. Moreover, increased hydrogen sulfide production results in 7-α-dehydroxylation stimulation and increased DCA production.65 No human taurine supplementation trial exists that assesses hydrogen sulfide and DCA production.
2. SECOND MICROBIAL BIOTRANSFORMATION
Microbial Enzyme: β-glucuronidase
Microbial Biotransformation: Deglucuronidation
Functional Result: Reversal of liver detoxification
Background
In the liver's phase II of detoxification, xenobiotic molecules such as drugs and pollutants as well as estrogens, androgens, bile acids, glucocorticoids, mineralocorticoids, retinoids, and fatty acid derivatives are made more hydrophilic by conjugation with glucuronic acid. This process, termed glucuronidation, allows excretion via the bile or urine (Figure 2). The class of responsible liver enzymes is termed UDP-glucuronyl transferase. The resulting molecules are termed glucuronides. Estrogens are metabolized primarily in the liver via conjugation, which includes glucuronidation.66,67 The resulting conjugated estrogens are not ligands for estrogen receptors and are excreted from the liver into the bile and later from the body in the stool.68 However, in the large intestine, these conjugated estrogens can be subjected to deconjugation of the added glucuronic acid, a complete reversal by intestinal bacteria of the hepatic detoxification.69 These deconjugated estrogens can then be reabsorbed through the mucosa and re-enter the circulation via the portal vein.70
Figure 2.
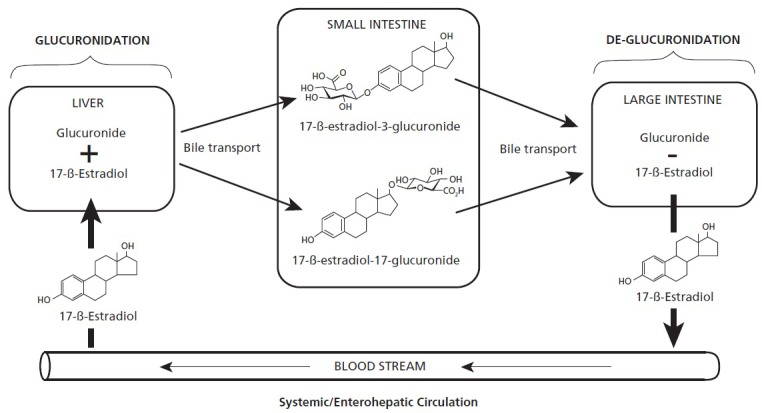
Hepatic glucuronidation of hormones and xenobiotics can be undone in the large intestine via bacterial beta-glucuronidase-mediated de-glucuronidation. The original products for disposal in the stool are then eligible for re-uptake and recirculation.
Pertinent Microbial Biotransformation
Reversal or deconjugation of liver glucuronidation depends upon the presence of the bacterial enzyme beta-glucuronidase.71 This enzyme hydrolyzes β-D-glucuronides to glucuronic acid and an aglycone.
The intestinal microbiome can vary significantly in its capacity to deconjugate hormones such as estrogens and xenobiotics like chemotherapy drugs and heterocyclic amines. Altering the microbiome by administration of antibiotics results in significant increases in fecal progesterone and estriol metabolites.72
Responsible Microbiota Bacteria
Numerous bacteria harbor genes for beta-glucuronidase activity including Firmicutes genera (Lactobacillus, Streptococcus, Clostridium, Ruminococcus, Roseburia, Faecalibacterium), the Proteobacteria genus Escherichia and in one species from the phyla Actinobacteria (Bifidobacterium dentium).73 Many of these bacteria are found within the Clostridium leptum group (cluster IV) and Lachnospiraceas (cluster XIVa).74
Laboratory Measurements
Fecal β-glucuronidase.
Representative Consequences for Cancer
Colon, Pancreatic, Ovarian, and Lung Cancers
The chemotherapeutic agents topotecan and irinotecan are pro-drugs that undergo transformation into the active drug SN-38 by hepatic carboxylesterases.75 SN-38 is then metabolized into the inactive metabolite SN38G by the liver via glucuronidation and excreted in the bile. Reactivation of SN38 in the intestines can occur via bacteria-mediated removal of the glucuronide group.76 SN38 does not appear to be subject to enterohepatic recirculation and remains active as a poison of human topoisomerase I. This means inhibition of both DNA replication and transcription with preferential activity in rapidly dividing cells, both malignant and normal.
With deconjugation and no enterohepatic recirculation, bacterial β-glucuronidase activity results in high concentrations of the activated forms of the chetmotherapeutic pro-drugs irinotecan and topotecan in the intestinal tract. These drugs harm rapidly dividing intestinal epithelial cells and cause tight junction defects and mucosal barrier dysfunction.77 The result can be dose-limiting, or even life-threatening, diarrhea. Inhibitors of bacterial β-glucuronidases protect mice from diarrhea without altering the microbiome or harming mammalian cells.78
Gastrointestinal Cancers
Heterocyclic aromatic amines are genotoxic and carcinogenic compounds formed in meat and fish during cooking.79,80 These are metabolized in the liver by UDP-glucuronysyl transferases to harmless glucuronidated derivates that are excreted via the bile. However, the presence of β-glucuronidase will reverse this. For example, in the digestive lumen in one animal model, the presence of β-glucuronidase increased the genotoxicity of heterocyclic amines by 300%.81 This phenomenon may explain why prebiotics, such as inulin and non-digestible oligosaccharides, both reduce β-glucuronidase concentrations and protect against carcinogenesis in animal models.82–84
Breast Cancer
With deconjugation and enterohepatic recirculation, β-glucuronidase bacterial activity can result in sustained elevation of sex hormone levels including estrogens. This is concerning because breast cancer risk for postmenopausal women in associated with the concentration of serum estrogens and androgens85,86 and circulating sex hormone concentrations are strongly associated with severely established risk factors for breast cancer.87
In a study of 51 male and female epidemiologists at the National Institutes of Health, fecal β-glucuronidase correlated inversely with fecal total estrogens, both conjugated and unconjugated, as well as serum estrone. The study documented that non-ovarian systemic estrogens were strongly and directly associated with all measures of fecal microbiome richness and Clostridia taxa. The authors concluded that intestinal microbial richness as well as β-glucuronidase influence the levels of non-ovarian estrogens via enterohepatic circulation.88
Possible Therapeutic Interventions: β-glucuronidase
Adoption of a plant-based low meat or vegetarian diet. β-glucuronidase activity is markedly increased in human volunteers on a high meat diet compared to a vegetarian diet.89 Omnivorous diets are associated with increased, and vegetarian diets are associated with reduced, concentrations of clostridial cluster XIVa bacteria.51,52
Adoption of a raw vegan diet. This change from a standard diet rapidly and significantly reduces β-glucuronidase activity.90
Avoidance of charred meat or fish (heterocyclic amines) plus ingestion of probiotics containing L casei,91 or L helveticus and S thermophilus groups or either Bifidobacterium animalis92 or B longum93 to bind carcinogenic heterocyclic amines generated with grilling meats and fish.94 Of note, heterocyclic amines are less genotoxic and carcinogenic in individuals who consume mainly plant-derived foods.95
Ingestion of cultured or fermented dairy products96 along with cruciferous vegetables and other prebiotics.97
Ingestion of a multispecies probiotic (that includes Lactobacillus GG)98 or Lactobacillus strain GG by itself.99
The Kampo (traditional Japanese herbal medicine) formula termed Hangeshashinto may alter intestinal ecology and reduce irinotecan and topotecan diarrhea.100 This formula contains root of the herb Scutellaria baicalensis (Skullcap) that contains the beta-glucuronidase inhibitor baical.101
Ingestion of Kanjika, a rice-based Ayurvedic fermented food or the probiotic L. plantarum along with prebiotic fructooligosaccharides (FOS).102
Ingestion of blackcurrant. Consumption of blackcurrant products (First Leaf, composed of blackcurrant extract powder, lactoferrin, and lutein, or Cassis Anthomix, blackcurrant extract powder) by healthy volunteers resulted in significant reduction in activity, significant reduction in pH, significant increases in lactobacilli and bifidobacteria, with significant reductions in Clostridium and Bacteroides species.103
3. THIRD MICROBIAL BIOTRANSFORMATION
Microbial Enzymes: Butyrogenic Pathway (6 enzymes)
Microbial Biotransformation: Dietary Fiber to Acetyl-CoA to Butyrate
Functional Result: Colonocyte energy production
Background
Butyrate is a four-carbon short-chain fatty acid (SCFA) produced in the colon by bacterial fermentation (anaerobic respiration) of dietary fiber, complex carbohydrates that are unable to be either digested or absorbed in the small intestine. We depend upon saccharolytic anaerobic bacteria in our intestines for providing the enzymes necessary to break down such dietary fiber. Unlike the human genome, the intestinal microbiome is highly enriched with genes for digestion of dietary fiber.104 Moreover, by comparative analysis of microbial genes via the COG (clusters of othologous groups) database, the colon's microbiome is enriched with genes for the production of small chain fatty acids especially butyrate kinase,105 the last of six enzymes in the production of butyrate from acetyl-CoA (Figure 3).106
Figure 3.
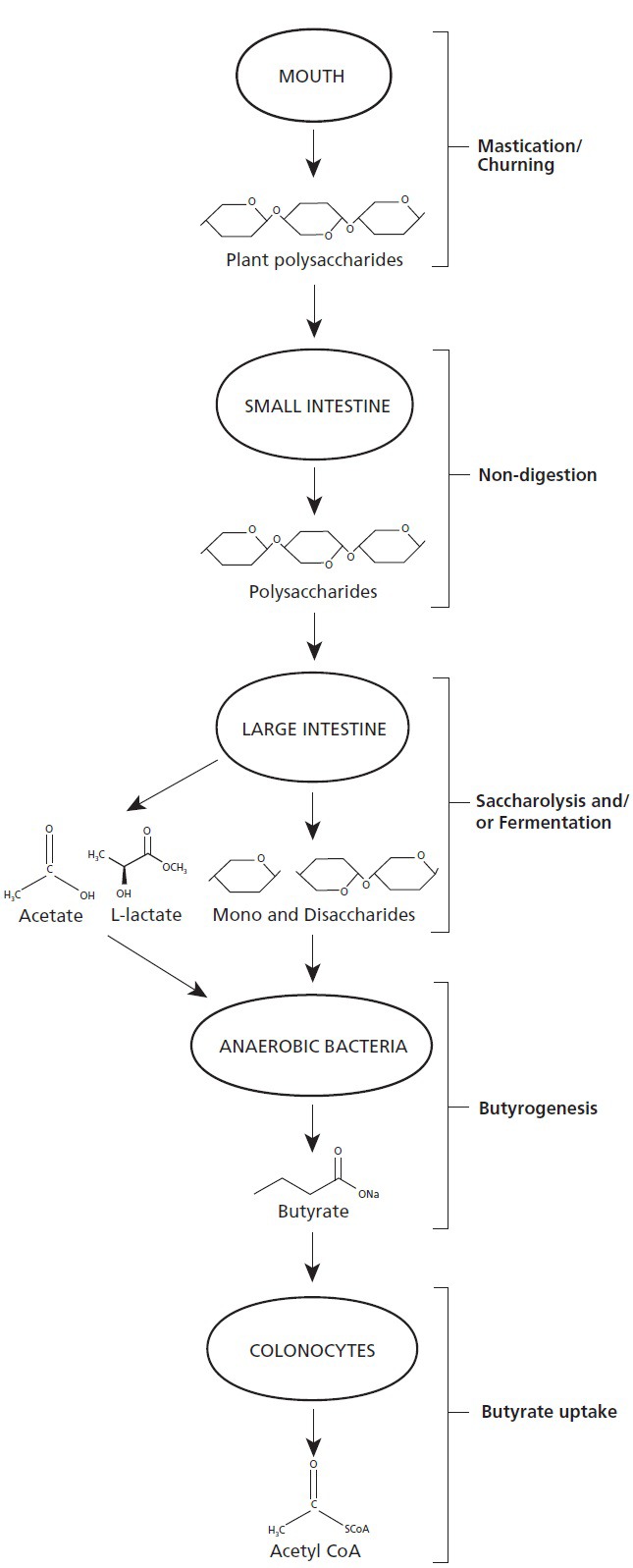
Ingested complex plant polysaccharides undergo significant digestion not in the small intestine, as expected, but instead undergo saccharolysis and fermentation in the large intestine. The mono- and dissacharides produced, as well as products of fermentation, are then transformed by subsets of anaerobic bacteria into butyrate, the predominant energy source for colonocytes. This is one example of cross-kingdom mutualism in the human intestinal tract.
Butyrate is the preferred substrate and the major source of energy for human colonocytes.107 We do not provide our colonocytes with this nourishment: our bacteria do. Butyrate is actively transported by two means (CMT1 and MCT1) into colonocytes108 whose expression is reduced in colonic epithelial tumor cells.109 Within colonocyte mitochondria, butyrate undergoes beta-oxidation into acetyl CoA and then enters the Kreb's cycle with subsequent oxidative phosphorylation for ATP production.110 Unless energy production from butyrate is maximized, very little accumulates in either the cytoplasm or the nucleus.111 Butyrate produced in the gut can also be found in the systemic circulation.
Interest in butyrate follows from its capacity to effect histone acetylation, an epigenetic modification that is regulated by two classes of enzymes: histone deacetylases (HDACs) and acetylases (HATs). Control of acetylation means control of gene expression.112 Butyrate was the first HDAC inhibitor to be discovered.113
Acetylation is important for regulation of chromatin, the tight complex of DNA and associated proteins that enable DNA to fit inside the nucleus. The fundamental units of chromatin are nuclesomes, 147 bp of DNA wrapped 1.65 times around an octamer of core histone proteins. Acetylation of these histones decreases chromatin's electrostatic interaction. This relaxes the tight structure of chromatin and allows transcription factors access to DNA to target gene promoters.114 In brief, acetylation induces transcription, and deacetylation represses transcription.
Butyrate effects acetylation by inhibiting HDAC deacetylation as well as supporting HAT acetylation. The latter is effected via butyrate undergoing beta-oxidation to acetyl CoA in the mitochondria and then combining with oxaloacetic acid in the first step of the Kreb's cycle to yield citrate. The citrate shuttle transports citrate out of the mitochondria where it is converted by ATP citrate lyase (ACL) back to acetyl CoA and oxaloacetate. ACL is found in the nucleus, produces acetyl CoA, and regulates histone acetylation.115 ACL is in turn upregulated by glucose-induced signal transduction, an important finding for cancerous cells.116
Butyrate paradoxically has different effects in normal and cancerous cells. In normal colonocytes, butyrate metabolism results in oxidative phosphorylation to produce ATP. In tumor cells, however, glucose rather than butyrate is the primary source of energy. This means that the active transport of butyrate results in levels that exceed metabolic capacity for utilization. This results in elevated butyrate levels in the nucleus where key genes are regulated via butyrate-mediated HDAC inhibition. Additionally, in cancer cells, upregulation of ACL by glucose means that butyrate functions as an acetyl-CoA donor and also stimulates HAT-mediated histone acetylation. The result is upregulated expression of downstream target genes including genes for cell cycle arrest/cellular proliferation, differentiation, and apoptosis.117
Specifically, butyrate's anti-cancer activities include cell cycle arrest via upregulation of p21118 and downregulation of cyclin D1 plus numerous pro-apoptotic mechanisms including WAF1, downregulation of apoptotic regulator Neuropilin-1 (NRP-1),119 upregulation of BAK,120 downregulation of Bcl-xL and cyclin D1,121 activation of the JNK MAP kinase pathway,122 upregulation of membrane death receptors (DR4/5), higher-level and activation of Smad3 protein in TGF-beta-dependent apoptotic pathway, and activation of proapoptotic tBid protein, as well as lower levels of antiapoptotic proteins (cFLIP, XIAP).123
Independently of histone acetylation, butyrate is also an agonist for G protein-coupled receptors including GPR109A. This receptor is silenced in colorectal, breast, and other cancers but in the presence of butyrate is reexpressed. Butyrate binding results in induction of apoptosis via downregulation of Bcl-2, Bcl-xL, and cyclin D1 and upregulation of the death receptor pathway as well as suppression of nuclear factor-kappaB activation.124
Butyrate's anti-cancer effects include suppression of NF-kappaB activation and thus gene expression for pro-inflammatory cytokines, inflammation-inducing enzymes, adhesion molecules, growth factors, heat shock proteins, and immune receptors.125,126 Butyrate also induces the expression of adhesion molecules including ICAM-1, V-CAM, and E-selectin.127,128
Moreover, butyrate may potentiate radiation therapy129 and chemotherapy with cisplatin,130,131 ARA-C, vincristine and etoposide132 as well as celecoxib.133
Pertinent Microbial Biotransformation
Complex carbohydrates are subjected in the colon to unique bacterial digestive enzymes not available from the human genome. Digestion results in glucose that can undergo anaerobic glycolysis to acetyl-CoA, the terminal oxidation product of glycolysis.
In obligate anaerobic bacterial cells, acetyl-CoA can then proceed through the butyrogenic pathway of six enzymes, resulting in butyrate production and ATP. For maintaining redox balance with ATP production, butyrate is the terminal electron acceptor.
In aerobic colonocytes, bacterially produced butyrate is subjected to beta-oxidation and returned to acetyl-CoA, which can then undergo oxidative phosphorylation with production of significant ATP.
Responsible Microbiota Bacteria
Gene-encoding enzymes for this pathway are widespread in genome-sequenced clostridia and related species.134
Laboratory Measurement
Fecal N-butyrate.
Representative Consequences for Cancer
Colorectal
Increased butyrate stool concentration may optimize cancerous colonocyte apoptosis. Numerous studies in multiple colorectal cell lines have demonstrated that butyrate inhibits cell proliferation and stimulates apoptosis.135–137 In addition to the means noted above, sodium butyrate upregulates expression of annexin A1 (ANXA1) in human colon adenocarcinoma cells. Annexins are proteins that are important as factors in the invasiveness and proliferation of cancer cells.138 ANXA1 specifically is involved in both proliferation and apoptosis. Expression of ANXA1 appears relevant to outcomes in gastrointestinal cancers139,140 but not in other cancers, including breast cancer.141 At this time, the butyrate hypothesis has not been subject to any human trials.
Esophageal
Butyrate may augment the efficacy of fractionated ionizing radiation (IR) therapy. In KYSE-150R radioresistant cells, butyrate increased radio-sensitivity, IR-induced ROS generation, and IR-induced G2/M arrest and apoptosis. Butyrate also increased p21 and inhibited Bmi-1 expression.142 Bmi-1 plays a key role in the functioning of endogenous stem cells and cancer stem cells.
Breast
The colonic SCFA butyrate appears to inhibit breast tumorigenesis.143 Multiple mechanisms appear relevant. In MCF-7 cells, butyrate induced P53-independent, Fas-mediated apoptosis.144 The G Protein–coupled receptor GPR109A activation inhibits genes relevant to cell survival and pro-apototic signaling. Specifically, activation potentiates anti-inflammatory pathways, decreases cyclic AMP production, induces apoptosis, and blocks colony formation and breast tumor growth. This receptor is expressed in normal mammary tissue irrespective of hormone receptor status and silenced in multiple breast cancer cell lines. Evidence supports GPR109A as a tumor suppressor in breast tissue. As with colon cancer, its re-expression and butyrate binding may induce apoptosis. In the MMTV/neu mouse model of spontaneous breast cancer, deletion of GPR109A increased tumor incidence, triggered early onset of tumorigenesis, and increased lung metastasis.145 In ER-positive breast cancer cell lines, butyrate was more potent than the steroidal anti-estrogen ICI in regulating expression of cell cycle proteins and cell growth. Specifically, butyrate-mediated transcriptional and post-transcriptional regulation and ERα phosphorylation resulted in marked depletion of ERα expression. Butyrate appears to antagonize E2-dependent responses.146 In HER2/neu overexpressing cell lines, butyrate functioned synergistically with trastuzumab.147 In both differentiation-induced (HT-29) and cell death–induced (HeLa) cell lines, pretreatment of cells with butyrate followed by butyrate + paclitaxel resulted in increased therapeutic results in the HT-29 cells but proved detrimental in HeLa cells.148
Possibly Therapeutic Interventions: Butyrate
- Prebiotic diet
- High plant-based diet for fiber from difficult-to-digest plant polysaccharides (cellulose, hemicellulose, and lignins).
- Resistant starch (green bananas, raw potato, cooked then cooled potato)149
- Inulin-containing foods (wheat, onion, bananas, garlic, asparagus, and chicory).
- Pectin-containing foods (dried citrus peels and apples, carrots, guavas, gooseberries, oranges, pears, plums, quince)
- Oligosaccharides: Fructooligosaccharides (FOS) include asparagus, bananas, barley, chicory, Jerusalem artichoke, jicama, leeks, wheat, and yacón. Galactooligosaccharides (GOS) include soybeans and bovine lactose derivatives.
Citrus pectin as a supplement.
Italian pecorino, Greek feta and other sheep cheeses made from lamb rennet paste150,151 as well as butter152 are rich in butyrate or its prodrug form tributyrin.153 Of note, the term butyrate comes from the word butter, its best-known dietary source.
Butyrate supplementation as an enteric coated tablet such as ButyrEn (Allergy Research Group, Alameda, California). Oral butyrate has both a short half-life and is subject to first-pass hepatic clearance. Multigram doses are needed to achieve therapeutic concentrations in vivo.154,155 Side effects with oral use include headache, nausea, and anorexia.
Green tea ingestion.156 EGCG, an active ingredient of green tea, appears to work synergistically with butyrate in promoting apoptosis including cell cycle arrest and DNA damage in colorectal cancer cells.
DHA supplementation.157 DHA, one of the two long-chain fatty acids found in fish and krill oils, appears to work synergistically with butyrate to induce apoptosis.
Precaution
Glutamine supplementation: This is widely marketed for N-butyrate production support. However, many cancers actually depend upon glutamine for mitochondrial function, carbon and nitrogen donation, and NADH production for redox control and macromolecule synthesis.158 Cancer cell lines can consume 10 times greater rates of glutamine than any other amino acid.159 Many types of cancer cells are sensitive to glutamine deprivation. Pancreatic, lung, and glioma cancer cells, for example, cannot maintain viability, much less proliferate, in the absence of glutamine. Likewise, lymphoma cells can use glutamine for ATP production even in the absence of either oxygen or glucose.160
CONCLUSION
This article's explicit intention was to describe three biotransformations by intestinal microbiota relevant to both gastrointestinal and non-gastrointestinal cancers. Unintentionally, this review strengthens the biological importance of a diet low in animal fat and high in vegetables. In all three microbial transformations discussed here, a low–animal fat/high-vegetable diet is associated with beneficial ecological profiles including decreased production of toxic bile acids, decreased de-glucuronidation, and increased butyrate production.
Laboratory technologies now allow both clinicians and researchers to measure bacterial factors very relevant to cancer prevention and treatment. Specifically, quantification of deoxycholic acid, β-glucuronidase, and butyrate provides clinically relevant data that can guide dietary interventions and monitor clinical progress toward goals. Increased testing of these bacterial factors could lead to improved dietary adherence and enhanced supplementation strategies that reduce costs and improve outcomes for many conditions, including cancer.
The benefits for researchers appear to be quite promising as well. First, quantification of these three factors may help researchers define “super donors” for fecal microbial transplant. Second, use of these measures will help define the functional consequences to the intestinal ecology of antibiotics, surgery, and chemotherapy. Third, these functional measures may provide greater insight into the potential confounders of clinical trials including chemotherapy trials.
The rapid emergence of a robust basic science literature on the intestinal microbiota in cancer means that many exciting hypotheses are being generated that need to be tested. The existing evidence suggests that the microbiota might be managed via diet, prebiotics, probiotics and even targeted antibiotics to minimize risk or optimize therapies. Most importantly, these three biotransformations represent microbial factors that are easily accessible, measurable, and modifiable. As such, fecal testing to quantify these will likely play significant roles in future cancer prevention and treatment.
Disclosure The author completed the ICMJE Form for Disclosure of Potential Conflicts of Interest and had no conflicts to disclose.
REFERENCES
- 1.Starnes CO. Coley's toxins in perspective. Nature. 1992; 357: 11–12 [DOI] [PubMed] [Google Scholar]
- 2.Lofgren JL, Whary MT, Ge Z, et al. Lack of commensal flora in Helicobacter pylori-infected INS-GAS mice reduces gastritis and delays intraepithelial neoplasia. Gastroenterology. 2011;140(1):210–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Peek RM, Blaser MJ. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nature Rev Cancer. 2002;2: 28–37 [DOI] [PubMed] [Google Scholar]
- 4.Islami F, Kamangar F. Helicobacter pylori and esophageal cancer risk: a meta-analysis. Cancer Prev Rev (Phila). 2008; 1: 329–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vannucci L, Stepankova R, Kozakova H, Fiserova A, Rossmann P, Tlaskalova-Hogenova H. Colorectal carcinogenesis in germ-free and conventionally reared rats: different intestinal environments affect the systemic immunity. Int J Oncol. 2008;32(3):609–17 [PubMed] [Google Scholar]
- 6.Dapito DH, Mencin A, Gwak GY, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell. 2012;21(4):504–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grivennikov SI, Wang K, Mucida D, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 2012;491(7423):254–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yu LX, Yan HX, Liu Q, et al. Endotoxin accumulation prevents carcinogen-induced apoptosis and promotes liver tumorigenesis in rodents. Hepatology. 2010;52(4):1322–33 [DOI] [PubMed] [Google Scholar]
- 9.Zackular JP, Baxter NT, Iverson KD, et al. The gut microbiome modulates colon tumorigenesis. MBio. 2013;4(6):e00692–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Viaud S, Saccheri F, Mignot G, et al. The intestinal microbiota modulates the anticancer immune effects of cyclophosphamide. Science. 2013;342(6161):971–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iida N, Dzutsev A, Stewart CA, et al. Commensal bacteria control cancer response to therapy by modulating the tumor microenvironment. Science. 2013;342(6161):967–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Russell DW. The enzymes, regulation and genetics of bile acid synthesis. Ann Rev Biochem. 2003; 72; 137–74 [DOI] [PubMed] [Google Scholar]
- 13.Fiorucci S, Cipriani A, Mencarelli A, Renga B, Distrutti E, Baldellia F. Counter-regulatory role of bile acid activated receptors in immunity and inflammation. Curr Mol Med. 2010; 10: 579–595 [DOI] [PubMed] [Google Scholar]
- 14.Hirano S, Nakama R, Tamiki M, Masuda N, Oda H. Isolation and characterization of thirteen intestinal microorganisms capable of 7-α-dehydroyxlating bile acids. Appl Environ Microbiol. 1981; 41: 737–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ridlon JM, Kang DJ, Hylemon PB. Bile salt transformations by human intestinal bacteria. J Lipid Res. 2006; 47:241–59 [DOI] [PubMed] [Google Scholar]
- 16.Reddy BS, Narasawa T, Weisburger JH, Wynder EL. Promoting effect of sodium deoxycholate on colon adenocarcinomas in germfree rats. J Natl Canc Inst. 1976; 56: 441–442 [DOI] [PubMed] [Google Scholar]
- 17.Narisawa T, Magadia NE, Weisburger JH, Wynder EL. Promoting effect of bile acids on colon carcinogenesis after intrarectal instillation of N-methyl-N'-nitro-N-nitrosoguanidine in rats. J Natl Canc Inst. 1974; 53: 1093–1097 [DOI] [PubMed] [Google Scholar]
- 18.Baptissart M, Vega A, Maqdasy Set al. Bile acids: From digestion to cancers. Biochimie. 2013; 95: 504–517 [DOI] [PubMed] [Google Scholar]
- 19.Zeng H, Botnen JH, Briske-Anderson M. Deoxycholic acid and selenium metabolite methylselenol exert common and distinct effects on cell cycle, apoptosis, and MAP kinase pathway in HCT116 human colon cancer cells. Nutr Cancer. 2010; 62(1): 85–92 [DOI] [PubMed] [Google Scholar]
- 20.Baeuerle PA, Baltimore D. I kappa B: a specific inhibitor of the NF-kappa B transcription factor. Science. 1988; 242(4878): 540–6 [DOI] [PubMed] [Google Scholar]
- 21.Schmidt DR, Mangelsdorf DJ. Nuclear receptors of the enteric tract: guarding the frontier. Nutr Rev. 2008; 66; S88–S97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Matsubara T. Li F, Gonzalez FJ. FXR signaling in the enterohepatic system. Mol Cell Endocrinol. 2013; 368 (1-2): 17–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vaquero J, Monte MJ, Dominguez M, Muntané J, Marin JJ. Differential activation of the human farnesoid X receptor depends on the pattern of expressed isoforms and the bile acid pool composition. Biochem Pharmacol. 2013; 86(7): 926–39 [DOI] [PubMed] [Google Scholar]
- 24.van Munster IP, Tanterman A, Nagengast FM. Effect of resistant starch on colonic fermentation, bile acid metabolism, and mucosal proliferation. Dig Dis Sci. 1994; 39(4): 834–42 [DOI] [PubMed] [Google Scholar]
- 25.Reddy S, Sanders TA, Owen RW, Thompson MH. Faecal pH, bile acid and sterol concentrations in premenopausal Indian and white vegetarians compared with white omnivores. Br J Nutr. 1998; 79(6): 495–500 [DOI] [PubMed] [Google Scholar]
- 26.Christl SU, Bartram HP, Paul A, Kelber E, Scheppach W, Kasper H. Bile acid metabolism by colonic bacteria in continuous culture: effects of starch and pH. Ann Nutr Metab. 1997;41(1):45–51 [DOI] [PubMed] [Google Scholar]
- 27.Wells JE, Berr F, Thomas LA, Dowling RF, Hylemon PB. Isolation and characterization of cholic acid 7-α-dehydroxylating fecal bacteria from cholesterol gallstone patients. J Hepatol. 2000; 32: 4–10 [DOI] [PubMed] [Google Scholar]
- 28.Nehra D, Howell P, Williams CP, Pye JK, Beynon J. Toxic bile acids in gastroesophageal reflux disease: influence of gastric acidity. Gut. 1999: 44: 598–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jürgens S, Meyer F, Spechler SJ, Souza R. The role of bile acids in the neoplastic progression of Barrett's esophagus—a short representative overview. Z Gastroenterol. 2012; 50(9): 1028–34 [DOI] [PubMed] [Google Scholar]
- 30.Sital RR, Kusters JG, DeRooji FW, Kuipers EJ, Siersema PA. Bile acids and Barrett's oesophagus: a sine qua non or coincidence? Scand J Gastroenterol Suppl. 2006; 243: 11–17 [DOI] [PubMed] [Google Scholar]
- 31.Theisen J, Nehra D, Citron D, et al. Suppression of gastric acid secretion in patients with gastroesophageal reflux disease results in gastric bacterial over-growth and deconjugation of bile acids. J Gastrointest Surg. 2000; 4(1): 50–4 [DOI] [PubMed] [Google Scholar]
- 32.McQuaid KR, Laine L, Fennerty MB, Souza R, Spechler SJ. Systematic review: the role of bile acids in the pathogenesis of gastroesophageal reflux disease and related neoplasia. Aliment Pharmacol Ther. 2011; 34: 146–65 [DOI] [PubMed] [Google Scholar]
- 33.Guy NC, Garewal H, Holubec H, et al. A novel dietary-related model of esophagitis and barrett's esophagus, a premalignant lesion. Nutr Cancer. 2007; 59: 217–227 [DOI] [PubMed] [Google Scholar]
- 34.De Gottardi A, Dumonceau JM, Bruttin F, et al. Expression of the bile acid receptor FXR in Barrett's esophagus and enhancement of apoptosis by guggulsterone in vitro. Mol Cancer. 2006; 5: 48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guan B, Li H, Yang Z, Hogue A, Xu X. Inhibition of farnesoid X receptor controls esophageal cancer cell growth in vitro and in nude mouse xenografts. Cancer. 2013; 119(7): 1321–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hill MJ, Goddard P, Williams RE. Gut bacteria and aetiology of cancer of the breast. Lancet. 1971; 2(7722): 472–3 [DOI] [PubMed] [Google Scholar]
- 37.Raju U, Katz J, Levitz M. Effect of bile acids and estradiol on thymidine incorporation into DNA in MCF-7 and MCF-10A breast cell lines. Steroids. 1997; 62(10): 643–6 [DOI] [PubMed] [Google Scholar]
- 38.Javitt NB, Budai K, Miller DG, Cahan AC, Raju U, Levitz M. Breast-gut connection: origin of cehnodeoxycholic acid in breast cyst fluid. Lancet. 1994; 343(8898): 633–5 [DOI] [PubMed] [Google Scholar]
- 39.Costarelli V, Sanders TA. Acute effects of dietary fat composition on postprandial plasma bile acid and cholecystokinin concentrations in healthy premenopausal women. Br J Nutr. 2001; 86(4): 471–7 [DOI] [PubMed] [Google Scholar]
- 40.Swales KE, Korbonits M, Carpenter R, Walsh DT, Warner TD, Bishop-Bailey D. The farnesoid x receptor is expressed in breast cancer and regulates apoptosis and aromatase expression. Cancer Res. 2006; 66(20): 10120–6 [DOI] [PubMed] [Google Scholar]
- 41.Giordana C, Catalano S, Panza Set al. Farnesoid X receptor inhibits tamoxifen-resistant MCF-7 breast cancer cell growth through downregulation of HER2 expresssion. Oncogene. 2011; 30(39): 4129–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Journe F, Durbecq V, Chaboteaux C, et al. Association between farnesoid-x-receptor expression and cell proliferation in estrogen-receptor positive luminal-like breast cancer from postmenopausal patients. Breast Cancer Res Tret. 2009; 115(3): 532–35 [DOI] [PubMed] [Google Scholar]
- 43.Journe F, Laurent G, Chaoteaux Cet al. Farnesol, a mevalonate pathway intermediate, stimulates MCF-7 breast cancer cell growth through farnesoid-x-receptor mediated estrogen receptor activation. Breast Cancer Res Treat. 2008; 107(1): 49–61 [DOI] [PubMed] [Google Scholar]
- 44.Krishnamurthy K, Wang G, Rokhfeld D, Bieberich E. Deoxycholate promotes survival of breast cancer cells by reducing the level of pro-apoptotic ceramide. Breast Cancer Res. 2008; 10(6): R106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Silva J, Dasgupta S, Wang G, Krishnamurthy K, Ritter E, Bieberich E. Lipids isolated from bone induce the migration of human breast cancer cells. J Lipid Res. 2006; 47: 724–733 [DOI] [PubMed] [Google Scholar]
- 46.Spassieva S, Bieberich E. The gut-to-breast connection—Interdependence of sterols and shingolipids in multi-drug resistance and breast cancer therapy. Anti-Cancer Agents Med Chem. 2011; 11: 882–890 [DOI] [PubMed] [Google Scholar]
- 47.Costarelli V, Sanders TA. Plasma deoxycholic acid concentration is elevated in postmenopausal women with newly diagnosed breast cancer. Eur J Clin Nutr. 2002; 56(9): 925–7 [DOI] [PubMed] [Google Scholar]
- 48.McGarr SE, Ridlon JM, Hylemon PB. Diet, anaerobic bacterial metabolism, and colon cancer: a review of the literature. J Clin Gastroenterol. 2005; 39(2): 98–109 [PubMed] [Google Scholar]
- 49.Bajor A, Gillberg PG, Abrahamson H. Bile acids: short and long term effects in the intestine. Scand J Gastroenterol. 2010; 45(6): 645–64 [DOI] [PubMed] [Google Scholar]
- 50.O'Keefe SJ, Chung D, Mahmoud N, et al. Why do African Americans get more colon cancer than Native Africans? J Nutr. 2007; 137(1suppl): 175S–182S [DOI] [PubMed] [Google Scholar]
- 51.Matijašić BB, Obermajer T, Lipoglavšek L, Grabnar I, Avguštin G, Rogelj I. Association of dietary type with fecal microbiota in vegetarians and omnivores in Slovenia. Eur J Nutr. 2013October13 Epub ahead of print [DOI] [PubMed]
- 52.Kabeerdoss J, Devi RS, Mary RR, Ramakrishna BS. Faecal microbiota composition in vegetarians: comparison with omnivores in a cohort of young women in southern India. Br J Nutr. 2012; 108(6): 953–7 [DOI] [PubMed] [Google Scholar]
- 53.Rawat N, Alhamadani A, McAdam E, et al. Curcumin abrogates bile-induced NF-kappaB activity and DNA damage in vitro and suppresses NF-kappaB activity whilst promoting apoptosis in vivo, suggesting chemopreventive potential in Barrett's oesophagus. Clin Transl Oncol. 2012; 14(4): 302–11 [DOI] [PubMed] [Google Scholar]
- 54.Urizar NL, Liverman AB, Dodds DTet al. A natural product that lowers cholesterol as an antagonist ligand for FXR. Science. 2002; 296(5573): 1703–6 [DOI] [PubMed] [Google Scholar]
- 55.Jiang G, Xiao X, Zeng Y, Nagabhushanam K, Majeed M, Xiao D. Targeting beta-catenin signaling to induce apoptosis in human breast cancer cells by z-guggulsterone and Gugulipid extract of Ayurvedic medicine plant Commiphora mukul. BMC Complement Altern Med. 2013; 13:203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Silva J, Dasgupta S, Wang G, Krishnamurthy K, Ritter E, Bieberich E. Lipids isolated from bone induce the migration of human breast cancer cells. J Lipid Res. 2006; 47(4): 724–33 [DOI] [PubMed] [Google Scholar]
- 57.Xiao M, Xiao D. Gugulipid, an Extract of Ayurveda Medicine Plant Commiphora Mukul as a potent agent for cancer chemoprevention and cancer chemotherapy. Med Chem. 2012;13(6):1000e105 [Google Scholar]
- 58.Bernardes-Silva CF, Damião AO, Sipahi AM, et al. Ursodeoxycholic acid ameliorates experimental ileitis counteracting intestinal barrier dysfunction and oxidative stress. Dig Dis Sci. 2004; 49(10): 1569–74 [DOI] [PubMed] [Google Scholar]
- 59.Rodrigues CM, Fan G, Wong PY, Kren BT, Steer CJ. Ursodeoxycholic acid may inhibit deoxycholic acid-induced apoptosis by modulating mitochondrial transmembrance potential and reactive oxygen species production. Mol Med. 1998; 4(3): 165–78 [PMC free article] [PubMed] [Google Scholar]
- 60.Loddenkemper C, Keller S, Hanski ML, et al. Prevention of colitis-associated carcinogenesis in a mouse model by diet supplementation with ursodeoxycholic acid. Int J Cancer. 2006; 118(11): 2750–7 [DOI] [PubMed] [Google Scholar]
- 61.Kurdi P, van Veen HW, Tanaka H, et al. Cholic acid is accumulated spontaneously, driven by membrane 8pH, in many lactobacilli. J Bacteriol. 2000; 182: 6525–6528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kurdi P, Tanaka H, Van Veen HW, Asano K, Tomita F, Yokota A. Cholic acid accumulation and its diminution by short-chain fatty acids in bifidobacteria. Microbiology. 2003; 149: 2031–37 [DOI] [PubMed] [Google Scholar]
- 63.Islam KB, Fukiya S, Hagio M, et al. Bile acid is a host factor that regulates composition of the cecal microbiota in rats. Gastroenterology. 2011; 141: 1773–81 [DOI] [PubMed] [Google Scholar]
- 64.Ridlon JM, Alves JM, Hylemon PB, Bajaj JS. Cirrhosis, bile acids and gut microbiota: unraveling a complex relationship. Gut Microbes. 2013; 4(5): 382–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Van Eldere J, Celis P, De Pauw G, Lesaffre E, Eyssen H. Tauroconjugation of cholic acid stimulates 7-α-dehydroxylation by fecal bacteria. Appl Environ Microb. 1996; 62: 656–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Raftogianis R, Creveling C, Weinsshilboum R, Weisz J. Estrogen metabolism by conjugation. J Natl Canc Inst Monogr. 2000: 113–24 [DOI] [PubMed]
- 67.Roberts MS, Magnusson BM, Burczynski FJ, Weiss M. Enterohepatic circulation: physiological, pharmacokinetic and clinical implications. Clin Pharmaco. 2002; 41(10): 751–790 [DOI] [PubMed] [Google Scholar]
- 68.Adlercreutz H, Martin F. Biliary excretion and intestinal metabolism of progesterone and estrogens in man. J Steroid Biochem. 1980;13:231–244 [DOI] [PubMed] [Google Scholar]
- 69.Sandberg AA, Slaunwhite WR. Studies on phenolic steroids in human subjects. II. The metabolic fate and hepato-biliary-enteric circulation of C14-estrone and C14-estradiol in women. J Clin Invest. 1957;36:1266–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lombardi P, Goldin B, Boutin E, Gorbach SL. Metabolism of androgens and estrogens by human fecal microorganisms. J Steroid Biochem. 1978; 9(8): 795–801 [DOI] [PubMed] [Google Scholar]
- 71.Flores R, Shi J, Fuhrman Bet al. Fecal microbial determinants of fecal and systemic estrogens and estrogen metabolites: a cross-sectional study. J Transl Med. 2012; 10: 253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Martin F, Peltonen J, Laatikainen T, Pulkkinen M, Adlercreutz H. Excretion of progesterone metabolites and estriol in faeces from pregnant women during ampicillin administration. J Steroid Biochem. 1975;6:1339–1346 [DOI] [PubMed] [Google Scholar]
- 73.Gloux K, Berteau O, El oumami H, et al. A metagenomic beta-glucuronidase uncovers a core adaptive function of the human intestinal microbiome. Proc Natl Acad Sci. 2011; 108 (suppl 1): 4539–4546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dabek M, McRae SI, Stevens VJ, Duncan SH, Louis P. Distribution of beta-glucosidase and beta-gluronidase activity and of beta-glucuronidase gene gus in human colonic bacteria. FEMS Microbiol Ecol. 2008; 66: 487–495 [DOI] [PubMed] [Google Scholar]
- 75.Smith NF, Figg WD, Sparreboom A. Pharmacogenetics of irinotecan metabolism and transport: an update. Toxicol In Vitro. 2006; 20: 163–175 [DOI] [PubMed] [Google Scholar]
- 76.Ngar S, Blanchard RL. Pharmacogenetics of uridine diphosphoglucuronosyltransferease (UGT) 1A family members and its role in patient response to irinotecan. Drug Metab Rev. 2006; 38: 393- [DOI] [PubMed] [Google Scholar]
- 77.Wardill HR, Bowen JM, Al-Dasooqi Net al. Irinotecan disrupts tight junction proteins within the gut: implications for chemotherapy-induced gut toxicity. Cancer Biol Ther. 2013; 15(2): Epub ahead of print [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wallace BD, Wang H, Lane KT, et al. Alleviating cancer drug toxicity by inhibiting a bacterial enzyme. Science. 201; 330: 831–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Augustsson K, Skog K, Jägerstad M, Dickman PW, Steineck G. Dietary hetero-cyclic amines and cancer of the colon, rectum, bladder and kidney: a population-based study. Lancet. 1999; 353: 703–7 [DOI] [PubMed] [Google Scholar]
- 80.Hirayama K, Baranczewski P, Akerlund JE, Midtvedt T, Möller L, Rafter J. Effects of human intestinal flora on mutagenicity of and DNA adduct formation from food and environmental mutagens. Carcinogenesis. 2000; 21: 2105–11 [DOI] [PubMed] [Google Scholar]
- 81.Humblot C, Murkovic M, Rigottier-Gois L, et al. Beta-glucuronidase in human intestinal microbiota is necessary for the colonic genotoxicity of the food-borne carcinogen 2-amino-3-methylimidazo[4,5-f]quinoline in rats. Carcinogenesis. 2007;28(11):2419–25 [DOI] [PubMed] [Google Scholar]
- 82.Van Loo J, Cummings J, Delzenne N, et al. Functional food properties of non-digestible oligosaccharides: a consensus report from the ENDO project (DGXII AIRII-CT94-1095). Br J Nutr. 1999; 81(2): 121–32 [DOI] [PubMed] [Google Scholar]
- 83.Reddy BS. Possible mechanisms by which pro- and prebiotics influence colon carcinogenesis and tumor growth. J Nutr. 1999;129(7 Suppl):1478S–82S [DOI] [PubMed] [Google Scholar]
- 84.Hijová E, Szabadosova V, Štofilová J, Hrčková G. Chemopreventive and metabolic effects of inulin on colon cancer development. J Vet Sci. 2013;14(4):387–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Endogenous Hormones and Breast Cancer Collaborative Group. Endogenous hormones and breast cancer in postmenopausal women: reanalysis of nine prospective studies. J Natl Cancer Inst. 2002; 94: 606–616 [DOI] [PubMed] [Google Scholar]
- 86.Missmer SA, Eliassen AH, Barbieri RL, Hankinson SE. Endogenous estrogen, androgen, and progesterone concentrations and breast cancer risk among post-menopausal women. J Natl Cancer Inst. 2004; 96: 1856–1865 [DOI] [PubMed] [Google Scholar]
- 87.Endogenous Hormones and Breast Cancer Collaborative Group. Circulating sex hormones and beast cancer risk factors I postmenopausal women: reanalysis of 13 studies. Br J Canc. 2011; 105: 709–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Flores R, Shi X, Fuhrman B, et al. Fecal microbial determinants of fecal and systemic estrogens and estrogen metabolites: a cross-sectional study. J Transl Med. 2012; 10: 253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Reddy BS, Weisburger JH, Wynder EL. Fecal bacterial beta-glucuronidase: control by diet. Science. 1974; 183(4123): 416–7 [DOI] [PubMed] [Google Scholar]
- 90.Ling WH, Hänninen O. Shifting from a conventional diet to an uncooked vegan diet reversibly alters fecal hydrolytic activities in humans. J Nutr. 1992; 122(4): 924–30 [DOI] [PubMed] [Google Scholar]
- 91.Nowak A, Libudzisz Z. Ability of probiotic Lactobacillus casei DN 114001 to bind or/and metabolise heterocyclic aromatic amines in vitro. Eur J Nutr. 2009; 48(7): 419–27 [DOI] [PubMed] [Google Scholar]
- 92.Tavan E, Cayuela C, Antoine JM, Trugnan G, Chaugier C, Cassand P. Effects of dairy products on hetercyclic aromatic amin-induced rat colon carcinogenesis. Carcinogenesis. 2002; 23: 477–483 [DOI] [PubMed] [Google Scholar]
- 93.Zsivkovits M, Fekadu K, Sontag G, Nabinger U, Huber WW, Kundi M, Chakraborty A, Foissy H, Knasmüller S. Prevention of heterocyclic amine-induced DNA damage in colon and liver of rats by different lactobacillus strains. Carcinogenesis. 2003; 24(12): 1913–8 [DOI] [PubMed] [Google Scholar]
- 94.Hirayama K, Baranczewski P, Akerlund JE, Midtvedt T, Möller L, Rafter J. Effects of human intestinal flora on mutagenicity of and DNA adduct formation from food and environmental mutagens. Carcinogenesis. 2000; 21: 2105–11 [DOI] [PubMed] [Google Scholar]
- 95.Kassie F, Lhoste EF, Bruneau A, et al. Effect of intestinal microfloras from vegetarians and meat eaters on the genotoxicity of 2-amino-3-methylimidazo[4,5-f] quinoline, a carcinogenic heterocyclic amine. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;802(1):211–5 [DOI] [PubMed] [Google Scholar]
- 96.De Moreno de LeBlanc A, Perdigón G. Reduction of beta-glucuronidase and nitroreductase activity by yoghurt in a murine colon cancer model. Biocell. 2005; 29(1): 15–24 [PubMed] [Google Scholar]
- 97.Humblot C, Lhoste E, Knasmüller S, et al. Protective effects of Brussels sprouts, oligosaccharides and fermented milk towards 2-amino-3-methylimidazo[4,5-f] quinoline (IQ)-induced genotoxicity in the human flora associated F344 rat: role of xenobiotic metabolising enzymes and intestinal microflora. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;802(1):231–7 [DOI] [PubMed] [Google Scholar]
- 98.Kajander K, Krogius-Kurikka L, Rinttilä T, Karjalainen H, Palva A, Korpela R. Effects of multispecies probiotic supplementation on intestinal microbiota in irritable bowel syndrome. Aliment Pharmacol Ther. 2007; 26(3): 463–73 [DOI] [PubMed] [Google Scholar]
- 99.Ling WH, Korpela R, Mykkänen H, Salminen S, Hänninen O. Lactobacillus strain GG supplementation decreases colonic hydrolytic and reductive enzyme activities in healthy female adults. J Nutr. 1994; 124(1): 18–23 [DOI] [PubMed] [Google Scholar]
- 100.Okumi H, Koyama A. Kampo medicine for palliative care in Japan. Biopsychosoc Med. 2014; 8(1):6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Narita M, Nagal E, Hagiwara H, Aburada M, Yokoi T, Kamataki T. Inhibition of beta-glucuronidase by natural glucuronides of Kampo medicines using glucuronide of SN-38 (7-ethyl-10-hydroxycamptothecin) as a substrate. Xenobiotica. 1993; 23(1): 5–10 [DOI] [PubMed] [Google Scholar]
- 102.Ningegowda MA, Gurudutt PS. In vitro fermentation of prebiotics by Lactobacillus plantarum CFR 2194: selectivity, viability and effect of metabolites on Beta-glucuronidase activity. World J Microbiol Biotechnol. 2012; 28: 901–8 [DOI] [PubMed] [Google Scholar]
- 103.Molan AL, Liu Z, Rlimmer G. Evaluation of the effect of blackcurrant products on gut microbiota and on markers of risk for colon cancer in humans. Phytother Res. 2013; May15 (Epub ahead of print) [DOI] [PubMed]
- 104.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature. 2006;444(7122):1022–3 [DOI] [PubMed] [Google Scholar]
- 105.Gill SR, Pop M, Deboy RT, et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006; 312: 1355–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hartmanis MGN. Butyrate kinase from Clostridium acetobutylicum. J Biol Chem. 1987; 262(2): 617–21 [PubMed] [Google Scholar]
- 107.Roediger WE. Role of anaerobic bacteria in the metabolic welfare of the colonic mucosa in man. Gut. 1980; 21: 793–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gonçalves P, Martel F. Butyrate and colon cancer: the role of butyrate transport. Curr Drug Metab. 2013; 14(9): 994–1008 [DOI] [PubMed] [Google Scholar]
- 109.Goncales P, Martel F. Butyrate and colorectal cancer: the role of butyrate transport. Curr Drug Metab. 2013; 14(9): 994–1008 [DOI] [PubMed] [Google Scholar]
- 110.Macfarlane GT, Macfarlane S. Fermentation in the human large intestine: its physiologic consequences and the potential contribution of prebiotics. J Clin Gastroenterol. 2011; 45(Suppl.): S120–127 [DOI] [PubMed] [Google Scholar]
- 111.Donohoe DR, Garge N, Zhang X, Sun W, O'Connell TM, Bunger MK, et al. The microbiome and butyrate regulate energy metabolism and autophagy in the mammalian colon. Cell Metab. 2011; 13: 517–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Pham TX, Lee J. Dieatry regulation of histone acetylases and deactylases for the prevention of metabolic diseases. Nutrient. 2012; 4: 1868–1886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Davie JR. Inhibition of histone deactylase activity by butyrate. J Nutr. 2003; 133Suppl 7: 2485–93 [DOI] [PubMed] [Google Scholar]
- 114.Das C, Tyler JK. Histone exchange and histone modifications during transcription and aging. Biochim Biophys Acta. 2013Mar-Apr;1819(3-4):332–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wellen KE, Hatzivassiliou G, Sachdeva UM, Bui TV, Cross JR, Thompson CB. Science. ATP-citrate lyase links cellular metabolism to histone acetylation. 2009;324(5930):1076–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Berwick DC, Hers I, Heesom KJ, Moule SK, Tavare JM. The identification of ATP-citrate lyase as a protein kinase B (Akt) substrate in primary adipocytes. J Biol Chem. 2002September13;277(37):33895–900 [DOI] [PubMed] [Google Scholar]
- 117.Donohoe DR, Collins LB, Wali A, Bigler R, Sun W, Bultman SJ. The Warburg effect dictates the mechanism of butyrate-mediated histone acetylation and cell proliferation. Mol Cell. 2012;48(4):612–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Crim KC, Sanders LM, Hong MY, et al. Upregulation of p21Waf1/Cip1 expression in vivo by butyrate administration can be chemoprotective or chemopro-motive depending on the lipid component of the diet. Carcinogenesis. 2008;29(7):1415–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.YU DC, WAby JS, Chirakkal H, et al. Butyrate suppresses expression of neuropilin I in colorectal cell lines through inhibition of Sp1 transactivation. Mol Caner. 2010; 9: 276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Chirakkal H, Leech SH, Brookes KE, Prais AL, Waby JS, Corfe BM. Upregulation of BAK by butyrate in the colon is associated with increased Sp3 binding. Oncogene. 2006;25(54):7192–7200 [DOI] [PubMed] [Google Scholar]
- 121.Maier S, Daroqui MC, Scherer S, et al. Butyrate and vitamin D3 induce transcriptional attenuation at the cyclin D1 locus in colonic carcinoma cells. J Cell Physiol. 2009March;218(3):638–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zhang Y, Zhou L, BAo YL, et al. Butyrate induces cell apoptosis through activation of JNK MAP kinase pathway in human colon cancer RKO cells. Chem-Biol Interact. 2010; 185: 174–181 [DOI] [PubMed] [Google Scholar]
- 123.Pajak B, Orzechowski A, Gajkowska B. Molecular basis of sodium butyrate-dependent proapoptotic activity in cancer cells. Adv Med Sci. 2007;52:83–8 [PubMed] [Google Scholar]
- 124.Thangaraju M, Cresci GA, Liu K, et al. GPR109A is a G-protein-coupled receptor for the bacterial fermentation product butyrate and functions as a tumor suppressor in colon. Cancer Res. 2009;69(7):2826–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Vinolo MA, Rodrigues HG, Hatanaka E, Sato FT, Sampaio SC, Curi R. Suppressive effect of short-chain fatty acids on production of proinflammatory mediators by neutrophils. J Nutr Biochem. 2011September;22(9):849–55 [DOI] [PubMed] [Google Scholar]
- 126.Quivy V, Van Lint C. Regulation at multiple levels of NF-kappaB-mediated transactivation by protein acetylation. Biochem Pharmacol. 2004;68(6):1221–9 [DOI] [PubMed] [Google Scholar]
- 127.Zapolska-Downar D, Siennicka A, Kaczmarczyk M, Kołodziej B, Naruszewicz M. Butyrate inhibits cytokine-induced VCAM-1 and ICAM-1 expression in cultured endothelial cells: the role of NF-kappaB and PPARalpha. J Nutr Biochem. 2004April;15(4):220–8 [DOI] [PubMed] [Google Scholar]
- 128.Miller SJ, Zaloga GP, Hoggatt AM, Labarrere C, Faulk WP. Short-chain fatty acids modulate gene expression for vascular endothelial cell adhesion molecules. Nutrition. 2005;21(6):740–8 [DOI] [PubMed] [Google Scholar]
- 129.Koprinarova M, Botev P, Russev G. Histone deacetylase inhibitor sodium butyrate enhances cellular radiosensitivity by inhibiting both DNA nonhomologous end joining and homologous recombination. DNA Repair (Amst). 2011;10(9):970–7 [DOI] [PubMed] [Google Scholar]
- 130.Koprinarova M, Markovska P, Iliev I, Anachkova B, Russev G. Sodium butyrate enhances the cytotoxic effect of cisplatin by abrogating the cisplatin imposed cell cycle arrest. BMC Mol Biol. 2010;11:49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Maruyama T, Yamamoto S, Qiu J, et al. Apoptosis of bladder cancer by sodium butyrate and cisplatin. J Infect Chemother. 2012;18(3):288–95 [DOI] [PubMed] [Google Scholar]
- 132.Ramos MG, Rabelo FL, Brumatti G, Bueno-da-Silva AE, Amarante-Mendes GP, Alvarez-Leite JI. Butyrate increases apoptosis induced by different antineoplastic drugs in monocytic leukemia cells. Chemotherapy. 2004;50(5):221–8 [DOI] [PubMed] [Google Scholar]
- 133.Kang SN, Hong SS, Lee MK, Lim SJ. Dual function of tributyrin emulsion: solubilization and enhancement of anticancer effect of celecoxib. Int J Pharm. 2012;428(1-2):76–81 [DOI] [PubMed] [Google Scholar]
- 134.Aboulnaga el-H, Pinkenburg O, Schiffels J, El-Refai A, Buckel W, Selmer T. Effect of an oxygen-tolerant bifurcating butyryl coenzyme A dehydrogenase/electron-transferring flavoprotein complex from Clostridium difficile on butyrate production in Escherichia coli. J Bacteriol. 2013;195(16):3704–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Leonel AJ, Alvarez-Leite JI. Butyrate: implications for intestinal function. Curr Opin Clin Nutr Metab Care. 2012;15(5):474–9 [DOI] [PubMed] [Google Scholar]
- 136.Comalada M, Bailón E, de Haro O, et al. The effects of short-chain fatty acids on colon epithelial proliferation and survival depend on the cellular phenotype. J Cancer Res Clin Oncol. 2006;132(8):487–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hinnebusch BF, Meng S, Wu JT, Archer SY, Hodin RA. The effects of short-chain fatty acids on human colon cancer cell phenotype are associated with histone hyperacetylation. J Nutr. 2002;132(5):1012–7 [DOI] [PubMed] [Google Scholar]
- 138.Shimizu T, Kasamatsu A, Yamamoto A, et al. Annexin A10 in human oral cancer: Biomarker for tumoral growth via G1/S transition by targeting MAPK signaling pathways. PLOS ONE. 2012; 7: e45510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang KL, Wu TT, Resetkova E, et al. Expression of annexin A1 in esophageal and esophogastric junction adenocarcinomas: association with poor outcome. Clinical Cancer res. 2006; 12: 4598–4604 [DOI] [PubMed] [Google Scholar]
- 140.Bai XF, NI XG, Zhao P, et al. Overexpression of annexin 1 in pancreatic cancer and its clinical significance. W J Gastroenterol. 2003; 10: 1466–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Shen D, ooraie F, Elshimali Y, et al. Decreased expression of annexin A1 is correlated with breast cancer development and progression as determined by a tissue microarray analysis. Hum Pathol. 2006; 37: 1583–1591 [DOI] [PubMed] [Google Scholar]
- 142.Dong Q, Sharma S, Liu H, et al. HDAC inhibitors reverse acquired radio resistance of KYSE-150R esophageal carcinoma cells by modulating Bmi-1 expression. Toxicol Lett. 2014;224(1):121–9 [PubMed] [Google Scholar]
- 143.Belobrajdic DP, McIntosh GH. Dietary butyrate inhibits NMU-induced mam-mary cancer in rats. Nutr Cancer. 2000;36(2):217–23 [DOI] [PubMed] [Google Scholar]
- 144.Chopin V, Toillon RA, Jouy N, Bourhis XL. Sodium butyrate induces P53-independent, Fas-mediated apoptosis in MCF-7 human breast cancer cells. Br J Pharamcol. 2009; 135: 79–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Elangovan S, Pathania R, Ramachandran S, et al. The Niacin/Butyrate Receptor GPR109A Suppresses Mammary Tumorigenesis by Inhibiting Cell Survival. Cancer Res. 2014;74(4):1166–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.De los Santos M, Martínez-Iglesias O, Aranda A. Anti-estrogenic actions of his-tone deacetylase inhibitors in MCF-7 breast cancer cells. Endocr Relat Cancer. 2007;14(4):1021–8 [DOI] [PubMed] [Google Scholar]
- 147.Chen W, Wei F, Xu J, et al. Trastuzumab enhances the anti-tumor effects of the histone deacetylase inhibitor sodium butyrate on a HER2-overexpressing breast cancer cell line. Int J Mol Med. 2011;28(6):985–91 [DOI] [PubMed] [Google Scholar]
- 148.Rivkin L, Cohen K, Bod T, Argov M, Margalit R. Cancer cell sensitization and iparoved treatment efficacy by combined butyrate and paclitaxel formulations is cancer-type specific. In J Pharm. 2104; 461(1-2): 437–47 [DOI] [PubMed] [Google Scholar]
- 149.Cummings JH, Macfarlane GT, Englyst HN. Prebiotic digestion and fermentation. Am J Clin Nutr. 2001;73(2 Suppl):415S–420S [DOI] [PubMed] [Google Scholar]
- 150.Addis M, Piredda G, Pirisis A. The use of lamb rennet paste in traditional sheep milk production. Small Ruminant Res. 2008; 79:2–10 [Google Scholar]
- 151.Santillo A, Albenzio M, Quinto M, et al. Probiotic in lamb rennet paste enhances rennet lipolytic activity, and conjugated linoleic acid and linoleic acid content in pecorino cheese. J Dairy Sci. 2009; 92: 1330–1337 [DOI] [PubMed] [Google Scholar]
- 152.Grummer RR. Effect of feed on the composition of milk fat. J Dairy Sci. 1991; 74(9): 3244–57 [DOI] [PubMed] [Google Scholar]
- 153.Gaschott T, Steinhilber D, Milovic V, Stein J. Tributyrin, a stable and rapidly absorbed prodrug of butyric acid, enhances antiproliferative effects of dihydroxycholecalciferol in human colon cancer cells. J Nutr. 2001June;131(6):1839–43 [DOI] [PubMed] [Google Scholar]
- 154.Riester D, Hildmann C, Schwienhorst A. Histone deacetylatase inhibiotrs— turning epigenetic mechanisms of gene regulation into tools of therapeutic intervention in malignant and other diseases. Appl Microbiol Biotechnol. 2007; 75: 499–514 [DOI] [PubMed] [Google Scholar]
- 155.Kuroiwa-Trzmielina J, de Conti A, Scolastici C, et al. Chemoprevention of rat hepatocarcinogenesis with histone deacetylase inhibitors: Efficacy of tributyrin, a butyric acid prodrgu. In J Cancer. 2009; 124: 2520–2527 [DOI] [PubMed] [Google Scholar]
- 156.Saldanha SN, Kala R, Tollefsbol TO. Molecular mechanisms for inhibition of colon cancer cells by combined epigenetic-modulating epigallocatechin gallate and sodium butyrate. Exp Cell Res. 2014February8 [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- 157.Cho Y, Turner ND, Davidson LA, Chapkin RS, Carroll RJ, Lupton JR. Colon cancer cell apoptosis is induced by combined exposure to the n-3 fatty acid docosahexaenoic acid and butyrate through promoter methylation. Exp Biol Med (Maywood) 2014February4 [Epub ahead of print] [DOI] [PMC free article] [PubMed]
- 158.Wise DR, Thompson CB. Trends Glutamine addiction: a new therapeutic target in cancer. Biochem Sci. 2010;35(8):427–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kovacević Z, Morris HP. The role of glutamine in the oxidative metabolism of malignant cells. Cancer Res. 1972;32(2):326–33 [PubMed] [Google Scholar]
- 160.Le A, Lane AN, Hamaker M, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15(1):110–21 [DOI] [PMC free article] [PubMed] [Google Scholar]